
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox-responsive supramolecular polymeric networks having double-threaded inclusion complexes†
Hikaru
Aramoto
a,
Motofumi
Osaki
 a,
Subaru
Konishi
a,
Chiharu
Ueda
a,
Yuichiro
Kobayashi
a,
Yoshinori
Takashima
*ab,
Akira
Harada
*c and
Hiroyasu
Yamaguchi
*a
a,
Subaru
Konishi
a,
Chiharu
Ueda
a,
Yuichiro
Kobayashi
a,
Yoshinori
Takashima
*ab,
Akira
Harada
*c and
Hiroyasu
Yamaguchi
*a
aDepartment of Macromolecular Science, Graduate School of Science, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan. E-mail: hiroyasu@chem.sci.osaka-u.ac.jp
bInstitute for Advanced Co-Creation Studies, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan. E-mail: takasima@chem.sci.osaka-u.ac.jp
cThe Institute of Scientific and Industrial Research, Osaka University, 8-1 Mihogaoka, Ibaraki, Osaka 567-0047, Japan. E-mail: harada@chem.sci.osaka-u.ac.jp
First published on 16th March 2020
Abstract
Stimuli-responsive hydrogels have attracted attention as soft actuators that act similarly to muscles. In this work, hydrogel actuators controlled by host–guest interactions have been developed. The introduction of a 1:1 inclusion complex into a hydrogel is a popular design for achieving a change in cross-linking density. To realize faster and larger deformation properties, the introduction of a 1:2 inclusion complex is effective because the alteration in cross-linking density in a hydrogel with 1:2 complexes is larger than that in a hydrogel with 1:1 complexes. A redox-responsive hydrogel actuator cross-linked with 1:2 inclusion complexes is designed, where γ-cyclodextrin (γCD) and viologens modified with an alkyl chain derivative (VC11) were employed as the host and guest units, respectively. γCD includes two VC11 molecules in its cavity. The obtained γCD–VC11 hydrogel cross-linked with the 1:2 complex showed faster and larger deformation behaviour than the αCD–VC11 and the βCD–VC11 hydrogels cross-linked with a 1:1 complex. The deformation ratio and response speed of the γCD–VC11 hydrogel, which forms a supramolecular cross-linking structure by stimuli, are 3 and 11 times larger, respectively, than those of our previous hydrogel consisting of a βCD/ferrocene 1:1 inclusion complex.
1. Introduction
Stimuli-responsive polymeric materials have attracted much attention because of their flexibility and macroscopic motion when used as actuators in a similar manner to a muscle.1 There are some principles and material designs for preparing stimuli-responsive soft materials.2–5 Dielectric elastomers have typically been used to prepare dielectric actuators, which are driven by generated charges in polymer networks near electrodes.6–8 The orientation of a crystal lattice or a liquid crystal has been utilized for electrically responsive and photo-responsive materials, where the controlled orientation of the responsive molecules in materials leads to macroscopic deformations in the materials.9–13 Another method to design responsive materials, control of the cross-linking density or distance between cross-linking points in polymer networks, caused the deformation of materials. Fig. 1 shows those network design concepts of stimuli-responsive supramolecular hydrogels. To achieve stimuli-responsive deformations, supramolecular chemists have chosen certain kinds of external stimuli, such as temperature, chemicals, pH, ionic strength, electric field/voltage/current, and light intensity.14–24 Herein, we focus on redox-responsive materials based on the mechanism of controlling the cross-linking density by proposing another network design concept that uses an inclusion complex on the polymer side chain (Fig. 1b).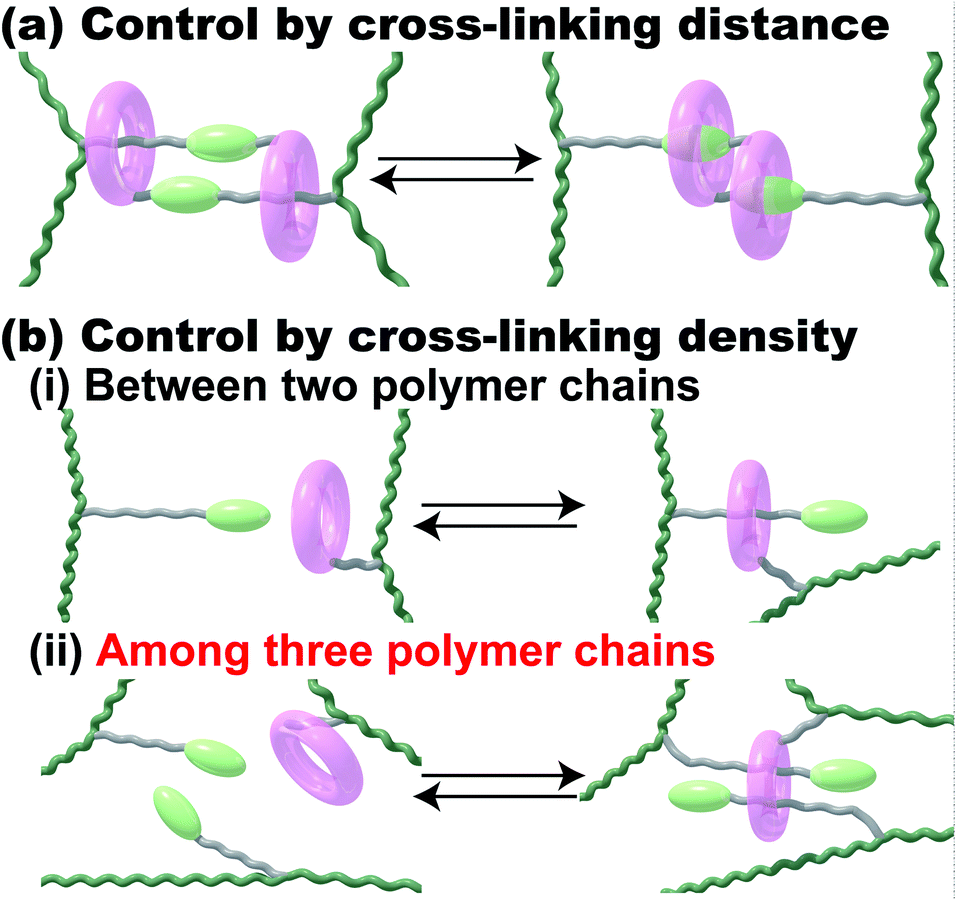 | ||
| Fig. 1 Network design concepts of stimuli-responsive supramolecular hydrogels driven by a change in the cross-linking distance (a) and cross-linking density (b). Pink ring: macrocyclic host molecule. Pale green elliptical sphere: stimuli-responsive barrier for the host. Green line: polymer chain. Grey line: linker connecting the host/guest moieties to the polymer chain. The grey linker acts as a station where the host molecule stays. We can control the cross-linking density by using a 1:1 inclusion complex (i) or a 1:2 inclusion complex (ii). | ||
Previously, we reported supramolecular polymeric hydrogels showing deformation based on the control of cross-linking density, where the host–guest interactions serving as cross-linking points are controlled by photo-25–29 and redox-stimuli.30,31 To realize actuators driven by host–guest interaction, we chose the combination between α-cyclodextrin (αCD) and azobenzene or between β-cyclodextrin (βCD) and ferrocene (Fc) as the 1:1 inclusion complex.26,30 The 1:1 complex system involves two polymer chains (Fig. 1b(i)). The external stimuli control the association and dissociation of the 1:1 inclusion complex to generate the macroscopic expansion and contraction behaviours of the polymeric materials.5,14 The change ratio of the cross-linking density resulted in a larger amount of displacement in the supramolecular polymeric hydrogel. Considering these results, the ratio of the host and guest units was increased to achieve larger changes in the cross-linking density. However, the βCD–Fc hydrogels with over 5 mol% of the host and guest cross-linking units showed too high Young's moduli to bear the increasing displacement, effectively.30 Although polymer networks having a 1:1 inclusion complex are one of the established designs for achieving macroscopic motion, additional new network designs for supramolecular polymeric hydrogels should be proposed.
To realize larger deformation, we introduced a 1:2 double-threaded inclusion complex into the polymer network because three polymer chains are involved in the 1:2 inclusion complex systems (Fig. 1b(ii)). Compared to a 1:1 inclusion complex, a 1:2 inclusion complex can bind three polymer chains together through the association and dissociation of the complexes to bring about a larger change in the cross-linking density. Of course, some excellent supramolecular gels using a 1:2 inclusion complex with two kinds of guest molecules and cucurbit[8]uril as a host molecule were reported.32–38 These polymeric host–guest materials having two kinds of guest molecules successfully achieved hydrogelation and modulation in the presence of cucurbit[8]uril. The 1:1 inclusion complex in these systems can be applied as a cross-linking point between two polymer chains.
If we use a 1:2 inclusion complex with three polymer chains, we will observe a larger change in cross-linking density and volume change. Based on this hypothesis, herein, we focus on redox-responsive materials, the cross-linking density of which can be controlled. We chose the combination of cyclodextrins (CDs) and viologens tethered to an undecyl unit (VC11; 4-undecyl-4′-methyl-bipyridinium dichloride) as host and guest units, respectively, as the 1:2 inclusion complex with three polymer chains. Here, we used α-, β- and γ-cyclodextrin (αCD, βCD and γCD) that consist of 6, 7 and 8 glucose units, respectively, with different cavity sizes (α-CD < β-CD < γ-CD).
2. Results and discussion
2.1. Preparation of host–guest hydrogels
We use three kinds of redox-responsive host–guest hydrogels, αCD–VC11(x,y), βCD–VC11(x,y) and γCD–VC11(x,y) hydrogels (Fig. 2a–c). Both the undecyl unit and the one-electron reduced viologen (monocation radical) unit in VC11 can act as guest units for the CD host.39–41 While shorter alkyl chains such as hexyl groups also act as guest units for CDs, the undecyl group was found to form a more stable complex with CDs.42 In contrast, the oxidized viologen unit (dication) in VC11 does not form an inclusion complex with CDs because of the electrostatic repulsion between the inner cavity of the CDs and the oxidized viologen unit.43–47 The electrostatic repulsion at the ends of the alkyl chain can function as an electric barrier to prevent the dissociation of the inclusion complex between the alkyl chain-tethered viologen guests and the CD hosts.42 Therefore, the viologen unit can be used to kinetically control the association and dissociation of the complex. In this work, we found that the responsiveness (displacement and speed of the macroscopic deformation) of the γCD–VC11(x,y) hydrogel is greater than that of the αCD–VC11(x,y), βCD–VC11(x,y) and chemically cross-linked control hydrogels (the VC11(0.2,y), γCD(x) and polyacrylamide (pAAm) hydrogels in Fig. 2d–f). The covalent cross-linking by MBAAm often gave hard hydrogels, compared to host–guest cross-linking hydrogels. Hardness has some potential to affect the other physical properties. Therefore, the molar fraction of MBAAm was optimized to be 0.2 mol% to adjust the Young's modulus of the VC11 hydrogel to those of the CD–VC11 hydrogels.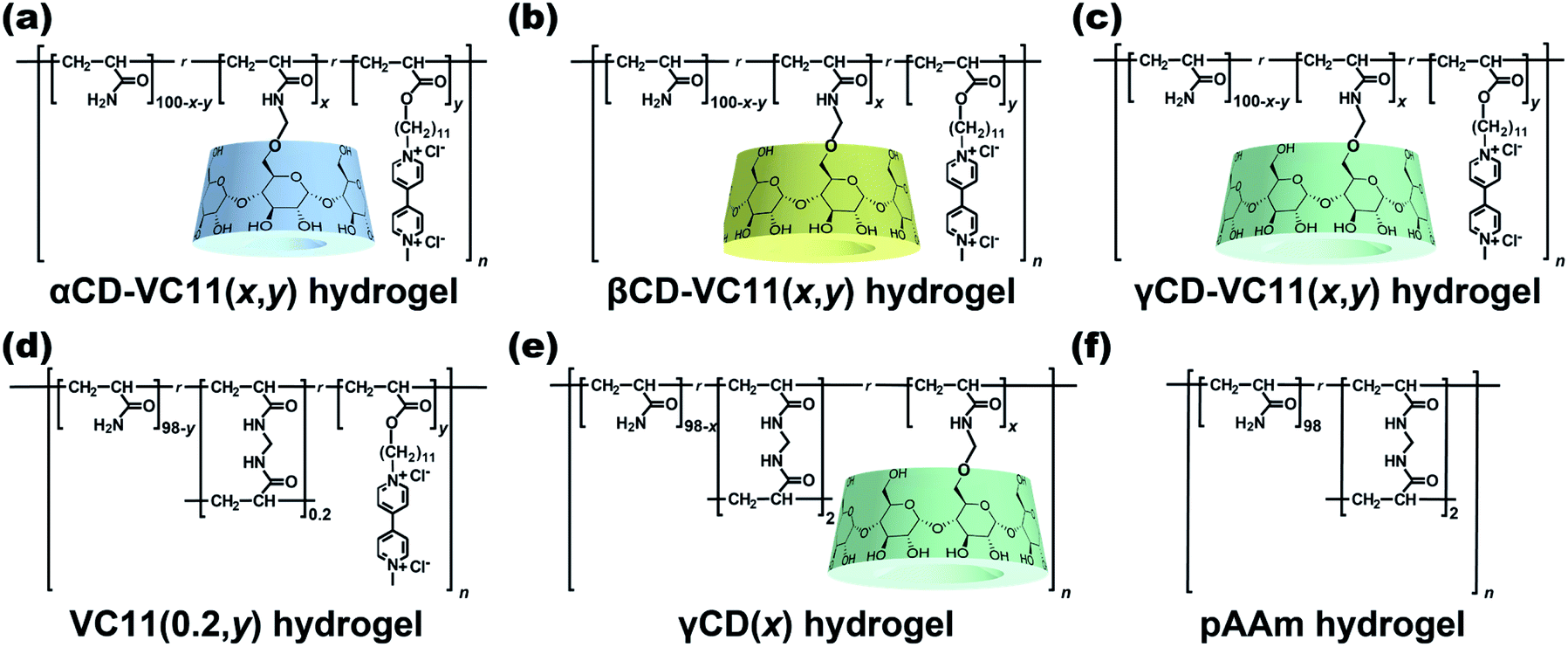 | ||
| Fig. 2 Chemical structures of the αCD–VC11(x,y) hydrogels (a), βCD–VC11(x,y) hydrogels (b), and γCD–VC11(x,y) hydrogels (c) as supramolecular host–guest hydrogels. The VC11(0.2,y) hydrogels (d), γCD(x) hydrogels (e), and pAAm hydrogels (f) are covalently cross-linked control hydrogels. | ||
Prior to polymerization, the complex formation behaviours and stoichiometric proportions of the CDs and VC11 units were investigated. Fig. S3, S5 and S7† show the inclusion complexes of the CDs with VC11 observed by acquiring 2-dimensional rotating frame Overhauser effect spectroscopy (2D ROESY) nuclear magnetic resonance (NMR) spectra in D2O. These spectra demonstrated that the inner protons of the CDs were correlated with the protons of the undecyl unit of the VC11 monomer, indicating that the CD and VC11 monomers form inclusion complexes. The stoichiometry of the CD/VC11 complexes was determined by 1D 1H NMR and isothermal titration calorimetry (ITC) measurements. α and βCDAAmMe each form a 1:1 inclusion complex with the VC11 monomer (Fig. S4 and S6†). On the other hand, 1:2 complexation was observed between γCDAAmMe and the two VC11 monomers in the Job plot and ITC (Fig. S8†).
αCD–VC11(x,y), βCD–VC11(x,y), γCD–VC11(x,y), VC11(0.2,y), γCD(x), and pAAm hydrogels (Fig. 2) were prepared by using conventional radical copolymerization in water (total monomer concentration Cm = 2 mol kg−1), in which potassium dithionite (K2S2O4) and N,N,N′,N′-tetramethyl ethylenediamine (TEMED) were used as redox initiators (Schemes S2–S7 and Tables S1–S6†). Three types of CD vinyl monomers with different sizes of CD cavities (α, β and γCDAAmMe, Scheme S1, Fig. S1 and S2†) were employed. 4-(11-Acryloyloxyundecyl)-4′-(methyl)-bipyridinium dichloride (the VC11 monomer) was prepared as the guest monomer (see the ESI†).
The radical copolymerization of acrylamide (AAm), γCDAAmMe and the VC11 monomer in an aqueous solution yielded γCD–VC11(x,y) hydrogels. Fourier transform infrared (FT-IR) spectra of the γCD–VC11(x,y) hydrogels exhibited the characteristic peaks of both the γCD and VC11 moieties (Fig. S9†). The number of γCD and VC11 units in the obtained hydrogels was determined by solid-state 1H field gradient magic angle spinning (FGMAS) NMR measurements of the hydrogels (Fig. S12†). The other host–guest hydrogels in Fig. 1 were also characterized by spectroscopy (Fig. S9–S11†). In these gel-state FGMAS NMR spectra (Fig. S10–S12†), the integral values of each signal indicate that the obtained host–guest hydrogels contain 2 mol% of the CD and VC11 residues as prepared. The αCD–VC11(2,2) and βCD–VC11(2,2) hydrogels showed signals derived from the methylene protons in the VC11 unit at δ = 1.29 and 1.30 ppm, respectively. On the other hand, the corresponding signals in the γCD–VC11(2,2) hydrogel appeared at δ = 1.24 ppm (shifted to a higher magnetic field), suggesting that the methylene chains of the VC11 units in γCD–VC11(2,2) with the 1:2 host–guest complex are closely packed in the γCD cavity to show a higher shielding effect, compared to αCD–VC11(2,2) and βCD–VC11(2,2). In former reports,48,49 the polymerization of a host–guest inclusion complex effectively introduces the complex into polymeric materials. The complex structure is maintained even in the polymeric materials. According to the previous studies, the CD–VC11 complexes should maintain the 1:1 or 1:2 host–guest complex structures even in the hydrogel states.
2.2. Deformation behaviours of the host–guest hydrogels
Fig. 3 shows the deformation behaviours of the αCD–VC11(2,2), βCD–VC11(2,2), γCD–VC11(2,2) and VC11(0.2,2) hydrogels. After immersion in 0.5 M phosphate buffer (pH 7.0) with 0.1 M sodium dithionite (Na2S2O4) as a reductant, the colour of the hydrogels changed from light yellow to deep purple (Fig. 3a and S13†). This colour change indicates the formation of the radical cation species of the viologen moiety due to one-electron reduction. Fig. 3b shows the time-course of the volume change in the hydrogel in the reductant solution. The normalized lengths of the hydrogels (r) were determined by the ratio r = Lt/L0 (Lt, length of the hydrogel during the volume change; L0, length of the hydrogel in phosphate buffer before the reduction, which is the equilibrium state). The size of the CD–VC11(2,2) hydrogels was decreased by the reduction. The displacement and speed of the contraction were different depending on the cavity size of the CDs. The γCD–VC11(2,2) hydrogel showed a markedly larger volume change than the αCD–VC11(2,2), βCD–VC11(2,2), and VC11(0.2,2) hydrogels (Fig. 3c). The αCD–VC11(2,2) and βCD–VC11(2,2) hydrogels showed an increase of the r values at longer time (t > 30 min). A change in osmotic equilibrium in the reducing reagents is supposed to contribute to an increase in the volume of the αCD–VC11(2,2) and βCD–VC11(2,2) hydrogels at t > 30 min. The other control samples, the γCD(2) and pAAm hydrogels, did not change their colour or volume at all. If only the change in the charge of viologens affected the equilibrium of the polymer network's form, the degree of the volume change should be the same irrespective of the cavity size of CDs. Therefore, the difference in the volume change (Fig. 3d) indicates that complex formation between the γCD and VC11 units plays some role in the deformation.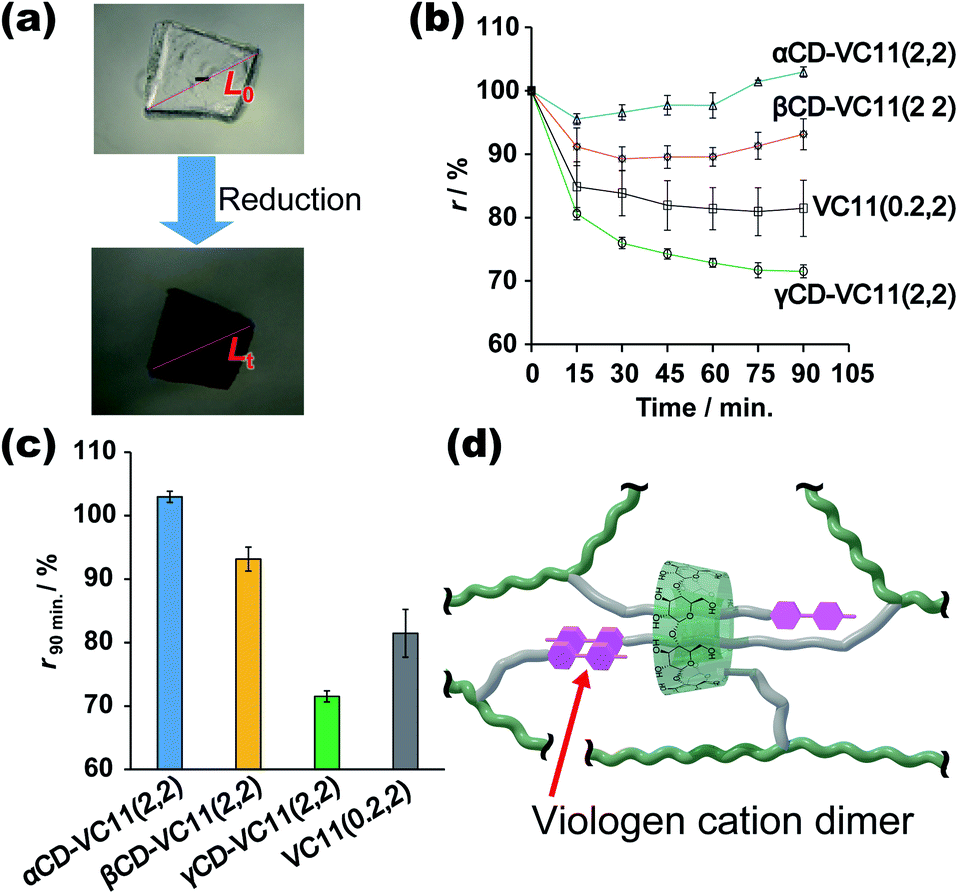 | ||
| Fig. 3 (a) Photographs of the γCD–VC11(2,2) hydrogel before and after immersion in 0.5 M phosphate buffer (pH 7.0) with Na2S2O4 (0.1 M) as a reductant. (b) Time-course of the normalized length (r) of the αCD–VC11(2,2), βCD–VC11(2,2), γCD–VC11(2,2) and VC11(0.2,2) hydrogels during immersion in the reductant solution. The error bars were obtained through three experiments. (c) The change in the r values of the hydrogels immersed in the reductant solution for 90 min. (d) The proposed structure of the γCD–VC11(x,y) hydrogel in a reduced state. Green line: the pAAm main chain in the polymer network. Green ring: γCD unit. Purple unit: the reduced viologen moiety in VC11. Grey line: the undecyl linker in VC11. The grey linker acts as a station where the host molecule stays. The reduced viologen units (purple) form a cationic dimer via cation–π interactions. The viologen dimer can be included by another γCD unit. | ||
UV-Vis absorption measurements on the hydrogels revealed the details about the state of the viologen unit during the reduction (Fig. S14†). In the oxidized state (initial state), the absorption band derived from the dicationic species of the viologen unit was observed at approximately 400–450 nm. After the reduction, other new bands at approximately 480–650 nm appeared, indicating a radical cation species. The radical cation's band can be divided into two absorption modes, which are derived from the radical cation monomer and its dimer (Fig. S15†). The molar ratio of the dimer in the hydrogels ([dimer]/([dimer] + [monomer])) was evaluated by waveform separation of the UV/Vis spectra. The order of the dimmer's molar fraction is γCD–VC11(2,2) hydrogel (85%) > αCD–VC11(2,2) hydrogel (75%) ≥ βCD–VC11(2,2) hydrogel (74%) > VC11(0.2,2) hydrogel (67%) (Table S7†). Moreover, the radical cation dimer of the γCD–VC11(2,2) hydrogel showed bimodal peaks (Fig. S15c†), whereas the unimodal peak of the dimer was observed in the other hydrogels. The bimodal peaks in the γCD–VC11(2,2) hydrogel are derived from the dimers isolated in the solution and included in the γCD cavity, respectively. The wave separation analysis also indicates that 52% of viologens are included in the γCD cavity (Table S7†). These results indicate that the VC11 units in the γCD–VC11(2,2) hydrogel effectively form radical cation dimers in the reduction reaction. This finding coincides with the results of the stoichiometric study mentioned above. The γCD unit includes the two undecyl residues in the VC11 unit and should also form a stable double-threaded 1:2 inclusion complex with the oxidized viologen dimer in the VC11 units, leading to the formation of radical cation dimers of the viologen residues (Fig. 3d). These results suggest that this double-threaded structure contributes to a larger deformation of the γCD–VC11(2,2) hydrogel than the other hydrogels following the reduction.
2.3. Change in the mechanical properties of the host–guest hydrogels through redox reactions
We investigated the reductive/oxidative state of the VC11 unit and the internal molecular state in the CD–VC11(x,y) hydrogels. The reduced γCD–VC11(2,2) hydrogel was immersed in 0.5 M phosphate buffer (pH 7.0) with potassium nitrite (KNO2, 0.1 M) as an oxidant. The reduced γCD–VC11(2,2) hydrogel was restored to its initial transparent state (see Movie S1†), suggesting that the viologen unit returned to its original dicationic state (Fig. 4a). UV-Vis spectroscopy also supports the oxidation of the reduced viologen units (Fig. S16†). The UV/Vis absorption bands corresponding to the radical cation monomer and dimer in the reduced γCD–VC11(2,2) hydrogel disappeared after oxidation. In addition, the band of the dicationic species appeared again.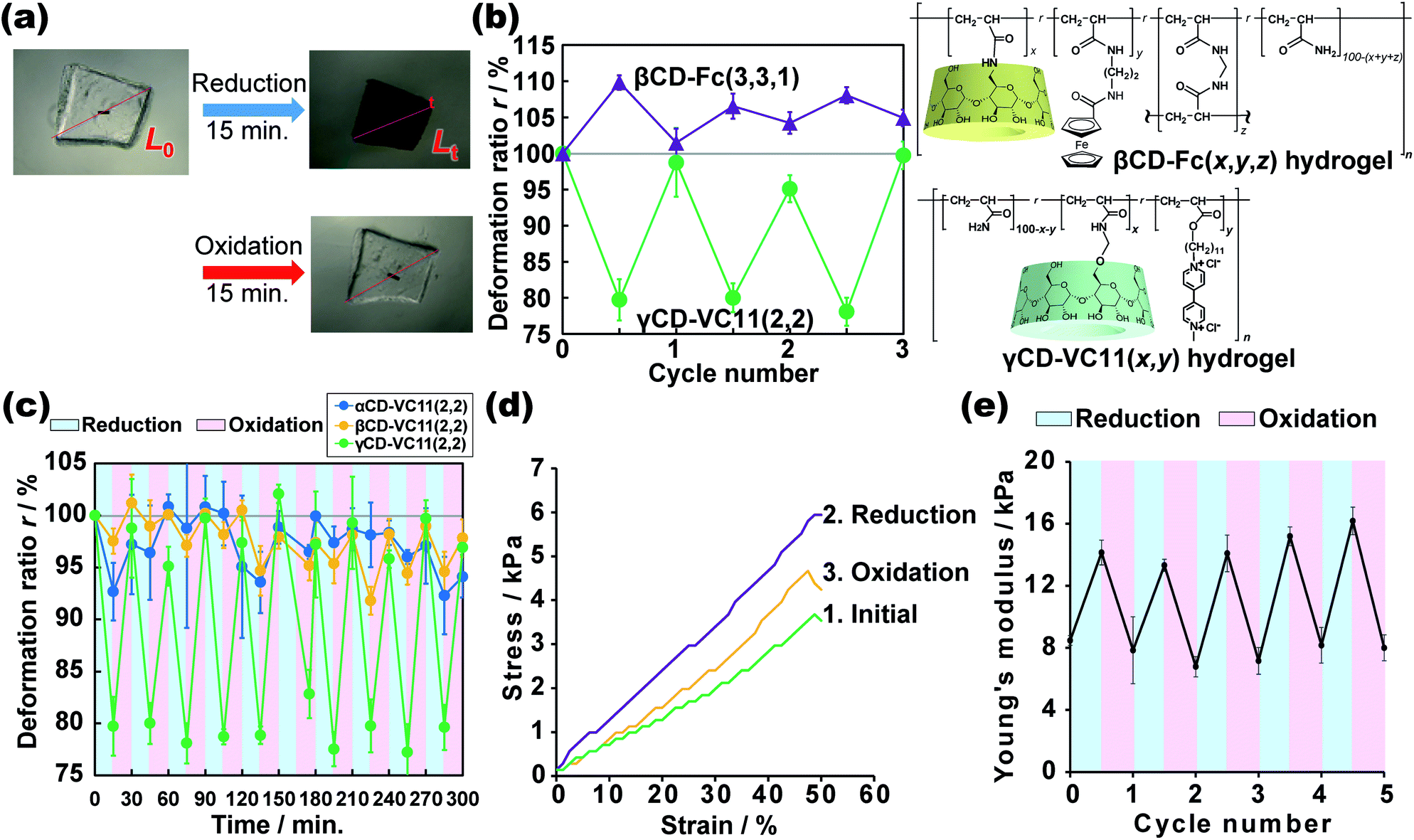 | ||
| Fig. 4 (a) Photographs of the γCD–VC11(2,2) gel [initial (upper left); after reduction (upper right); after oxidation (lower right)]. (b) Plot of the normalized length (r) of the γCD–VC11(2,2) and the βCD–Fc(3,3,1) hydrogels during redox cycling. The data of βCD–Fc(3,3,1) were newly obtained in this work to examine under the same redox conditions. (c) The results of the repeated redox cycling test of the αCD–VC11(2,2), βCD–VC11(2,2), and γCD–VC11(2,2) hydrogels. The r values are plotted based on the redox cycles. (d) Stress–strain curve of the γCD–VC11(2,2) hydrogel in compression tests (green line: the initial oxidized state; purple line: after treatment in reducing agent solution; yellow line: the oxidized hydrogel after the first reduction. The reduced γCD–VC11(2,2) hydrogel was treated with an oxidative reagent. At larger strain λ > 40%, the standard deviation of the stress value is >18%). (e) Repeated testing of the Young's modulus of the γCD–VC11(2,2) hydrogel over multiple reduction/oxidation cycles. The error bars were obtained through three experiments under the same conditions. | ||
The volume of the reduced γCD–VC11(2,2) hydrogel was also recovered to the volume of the hydrogel in its initial state by oxidation. Note that this contraction and expansion behaviour of the γCD–VC11(2,2) hydrogel is exactly the opposite of that of the βCD–Fc(3,3,1) hydrogel under the same redox conditions (the βCD–Fc(3,3,1) hydrogel was prepared by the same method in our previous work30) (Fig. 4b). The volume of gels generally increases upon decreasing their cross-linking density. While the neutral Fc molecule is included by the βCD host with a high association constant Ka = 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1, the oxidized monocationic Fc+ shows a low affinity for βCD (Ka = 15 M−1) owing to the electric instability of the inclusion complex.50–52 Thus, the addition of an oxidant to the inclusion complex of βCD with Fc results in the dissociation of the complex. A reductant leads to the reassociation of the complex. In the βCD–Fc(3,3,1) hydrogel, reduction/oxidation triggered association/dissociation between the βCD and Fc units, resulting in an increase/decrease in the cross-linking density. Therefore, after the oxidation of the Fc unit, the cross-linking density of the βCD–Fc(3,3,1) hydrogel decreased, leading to an increase in the volume.30 On the other hand, the initial state of the γCD–VC11(2,2) hydrogel was the oxidative state. Through the reduction of the viologen unit, the cross-linking density of the γCD–VC11(2,2) hydrogel should increase, leading to a decrease in volume. The displacement of deformation in the γCD–VC11(2,2) hydrogel is two times higher than that in the βCD–Fc(3,3,1) hydrogel. Additionally, the γCD–VC11(2,2) hydrogel still showed a larger volume change than the βCD–Fc(3,3,1) hydrogel in third cycles (Fig. 4b). Although Fc is generally a stable molecule against oxidation, some acylated Fc was gradually decomposed with oxidation.53 The smaller deformation through the repeated oxidation/reduction reaction was derived from the decomposition of the Fc unit in the βCD–Fc(3,3,1) hydrogel.
000 M−1, the oxidized monocationic Fc+ shows a low affinity for βCD (Ka = 15 M−1) owing to the electric instability of the inclusion complex.50–52 Thus, the addition of an oxidant to the inclusion complex of βCD with Fc results in the dissociation of the complex. A reductant leads to the reassociation of the complex. In the βCD–Fc(3,3,1) hydrogel, reduction/oxidation triggered association/dissociation between the βCD and Fc units, resulting in an increase/decrease in the cross-linking density. Therefore, after the oxidation of the Fc unit, the cross-linking density of the βCD–Fc(3,3,1) hydrogel decreased, leading to an increase in the volume.30 On the other hand, the initial state of the γCD–VC11(2,2) hydrogel was the oxidative state. Through the reduction of the viologen unit, the cross-linking density of the γCD–VC11(2,2) hydrogel should increase, leading to a decrease in volume. The displacement of deformation in the γCD–VC11(2,2) hydrogel is two times higher than that in the βCD–Fc(3,3,1) hydrogel. Additionally, the γCD–VC11(2,2) hydrogel still showed a larger volume change than the βCD–Fc(3,3,1) hydrogel in third cycles (Fig. 4b). Although Fc is generally a stable molecule against oxidation, some acylated Fc was gradually decomposed with oxidation.53 The smaller deformation through the repeated oxidation/reduction reaction was derived from the decomposition of the Fc unit in the βCD–Fc(3,3,1) hydrogel.
The deformation process of the γCD–VC11(2,2) hydrogel is reversible over more than ten oxidation/reduction cycles without hysteresis (Fig. 4c), indicating that the use of a viologen induces superior reversibility to the βCD–Fc(3,3,1) hydrogel. The γCD–VC11(2,2) hydrogel maintained its purple colour in the reductant solution at least for a month. The reduced γCD–VC11(2,2) hydrogel was able to recover the original colourless transparent state by the oxidation. These results show the reversibility and stability of the γCD–VC11(2,2) hydrogel. Fig. 4c also shows the results of the αCD–VC11(2,2) and βCD–VC11(2,2) hydrogels, which showed much lower volume change compared with the γCD–VC11(2,2) hydrogel. These results indicate that the molecular design with 1:2 complex cross-linking is more effective to achieve large deformation than that with the 1:1 complex.
We can clearly examine the change in the cross-linking density of the γCD–VC11(2,2) hydrogel in response to redox stimuli based on its Young's modulus. Fig. 4d shows the stress–strain curves, which were obtained by compression tests, of the γCD–VC11(2,2) hydrogel before and after reduction/oxidative reactions. After the reduction reaction, the stress of the γCD–VC11(2,2) hydrogel increased, and with the oxidation reaction, the stress of the reduced γCD–VC11(2,2) hydrogel under the same strain decreased. The stress decreased by another oxidation reaction. We calculated the Young's modulus as the initial gradients of these curves in the range between 1% and 10% strain. In a typical polymer network material, its cross-linking density (the number of entropic springs between the cross-linking points) is proportional to its Young's moduli. Fig. 4e shows the change in the Young's modulus of the γCD–VC11(2,2) hydrogel in response to redox stimuli. The reduction reaction increased the Young's modulus of the γCD–VC11(2,2) hydrogel, indicating an increase in the cross-linking density due to formation of a complex between the double-threaded structure and the radical cation dimer. The Young's modulus of the γCD–VC 11(2,2) hydrogel was restored with the dissociation of these complexes in response to oxidative stimuli. The change in the Young's modulus was observed over more than 5 reduction/oxidation cycles. These results supported our proposed scheme: the deformation behaviour is derived from changing the cross-linking density based on supramolecular complex formation between the CD and VC11 units.
On the other hand, the Young's moduli of the αCD–VC11(2,2) and βCD–VC11(2,2) hydrogels before and after immersion in the reductant solution were comparable (Fig. S19†), although the Young's modulus of the VC11(0.2,2) hydrogel, which has no CD moiety, slightly increased after the reduction due to the formation of the radical cation dimer. These results support the large deformation of the γCD–VC11(2,2) hydrogel driven by the 2:1 inclusion complexation between the two VC11 moieties and the γCD unit with a large cavity size.
2.4. Mechanism of deformation of the host–guest hydrogels
Fig. 5 shows a proposed mechanism for the deformation of the γCD–VC11(x,y) hydrogels. In the oxidized state, the γCD moiety and two VC11 residues form a 1:2 inclusion complex to make the double-threaded structure, where γCD includes the two alkyl chains in the VC11 unit. The reduction of the viologen unit triggers the dimerization of the monocation radical of the viologen units through cation–π electrostatic interactions, which increases the cross-linking density of the polymer network. Furthermore, UV/Vis and NMR spectroscopies of the VC11 monomer support the formation of the monocationic viologen radical dimer included in the γCD cavity (Fig. S17 and S18†). This inclusion behaviour between the cation dimer and γCD contributes to the formation of additional cross-linking points (Fig. 5). The 1:2 inclusion complexes of γCD and viologen monocation dimer connect the original cross-linking points to make a supramolecular cascade structure. The molar ratios of the viologens in the γCD–VC11(2,2) hydrogel are xmonomer = 15%, xdimer = 33%, and xCD-dimer = 52% (Fig. S15 and Table S7†), indicating that average number of viologen dimers is 5.7, and that of CD units in the single cascade structure is 10.3 (Fig. 5). Thus, the γCD–VC11(x,y) hydrogel should show a larger displacement of deformation than the other hydrogels. As these processes occurring via the reduction/oxidation of the viologen residues are reversible, the hydrogel can show repeated deformation more than ten times. The large Young's moduli change of the γCD–VC11(x,y) hydrogels (Fig. S19†) also supports that the large deformation of the hydrogel is driven by the 2:1 inclusion complexation between the two VC11 moieties and the γCD unit. The αCD–VC11(2,2) and βCD–VC11(2,2) hydrogels did not show such change in the Young's moduli.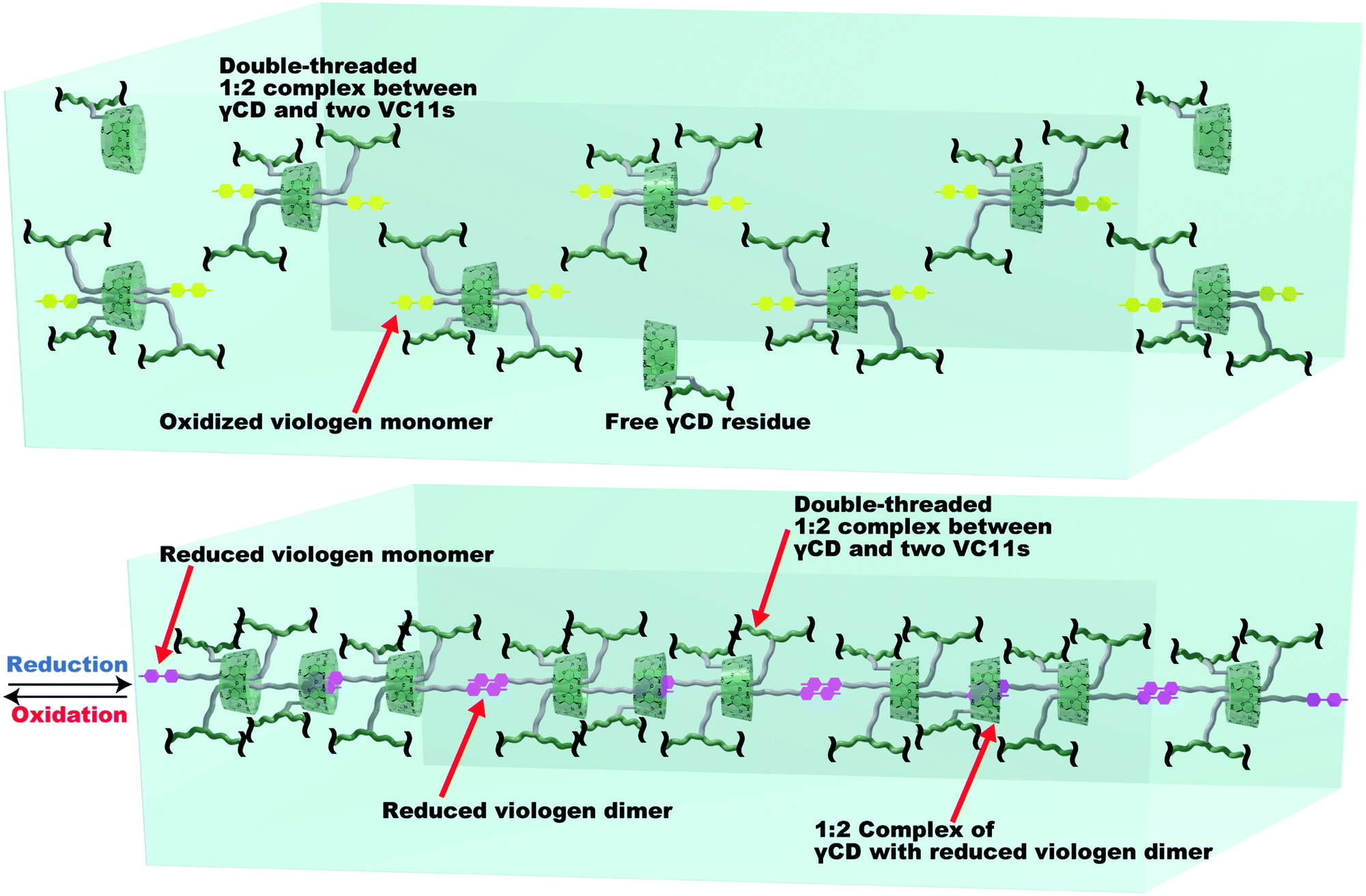 | ||
| Fig. 5 The proposed mechanism of the association/dissociation behaviours in the γCD–VC11(x,y) hydrogels leading to large volume changes. Green line: the pAAm main chain in the polymer network. Green ring: γCD unit. Yellow unit: the oxidized viologen moiety in VC11. Purple unit: the reduced viologen moiety in VC11. Grey line: the undecyl linker in VC11. The reduced monocation radical viologen unit (purple) can form a stacked dimer structure via cation–π electrostatic interactions. The viologen dimer is able to be included by another γCD unit to make a new cross-linking point. These two types of host–guest interactions result in the formation of supramolecular cross-linking, which contributes to the large deformation of the γCD–VC11(2,2) hydrogel. The xmonomer, xdimer, xCD-dimer values in Table S7† indicate the number of inclusion complexes in the supramolecular cross-linking, where 5.7 viologen dimers are formed in the single supramolecular cross-linking. 10.3 inclusion complexes of γCD with the undecyl units or with the viologen dimers are connected to form a supramolecular cascade structure. | ||
In the case of αCD–VC11(2,2) and βCD–VC11(2,2) hydrogels, the αCD or βCD unit and the undecyl moiety in VC11 form a 1:1 inclusion complex that functions as a cross-linking point between the polymer chains. However, this 1:1 inclusion complex does not contribute to the formation of radical cation dimers, even in the presence of a reducing agent, because the αCD and βCD units do not largely promote the formation of the radical cationic dimer due to steric hindrance (αCD and βCD with small cavities are not able to include to stabilize the radical cationic dimer). Therefore, the αCD–VC11(2,2) and βCD–VC11(2,2) hydrogels did not show the cross-linking formation with radical cationic dimers of the viologen units. This is one of the reasons that unlike the γCD–VC11(2,2) hydrogel, the αCD–VC11(2,2) and βCD–VC11(2,2) hydrogels did not exhibit large deformations.
Different swelling mechanisms of the αCD–VC11(2,2), βCD–VC11(2,2), and γCD–VC11(2,2) hydrogels were expected based on the Young's modulus. We defined the degree of change in the Young's modulus f = Ereduction/Einitial, and degree of change in the volume φ = φreduction/φinitial, respectively (Fig. S20†). The f values of the αCD–VC11(2,2) and βCD–VC11(2,2) hydrogels almost did not change at all during the oxidation. On the other hand, the f value of the 1:2 inclusion complex hydrogel significantly increased as the φ value increased during the oxidation, indicating that the swelling mechanism of the γCD–VC11(x,y) hydrogel was different from that of the αCD–VC11(x,y) and βCD–VC11(x,y) hydrogels. The two kinds of association and dissociation behaviours of the double-threaded 2:1 complex (γCD with the alkyl chain of VC11 and γCD with the reduced viologen dimer) form cross-linked structures consisting of the supramolecular polymers (Fig. 5), resulting in a large change in the f value.
Dynamic mechanical measurements (DMA) support the association–dissociation behaviours of the double-threaded dimer and the radical cation dimer, which were observed by using a dynamic shear rheometer (Fig. S21 and Table S8†). Some relaxation modes with relaxation time (τ) were observed in the hydrogels. In the reduced initial state, the VC11(0.2,2) hydrogel showed a very short shear relaxation time (τ < 10−3), indicating that there is no cross-linking mode within the frequency range in the DMA measurement. The τ values of the αCD–VC11(2,2), βCD–VC11(2,2), and γCD–VC11(2,2) hydrogels are 4000, 0.16, and 0.10 s, respectively. The relaxation modes are derived from threading movement of the CD ring onto the VC11 unit in the hydrogels. In particular, the very long relaxation time of the αCD–VC11(2,2) hydrogels is contributed by the electric trap effect of the viologen because of the narrow αCD cavity.46 The higher G′ value of the αCD–VC11(2,2) hydrogels than those of βCD–VC11(2,2), and γCD–VC11(2,2) hydrogels also agrees with these analyses.
The reductant decreased the relaxation times of the αCD–VC11(2,2) and βCD–VC11(2,2) hydrogels, indicating that αCD and βCD move faster through the reduced monocationic viologen barrier than through the oxidized dicationic viologen barrier. In contrast, the τ value of the γCD–VC11(2,2) hydrogel increased with the reduction, suggesting that a new cross-linking mode appears in the reduced γCD–VC11(2,2) hydrogel. The inclusion complex of γCD with a radical cation dimer is supposed to prevent the threading and dethreading motion of the inclusion complexes. Thus, only the γCD–VC11(2,2) hydrogel network should form a kinetically stable supramolecular cross-linking structure in response to reducing stimuli (Fig. 5). This structural change should enable the γCD–VC11(2,2) hydrogel to show a larger deformation than the αCD–VC11(2,2) and βCD–VC11(2,2) hydrogels.
2.5. Energy conversion by the host–guest hydrogels
We evaluated the mechanical work done by the chemical reactions of the γCD–VC11(2,2) hydrogel and the control hydrogels. We compared the γCD–VC11(2,2) hydrogel (supramolecular host–guest hydrogel) and the VC11(0.2,2) hydrogel (chemically cross-linked hydrogel) (Fig. 6c) and investigated the redox responsiveness of the γCD–VC11(2,2) hydrogel (Fig. 6d), the relationship between the thickness of the hydrogel and the deformation rate (Fig. 6e), and the relationship between the mechanical work and the ratio of γCD–VC11 units introduced into the hydrogels (Fig. 6f).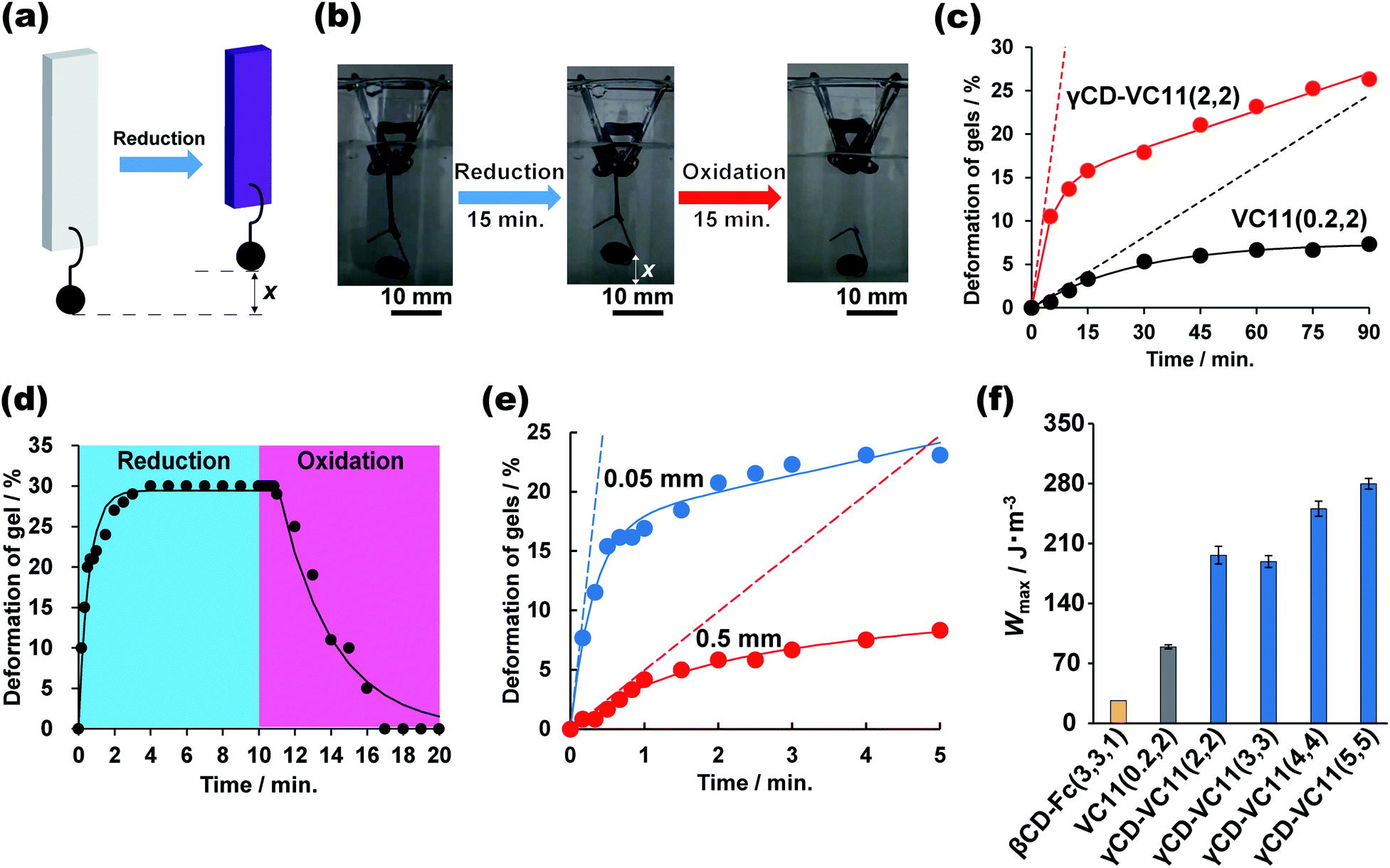 | ||
| Fig. 6 (a) Schematic illustration of hydrogel actuation with a weight (422 mg) through the reduction reaction. “x” is the displacement of the lifted weight. (b) Photographs of the γCD–VC11(2,2) hydrogel responding to redox stimuli for 15 min. Scale bar indicates 10 mm. (c) Plot of the normalized position of the weight versus the immersion time in the case of γCD–VC11(2,2) and VC11(0.2,2) hydrogels (0.05 mm (thickness) × 5 mm (width) × 10 mm (length)) with the same weight (422 mg). (d) Plot of the normalized position of the weight versus the immersion time in the case of the γCD–VC11(5,5) hydrogel (0.05 mm (thickness) × 5 mm (width) × 10 mm (length)) responding to redox stimuli. (e) The relationship between the deformation rate and the thickness of the γCD–VC11(2,2) hydrogel. The red circles indicate the results for the γCD–VC11(2,2) hydrogel with a thickness of 0.5 mm, and the blue circles indicates the results for the γCD–VC11(2,2) hydrogel with a thickness of 0.05 mm and the same width and length (5 mm (width) × 10 mm (length)). (f) The relationship between the work performed in the deformation of the hydrogels per volume (W) and the mol% contents of the host/guest units in the γCD–VC11(x,y) hydrogel and the control hydrogels. | ||
Fig. 6a shows the experimental setup used to analyse the energy conversion from chemical energy to mechanical work. A weight (422 mg) is hung at the bottom end of rectangular hydrogels (0.5 mm (thickness) × 5 mm (width) × 10 mm (length)). The hydrogel was immersed in 0.5 M phosphate buffer (pH 7.0) containing Na2S2O4 (0.10 M) as a reductant. To observe the reverse deformation, the hydrogel was subsequently immersed in 0.50 M phosphate buffer (pH 7.0) with KNO2 (0.10 M) as an oxidant. In the case of the γCD–VC11(2,2) hydrogel, the hydrogel was contracted by the reductant, vertically lifting the weight up. The reduced γCD–VC11(2,2) hydrogel was expanded by the oxidant, which restored the original position of the weight (Fig. 6b, d and Movie S1†).
In comparison with the γCD–VC11(2,2) hydrogel, the VC11(0.2,2) hydrogel showed a smaller change in the normalized position of the weight per unit time (Fig. 6c). The initial slope of the displacement, which was calculated by fitting a nonlinear curve to the data, indicates that the γCD–VC11(2,2) hydrogel shows 10 times faster deformation (3.3% min−1) than the VC11(0.2,2) hydrogel (0.27% min−1). These behaviours were in agreement with the degree of volume change in each hydrogel, as described above. Fig. 6d shows the time-course of the normalized position of the weight hung on the γCD–VC11(5,5) hydrogel in response to redox stimuli. The displacement of the weight increased with the reduction reaction within 4 minutes. Subsequently, the oxidation reaction completely returned the weight to its initial position in 7 minutes.
The deformation rate of the γCD–VC11(x,y) hydrogel was closely related to the thickness of the hydrogel. Fig. 6e shows the thickness dependence of the deformation of the γCD–VC11(2,2) hydrogels (thickness: 0.5 mm or 0.05 mm). The width and length of each hydrogel are the same (5 mm (width) × 10 mm (length)). Nonlinear least-squares curve fitting revealed that the initial deformation rate of the γCD–VC11(2,2) hydrogel sample with 0.05 mm thickness was 57% min−1, which is eleven times faster than that of the γCD–VC11(2,2) hydrogel sample with 0.5 mm thickness (4.9% min−1).
The mechanical work was determined by using the equation W = (m − ρV)gx (m: mass of the weight 422 mg, ρ: density of the buffer 1007 kg m−3, V: volume of the weight 48 μL, g: acceleration of gravity 9.80665 m s−2, and x: length of the weight that was lifted). The energy conversion of the γCD–VC11(x,y) hydrogel was calculated to be 0.92% based on the W value and sum of the redox potential of the viologen units (ΔG = −0.4 V) in the γCD–VC11(x,y) hydrogel. Fig. 6f shows the W values of the γCD–VC11(x,y) hydrogels and the control hydrogels per volume. The W of the γCD–VC11(x,y) hydrogel increased with the mol% content of the γCD and VC11 moieties. Moreover, among the hydrogels tested, the γCD–VC11(5,5) hydrogel showed the largest W value, indicating that the energy conversion of the γCD–VC11(x,y) hydrogel depends on the molar content of the VC11 unit. In addition, the W of the γCD–VC11(2,2) hydrogel was 1.8 times higher than that of the VC11(0.2,2) hydrogel. Notably, the W value of the γCD–VC11(3,3) hydrogel is also 7.4 times higher than that of the βCD–Fc(3,3,1) hydrogel in our previous work.30 These results show that the formation of the complex between the double-threaded structure and the radical cation dimer contributes to the large deformation and work done by the γCD–VC11(x,y) hydrogel. Work rates are also estimated from the initial slope values of Fig. 6c. The γCD–VC11(2,2) hydrogel shows a power P = 470 mW m−3, which is 10 times higher than that of the control VC11(0.2,2) hydrogel (P = 48 mW m−3). These results indicate that the design of 1:2 complexation is useful not only for the displacement but also the rate of deformation. The polymeric design of the γCD–VC11 hydrogel supports the superiority of its deformation ability in response to redox stimuli.
3. Conclusion
In conclusion, γCD–VC11(x,y) hydrogels showed effective contraction and expansion properties. The γCD and VC11 unit in the polymer network form a double-threaded 1:2 inclusion complex, which binds three polymer chains through a host–guest cross-linking point. When the γCD–VC11(x,y) hydrogel was reduced in solution, the radical cation dimer of the viologen moieties is formed as a new cross-linking point. The dimer is included by another γCD unit to form supramolecular cross-linking structures. These two modes, double-threaded 1:2 inclusion complexation and radical cation dimerization inside the γCD cavity, enable the γCD–VC11(x,y) hydrogels to show the largest deformation among all the hydrogels investigated in this work (the αCD–VC11(x,y), βCD–VC11(x,y), and VC11(0.2,y) hydrogels) and our previous work (βCD–Fc(3,3,1) hydrogel). The mechanism achieved by using two kinds of 2:1 complexation is a new design concept for creating stimuli-responsive materials. In the future, swelling hydrogel actuators triggered by external stimuli could be developed by using new concepts inspired by biomacromolecules (sponge or sarcomere filament) or artificial topological polymer networks.4. Experimental section
To prepare the γCD–VC11(2,2) hydrogel (a host–guest hydrogel containing γCD and VC11 moieties), γCDAAmMe (166 mg, 0.12 mmol), VC11 monomer (56 mg, 0.12 mmol), and AAm (410 mg, 5.8 mmol) were copolymerized by radical copolymerization initiated by K2S2O4 (16 mg, 0.06 mmol) and TEMED (8.9 μL, 0.06 mmol) in water (3.0 mL). Fig. 2c and Scheme S4† depict the synthetic scheme. The syntheses of the other hydrogels in this work are also described in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by a Grant-in-Aid of Scientific Research (B) (No. JP26288062 & JP18H02035) from MEXT, Scientific Research on Innovative Area Grant Number JP19H05721 from JSPS of Japan, JST-Mirai Program Grant Number JPMJMI18E3, the Ogasawara Foundation for the Promotion of Science & Engineering, and the Tonen General Sekiyu Research/Development Encouragement & Scholarship Foundation. We wish to thank Dr N. Inazumi of Osaka University for technical assistance with NMR.Notes and references
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, 2008 Search PubMed.
- M. W. Urban, Handbook of stimuli-responsive materials, John Wiley & Sons, 2011 Search PubMed.
- K. Bertoldi, V. Vitelli, J. Christensen and M. van Hecke, Nat. Rev. Mater., 2017, 2, 17066 CrossRef CAS.
- J. Yang, R. Bai, B. Chen and Z. Suo, Adv. Funct. Mater., 2019, 1901693 Search PubMed.
- Y. Gu, J. Zhao and J. A. Johnson, Trends Chem., 2019, 1, 318–334 CrossRef.
- P. Brochu and Q. Pei, Macromol. Rapid Commun., 2010, 31, 10–36 CrossRef CAS PubMed.
- Y. Zhang, C. Ellingford, R. N. Zhang, J. Roscow, M. Hopkins, P. Keogh, T. McNally, C. Bowen and C. Y. Wan, Adv. Funct. Mater., 2019, 29, 1808431 CrossRef.
- C. Christianson, N. N. Goldberg, D. D. Deheyn, S. Q. Cai and M. T. Tolley, Sci. Robot., 2018, 3, eaat1893 CrossRef.
- T. Ikeda and O. Tsutsumi, Science, 1995, 268, 1873–1875 CrossRef CAS PubMed.
- Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS PubMed.
- T. Ikeda, J. Mamiya and Y. Yu, Angew. Chem., Int. Ed., 2007, 46, 506–528 CrossRef CAS PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS PubMed.
- A. M. Rosales and K. S. Anseth, Nat. Rev. Mater., 2016, 1, 15012 CrossRef CAS PubMed.
- G. Schill, Catenanes, rotaxanes, and knots, Elsevier, 2017 Search PubMed.
- C. O. Dietrich-Buchecker and J. P. Sauvage, Chem. Rev., 1987, 87, 795 CrossRef CAS.
- D. B. Amabilino and J. F. Stoddart, Chem. Rev., 1995, 95, 2725–2828 CrossRef CAS.
- F. Niess, V. Duplan and J. P. Sauvage, Chem. Lett., 2014, 43, 964–974 CrossRef CAS.
- P. R. Ashton, I. Baxter, S. J. Cantrill, M. C. T. Fyfe, P. T. Glink, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 1294–1297 CrossRef CAS PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed.
- A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19 RSC.
- J. Rotzler and M. Mayor, Chem. Soc. Rev., 2013, 42, 44 RSC.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- T. Kudernac, N. Katsonis, W. R. Browne and B. L. Feringa, J. Mater. Chem., 2009, 19, 7168–7177 RSC.
- S. Tamesue, Y. Takashima, H. Yamaguchi, S. Shinkai and A. Harada, Angew. Chem., Int. Ed., 2010, 49, 7461–7464 CrossRef CAS PubMed.
- Y. Takashima, S. Hatanaka, M. Otsubo, M. Nakahata, T. Kakuta, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2012, 3, 1270 CrossRef PubMed.
- K. Iwaso, Y. Takashima and A. Harada, Nat. Chem., 2016, 8, 625–632 CrossRef CAS PubMed.
- Y. Takashima, Y. Hayashi, M. Osaki, F. Kaneko, H. Yamaguchi and A. Harada, Macromolecules, 2018, 51, 4688–4693 CrossRef CAS.
- S. Ikejiri, Y. Takashima, M. Osaki, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2018, 140, 17308–17315 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, A. Hashidzume and A. Harada, Angew. Chem., Int. Ed., 2013, 52, 5731–5735 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- J. Liu, Y. Lan, Z. Yu, C. S. Y. Tan, R. M. Parker, C. Abell and O. A. Scherman, Acc. Chem. Res., 2017, 50, 208 CrossRef CAS PubMed.
- Z. Yu, J. Liu, C. S. Y. Tan, O. A. Scherman and C. Abell, Angew. Chem., Int. Ed., 2018, 57, 3079–3083 CrossRef CAS PubMed.
- E. A. Appel, X. J. Loh, S. T. Jones, F. Biedermann, C. A. Dreiss and O. A. Scherman, J. Am. Chem. Soc., 2012, 134, 11767–11773 CrossRef CAS PubMed.
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- A. Tabet, R. A. Forster, C. C. Parkins, G. Wu and O. A. Scherman, Polym. Chem., 2019, 10, 467–472 RSC.
- J. Liu, C. S. Y. Tan, Z. Yu, Y. Lan, C. Abell and O. A. Scherman, Adv. Mater., 2017, 29, 1604951 CrossRef PubMed.
- J. Liu, C. S. Y. Tan, Z. Yu, N. Li, C. Abell and O. A. Scherman, Adv. Mater., 2017, 29, 1605325 CrossRef PubMed.
- A. Yasuda, H. Kondo, M. Itabashi and J. Seto, J. Electroanal. Chem. Interfacial Electrochem., 1986, 210, 265–275 CrossRef CAS.
- A. Yasuda, H. Mori and J. Seto, J. Appl. Electrochem., 1987, 17, 567–573 CrossRef CAS.
- C. Lee, M. S. Moon and J. W. Park, J. Inclusion Phenom. Mol. Recognit. Chem., 1996, 26, 219–232 CrossRef CAS.
- Y. Takashima, K. Otani, Y. Kobayashi, H. Aramoto, M. Nakahata, H. Yamaguchi and A. Harada, Macromolecules, 2018, 51, 6318–6326 CrossRef CAS.
- H. Yonemura, M. Kasahara, H. Saito, H. Nakamura and T. Matsuo, J. Phys. Chem., 1992, 96, 5765–5770 CrossRef CAS.
- H. Yonemura, H. Nakamura and T. Matsuo, Chem. Phys., 1992, 162, 69–78 CrossRef CAS.
- W. Herrmann, B. Keller and G. Wenz, Macromolecules, 1997, 30, 4966–4972 CrossRef CAS.
- Y. Kawaguchi and A. Harada, J. Am. Chem. Soc., 2000, 122, 3797–3798 CrossRef CAS.
- G. Wenz, C. Gruber, B. Keller, C. Schilli, T. Albuzat and A. Müller, Macromolecules, 2006, 39, 8021–8026 CrossRef CAS.
- T. Kakuta, Y. Takashima and A. Harada, Macromolecules, 2013, 46, 4575–4579 CrossRef CAS.
- M. Nakahata, Y. Takashima and A. Harada, Macromol. Rapid Commun., 2016, 37, 86–92 CrossRef CAS PubMed.
- A. Harada and S. Takahashi, J. Inclusion Phenom., 1984, 2, 791–798 CrossRef CAS.
- W. Ji-Shi, T. Kei, T. Akira and S. Isao, Bull. Chem. Soc. Jpn., 1998, 71, 1615–1618 CrossRef.
- A. U. Moozyckine, J. L. Bookham, M. E. Deary and D. M. Davies, J. Chem. Soc., Perkin Trans. 2, 2001, 1858–1862, 10.1039/b008440i.
- T. Hayashi, Y. Okada and T. Yamashita, Bull. Chem. Soc. Jpn., 1991, 64, 485–489 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05589d |
| This journal is © The Royal Society of Chemistry 2020 |