
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeted cell imaging properties of a deep red luminescent iridium(III) complex conjugated with a c-Myc signal peptide†
Adam H.
Day‡
a,
Martin H.
Übler‡
a,
Hannah L.
Best
b,
Emyr
Lloyd-Evans
b,
Robert J.
Mart
a,
Ian A.
Fallis
 a,
Rudolf K.
Allemann
a,
Eman A. H.
Al-Wattar
a,
Nathaniel I.
Keymer
a,
Niklaas J.
Buurma
a and
Simon J. A.
Pope
*a
a,
Rudolf K.
Allemann
a,
Eman A. H.
Al-Wattar
a,
Nathaniel I.
Keymer
a,
Niklaas J.
Buurma
a and
Simon J. A.
Pope
*a
aSchool of Chemistry, Cardiff University, Main Building, Cardiff, CF10 3AT, UK. E-mail: popesj@cardiff.ac.uk
bSchool of Biosciences, Cardiff University, Sir Martin Evans Building, Cardiff, UK
First published on 8th January 2020
Abstract
A nuclear localisation sequence (NLS) peptide, PAAKRVKLD, derived from the human c-Myc regulator gene, has been functionalised with a long wavelength (λex = 550 nm; λem = 677 nm) cyclometalated organometallic iridium(III) complex to give the conjugate Ir-CMYC. Confocal fluorescence microscopy studies on human fibroblast cells imaged after 18–24 h incubation show that Ir-CMYC concentrations of 80–100 μM promote good cell uptake and nuclear localisation, which was confirmed though co-localisation studies using Hoechst 33342. In comparison, a structurally related, photophysically analogous iridium(III) complex lacking the peptide sequence, Ir-PYR, showed very different biological behaviour, with no evidence of nuclear, lysosomal or autophagic vesicle localisation and significantly increased toxicity to the cells at concentrations >10 μM that induced mitochondrial dysfunction. Supporting UV-visible and circular dichroism spectroscopic studies show that Ir-PYR and Ir-CMYC display similarly low affinities for DNA (ca. 103 M−1), consistent with electrostatic binding. Therefore the translocation and nuclear uptake properties of Ir-CMYC are attributed to the presence of the PAAKRVKLD nuclear localisation sequence in this complex.
Introduction
A signal peptide is a short peptide sequence often present at the N-terminus of newly synthesised proteins that is responsible for intra- and extracellular translocation.1 When that translocation target is the cell nucleus such a peptide can facilitate transport of macromolecules between the cytoplasm and the nucleus and is referred to as a nuclear localisation signal (NLS).2 The best characterised mechanisms for protein import across the nuclear envelope are described for the “classical” NLSs, which consists of either one (PKKKRKV) or two (KRPAATKKAGQAKKKK) amino acid sequences.3 Previous research has shown that >50% of nuclear proteins conform to these sequences.4We were interested in an alternative NLS peptide sequence (PAAKRVKLD) which is derived from the human c-Myc protein (residues 320–328) and was first identified in 1988 to be essential for nuclear localisation of the protein.5 The PAAKRVKLD sequence is somewhat unusual as a NLS because three of its residues are cationic at physiological pH.5 The c-Myc protein is the transcription regulator expressed from the c-Myc gene, whose constitutive expression in cancer is associated with increased expression of other genes involved in cell proliferation, contributing to cancer development.6 It is tightly regulated in non-cancerous cells, but is now understood to be a frequently deregulated oncogene.7 Therefore c-Myc is of interest when considering approaches to targeted cancer therapy.8 Fusion proteins of the c-Myc NLS with β-galactosidase have also been used as a positive control for the identification of other nuclear localisation sequences,9 and in the production of light-inducible nuclear localisation sequences for tracking protein dynamics in live cells.10 Recently, a study described how green fluorescent protein (GFP) loaded nanoparticulate assemblies have been used to deliver proteins directly to the cytosol of cells. Conjugation of GFP with a variety of NLSs enabled observable trafficking of the protein to the nucleus using confocal fluorescence microscopy.11 These results showed that the c-Myc NLS conjugate produced the highest relative intensity of fluorescence in the nucleus versus the cytosol.
In this study, we wished to visualise the cellular translocalisation characteristics of the c-Myc NLS (PAAKRVKLD) in human fibroblast cells. To do this we investigated the use of a deep red luminescent organometallic complex as an optical label for the c-Myc NLS, which we envisaged would be advantageous when applied to confocal fluorescence microscopy. The potential of luminescent transition metal complexes (e.g. based upon Ru(II), Re(I), Ir(III) and Pt(II)) for cellular bioimaging has been explored over the last decade.12 In some cases metal complexes functioning as probes for cellular dysfunction have also been investigated.13 However, in biological and bioimaging studies it is notoriously difficult to predict the intracellular localisation behaviour of such species because of the challenging interplay of charge, hydro- and lipophilicity, structure–function relationships, and cytotoxicities.14 This in turn makes the rational design of organometallic bioimaging probes extremely challenging. The use of targeting vectors is one way to address this challenge.
Only a small number of reports have detailed the use of signal peptide conjugates of luminescent coordination complexes. Examples include rhenium(I) bisquinoline complexes for targeting the folate receptor of cancer cells,15 and 99mTc(I) labelled compounds to investigate targeting of radioimaging (and therapeutic) agents.16 Polypyridine complexes of Ru(II) have been the most developed in this area. Keyes and co-workers have reported a cell permeable polyarginine–Ru(II) complex possessing triplet metal-to-ligand charge transfer (3MLCT) emission behaviour and associated oxygen quenching sensitivity.17 Subsequent studies by the same group have described multimodal variants,18 peptide bridged dinuclear Ru(II) complexes for monitoring oxygen concentration in cells,19 and nuclear targeting with a NLS (VQRKRQKLMP) Ru(II) conjugate20 leading to photoinduced DNA destruction.21 Related 1,4,5,8-tetraazaphenanthrene (TAP) ligands on Ru(II) have also been studied, because of their photooxidising properties that can induce significant DNA damage, although interestingly the phototoxicity can be strongly inhibited by peptide (VQRKRQKLMP) conjugation of the complex.22 The cell nucleus is clearly an attractive target when considering the delivery of phototherapeutic action and transition metal complexes, particularly those of the 4th and 5th rows, have long been investigated for applications to photodynamic therapy (PDT).23
Herein we present an example of a peptide functionalised luminescent Ir(III) complex, and, to the best of our knowledge, the first small molecule luminescent moiety to feature a c-Myc inspired NLS. Together with an appropriately designed structural variant that lacks the NLS, this study demonstrates that the c-Myc NLS enables transport of the Ir(III) conjugate into the nucleus of human fibroblast cells.
Results and discussion
Synthesis of the NLS peptide and the Ir(III) complexes
The adopted strategy was to conjugate an organometallic iridium(III) complex entity to the N-terminus of the peptide via an amide bond whilst retaining the intrinsic luminescent properties of the complex. The cyclometalated iridium(III) complexes (Scheme 1) were synthesised according to previous methods described for related systems.24 Thus, the difluorinated quinoxaline ligand (6,7-difluoro-2-methyl-3-phenylquinoxaline) was used as a cyclometalating ligand to form the dimetallic precursor [(L)2Ir(μ-Cl)2Ir(L)2], which was then split using 4-methyl-2,2′-bipyridine-4-carboxylic acid to give monometallic Ir-COOH.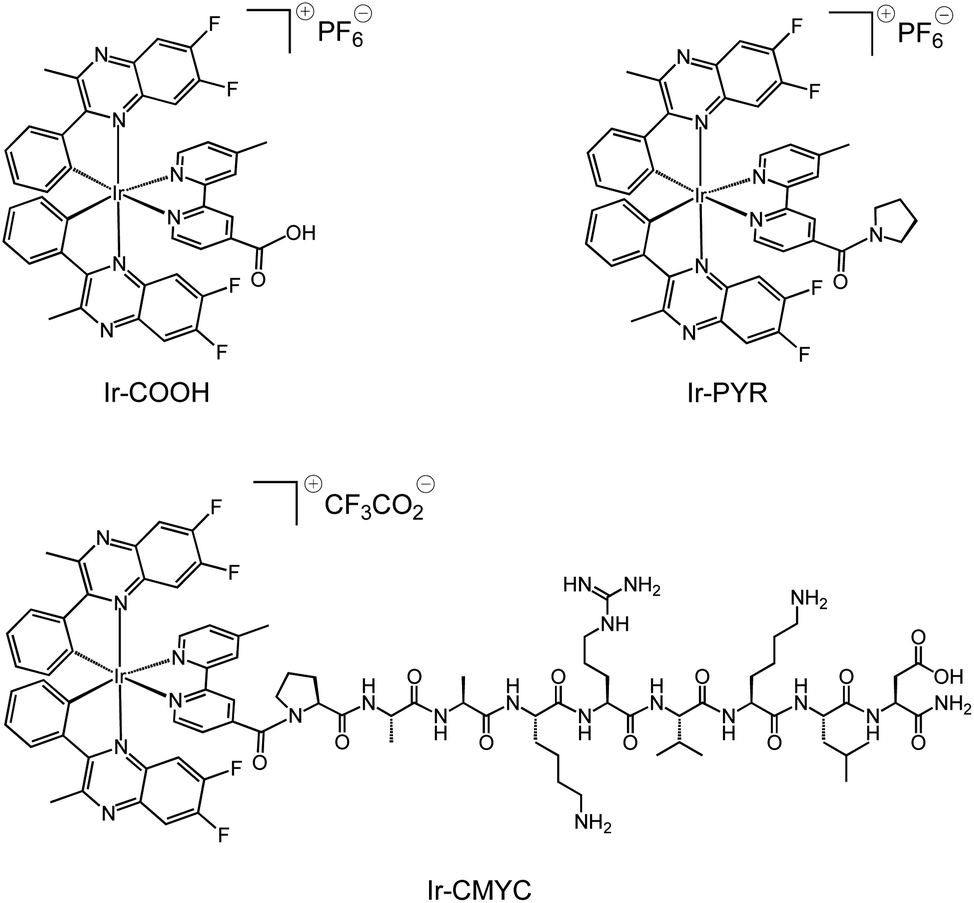 | ||
| Scheme 1 Structures of the luminescent complexes Ir-COOH, Ir-PYR and Ir-CMYC, investigated during this study. | ||
For the PAAKRVKLD sequence, Fmoc-solid phase peptide synthesis was performed using the Rink amide resin (0.19 mmol g−1 loading) at 0.1 mmol scale using standard techniques. Coupling was performed using 5.0 eq. O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate, 5.0 eq. 1-hydroxybenzotriazole, N,N-diisopropylethylamine and 4.0 eq. of amino acid. Single coupling was performed for 60 min, while double coupling was carried out for 45 min per cycle for arginine and valine and their direct successive amino acids. Subsequent Fmoc deprotection was achieved using standard conditions (see Experimental) to afford the crude peptide attached to the resin. Ir-COOH was then coupled in identical conditions to afford the crude iridium peptide complex. The crude iridium–peptide complex was cleaved off the resin by stirring in (v/v/v/v) 92.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5:2.5 TFA/water/DODT/TIS (where TFA = trifluoroacetic acid, DODT = 2,2′-(ethylenedioxy)diethanethiol, TIS = triisopropylsilane) for 2 hours at room temperature to yield 5 mg of crude conjugate. LC-MS analysis using a linear gradient of 10% to 95% acetonitrile–water (+0.1% TFA) over 13 min revealed the crude Ir-CMYC which eluted at 8.0 min, with the molecular composition of the iridium–peptide complex confirmed by LC-MS with observed m/z = 947 and 632, corresponding to the [M + H]2+ and [M + 2H]3+ of Ir-CMYC, respectively. The crude conjugate was purified via preparatory HPLC (Fig. S2 and S3, ESI†) under an identical gradient eluting at 28.6 min, prior to lyophilization yielding 1.5 mg of pure Ir-CMYC. The lyophilized Ir-CMYC was subsequently used for all characterization and biological testing.
:2.5:2.5:2.5 TFA/water/DODT/TIS (where TFA = trifluoroacetic acid, DODT = 2,2′-(ethylenedioxy)diethanethiol, TIS = triisopropylsilane) for 2 hours at room temperature to yield 5 mg of crude conjugate. LC-MS analysis using a linear gradient of 10% to 95% acetonitrile–water (+0.1% TFA) over 13 min revealed the crude Ir-CMYC which eluted at 8.0 min, with the molecular composition of the iridium–peptide complex confirmed by LC-MS with observed m/z = 947 and 632, corresponding to the [M + H]2+ and [M + 2H]3+ of Ir-CMYC, respectively. The crude conjugate was purified via preparatory HPLC (Fig. S2 and S3, ESI†) under an identical gradient eluting at 28.6 min, prior to lyophilization yielding 1.5 mg of pure Ir-CMYC. The lyophilized Ir-CMYC was subsequently used for all characterization and biological testing.
An additional iridium(III) complex lacking the peptide sequence, Ir-PYR, was also synthesised for investigation as a photophysical analogue of Ir-CMYC. Thus, 4-methyl-2,2′-bipyridine-4-carboxylic acid was simply converted to 4′-methyl-(2,2′-bipyridine)-4-(pyrrolidin-1-yl)methanone to give a new bipyridine derivative (see ESI† for experimental procedure), and then in turn reacted with [(L)2Ir(μ-Cl)2Ir(L)2] to give the pyrrolidine terminated species Ir-PYR. Ir-PYR was purified using preparatory HPLC (Fig. 1, ESI) and characterised using the standard array of techniques: 1H NMR data was particularly insightful, revealing the three unique methyl proton environments (two from the inequivalent quinoxalines, one from the bipyridine), as well as the pyrrolidine protons at 3.2–1.8 ppm. High resolution MS was also obtained confirming the parent cation with the correct isotopic distribution at m/z = 968.2446 (Fig. S1, ESI†).
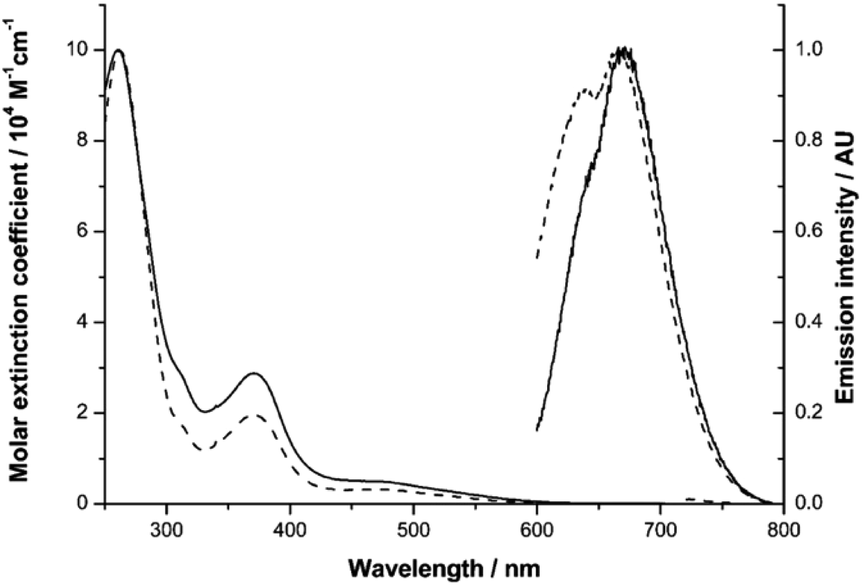 | ||
| Fig. 1 UV-vis absorption and emission spectra of Ir-PYR (dashed line) and Ir-CMYC (solid line). | ||
Photophysical characterisation of Ir-PYR and Ir-CMYC
Our previous studies on Ir(III) complexes have shown that the use of substituted, cyclometalated quinoxaline ligands can impart red luminescence characteristics.25 Firstly, the UV-vis absorption and emission spectral characteristics of Ir-PYR were determined. The absorption spectrum was obtained in water and showed a composite of bands between 200–600 nm; the appearance of the spectrum is reminiscent of closely related [Ir(C^N)2(N^N)]+ complexes of this type.26 By inference, ligand-centred transitions are assumed to dominate below 350 nm, with both quinoxaline and bipyridine located π → π* contributing to the spectrum. At 350–450 nm it is likely that spin-allowed metal-to-ligand (5d-to-π*) charge transfers (1MLCT) are present, with a weaker (ε < 1000 M−1 cm−1) shoulder feature at 450–600 nm, which is presumably due to a spin forbidden (S0 → T1) 3MLCT absorption. The corresponding absorption spectrum for the peptide functionalised species Ir-CMYC shares the same features with Ir-PYR, showing that the addition of the peptide sequence does not dramatically alter the intrinsic electronic properties of the complex. To assess longer term stability in water, UV-vis absorption spectra of an aqueous solution (stored at 25 °C in light) of Ir-CMYC were recorded at 0, 24, 48 and 72 h (Fig. S4, ESI†). The resultant spectra were superimposable suggesting not only good chemical stability, but also excellent solubility.The luminescence properties of the Ir(III) complexes (Fig. 1) were determined in dilute aerated aqueous solvent. Initially, an excitation wavelength of 550 nm was adopted which is selective for the MLCT absorption bands of the complex, and resulted in a broad emission peak (λem) for Ir-PYR centred at 674 nm (cf.Ir-COOHλem = 673 nm). The long wavelength emission characteristics are consistent with our previous studies on cyclometalated quinoxaline [Ir(C^N)2(N^N)]+ complexes of Ir(III).25 The modest bathochromic shift is likely induced by the polar nature of the water solvent, which in turn suggests a 3MLCT contribution to the emitting state. For Ir-PYR time-resolved measurements gave the corresponding lifetime (τobs) at 42 ns, and a quantum yield assessment gave ca. 3%, which is typical for this class of compound in aerated solvent. The phosphorescent nature of Ir-PYR was demonstrated by a recorded lifetime of 353 ns in MeCN (Fig. S5, ESI†). Luminescence spectroscopy on Ir-CMYC gave comparable data (Fig. 1), with λem = 677 nm (τobs = 38 ns), thus confirming that the photophysical properties of the Ir(III) complex moiety are retained after functionalisation with the peptide sequence.
Cell imaging with Ir-PYR and Ir-CMYC
The non-peptide functionalised complex Ir-PYR was investigated at a range of concentrations (1, 5, 20, 50, 80, 100 μM) with respect to cell uptake, localization and bioimaging capability. Human fibroblast cells were incubated with the different concentration solutions of Ir-PYR and imaged after 18 hours using confocal fluorescence microscopy. With long wavelength excitation and emission parameters (λex = 480 nm; λem = 645 nm) compatible with the spectral properties of the complex, it was clearly evident (Fig. 2) that the deep red emission of Ir-PYR was highly amenable to confocal fluorescence microscopy.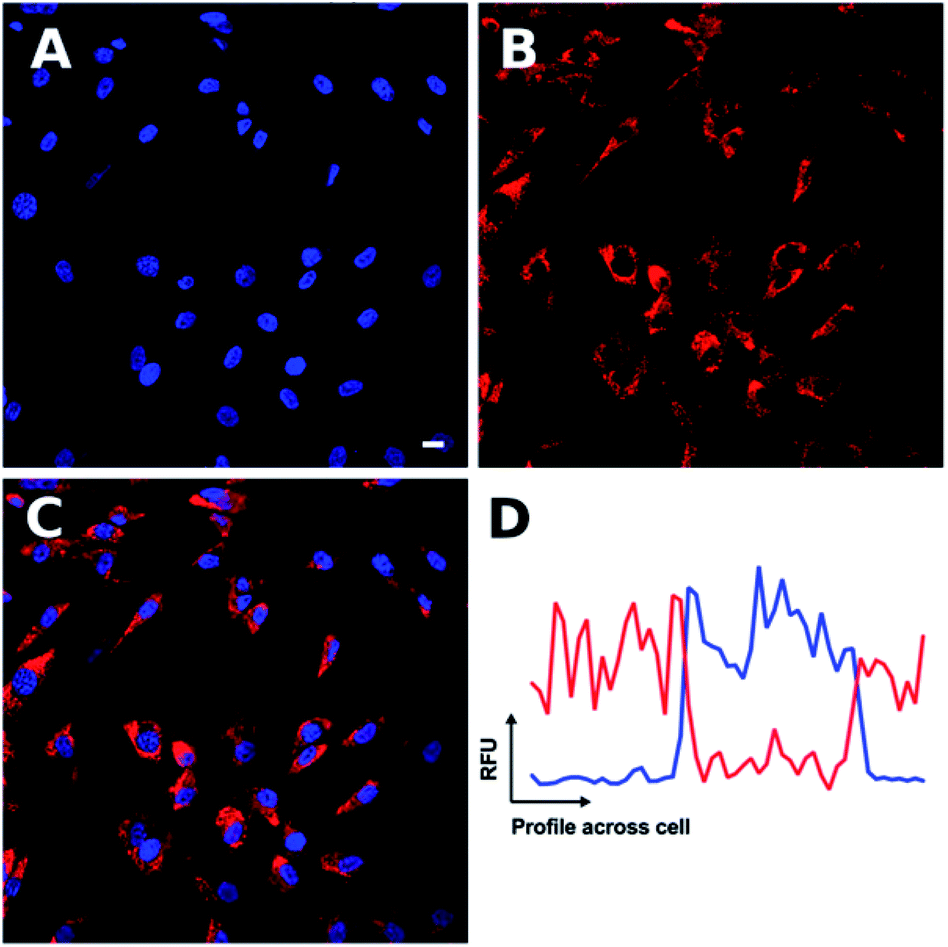 | ||
| Fig. 2 Confocal microscopy shows Ir-PYR to produce cytoplasmic punctate staining. Ir-PYR (red, (B)) was incubated with human fibroblasts at a concentration of 5 μM for 18 hours, and the nuclei with Hoechst 33342 (blue, (A)). Merged channels (C) demonstrate the lack of Ir-PYR nuclear co-localization, as represented in a sample co-localization plot (D) showing the relative fluorescent units (RFU) of both Ir-PYR (red) and Hoechst 33342 (blue) across a cell. Representative scale bar in (A) is equal to 7.5 μm. | ||
At lower concentrations Ir-PYR showed punctate staining of the cells, but with no evidence for nuclear localization (Fig. 2), whereas at 50–100 μM the compound appeared to be very toxic, as evidenced by reduced cell number and mitochondrial dysfunction (Fig. 3).27 A series of co-localization imaging experiments revealed that Ir-PYR does not localise in the nucleus, lysosomes or autophagic vesicles (Fig. 4).
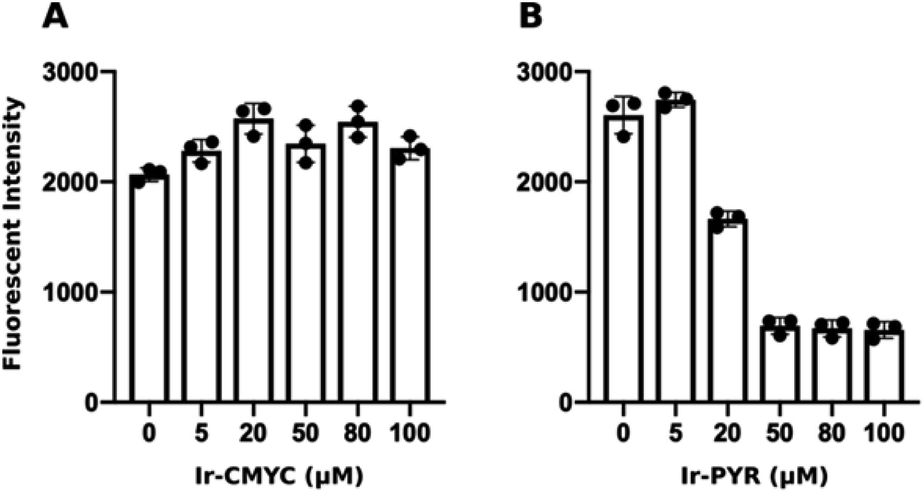 | ||
| Fig. 3 Ir-PYR is cytotoxic at 20 μM and above. Human fibroblasts were incubated with various concentrations (0 or DMSO only, 5, 20, 50, 80, 100 μM) of either Ir-CMYC (A) or Ir-PYR (B) for 18 hours. Resazurin was added to cells, and after a 4 hour incubation the relative fluorescence intensity was measured (λex = 530 nm; λem = 580 nm). Ir-PYR treated cells (>20 μM) show reduced conversion of resazurin to the fluorescent resorufin indicating reduced cell viability. No cytotoxicity was observed at any Ir-CMYC concentration. | ||
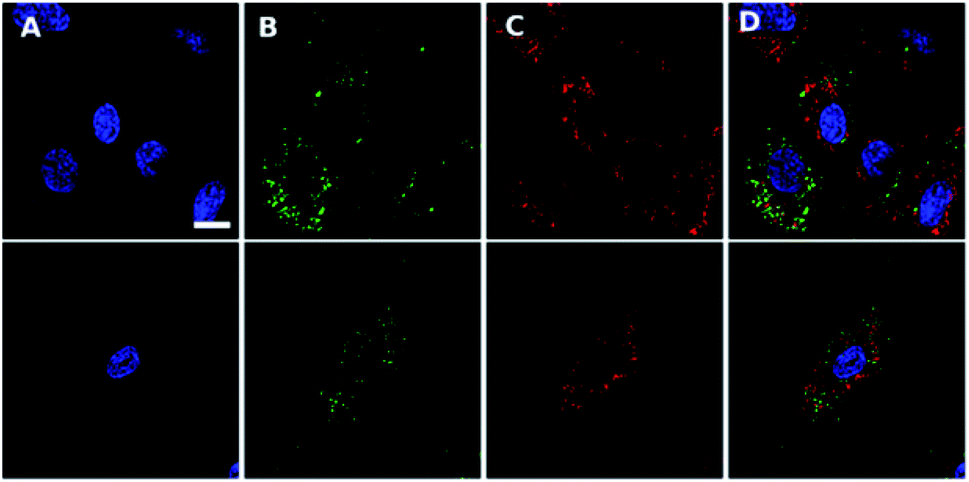 | ||
| Fig. 4 Confocal scanning microscopy shows Ir-PYR does not co-localize with nuclei, lysosomes or autophagic vesicles. Ir-PYR (red, (C)) was incubated with human fibroblasts at 5 μM for 18 hours and co-stained with Hoechst 33342 (blue, (A)) and either LysoTracker green (top row, (B)) or CYTO-ID (second row, (B)). Merged channels (D) demonstrate the lack of Ir-PYR co-localization with any utilised counterstain. Representative scale bar in (A) is equal to 7.5 μm. | ||
Having demonstrated that Ir-PYR does not localize in the cell nucleus, further imaging studies were then conducted on the peptide-functionalised analogue, Ir-CMYC. Again, a series of concentrations were used (1, 5, 20, 50, 80, 100 μM) for incubation with the cells. No uptake or staining was observed for 1–5 μM Ir-CMYC, but with increasing concentrations there was very clear uptake and staining evident (Fig. 5).
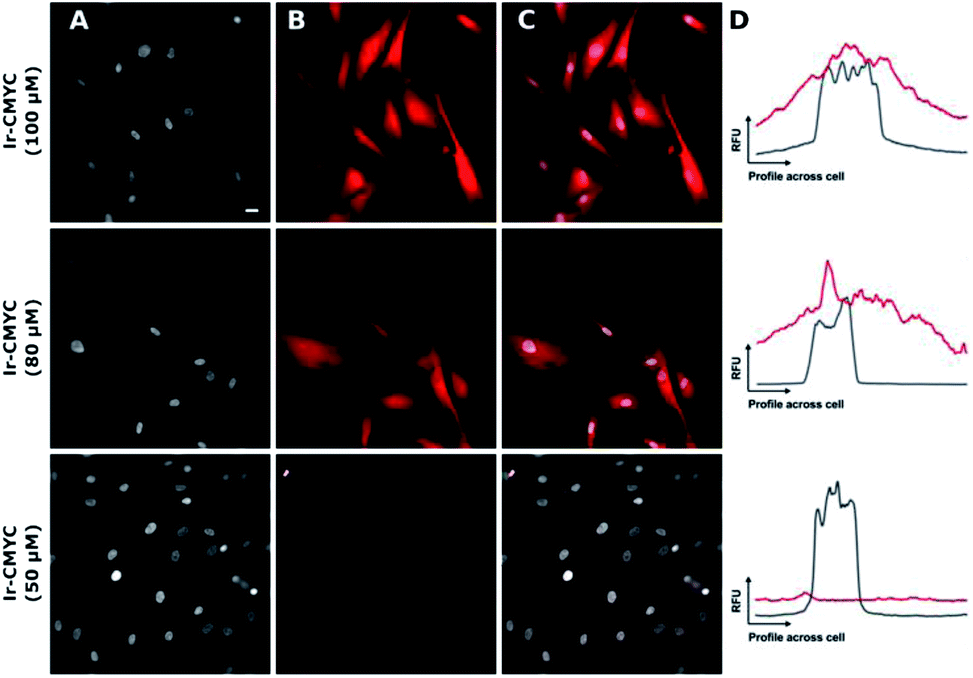 | ||
| Fig. 5 The addition of the c-Myc tag results in a significantly greater degree of nuclear co-localization. Ir-CMYC (red, (B)) was incubated with human fibroblast at a range of concentrations (50, 80 & 100 μM) for 18 hours prior to counterstaining with Hoechst 33342 (grey, (A)). Merged images (C) show nuclear co-localization at concentrations of 80 and 100 μM. Sample co-localization plots (D) show the intensity in relative fluorescent units (RFU) of Ir-CMYC (red) and Hoechst 33342 (grey) across a cell. Representative scale bar in (A) is equal to 7.5 μm, images acquired at (λex = 480 nm; λem = 645 nm). | ||
At 20–50 μM weak punctate staining was observed throughout the cytoplasm, and at 80–100 μM strong staining was seen throughout the cell, with clear nuclear localization. Co-localization experiments using Hoechst 33342 demonstrate that Ir-CMYC does enter and localize in the cell nucleus (Fig. 5). At 24 hours after Ir-CMYC incubation, there were no detectable changes in cellular morphology or mitochondrial activity (as monitored by a resazurin cell death assay) indicating no (or minimal) toxicity of Ir-CMYC to the cells.
In view of the nuclear localisation of Ir-CMYC, we also considered the potential role that affinity to DNA may play. To explore DNA binding of these Ir(III) complexes, UV-vis and circular dichroism titrations were carried out in buffer (25 mM MOPS, 50 mM NaCl, 1 mM EDTA) at 25 °C (Fig. S6, ESI†). To quantify the affinity of Ir-PYR for fish sperm DNA, titration curves at 400 and 493 nm were extracted from the circular dichroism data (Fig. 6). Similarly, for Ir-CMYC, titration curves at 373 and 493 nm were extracted from the circular dichroism data. The binding data were analysed globally (Table S1†) in terms of the multiple independent binding sites model, explicitly taking into account the dilution of the iridium complex during the titration.28 This analysis gave binding constants of (1.5 ± 0.7) × 103 M−1 and (4.5 ± 1.0) × 103 M−1 for Ir-PYR and Ir-CMYC, respectively (full fitting details in the ESI†). Although Ir-CMYC appears to bind a bit more strongly to DNA than Ir-PYR, the difference is less than an order of magnitude and both affinities are low. Indeed such low values are indicative of an interaction with DNA which is driven by electrostatics, suggesting that neither the c-Myc NLS or the cationic Ir(III) complex moiety strongly interact with the DNA structure.
 | ||
| Fig. 6 Ir-PYR and Ir-CMYC both interact weakly with DNA. Circular dichroism titration of Ir-PYR (top) with fish sperm DNA, 400 nm (●), 493 nm (▲); titration for Ir-CMYC (bottom), 373 nm (●), 493 nm (▲). | ||
To investigate whether the NLS was cleaved off Ir-CMYC prior to translocation to the nucleus, an immunoassay was trialled using an antibody raised against c-Myc. Following the immunocytochemistry procedure which permeabilises the cell membrane, the red emission from Ir-CMYC was no longer evident in the cells using fluorescence microscopy imaging. This suggests that permeabilisation of the cell membrane causes Ir-CMYC to leak out of the cell consistent with not being bound to DNA.
Taken together, these results suggest that the fundamental differences in nuclear localisation observed between Ir-PYR and Ir-CMYC cannot be explained in terms of an affinity for DNA: the nuclear localisation of Ir-CMYC is facilitated by the PAAKRVKLD sequence.
Conclusion
In summary, the synthesis, spectroscopic properties and cellular translocation characteristics of a deep red luminescent organometallic Ir(III) complex conjugated with a c-Myc NLS have been described. The NLS conjugated complex is essentially non-toxic, which contrasts strongly with the non-peptide complex analogue, which induces mitochondrial dysfunction and is toxic ≥20 μM. The conjugated complex was demonstrated, via UV-vis and circular dichroism titrations, to have a very weak affinity for DNA (∼4.5 × 103 M−1). Therefore, through a range of biological and spectroscopic studies it was demonstrated that the c-Myc nuclear localization sequence PAAKRVKLD promotes nuclear uptake of an organometallic iridium(III) complex in human fibroblast cells.Experimental
General considerations
All reagents and solvents were commercially available and were used without further purification if not stated otherwise. For the measurement of 1H, 31P, and 13C NMR spectra a Bruker Fourier300 (300 MHz), Bruker AVANCE HD III equipped with a BFFO SmartProbe™ (400 MHz) or Bruker AVANCE III HD with BBO Prodigy CryoProbe (500 MHz) was used. The obtained chemical shifts δ are reported in ppm and are referenced to the residual solvent signal. Spin–spin coupling constants J are given in Hz.Low-resolution mass spectra were obtained by the staff at Cardiff University. High resolution mass spectral (HRMS) data were obtained on a Waters MALDI-TOF mx at Cardiff University or on a Thermo Scientific LTQ Orbitrap XL by the EPSRC UK National Mass Spectrometry Facility at Swansea University. IR spectra were obtained from a Shimadzu IR-Affinity-1S FTIR. Reference to spectroscopic data are given for known compounds. Analytical HPLC was performed using an Agilent 1260 infinity II equipped with a Zorbax SB-C18 2.1 × 50 mm column and an Agilent 6120 Quadrupole LC/MS detector over a 13 minute gradient elution. Preparatory HPLC was performed using a Dionex UltiMate 3000 equipped with a YMC-Triart Prep C18-S 250 × 19 mM column over a 55 min gradient elution. UV-vis studies were performed on a Shimadzu UV-1800 spectrophotometer as MeCN solutions (2.5 or 5 × 10−5 M). Photophysical data were obtained on a JobinYvon–Horiba Fluorolog spectrometer fitted with a JY TBX picosecond photodetection module as MeCN solutions. Emission spectra were uncorrected and excitation spectra were instrument corrected. The pulsed source was a Nano-LED configured for 459 nm output operating at 1 MHz. Luminescence lifetime profiles were obtained using the JobinYvon–Horiba FluoroHub single photon counting module and the data fits yielded the lifetime values using the provided DAS6 deconvolution software. Quantum yield measurements were obtained on aerated MeCN solutions of the complexes using [Ru(bpy)3](PF6)2 in aerated MeCN as a standard (Φ = 0.016).
Human fibroblast primary cell culture
Human derived skin fibroblast cells lines were obtained from the Coriell Cell Repository (GM05399, Camden, NJ). Cells were cultured as monolayers in Dulbecco's modified Eagles medium (DMEM, Sigma, UK) supplemented with 10% fetal bovine serum (FBS, PAN-Biotech, Aidenbach, Germany) and 1% L-glutamine in a humidified incubator with 5% CO2 at 37 °C. For all imaging experiments low passage cells (P10-15) were seeded in 8-well chamber slides (Thistle, IB-80826) at a density of 15000 cells in 250 μL of media.
Cell staining with intracellular probes
To generate stock solutions, Ir-CMYC and Ir-PYR were resuspended in DMSO (final concentration 10 mM) and stored at −20 °C (it was noted that 3 freeze–thaws cycles did not appear to reduce the compound signal). Stock solutions were diluted to the stated concentrations in cell culture medium for incubation with fibroblasts. The equivalent amount of DMSO (<0.5% total volume) was added to controls for comparison. For nuclear visualisation cells were counterstained for 10 minutes with Hoechst 33342 diluted in cell culture media (1 μL mL−1, Thermo Fisher, UK). For non-live imaging, cells were fixed in cold 4% paraformaldehyde for 10 min at room temperature and rinsed with PBS. For live-cell colocalization studies, cells were incubated with Ir-PYR for 12 hours prior to counterstaining with Hoechst 33342, and either LysoTracker-green DND-26 (L7528, Thermo Fisher, UK), or CYTO-ID Autophagy detection kit 2.0 (175–0050, Enzo Life Sciences, UK), as per the manufacturer's instructions. Briefly, LysoTracker was diluted in cell culture media to a final concentration of 200 nM and left for 15 min at 37 °C. CYTO-ID was diluted by adding 2 μL of CYTO-ID green reagent in 1 mL of assay buffer and incubated for 30 min at 37 °C. After organelle staining, cells were washed twice in DPBS and immediately imaged in FluoroBrite DMEM (A1896701, Thermo Fisher, UK) supplemented with 10% FBS.Monitoring cellular metabolic activity
Human fibroblasts were plated at 10000 cells per well of a 96-well plate (Cellbind) and treated with the indicated concentrations of Ir-PYR, Ir-CMYC or DMSO. As a measure of cell viability and metabolic activity, at 18 hours post compound addition culture medium was removed and replaced with culture medium containing 10 μg mL−1 resazurin diluted in DPBS (10% v/v). After a 4 hour incubation at 37 °C, fluorescence was determined using a Molecular Devices Spectramax Gemini EM plate reader (λex = 445 nm; λem = 585 nm).
Imaging and localisation analysis
All widefield fluorescent microscopy was performed using a Zeiss 35 fitted with Cairn Optospin filter wheels at excitation and emission ports, an Orca flash 4.0/4.2, sCMOS camera, Exfo xCite multiband lightsource, and Micro-Manager software.29For confocal microscopy, live cells were imaged on an inverted Leica DMIRE2 with TCS SP2 AOBS confocal system using Leica AF software in combination with a 405 nm diode laser (Hoechst), a 458/476/488/514 nm argon multiline (LysoTracker and CYTO-ID), and a 594 nm HeNe (Ir-PYR). Images were captured using a 40× oil-immersion objective coupled to an additional 2× magnifying lens.30 During imaging, cells were maintained at 37 °C using an environmental chamber.
To generate intensity colocalization plots, images were imported into Fiji as separate channels,31 and a line drawn across an individual cell. Plot profile was used to generate intensity of gray values across the ROI. Plots using data from both channels was generated using Prism version 8.1.0 (GraphPad Software, La Jolla California, USA). All experimental cell data was repeated twice (n = 2), with three technical replicates per condition used in the individual experiment.
Monitoring cellular metabolic activity
Human fibroblasts were plated at 10000 cells per well of a 96-well plate and treated with the various concentrations of Ir-PYR, Ir-CMYC (5, 20, 80, 50, 100 μM) or the highest volume of equivalent of DMSO only. As a measure of cell viability and metabolic activity, at 18 hours post compound addition culture medium was removed and replaced with culture medium containing 10 μg mL−1 resazurin diluted in DPBS (10% v/v). Metabolically active cells reduce resazurin (blue and non-fluorescent) to resorufin (pink and fluorescent). After a 4 hour incubation at 37 °C, fluorescence was determined using a Molecular Devices Spectramax Gemini EM plate reader (λex = 530 nm; λem = 580 nm).
DNA-binding experiments
MOPS buffer was prepared by dissolving MOPS (3-(N-morpholino)propanesulfonic acid, CAS[1132-61-2]), NaCl, and EDTA (ethylenediaminetetraacetic acid disodium salt dihydrate, CAS[6381-92-6]) (all obtained from Fisher and used as supplied) in deionised water (Elga Purelab Flex), adjusting the pH to 7.0 using a NaOH solution (pH of the buffer was determined using a Hanna Instruments pH210 microprocessor pH meter with a VWR simple junction universal combined pH/reference electrode) and making up the solution to 0.5 litre.A stock solution of fish sperm DNA was prepared by dissolving approximately 0.1 g of fish sperm DNA in 10 mL of the buffer. The resulting solution was dialysed (3.5 kDa MWCO, Visking, Medicell International Ltd) against 0.5 litres of buffer. Following dialysis, the DNA concentration was determined using UV-visible spectroscopy (Shimadzu UV-1800 spectrophotometer) using an extinction coefficient ε260nm = 12800 M(bp)−1 cm−1.32
Stock solutions containing 10 mM Ir-PYR or Ir-CMYC were made as follows. For Ir-PYR, 1.01 mg was weighed out and dissolved in 104.1 μL of DMSO. For Ir-CMYC, 0.6 mg was weighed out and dissolved in 31.6 μL of DMSO.
Circular dichroism titrations were carried out using an Applied Photophysics Chirascan spectrophotometer thermostated at 25 °C. First, 2500 μL of buffer was placed in a 1 cm pathlength quartz cuvette (Hellma) and a spectrum was recorded between 700 and 230 nm. Next, between 10 and 15 μL (typically 12.5 μL) of the stock solution of the iridium complex in DMSO was added to the buffer and a spectrum was recorded. Subsequently, 5 aliquots of 20 μL, 4 aliquots of 50 μL and 2 aliquots of 100 μL (i.e. a cumulative added volume of 500 μL) of the DNA stock solution were added and a spectrum was recorded after every addition.
DNA binding – data analysis results
Spectra and titrations were plotted in OriginLab Origin 2019b. The titration data were analysed globally for each complex using an in-house written version of the multiple independent binding sites model which also explicitly takes ligand concentrations into account.28 Exploratory data analysis showed that binding affinity and binding site size could not be determined separately from this data, as is usual for relatively weak binding. The binding site size (n) was therefore set to three base pairs for both complexes. The molar ellipticity of the free ligand (θm,free/mdeg) was restricted to 0, i.e. no CD signal in the absence of DNA while the background ellipticity at the wavelength of interest (θbackground,x nm) was an optimisable parameter to address the effects of noise on the data. The data analysis thus provided optimised values for the equilibrium constant (K), the changes in molar ellipticity upon binding at the wavelengths of interest (Δθm,x nm) and the background ellipticities at the wavelengths of interest (θbackground,x nm). These values are summarised in Table S1.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ministry of Defence, UK and Prof. Petra Oyston and Dr Gurmit Bahra (Defence Science Technology Laboratory, UK). We also thank Cardiff University for support. We thank the staff of the EPSRC Mass Spectrometry National Service (Swansea University). BBSRC (BB/S002774/1) and the Royal Society (RG110215) are also thanked for funding. We thank The Ministry of Higher Education and Scientific Research of Iraq (E. A. H. A.-W.).Notes and references
- G. von Heijne, J. Membr. Biol., 1990, 115, 195 CrossRef CAS PubMed; H. Owji, N. Nezafat, M. Negahdaripour and A. Hajiebrehami, Eur. J. Cell Biol., 2018, 97, 422 CrossRef PubMed.
- M. Marfori, A. Mynott, J. J. Ellis, A. M. Mehdi, N. F. W. Saunders, P. M. Curmi, J. K. Forwood, M. Boden and B. Kobe, Biochim. Biophys. Acta, 2011, 1813, 1562 CrossRef CAS PubMed.
- C. Dingwall and R. A. Laskey, Trends Biochem. Sci., 1991, 16, 478 CrossRef CAS PubMed; A. Lange, R. E. Mills, C. J. Lange, M. Stewart, S. E. Devine and A. H. Corbett, J. Biol. Chem., 2007, 282, 5101 CrossRef PubMed.
- J. P. S. Makkerh, C. Dingwall and R. A. Laskey, Curr. Biol., 1996, 6, 1025 CrossRef CAS PubMed.
- C. V. Dang and W. M. Lee, Mol. Cell. Biol., 1988, 8, 4048 CrossRef CAS PubMed.
- S. N. Finver, K. Nishikura, L. R. Finger, F. G. Haluska, J. Finan, P. C. Nowell and C. M. Croce, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 3052 CrossRef CAS PubMed.
- Z. E. Stine, Z. E. Walton, B. J. Altman, A. L. Hsieh and C. V. Dang, Cancer Discovery, 2015, 1024 CrossRef CAS PubMed; C. V. Dang, Cell, 2012, 149, 22 CrossRef PubMed.
- H. Chen, H. Lui and G. Qing, Signal Transduction Targeted Ther., 2018, 3, 5 CrossRef PubMed.
- S. Singh and A. O. Gramolini, BMC Cell Biol., 2009, 10, 47 CrossRef PubMed.
- D. Niopek, D. Benzinger, J. Roensch, T. Draebing, P. Wehler, R. Eils and B. di Ventura, Nat. Commun., 2014, 5, 4404 CrossRef CAS PubMed.
- M. Ray, R. Tang, Z. Jiang and V. M. Rotello, Bioconjugate Chem., 2015, 26, 1004 CrossRef CAS PubMed.
- J. A. Thomas, Chem. Soc. Rev., 2015, 44, 4494 RSC; E. Baggaley, J. A. Weinstein and J. A. G. Williams, Struct. Bonding, 2015, 165, 205 CrossRef CAS; A. Zamora, G. Vigueras, V. Rodriguez, M. Dolores Santana and J. Ruiz, Coord. Chem. Rev., 2018, 360, 34 CrossRef; S. Sreedharan, M. R. Gill, E. Garcia, H. K. Saeed, D. Robinson, A. Byrne, A. Cadby, T. E. Keyes, C. Smythe, P. Pellett, J. B. de la Serna and J. A. Thomas, J. Am. Chem. Soc., 2017, 139, 15907 CrossRef PubMed; F. L. Thorp-Greenwood, R. G. Balasingham and M. P. Coogan, J. Organomet. Chem., 2012, 714, 12 CrossRef; K. K. Lo, M. Louie, K. Sze and J. Lau, Inorg. Chem., 2008, 47, 602 CrossRef PubMed; E. Baggaley, M. R. Gill, N. H. Green, D. Turton, I. V. Sazanovich, S. W. Botchway, C. Smythe, J. W. Haycock and J. A. Weinstein, Angew. Chem., Int. Ed., 2014, 53, 3367 CrossRef PubMed; E. Baggaley, S. W. Botchway, J. W. Haycock, H. Morris, I. V. Sazanovich, J. A. G. Williams and J. A. Weinstein, Chem. Sci., 2014, 5, 879 RSC; K. K.-W. Lo, C.-K. Chung, T. K.-M. Lee, L.-H. Lui, K. H.-K. Tsang and N. Zhu, Inorg. Chem., 2003, 42(21), 6886 CrossRef PubMed; A. J. Amoroso, M. P. Coogan, J. E. Dunne, V. Fernández-Moreira, J. Hess, A. J. Hayes, D. Lloyd, C. Millet, S. J. A. Pope and C. Williams, Chem. Commun., 2007, 3066 RSC; E. E. Langdon-Jones, N. O. Symonds, S. E. Yates, A. J. Hayes, D. Lloyd, R. Williams, S. J. Coles, P. N. Horton and S. J. A. Pope, Inorg. Chem., 2014, 53, 3788 CrossRef PubMed; E. E. Langdon-Jones, C. F. Williams, A. J. Hayes, D. Lloyd, S. J. Coles, P. N. Horton, L. M. Groves and S. J. A. Pope, Eur. J. Inorg. Chem., 2017, 5279 CrossRef; E. E. Langdon-Jones, A. B. Jones, C. F. Williams, A. J. Hayes, D. Lloyd, H. J. Mottram and S. J. A. Pope, Eur. J. Inorg. Chem., 2016, 759 Search PubMed; R. G. Balasingham, F. L. Thorp-Greenwood, C. F. Williams, M. P. Coogan and S. J. A. Pope, Inorg. Chem., 2012, 51, 1419 CrossRef PubMed; C. Li, M. Yu, Y. Sun, Y. Wu, C. Huang and F. Li, J. Am. Chem. Soc., 2011, 133, 11231 CrossRef PubMed; P.-K. Lee, W. H.-T. Law, H.-W. Liu and K. K.-W. Lo, Inorg. Chem., 2011, 50, 8570 CrossRef PubMed; A. Jana, E. Baggaley, A. J. Amoroso and M. D. Ward, Chem. Commun., 2015, 51, 8833 RSC.
- G.-J. Yang, W. Wang, S. W. F. Mok, C. Wu, B. Y. K. Law, X.-M. Miao, K.-J. Wu, H.-J. Zhong, C.-Y. Wong, V. K. W. Wong, D.-L. Ma and C.-H. Leung, Angew. Chem., Int. Ed., 2018, 57, 13091 CrossRef CAS PubMed; K. Vellaisamy, G. Li, W. Weng, C.-H. Leung and D.-L. Ma, Chem. Sci., 2018, 9, 8171 RSC; S. Zhang, M. Hosaka, T. Yoshihara, K. Negishi, Y. Iida, S. Tobita and T. Takeuchi, Cancer Res., 2010, 70, 11 Search PubMed.
- M. P. Coogan and V. Fernandez-Moreira, Chem. Commun., 2014, 50, 384 RSC; K. K. Lo, T. K. M. Le, J. S. Y. Lo, W. L. Poon and S. H. Cheng, Inorg. Chem., 2008, 47, 2000 CrossRef CAS PubMed.
- N. Viola-Villegas, A. E. Rabideau, J. Cesnavicious, J. Zubieta and R. P. Doyle, Dalton Trans., 2014, 43, 4287 RSC.
- H. He, J. E. Morely, E. Silva-Lopez, B. Bottenus, M. Montajano, G. A. Fugate, B. Twamley and P. D. Benny, Bioconjugate Chem., 2009, 20, 78 CrossRef CAS PubMed; R. La Bella, E. Garcia-Garayoa, M. Bahler, P. Blauenstein, R. Schibli, P. Conrath, D. Tourwe and P. A. Schubiger, Bioconjugate Chem., 2002, 13, 599 CrossRef PubMed; V. Polyakov, V. Sharma, J. L. Dahlheimer, C. M. Pica, G. D. Luker and D. Picwnica-Worms, Bioconjugate Chem., 2000, 11, 762 CrossRef PubMed.
- U. Neugebauer, Y. Pellegrin, M. Devocelle, R. J. Forster, W. Signac, N. Moran and T. E. Keyes, Chem. Commun., 2008, 5307 RSC.
- L. Cosgrave, M. Devocelle, R. J. Forster and T. E. Keyes, Chem. Commun., 2010, 46, 103 RSC.
- A. Martin, A. Byrne, C. S. Burke, R. J. Forster and T. E. Keyes, J. Am. Chem. Soc., 2014, 136, 15300 CrossRef CAS PubMed.
- L. Blackmore, R. Moriarty, C. Dolan, K. Adamson, R. J. Forster, M. Devocelle and T. E. Keyes, Chem. Commun., 2013, 49, 2658 RSC.
- C. S. Burke, A. Byrne and T. E. Keyes, J. Am. Chem. Soc., 2018, 140, 6945 CrossRef CAS PubMed.
- L. Marcelis, S. Kajou, J. Ghesquiere, G. Fettweis, I. Coupienne, R. Lartia, M. Surin, E. Defrancq, J. Piette, C. Moucheron and A. Kirsch-De Mesmaeker, Eur. J. Inorg. Chem., 2016, 2902 CrossRef CAS.
- S. A. Archer, A. Raza, F. Droge, C. Robertson, A. J. Auty, D. Chekulaev, J. A. Weinstein, T. Keane, A. J. H. M. Meijer, J. W. Haycock, S. MacNeil and J. A. Thomas, Chem. Sci., 2019, 10, 3502 RSC; O. J. Stacey and S. J. A. Pope, RSC Adv., 2013, 3, 25550 RSC; A. Colombo, M. Fontani, C. Dragometti, D. Roberto, J. A. G. Williams, R. Scotto di Perrotolo, F. Casagrande, S. Barozzi and S. Polo, Chem.–Eur. J., 2019, 25, 7948 CrossRef CAS PubMed.
- (a) E. E. Langdon-Jones, A. J. Hallett, J. D. Routledge, D. A. Crole, B. D. Ward, J. A. Platts and S. J. A. Pope, Inorg. Chem., 2013, 52, 448 CrossRef CAS PubMed; (b) K. A. Phillips, T. M. Stonelake, P. N. Horton, S. J. Coles, A. J. Hallett, S. P. O'Kell, J. M. Beames and S. J. A. Pope, J. Organomet. Chem., 2019, 893, 11 CrossRef CAS.
- R. A. Smith, E. C. Stokes, E. E. Langdon-Jones, J. A. Platts, B. M. Kariuki, A. J. Hallett and S. J. A. Pope, Dalton Trans., 2013, 42, 10347 RSC.
- K. A. Phillips, T. M. Stonelake, K. Chen, Y. Hou, J. Zhao, S. J. Coles, P. N. Horton, S. J. Keane, E. C. Stokes, I. A. Fallis, A. J. Hallett, S. O'Kell, J. M. Beames and S. J. A. Pope, Chem.–Eur. J., 2018, 24, 8577 CrossRef CAS PubMed.
- J. Yang, J.-X. Zhao, Q. Cao, L. Hao, D. Zhou, Z. Gan, L.-N. Ji and Z.-W. Mao, ACS Appl. Mater. Interfaces, 2017, 9, 13900 CrossRef CAS PubMed; M. Tian, J. Li, S. Zhang, L. Guo, X. He, D. Kong, H. Zhang and Z. Liu, Chem. Commun., 2017, 53, 12810 RSC; J. J. Li, M. Tian, Z. Tian, S. Zhang, C. Yan, C. Shao and Z. Liu, Inorg. Chem., 2018, 57, 1705 CrossRef PubMed; M.-H. Chen, F.-X. Wang, J.-J. Cao, C.-P. Tan, L.-N. Ji and Z.-W. Mao, ACS Appl. Mater. Interfaces, 2017, 9, 13304 CrossRef PubMed; L. He, Y. Li, C.-P. Tan, R.-R. Ye, M.-H. Chen, J.-J. Cao, L.-N. Ji and Z.-W. Mao, Chem. Sci., 2015, 6, 5409 RSC.
- L. Hahn, N. J. Buurma and L. H. Gade, Chem.–Eur. J., 2016, 22, 6314 CrossRef CAS PubMed.
- A. D. Edelstein, M. A. Tsuchida, N. Amodaj, H. Pinkard, R. D. Vale and N. Stuurman, Journal of Biological Methods, 2014, 1, e11 CrossRef PubMed.
- (a) E. Lloyd-Evans, A. J. Morgan, X. He, D. A. Smith, E. Elliot-Smith, D. J. Sillence, G. C. Churchill, E. H. Schuchman, A. Galione and F. M. Platt, Nat. Med., 2008, 14, 1247 CrossRef CAS PubMed; (b) J. H. Lee, M. K. McBrayer, D. M. Wolfe, L. J. Haslett, A. Kumar, Y. Sato, P. P. Lie, P. Mohan, E. E. Coffey, U. Kompella, C. H. Mitchell, E. Lloyd-Evans and R. A. Nixon, Cell Rep., 2015, 12, 1430 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2019, 9, 676 CrossRef PubMed.
- L. A. Mullice, R. H. Laye, L. P. Harding, N. J. Buurma and S. J. A. Pope, New J. Chem., 2008, 32, 2140 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental synthetic procedures, HPLC data, additional photophysical data and DNA binding data. See DOI: 10.1039/c9sc05568a |
| ‡ Equal contribution to the work. |
| This journal is © The Royal Society of Chemistry 2020 |