
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymes in a golden cage†
Yael
Baruch-Shpigler
and
David
Avnir
 *
*
Institute of Chemistry, The Center for Nanoscience and Nanotechnology, The Hebrew University of Jerusalem, Jerusalem 9190401, Israel. E-mail: David.Avnir@mail.huji.ac.il
First published on 28th March 2020
Abstract
We describe a general method for the entrapment of enzymes within bulk metallic gold. This is a new approach for the immobilization of enzymes on metals, which is commonly carried out by 2D adsorption or covalent biding, that is, the enzyme is in contact with the metal at a specific contact zone of the enzyme, while most of the rest of it remains exposed to the environment. The 3D metallic encaging of the enzymes is quite different: the enzyme is in contact with the metallic cage walls all around it and is well protected inside. The porous nature of the metallic matrix enables substrate molecules to diffuse inside, reach the active site, and let product molecules diffuse out. The generality of the approach was proven by the successful entrapment of five enzymes representing different classes and different bio- and medical applications: L-asparaginase (Asp), collagenase, horseradish peroxidase (HRP), laccase and glucose oxidase (GOx). GOx–gold conjugates have been of particular interest in the literature. The main challenge we had to solve was how to keep the enzyme active in the process of gold-synthesis from its cation – this required careful tailoring of reaction conditions, which are detailed in the paper. The gold entrapped enzymes gain thermal stability and protectability against harsh conditions. For instance, we could keep Asp alive at the extreme pH of 13, which normally kills the enzyme instantly. The entrapped enzymes obey the Michaelis–Menten kinetics, and activation energies were determined. Good recyclability for eight cycles was found. Multi-enzymatic reactions by combinations of the off-the-shelf bioactive enzyme@gold powders are possible, as demonstrated for the classical detection of GOx activity with HRP. Detailed material characterization and proposed mechanisms for the 3D protectability of the enzymes are provided. The new enzyme immobilization method is of wide potential uses in medicine, biotechnology, bio-fuel cells and enzymatic (electro)sensing applications.
Introduction
Immobilized enzymes are widely used as biosensors,1 as catalysts in organic synthesis,2 in biotechnological processes,3 in bio-fuel cells,4 in therapeutics,5 and in many other industrial processes.6 In general, immobilizations have been carried out in 0D architectures such nanoparticles,7 in 1D architectures such as nanofibers,8 in the common 2D architectures of adsorption or covalent attachment to surfaces,9 and in 3D architectures in which the enzyme is entrapped physically or chemically within a porous matrix.10,11 The latter has been of particular interest, mainly because 3D immobilization provides high surface areas in small volumes, because the internal regions of the immobilizing material provides efficient protection against denaturing processes – heat, extreme pH, destructive chemicals – and because this immobilization reduces the susceptibility of detachment from the support.12,13 Of the three large family of materials – inorganic, (bio)organic and metals – the largest libraries of 3D immobilization matrices methodologies belong to the first two, examples being metal-oxide matrices such as sol–gel family of materials,10,13–15 and organic matrices such as cross-linked polymers.16 The most common metal used in the immobilization context is gold, because of its inertness to most biological processes and its low toxicity,17 which have led to the use of gold in diagnostics and many other therapeutic fields.18 However, these studies employed mainly 2D architectures where enzymes have been anchored by physisorption, chemisorption or covalent bonding to the gold surface.19 Significantly less research has been carried out on the use of gold (or any other metal) for 3D immobilization of enzymes, the topic of this report. Relevant studies we are aware of are the dealloying of gold alloys followed by immersing the resulting porous gold in an enzyme solution;20,21 and potentiostatic deposition of gold.22 A closer look at these studies reveals that, in fact, even in these 3D architectures, the enzyme molecules are anchored to the surface of the gold, such that one side of it touches the metal, and the counter-side is exposed to the environment. The concept we wish to advance in this report is different: immobilization of the enzyme molecules within metallic gold cages of bulk gold, that is, an immobilization configuration where the protein molecule is in touch with the metal all around it, while still remaining accessible to reaction (Scheme 1, top). The possibility of exploring this direction has emerged from the intensive development of the materials methodology of molecular doping of metals.23,24 A wide variety of molecules including organometallic complexes,25 drug molecules,26 dyes,27 polymers,28 nanoparticles29 and more30 have been incorporated into various metals, including copper, silver, gold, platinum, gallium and more. The resulting molecule@metal materials have been utilized for a host of activities ranging from catalysis to drug release, and in many instances synergistic and improved activities were recorded. The mechanism of entrapment – described below (Results and discussion) – basically involves carefully tailored conditions for a one-pot reduction of the metal cation in the presence of the molecule to be entrapped. We were motivated to embark on the development of a general methodology for entrapping any desired enzyme in gold by a preliminary observation that it is possible to entrap by this materials methodology acid-phosphatase in silver and gold, keeping it alive.31 The generalization task was quite non-trivial because the challenge has been to find the right conditions where the enzyme survives the chemistry involved in the chemical encaging process while still retaining the needed gold porosity for the biochemical activity; this aspect is detailed below.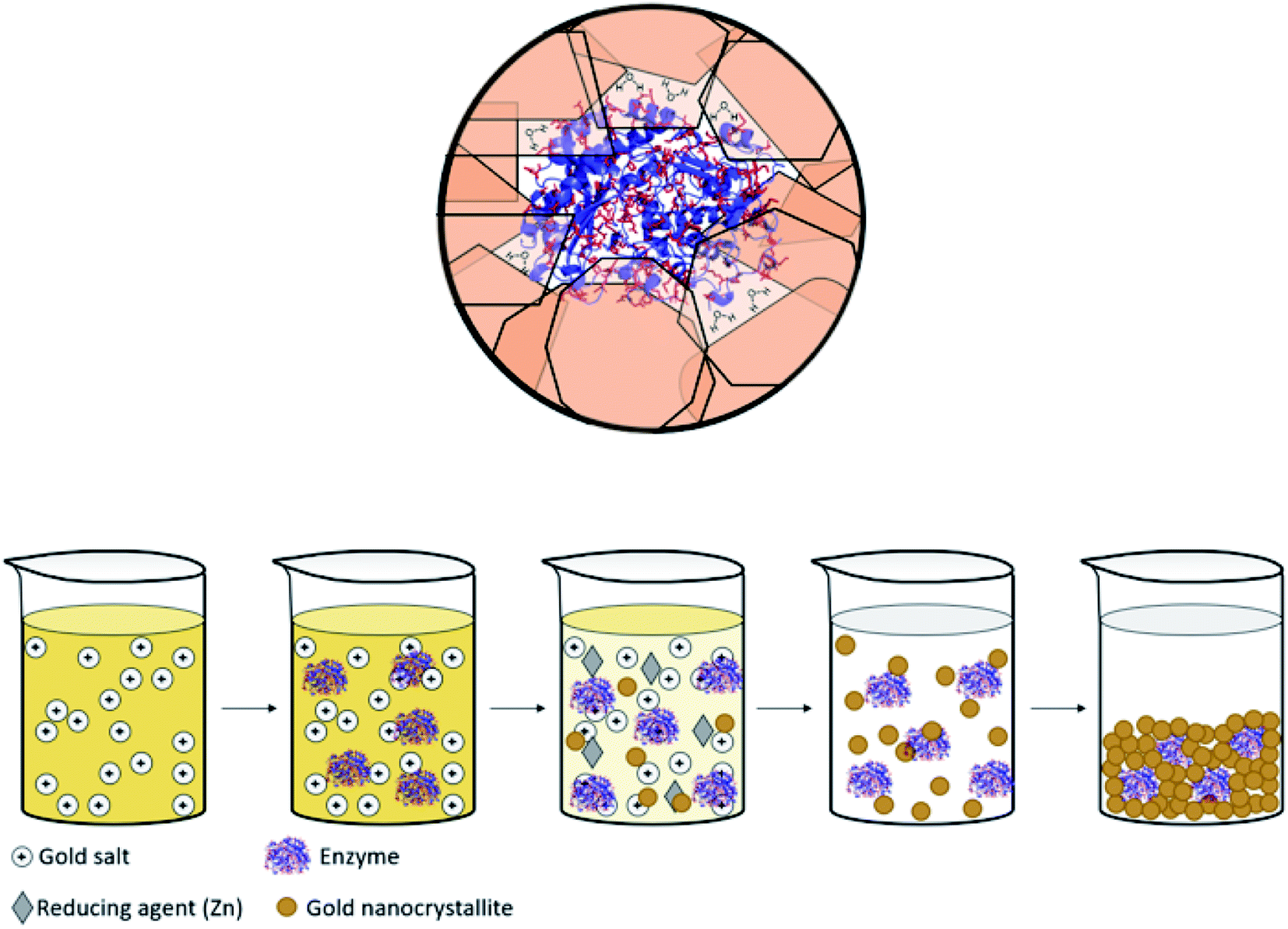 | ||
| Scheme 1 (Top) Entrapped enzyme (glucose oxidase, GOx) within a golden cage. In red – the surface amino acids of GOx which contain gold binding moieties (thiols, amines, imines and carboxylate anions, taken from PDB file 1CF3). (Bottom) The entrapment process – see also Scheme 2. | ||
The enzymes selected for this study are representatives of some prominent enzymatic classes (ESI Fig. S1†): glucose oxidase (GOx), an oxido-reductase which catalyzes the oxidation of D-glucose to D-gluconolactone which is extensively used in a variety of areas that extend from bio-diagnostics (glucose sensors) to food processing and preservatives, all through to enzymatic biofuel cells;4,32,33 collagenase (Col), a hydrolase and a metallopeptidase that breaks down the peptide bonds in collagen, used in therapeutics, for instance in wound healing (wound dressing and debridement)34 and in the treatment of Dupuytren's disease;35L-asparaginase (Asp), which catalyzes the hydrolysis of L-asparagine to L-aspartic acid and which is used in the treatment of acute lymphoblastic leukemia and lymphosarcoma,36 and in food processing;37 horseradish peroxidase (HRP) which utilizes hydrogen peroxide to catalyze the oxidation of many organic compounds, widely used in organic synthesis, coupled enzymatic assays, treatment of waste water38 and sensing of hydrogen peroxide;38 and laccase (Lac), which catalyzes the one-electron oxidation of mostly phenols, and which is used in a variety of processes such as textile bleaching, synthesis of antibiotics, and food processing of beer, wine and juice.39 In the context of conjunction of these enzymes with gold, GOx–Au conjugates were used for biosensing of glucose40 and for fuel-cell applications,41 in which the immobilization was carried out mainly by adsorption,40 by covalent binding,42 or by electrochemical adsorption.33L-Asp was reported to be used in combination with gold for targeting of cancer cells.43
Next is the description of the general procedure for the 3D encaging of enzymes in gold, followed by description of the bioactivity of the resulting enzymes@gold, and of the special features of these entrapments such as enhanced thermal stability and activity under extreme pH conditions.
Materials and methods
Chemicals, enzymes, reagents
Sodium tetrachloroaurate(III) dihydrate was purchased from Alfa Aesar. Zinc (granular, 20–30 mesh, ACS reagent, ≥99.8%) were purchased from Sigma Aldrich. Enzymes, their substrates and analytical reagents: glucose oxidase from A. niger (135 U mg−1) and o-dianisidine were purchased from Sigma Aldrich. Glucose was purchased from Honeywell Riedel-de Haën Research Chemicals. L-Asparaginase from E. coli (∼250 U mg−1) was purchased from Prospec Chemicals. L-asparagine was purchased from Sigma Aldrich. Nessler's reagent (K2HgI4) was purchased from Fisher Chemical. Collagenase Type I from C. histolyticum (0.25–1.0 U mg−1) was purchased from Sigma Aldrich. N-(3-[2-furyl]acryloyl)-Leu-Gly-Pro-Ala (FALGPA) was purchased from Apollo Scientific Ltd. Peroxidase from horseradish (∼200 U mg−1), hydrogen peroxide were purchased from Sigma Aldrich. Pyrogallol was purchased from TCI. Laccase from T. versicolor (∼1 U mg−1) and syringaldazine were purchased from Sigma Aldrich. Cetyltrimethylammonium bromide (CTAB) was purchased from Acros organics. Buffers (all of the weight values are for a volume of 500 mL of buffer): 50 mM sodium acetate buffer (pH = 5.1, 35 °C) was prepared for glucose oxidase and horseradish peroxidase by dissolving 2.0509 g of sodium acetate anhydrous in distilled water and adjusting the pH to 5.1 with 1 M HCl solution. 50 mM Tris–HCl buffer (pH 7.5, 25 °C) with 10 mM calcium chloride and 400 mM sodium chloride was prepared for collagenase by dissolving 3.0285 g of tris(hydroxymethyl)aminomethane, 0.5549 g of calcium chloride and 11.688 g of sodium chloride in distilled water and adjusting the pH to 7.5 with 1M HCl solution. 50 mM Tris–HCl buffer (pH 8.6, 37 °C) was prepared for asparaginase by dissolving 3.0285 g of tris(hydroxymethyl)aminomethane in distilled water and adjusting the pH to 8.6 with 1M HCl solution. 100 mM potassium phosphate buffer (pH 6.5, 30 °C) was prepared for the laccase assay, by dissolving 6.8045 g of potassium dihydrogen phosphate in distilled water and adjusting the pH to 6.5 with 1 M NaOH.Entrapment procedures (Scheme 1)
Bioactivity assays (Fig. S2, ESI†)
Additional activity characterizations
Solution method. The GOx@Au composite powder was placed in a polystyrene cuvette, along with 0.1 mL of 50 mM sodium acetate buffer solution. The cuvette was settled in a water bath, heated to the desired temperature, for 10 minutes. Then, 0.1 mL of horseradish peroxidase (∼200 U mg−1, 0.3 mg mL−1, in TDW), 2.4 mL of 0.21 mM o-dianisidine solution and 0.5 mL of 89.5 mM glucose solution were added to the cuvette and the activity of the composite was measured as previously described.
Dry powder method. Either free GOx solution, or the dry GOx@Au powder, were heated to the desired temperature (25 °C, 40 °C, 70 °C, 90 °C and 100 °C) for 10 minutes and their consequent activity was measured as previously described. Both methods were compared to the stability in solution. Similar procedures were applied for Col@Au.
Material characterizations
Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy analysis (EDAX) were carried out on a Sirion (FEI) high-resolution (HR) SEM instrument. Powder X-ray Diffraction (XRD) measurements were carried out using Phillips diffractometer (CuKα 1 (1.5406 Å) with a step scan mode 0.02 s−1). Specific surface areas were calculated from N2 adsorption/desorption isotherms obtained with a Micromeritics ASAP 2020 surface area analyzer, using the BET equation. Pore size distribution was determined from the BJH equation. UV-visible absorption spectroscopy was carried out with JASCO V-630 spectrophotometer.Results and discussion
The entrapment of the enzymes in gold
The general methodology of molecular doping of metals involves reduction of the metal cation in the presence of the molecules to be entrapped. Adapting this approach for the entrapment of enzymes, had posed a number of challenges: carrying out the reduction of the metal cation under conditions that do not denature the enzyme, that is that neither the reducing agent or its oxidized product will be harmful for the enzyme; obtaining the desired porosity of the entrapping metal such as to allow free in-diffusion of substrate molecules to the entrapped enzyme and out-diffusion of product molecules; having the final desired architecture in which the enzyme molecules reside within the gold matrix in porous metallic cages, on one hand, accessible to reaction and on the other hand, without being washed away into the solution; and avoiding phase separation resulting in separate metallic gold and the enzyme in solution. Addressing all of these issues required careful tailoring of all of the reaction parameters, until an optimal general procedure has been identified. The parameters included the reducing agent, the buffers, the pH, the concentrations and gold loading, and the time duration of the reactions. For instance, potassium phosphate buffer, which is commonly used for the Lac assay, stabilized the gold nanocrystallites which are formed in the reduction process, and hampered the matrix formation (see Experimental details for the optimal buffers). As for loading, using too much enzyme resulted in only partial entrapment, while the addition of too little enzyme resulted in a composite that was less active than optimal. An example of a reducing agent that had to be discarded is sodium hypophosphite which is almost the standard reducing agent in molecular entrapments in metals,23,24 because of the too-low pH induced by its oxidized form, phosphoric acid. A good reducing agent was found to be metallic zinc, a metal that is not only bio-friendly, but also an integral component in many metallo-enzymes.49 The sequence of reactions of the entrapment process involves, first, a hydrolysis equilibrium of the gold salt upon its dissolution| AuCl4− + 2H2O ⇌ AuCl3OH− + H3O+ + Cl−, | (1) |
| 2AuCl4− + 3Zn(s) → 2Au + 3Zn2+ + 8Cl− | (2) |
| 2AuCl3OH− + 3Zn(s) → 2Au(s) + 2Zn2+ + Zn(OH)2 + 6Cl−. | (3) |
Addition of sodium hydroxide solution to the entrapment mixture was crucial, because it was found that under the acidic conditions formed upon the hydrolysis (eqn (1), pH ∼ 2 (ref. 50 and 51)), entrapment does not occur.
The mechanism of entrapment is the following (Scheme 2): at time zero – mixing of the components – the anionic AuCl3OH− forms an adsorption equilibrium with the positively charged amino acids residues on the outer surface of the protein (namely lysine, arginine and histidine). The reducing agent is then acting both on the dissolved and the adsorbed gold ions, which are in very large excess over the protein. The former produces gold nanoparticles in the solution and the adsorbed ones are converted to gold atoms, some of which leave the protein and contribute to the growth of the gold nanocrystals in solution which continue to precipitate and thus entrap, while others form growth nuclei of gold on the protein surface. Continued reduction and formation of the gold nanocrystallites is followed by their aggregation, while adsorption–desorption of the enzyme molecules on the forming gold takes place in parallel, not only through the well-known thiol–gold bonding, but also interactions between amines and carboxylate groups on the surface of the proteins (Scheme 1 and Fig. S3†) – and the gold.52,53 Entrapment occurs if the residence time of the adsorbed protein molecules on the surface of the formed gold, is longer than the reduction–aggregation process which forms additional gold nanoparticles. If these are the conditions, then the forming gold nanocrystallites capture the adsorbed enzyme molecules, forming nanocages which surround them all around, while developing to the 3D porous gold structure of metallic gold laced with enzyme molecules.
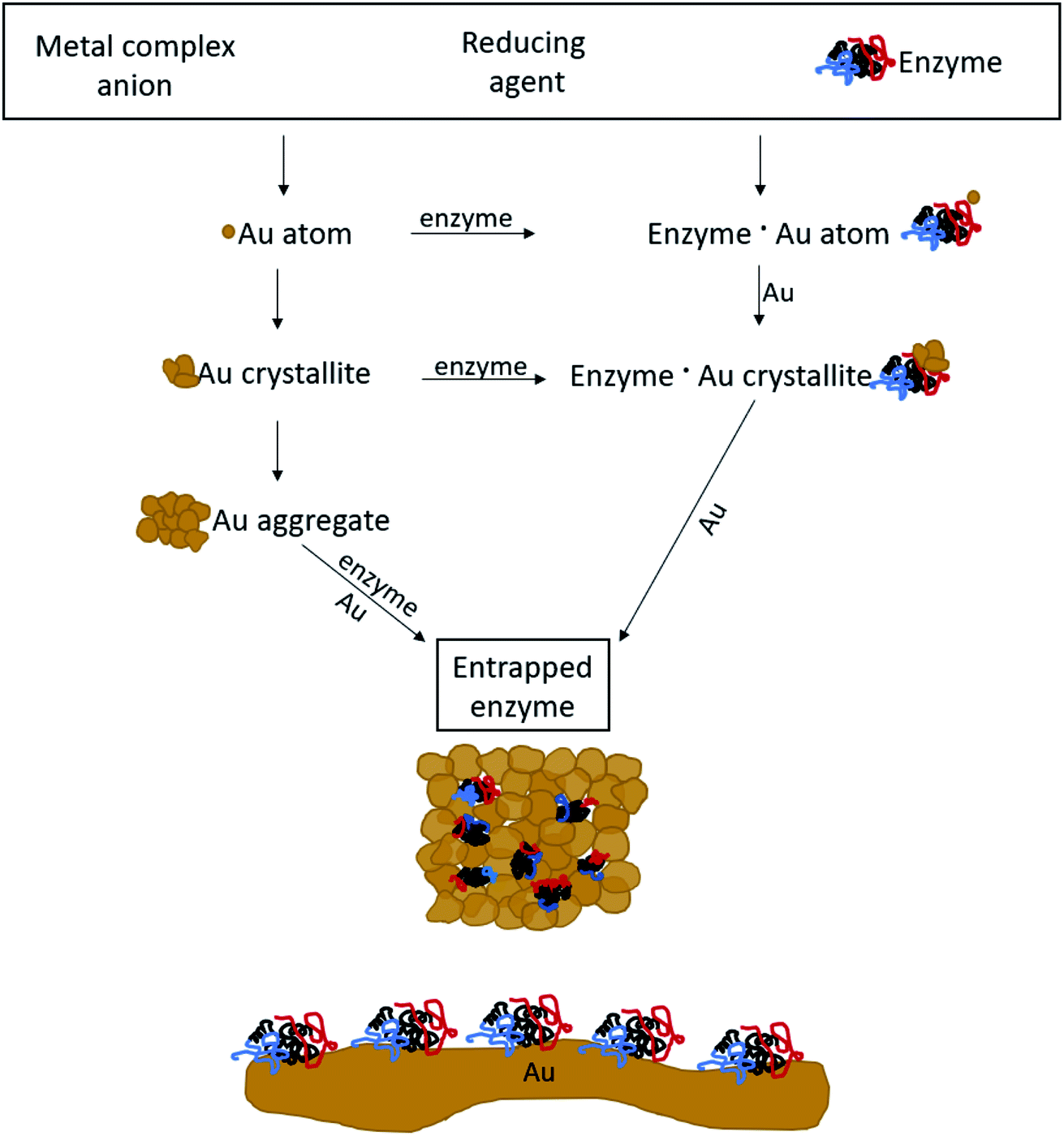 | ||
| Scheme 2 The mechanism of entrapment – see text for details. Bottom: 2D adsorption, a completely different architecture compared with the 3D entrapment. | ||
It is crucial to highlight the difference between adsorption or covalent bonding – two-dimensional processes (Scheme 2, bottom) – and entrapment, a three-dimensional process. Adsorption of proteins occurs through specific sites on the surface of the protein, where the energy gain of interaction between the relevant amino acid residues and the surface is maximal. Since adsorption is an equilibrium process, that interaction does not occur upon first collision between the protein and the surface, but dynamic adsorption/desorption takes place and the longest residence time on the surface is that of the maximal energy gain. In covalent anchoring the protein is placed at distance from the surface through the specific anchoring points, and if the spacer is flexible enough, the protein can sway and touch the surface. 3D encaging of the type described here, is very different: the protein is in contact with the metal all around its surface. Amino acid residues which are capable of interaction with gold abound everywhere on the protein surface with a variety of amino acid side chains (Scheme 1, top) and the protein is therefore in contact with the metallic cage all around it. As is evident from the results below, while the entrance to some of the actives sites of the entrapped enzyme molecules maybe blocked by this process, most remain accessible to reaction by being open to the pore network. The difference between 2D immobilization and 3D immobilization shows also in other properties: first, adsorbed proteins are easily washed away by solvents, while the entrapped enzymes do not leach out. Second, while in 2D architectures most the enzyme molecule is still exposed to the surrounding environment and therefore is sensitive to denaturing processes, the 3D caging enhances the stability to harsh chemicals and to heat, as we shall see below.
Properties of the gold biomaterials
HR-SEM imaging (GOx@Au – Fig. 1a; see more images in Fig. S4, ESI†) reveals that the resulting materials are hierarchically porous, built from elementary nanocrystals of gold. The average particle size of the gold nanocrystallites is influenced by the specific interactions of the outer surface of the protein with the growing gold particle. The smallest was found for collagenase – 8 nm – and the largest for laccase – 45 nm (Table S1, ESI†), as determined from the XRD analyses (Fig. 1b and S5†) by applying Scherrer's equation. As also seen in Fig. 1b, the typical fcc crystal packing of gold is retained upon entrapment. The porosity is nicely revealed by the N2 adsorption–desorption isotherms of GOx@Au (Fig. 1c) which fit a Type II classification54 isotherm, with a slight hysteresis at the higher pressures. Such isotherms are indicative of a wide pore size distribution, from nanopores (the sharp rise at the lower pressures) to meso- and macropores (the shape and hysteresis at the higher pressures). This is indeed seen in the pore size distribution in Fig. 1d: the population of pores is of wide distribution which originates from the aggregation of the metal nanocrystals creating interstitial pores and cages within which the enzymes are entrapped. The reproducibility of these parameters is around 10–12%. That wide distribution – from nanopores to tens of nm pores – assures, on one hand the efficient encaging of the enzymes, and on the other hand the needed routes which lead the substrate molecules to the active site, and the product molecules out to the surrounding solution. Fig. 1e – the BET analysis of the adsorption isotherm – provides more structural information: first, it shows an excellent compliance with the BET model (R = 0.999). The BET equation is known to be sensitive to chemical and geometrical heterogeneities and when these occur, then the basic equation is not obeyed, and instead one needs to resort, for instance, to a distribution analysis of local BET isotherms.55 This would happen if protein aggregates and gold would have formed different distinct zones (which are not seen by HR microscopy). The excellent compliance we found, suggests therefore uniformity. Second, it provides the nitrogen-accessible surface area, which stands at 30 m2 g−1 (compared with 23 m2 g−1 for pure gold prepared by the same method). This is an intermediate surface area, which again indicated a pore-size distribution of wide range. One should take into account that because of the convoluted geometry of the pores, the nitrogen surface area is not the surface area which is accessible to the enzyme substrates – these larger molecules “experience” a smaller effective surface area. Third, the BET analysis provides also the C-parameter which relates to the strength of the interaction with the surface. Here we obtained evidence for the existence of a dopant with many polar groups on the protein surface: while the C-parameter of the pure gold is 72 (mainly van-der-Waals N2–Au interactions), for GOx@Au it reaches 112, that is, the average surface is more polar. More direct evidence to the existence of the dopant is provided by EDX analysis (Fig. 1f): it clearly shows the gold matrix with evidence of the entrapped enzyme (the penetration depth is 200 nm) identified by the carbon, nitrogen and oxygen peaks (see Fig. S5† for additional EDX spectra).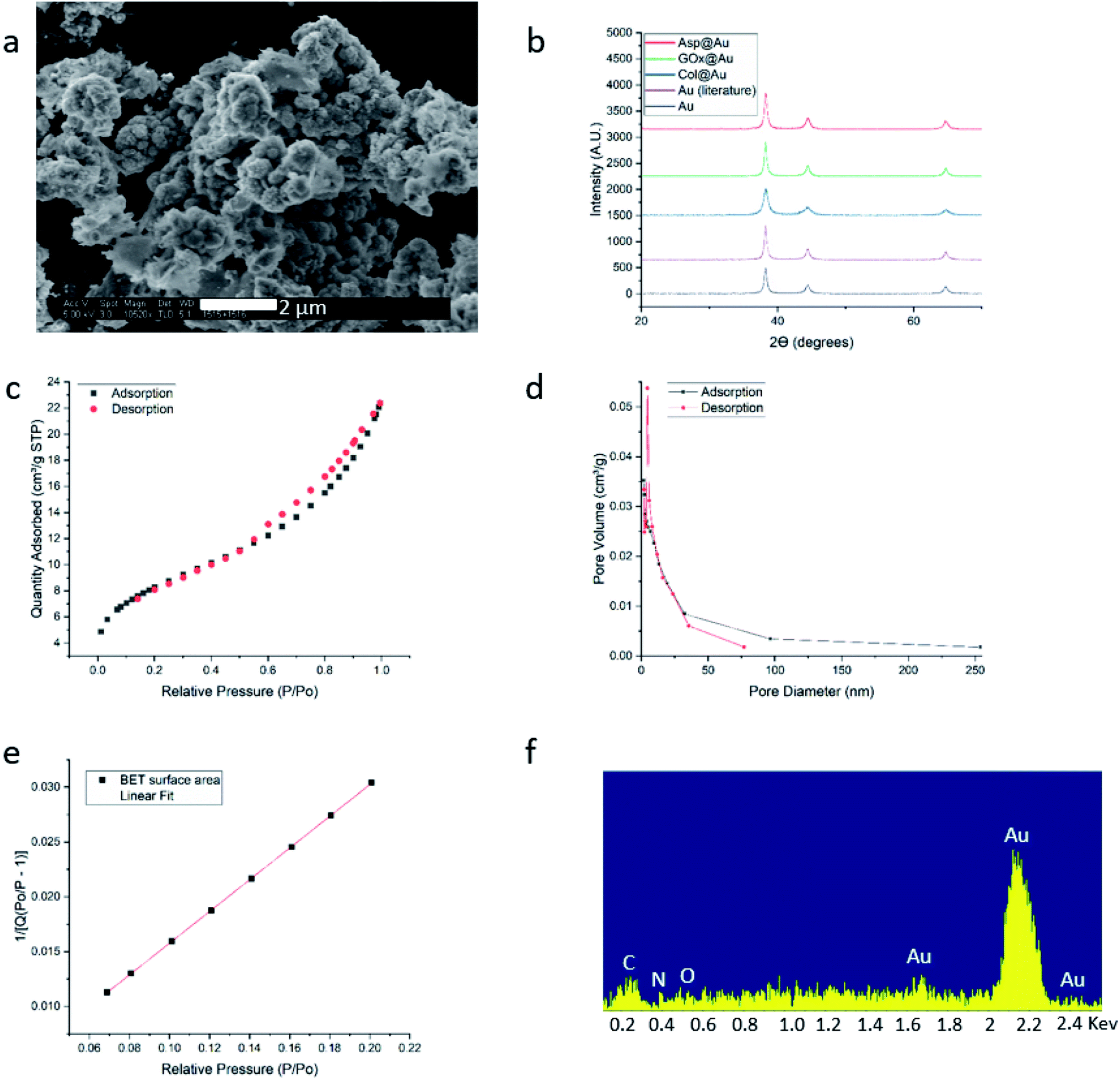 | ||
| Fig. 1 Analytical results for GOx@Au (a) HR-SEM image. For additional images of this and the other enzymes@Au, see Fig. S1, ESI.† (b) XRD graphs of GOx@Au compared to Asp@Au, to Col@Au, to home-made pure gold by the same method, and to literature gold (ICDD – http://www.icdd.com/ (accessed September 11, 2019)). (c) Adsorption–desorption isotherms of nitrogen, the type of which indicates mesoporosity. (d) The pore size distribution. (e) Compliance of the adsorption branch of (c) with the BET model (R = 0.999) and determination of the C-constant. (f) EDX analysis. For additional XRD and EDX spectra, see Fig. S5, ESI.† | ||
The enzymatic activities
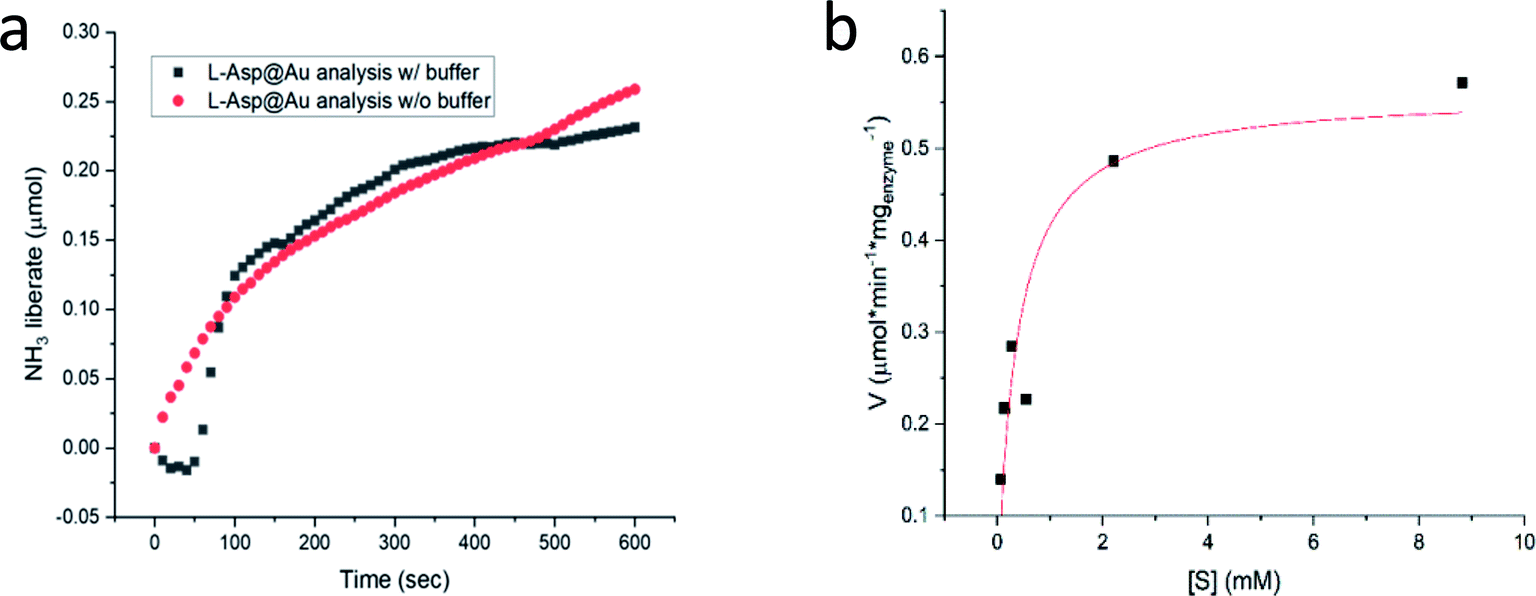
Following the carefully developed procedure described in the previous section and in the Experimental details section, the five enzymes selected for this study – glucose oxidase, collagenase, L-asparaginase, horseradish peroxidase and laccase – were successfully entrapped within gold matrices, biocatalyzing the reactions shown in Fig. S1 (ESI†). Fig. 2a shows the kinetics profile of glucose oxidase entrapped within gold (GOx@Au) confirming that the active site of the encaged enzyme is accessible through the pores network to glucose and oxygen, and that the hydrogen peroxide freely evolves from the matrix (as detected by the formation of the o-dianisidine). It is also seen that the bioactivity is due only to the entrapped enzyme and that no reaction was detected in the supernatant solution. A second blank test carried out here and for all other enzymes, was to make sure that the catalysis is not due to the gold itself, as indeed shown in the figure (Fig. 2a). Also shown in that figure is the inherent difference between adsorption and entrapment, explained in detail in the previous section, leading to very low activity of the former (about 7% of the entrapped architecture activity): under similar conditions, the adsorbed enzyme is largely washed away, while entrapment created the conditions to have the same amount of enzyme held tight by the gold without the need for covalent linkers. The activity of the entrapped enzyme follows the Michaelis–Menten model (Fig. 2b), providing Vmax and KM values of 1.81 U mg−1 (μmol min−1 mg−1) and 0.47 mM, respectively. The entrapment results in lower Vmax and KM values compared to solution (24.2 U mg−1; 21.7) (Fig. S6, ESI†): diffusional effects on Vmax and KM are a common feature of immobilized enzymes56–58 but this “cost” is paid for the many other advantages of 3D immobilizations, such as our next observation – the enhanced thermal stability.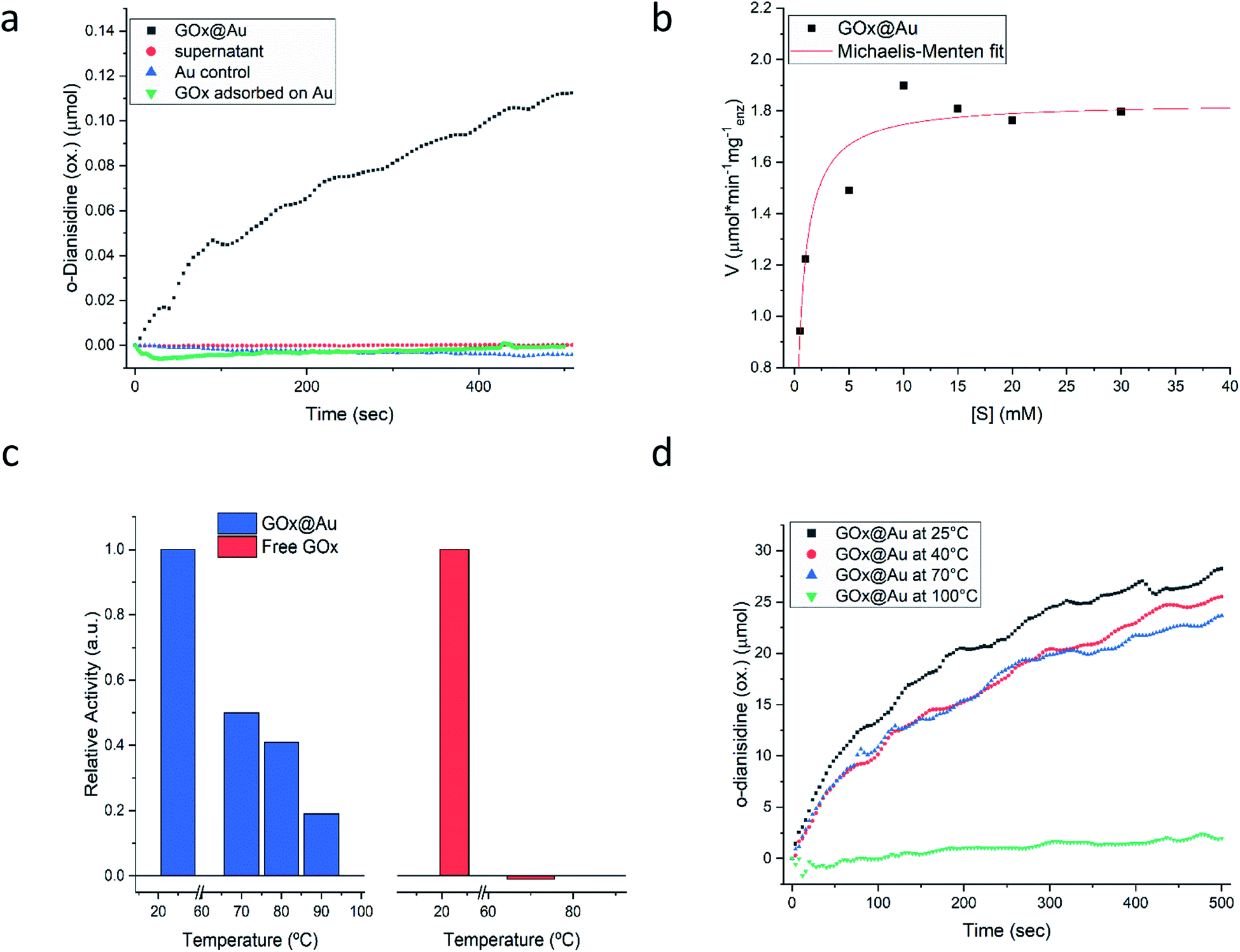 | ||
| Fig. 2 Glucose oxidase entrapped within gold (GOx@Au). (a) Kinetics of glucose oxidation, with three blank tests. (b) Fitting to the Michaelis–Menten model. (c) The thermal stability of the enzyme entrapped in gold compared to free in solution. (d) The activity after heating the dry powder of GOx@Au to the indicated temperature. | ||
As seen in Fig. 2c, free GOx activity drops to zero at 70 °C in solution (reflecting its reported irreversible thermal denaturation temperature is ∼56 °C59). GOx@Au, on the other hand, still retains activity at that temperature, and even at 90 °C about 20% of the room temperature activity is observed. We also set up to test the thermal stability of the dry GOx@Au powder. This experiment is relevant from two points of view: first, for evaluating the potential of the entrapment procedure as a long-term room-temperature storage option, replacing the costly current procedures of low-temperatures storage; and second, for evaluating the stability for applications of GOx–gold systems which require the dry form. It is seen – Fig. 2d – that heating the powder even to 70 °C, a temperature that scorches the free enzyme powder, leaves the entrapped enzyme alive; one needs to heat to as high as 100 °C, in order to kill the entrapped enzyme (Fig. 2d). The thermal stabilities of GOx@Au in solution or as a powder comprise a direct proof of the tight 3D cage in which the enzyme molecules reside: denaturing processes usually involve large conformational motions and rearrangements that need ample space and flexibility of the environment (water; polymeric support; exposure in 2D anchoring) in order to take place; the tight rigid metallic encompassing cage dramatically hinders denaturing conformational movements.
Is the developed entrapment procedure general, and is so the thermal protectability? The next enzyme, collagenase, was successfully entrapped as well (see Fig. S7a, ESI†), and it was found that the enhancement of the thermal stability of the of Col@Au even exceeds that of GOx@Au: the activity of the free enzyme in solution steadily declines upon heating and is totally denatured at 70 °C (Fig. 3a). However, when entrapped, its activity increases with temperature up to 60–70 °C, and even at 90 °C the activity is higher than at 25 °C (Fig. 3a), a behavior otherwise found only in thermophilic enzymes.60 This thermal stability has suggested that it may be possible to check if the temperature accelerates the activity in the classical Arrhenius-type way over a temperatures range. We found that this is indeed the case: the Arrhenius plot of the entrapped enzyme is shown in Fig. 3b, and compared to the Arrhenius plot of the free enzyme (Fig. 3c): the derived activation energies Col@Au and for the free collagenase are 21.5 kJ mol−1 and 28.9 kJ mol−1, respectively. The decrease in activation energy upon entrapment was expected, as it is well documented that various types of immobilization cause such a decrease, usually attributed to conformational changes upon interaction with the support.61 A large decrease is also observed for the Arrhenius exponential pre-factor, A, from 149 mM s−1 of the free enzyme in solution to 10.2 mM s−1 when entrapped. We recall that A is related to the number of collisions (fruitful or not) per second occurring with the proper orientation to react: the caging and the diffusional restrictions in the entrapped case, reduce this number. Dry powder thermal stability was found in this case as well and is described in the ESI (Fig. S7b†) – increase of activity with the treatment temperature was observed, apparently the result of some porosity opening by coalescence. The dry powder stability enables safe room temperature handling of the enzyme without loss of activity: while Col in solution becomes totally inactive after three days, Col@Au maintained over 90% of its activity.
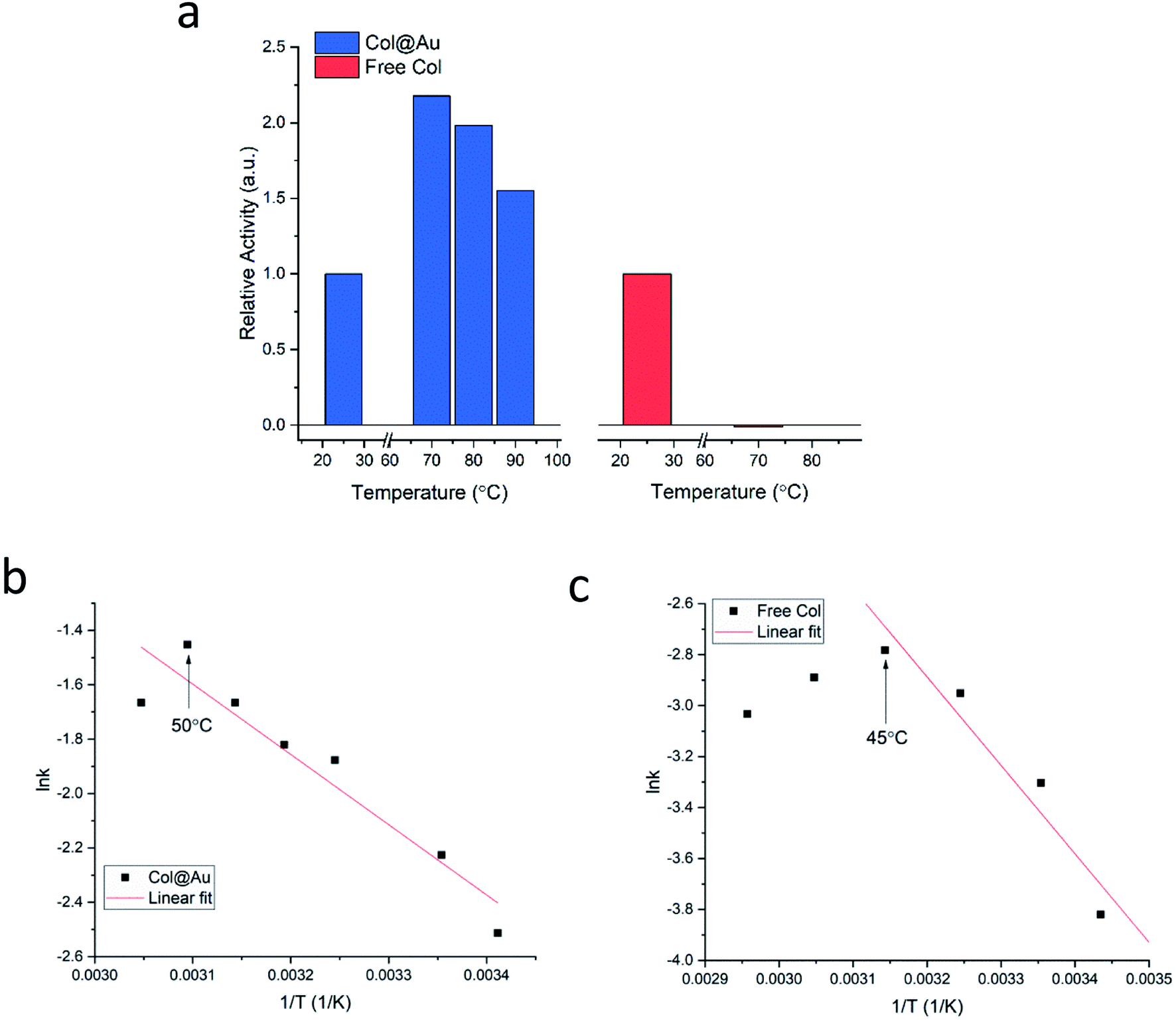 | ||
| Fig. 3 Col@Au. (a) Temperature effects on entrapped and free collagenase in solution. Arrhenius activation energy analysis of (b) entrapped and (c) free collagenase. Indicated by arrows are the temperatures beyond which denaturation begins. | ||
We return to Col@Au below, but first we show that the golden cage provides not only protection from elevated temperatures but also from extreme pH. For this purpose, L-asparaginase was taken: this ammonia producing enzyme (Fig. S1†) performs best at pH ∼ 8.6, but its activity drops quite drastically if the ammonia is not removed in an open system, leading to pH that exceeds 10. Furthermore, the standard assaying method for asparaginase uses Nessler's reagent that in itself has the extreme pH of over 13, which, when applied, kills the enzyme. For this reason, the recommended method45 for obtaining a kinetics plot requires a separate measurement for each time point in the plot (Fig. S8†). Asparaginase was successfully entrapped in gold (Fig. 4a), and amazingly, the Asp@Au turned out to be stable at this extreme pH of the Nessler reagent, which in solution kills the enzyme within a blink of an eye. This has allowed – for the first time – obtaining a continuous kinetic plot of Asp with this analytical method (Fig. 4a). Shown in the figure are the kinetics in either water suspension or in the Asp standard buffer solution (initial pH = 8.6, too weak to handle the extreme pH, anyway). Michaelis–Menten kinetics is obeyed as well – Fig. 4b (initial pH = 8.6) – with KM and Vmax values of 0.35 mM and 0.56 U mg−1 (compared to 0.14 mM and 1.8 U mg−1 in solution (Fig. S7.† Also seen there is that there is no enzyme activity in the supernatant solution, that is, again, all the activity is from the entrapped enzyme)).
 | ||
| Fig. 4 The activity of asparaginase@Au. (a) Shown is the activity at the extreme external pH = 13 of the analytic Nessler reagent. Red dots: reaction in water suspension. Black dots: reaction in the presence of the entrapping buffer (8.6; too weak to handle Nessler's pH). (b) The fit to Michaelis–Menten analysis of the kinetics. | ||
What then is the origin of the protectability against the extreme pH? We believe that it is a combination of two effects: the first is the above described rigid cage effect that operates also to enhance the thermal stability, that is, the restricted ability of conformational movements needed for the denaturing process. The second one is a more delicate argument, originally raised to explain pH stability of enzymes entrapped in sol–gel matrices.14 It is based on the often neglected fact that thermodynamic measurables, such as the dissociation constant of water and the pH scale which is based on it, collapse for very small assemblies, such as the number of water molecules which are co-entrapped in the nano-cage with the enzyme (Scheme 1), and which are part of the protein structure and of the solvation shell at the interface between the protein surface and the surface of the gold cage. Here is the argument in a nutshell: suppose that the external pH is 13, that is, the pOH is 1, namely [OH−] = 0.1 M, or, 1 hydroxyl in 550 water molecules. Suppose that the number of co-entrapped water molecules with the enzyme molecule within the gold cage, is of the same order, namely ∼500. This means that nominally, after equilibration between the bulk pH = 13 outside and the inside of the cage, that inside pH indicates only one or two hydroxyl ions per protein molecule per cage; the protein has absolutely no difficulty to take care of it by neutralizing these one or two hydroxyl ions with surface amino acids that have a carboxylic acid side chain. In solution or in 2D anchoring, the situation is totally different – here pH 13 means an Avogadro number of hydroxyls bombarding the protein from all sides.
The next enzyme that was successfully entrapped is horseradish peroxidase (HRP), Fig. 5a, adding to the proof of the generality of the developed method. One of the more common applications of HRP is its utilization in the assay of GOx activity (Fig. S2†), as indeed was practiced in the GOx@Au analyses described above. It is only natural then to test the possibility of using HRP@Au for that purpose62 (see Experimental details) – and yes, as seen in Fig. 5a, the activity of GOx@Au was detected by HRP@Au, by mixing the two doped gold biocomposites. One can envision therefore a library of enzymes@Au on the shelf, designing a network of enzymatic reactions at will.
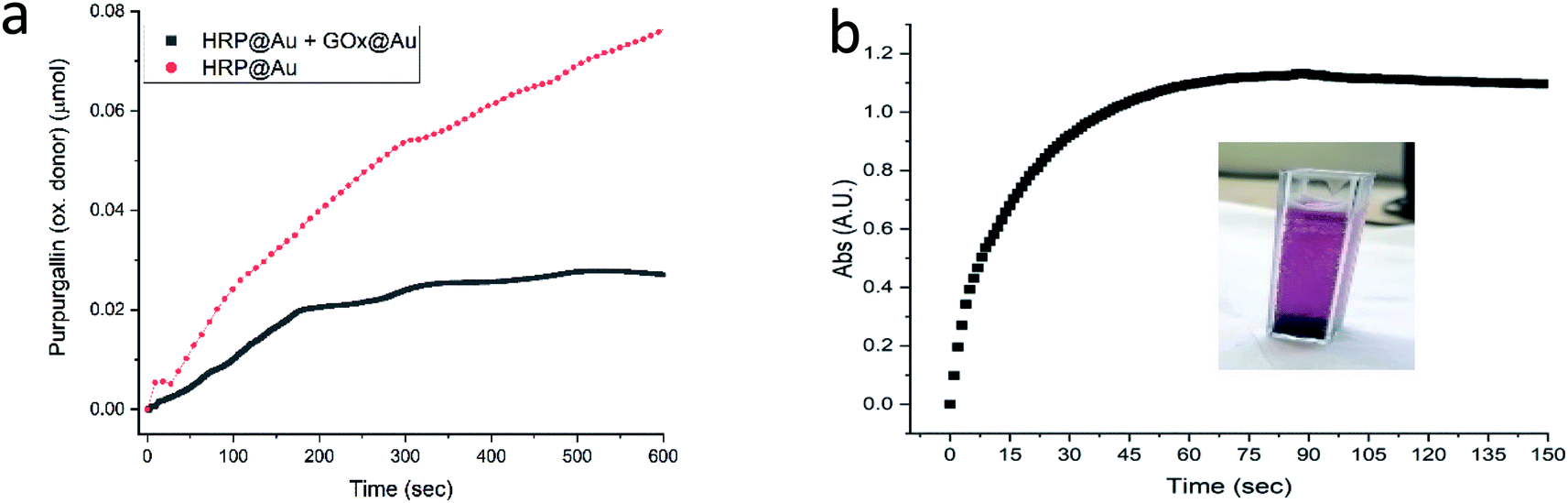 | ||
| Fig. 5 (a) The activity of HRP@Au (red) and of the combined powders of HRP@Au and GOx@Au (black). (b) The activity of the Lac@Au protected with CTAB. Inset: the formation of the product oxidative product (ESI Fig. S1†); Lac@Au is seen at the bottom of the cell. | ||
Only one of the enzymes we have tested – laccase (Lac) – required a modification of the entrapment procedure, because it appeared that the conformational changes induced in that enzyme upon direct entrapment, denatured it. However, since our purpose in this study has been to provide general solutions towards the preparation of enzymes@Au, we set up to design further conditions for such cases. The approach here was based on earlier observations we made regarding entrapped enzymes in sol–gel matrices,13,14 where we found that co-entrapping the enzyme with a surfactant, protects it quite efficiently. The isoelectric point of laccase is lower than 7,63 that is, in a neutral environment its surface is negatively charged. Therefore, the use of a cationic surfactant, such as cetyltrimethylammonium bromide (CTAB) would provide the desired protection. Indeed, under these conditions, Lac@Au is active (Fig. 5b).
Last but not least, for practical purposes, the question of recyclability is of relevance, and this was tested for Col@Au and for Lac@Au. It is seen for Col@Au (Fig. 6a) that after an increase in activity in cycles 2 and 3, the system stabilizes around 140% of the initial activity. This is a known observation in catalysts entrapped within porous matrices (such as in doped sol–gel catalysts),64 and has been attributed to equilibration of the material pore system and the cages which hold the (bio)catalyst molecules due to the first reaction and recycling manipulations; these apparently result in some widening of pores and in further removing of pore-blocking residues (no activity is observed in the supernatant) (Fig. S7a†). In any event, the overall picture of this test is stability for at least 8 cycles, which is an encouraging start for enzyme–gold devices designs. Lac@Au shows a similar pattern (Fig. 6b): increase of activity after the first cycle, and then activity of around 80% up to cycle 8.
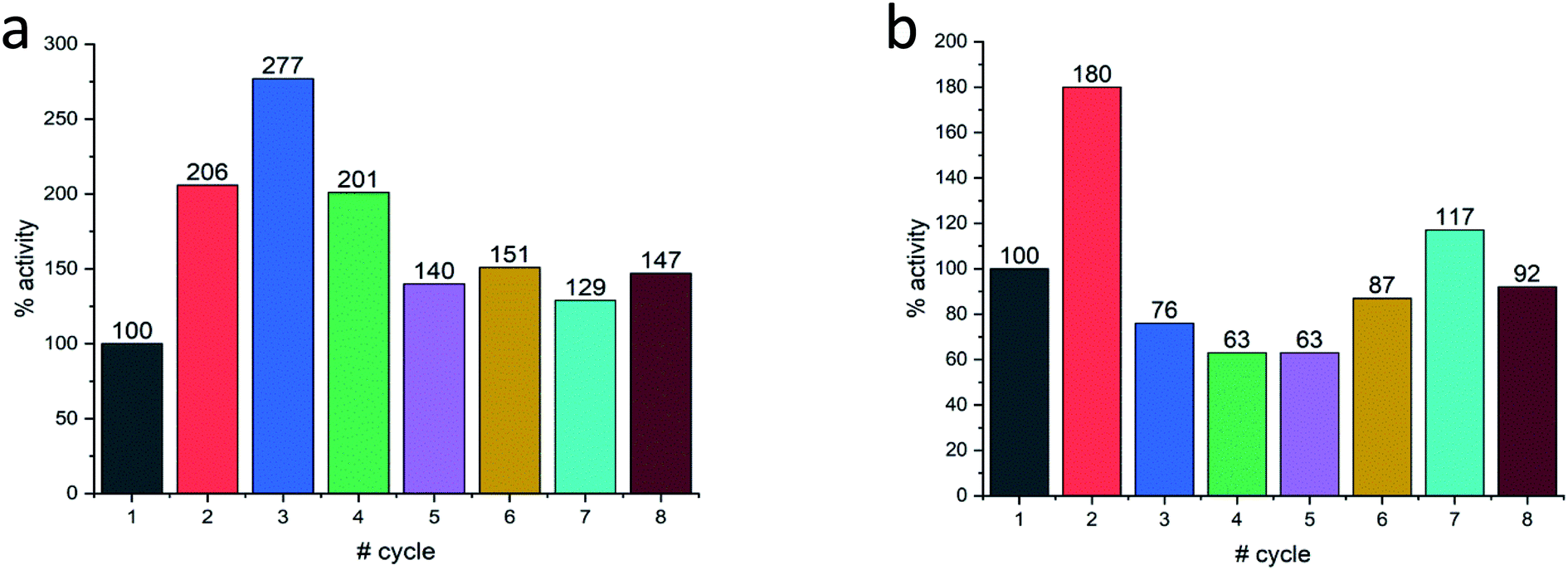 | ||
| Fig. 6 Recyclability of (a) Col@Au and of (b) Lac@Au. In both cases, acceleration at first, is observed. | ||
In conclusion, we present a general methodology for the 3D entrapment of enzymes in gold. It represents a new approach to the heterogenization of enzymes with metallic supports, which so far has been carried out by surface 2D anchoring methods. Five distinctly different enzymes were successfully entrapped within gold, with which various aspects of the enzymatic activity have been tested. Compliance with the Michaelis–Menten kinetics and with Arrhenius activation energy analysis, thermal stability, protectability against harsh chemical conditions, good recyclability and more, were all found for the enzymes@gold. The new enzyme immobilization method is of wide potential uses in medicine, biotechnology, bio-fuel cells and enzymatic (electro)sensing applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Supported by Supported by the PAZY Excellence in Science Foundation, by the US-Israel Binational Science foundation (BSF) grant # 2018622 in the framework of the US-National Science Foundation – BSF Collaborative Materials Program and by the Israel Ministry of Science, Technology and Space (Grant 3-12948). We thank the Center for Nanoscience and Nanotechnology, The Hebrew University, for analytical support of this study.References
- M. G. Wolfe and M. M. Ali,
et al., Enzymatic litmus test for selective colorimetric detection of C-C single nucleotide polymorphisms, Anal. Chem., 2019, 91, 4735–4740, DOI:10.1021/acs.analchem.9b00235
.
- G. M. Whitesides and C.-H. Wong, Enzymes as catalysts in synthetic organic chemistry [new synthetic methods (53)], Angew. Chem., Int. Ed. Engl., 1985, 24, 617–638, DOI:10.1002/anie.198506173
- M. K. Bhat, Cellulases and related enzymes in biotechnology, Biotechnol. Adv., 2000, 18, 355–383, DOI:10.1016/s0734-9750(00)00041-0
- G. Slaughter and T. Kulkarni, Enzymatic glucose biofuel cell and its application, J. Biochips Tissue Chips, 2015, 05 DOI:10.4172/2153-0777.1000110
-
R. A. Hatz and N. C. S. von Jan, et al., The Role of Collagenase in Wound Healing, in Proteolytic Enzym. Wound Heal., Springer Berlin Heidelberg, Berlin, Heidelberg, 1994, pp. 75–88, DOI:10.1007/978-3-642-78891-8_7
- A. Madhu and J. N. Chakraborty, Developments in application of enzymes for textile processing, J. Cleaner Prod., 2017, 145, 114–133, DOI:10.1016/j.jclepro.2017.01.013
- R. Chapman and M. H. Stenzel, All wrapped up: stabilization of enzymes within single enzyme nanoparticles, J. Am. Chem. Soc., 2019, 141(7), 2754–2769, DOI:10.1021/jacs.8b10338
- W.-C. Huang and W. Wang,
et al., Effective enzyme immobilization onto a magnetic chitin nanofiber composite, ACS Sustainable Chem. Eng., 2018, 6, 8118–8124, DOI:10.1021/acssuschemeng.8b01150
- A. Sassolas and L. J. Blum,
et al., Immobilization strategies to develop enzymatic biosensors, Biotechnol. Adv., 2012, 30, 489–511, DOI:10.1016/j.biotechadv.2011.09.003
- A. M. E. Smith and J. Fortuna,
et al., An automated materials screening approach for the development of sol-gel derived monolithic silica enzyme reactor columns, RSC Adv., 2014, 4, 15952–15960, 10.1039/c4ra00734d
- N. Serov and A. Prilepskii,
et al., Synthesis of plasmin-loaded fe 3 o 4 @caco 3 nanoparticles: towards next-generation thrombolytic drugs, ChemNanoMat, 2019, 5, 1267–1271, DOI:10.1002/cnma.201900359
- R. A. Sheldon and S. van Pelt, Enzyme immobilisation in biocatalysis: why, what and how, Chem. Soc. Rev., 2013, 42, 6223–6235, 10.1039/c3cs60075k
- N. Ganonyan and N. Benmelech,
et al., Entrapment of enzymes in silica aerogels, Mater. Today, 2020, 33, 24–35, DOI:10.1016/j.mattod.2019.09.021
- H. Frenkel-Mullerad and D. Avnir, Sol-gel materials as efficient enzyme protectors: preserving the activity of phosphatases under extreme ph conditions, J. Am. Chem. Soc., 2005, 127, 8077–8081, DOI:10.1021/ja0507719
- V. V. Vinogradov and D. Avnir,
et al., Enzyme renaturation to higher activity driven by the sol-gel transition: carbonic anhydrase, Sci. Rep., 2015, 5, 14411, DOI:10.1038/srep14411
-
S. A. Costa and H. S. Azevedo, et al., 7 Enzyme Immobilization in Biodegradable Polymers for Biomedical Applications, 2005, https://core.ac.uk/download/pdf/55614174.pdf, accessed April 29, 2019 Search PubMed
- R. A. Sperling and P. Rivera Gil,
et al., Biological applications of gold nanoparticles, Chem. Soc. Rev., 2008, 37, 1896, 10.1039/b712170a
- A. J. Mieszawska and W. J. M. Mulder,
et al., Multifunctional gold nanoparticles for diagnosis and therapy of disease, Mol. Pharm., 2013, 10, 831–847, DOI:10.1021/mp3005885
- H. du Toit and M. Di Lorenzo, Glucose oxidase directly immobilized onto highly porous gold electrodes for sensing and fuel cell applications, Electrochim. Acta, 2014, 138, 86–92, DOI:10.1016/j.electacta.2014.06.074
- L. Y. Chen and T. Fujita,
et al., Biofunctionalized nanoporous gold for electrochemical biosensors, Electrochim. Acta, 2012, 67, 1–5, DOI:10.1016/j.electacta.2011.12.132
- Q. Huajun and X. Caixia,
et al., Immobilization of laccase on nanoporous gold: comparative studies on the immobilization strategies and the particle size effects, J. Phys. Chem. C, 2009, 113, 2521–2525, DOI:10.1021/jp8090304
- H. Du Toit and M. Di Lorenzo, Glucose oxidase directly immobilized onto highly porous gold electrodes for sensing and fuel cell applications, Electrochim. Acta, 2014, 138, 86–92, DOI:10.1016/j.electacta.2014.06.074
- D. Avnir, Molecularly doped metals, Acc. Chem. Res., 2014, 47, 579–592, DOI:10.1021/ar4001982
- D. Avnir, Recent progress in the study of molecularly doped metals, Adv. Mater., 2018, 30, 1706804, DOI:10.1002/adma.201706804
- H.-P. Yang and Y.-N. Yue,
et al., Entrapment of a chiral cobalt complex within silver: a novel heterogeneous catalyst for asymmetric carboxylation of benzyl bromides with CO2, Chem. Commun., 2015, 51, 12216–12219, 10.1039/c5cc04554a
- B. Menagen and D. Avnir, Entrapment of drugs within metallic platinum and their delivery, ACS Biomater. Sci. Eng., 2019, 5, 2355–2364, DOI:10.1021/acsbiomaterials.9b00379
- H. Behar-Levy and D. Avnir, Entrapment of organic molecules within metals: dyes in silver, Chem. Mater., 2002, 14, 1736–1741, DOI:10.1021/cm011558o
- N. Ralbag and M. Mann-Lahav, et al., Composite materials with combined electronic and ionic properties, 2019, 1(4), 959–975, DOI:10.1016/j.matt.2019.04.007.
- N. Ralbag and I. Felner,
et al., Induction of enhanced magnetic behavior in gold, silver, and copper by doping with srf e 12 o 19 nanoparticles, Phys. Rev. B, 2019, 99, 064411, DOI:10.1103/physrevb.99.064411
- H.-P. Yang and Y.-N. Yue,
et al., Selective electrochemical reduction of CO2 to different alcohol products by an organically doped alloy catalyst, Green Chem., 2016, 18, 3216–3220, 10.1039/c6gc00091f
- R. Ben-Knaz and D. Avnir, Bioactive enzyme–metal composites: the entrapment of acid phosphatase within gold and silver, Biomaterials, 2009, 30, 1263–1267, DOI:10.1016/j.biomaterials.2008.11.026
- M. Rasmussen and S. Abdellaoui,
et al., Enzymatic biofuel cells: 30 years of critical advancements, Biosens. Bioelectron., 2016, 76, 91–102, DOI:10.1016/j.bios.2015.06.029
- H. du Toit and M. Di Lorenzo, Glucose oxidase directly immobilized onto highly porous gold electrodes for sensing and fuel cell applications, Electrochim. Acta, 2014, 138, 86–92, DOI:10.1016/j.electacta.2014.06.074
- C. T. Milne and A. Ciccarelli,
et al., A comparison of collagenase to hydrogel dressings in maintenance debridement and wound closure, Wounds, 2012, 24, 317–322 Search PubMed
- I. Degreef, Collagenase treatment in dupuytren contractures: a review of the current state versus future needs, Rheumatol. Ther., 2016, 3, 43–51, DOI:10.1007/s40744-016-0027-1
- K. B. McCredie and D. H. W. Ho,
et al., L-asparaginase for the treatment of cancer, Ca-Cancer J. Clin., 1973, 23, 220–227, DOI:10.3322/canjclin.23.4.220
- H. V. Hendriksen and B. A. Kornbrust,
et al., Evaluating the potential for enzymatic acrylamide mitigation in a range of food products using an asparaginase from aspergillus oryzae, J. Agric. Food Chem., 2009, 57, 4168–4176, DOI:10.1021/jf900174q
- N. C. Veitch, Horseradish peroxidase: a modern view of a classic enzyme, Phytochemistry, 2004, 65, 249–259, DOI:10.1016/j.phytochem.2003.10.022
- R. C. Minussi and G. M. Pastore,
et al., Potential applications of laccase in the food industry, Trends Food Sci. Technol., 2002, 13, 205–216, DOI:10.1016/s0924-2244(02)00155-3
- H. Qiu and L. Xue,
et al., Enzyme-modified nanoporous gold-based electrochemical biosensors, Biosens. Bioelectron., 2009, 24, 3014–3018, DOI:10.1016/j.bios.2009.03.011
- K. J. Stine, Enzyme immobilization on nanoporous gold: a review, Biochem. Insights, 2017, 10 DOI:10.1177/1178626417748607
- L. Y. Chen and T. Fujita,
et al., Biofunctionalized nanoporous gold for electrochemical biosensors, Electrochim. Acta, 2012, 67, 1–5, DOI:10.1016/j.electacta.2011.12.132
- G. Baskar and B. G. Garrick,
et al., Gold nanoparticle mediated delivery of fungal asparaginase against cancer cells, J. Drug Delivery Sci. Technol., 2018, 44, 498–504, DOI:10.1016/j.jddst.2018.02.007
- Enzymatic assay of glucose oxidase|Sigma-Aldrich, https://www.sigmaaldrich.com/technical-documents/protocols/biology/enzymatic-assay-of-glucose-oxidase.html, accessed September 8, 2019 Search PubMed.
- Enzymatic assay of l-asparaginase (ec 3.5.1.1), https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma/General_Information/2/asparaginase.pdf, accessed September 8, 2019 Search PubMed.
- Enzymatic assay of collagenase|Sigma-Aldrich, https://www.sigmaaldrich.com/technical-documents/protocols/biology/enzymatic-assay-of-collagenase-using-n-3-2furylacryloyl-leu-gly-pro-ala.html, accessed September 8, 2019 Search PubMed.
- Enzymatic assay of peroxidase (ec 1.11.1.7)|Sigma-Aldrich, https://www.sigmaaldrich.com/technical-documents/protocols/biology/enzymatic-assay-of-peroxidase.html, accessed September 8, 2019 Search PubMed.
- Enzymatic assay of laccase (ec 1.10.3.2)|Sigma-Aldrich, https://www.sigmaaldrich.com/technical-documents/protocols/biology/enzymatic-assay-of-laccase.html, accessed September 8, 2019 Search PubMed.
- K. A. McCall and C. Huang,
et al., Function and mechanism of zinc metalloenzymes, J. Nutr., 2000, 130, 1437S–1446S, DOI:10.1093/jn/130.5.1437s
- L. Carlsson and G. Lundgren, On the hydrolysis of the tetrachloroaurate(iii) ion, Acta Chem. Scand., 1967, 21, 819 CrossRef CAS
- F. H. Fry and G. A. Hamilton,
et al., The kinetics and mechanism of hydrolysis of tetrachloroaurate(iii), Inorg. Chem., 1966, 5, 1943–1946, DOI:10.1021/ic50045a024
- C. Subramaniam, R. T. Tom and T. Pradeep, On the formation of protected gold nanoparticles from AuCl4− by the reduction using aromatic amines, J. Nanopart. Res., 2005, 7, 209–217, DOI:10.1007/s11051-005-0315-0
- J. D. S. Newman and G. J. Blanchard, Formation of gold nanoparticles using amine reducing agents, Langmuir, 2006, 22, 5882–5887, DOI:10.1021/la060045z
-
M. Thommes and K. Kaneko, et al., IUPAC technical report physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC technical report), 2015, DOI:10.1515/pac-2014-1117
- B. G. Ershov and E. V. Abkhalimov,
et al., Gold nanoparticles in aqueous solutions: influence of size and ph on hydrogen dissociative adsorption and au(iii) ion reduction, Phys. Chem. Chem. Phys., 2016, 18, 13459–13466, 10.1039/c6cp01996j
- H. Shindo, D. Watanabe, T. Onaga, M. Urakawa, O. Nakahara and Q. Huang, Adsorption, activity, and kinetics of acid phosphatase as influenced by selected oxides and clay minerals, Soil Sci. Plant Nutr., 2002, 48(5), 763–767, DOI:10.1080/00380768.2002.10409268
-
A. Dwevedi, Enzyme immobilization: advances in industry, agriculture, medicine, and the …, alka dwevedi, Google books, https://books.google.co.il/books?id=3kgWDQAAQBAJ%26pg=PA28%26lpg=PA28%26dq=vmax+change+immobilization%26source=bl%26ots=zprPPZb689%26sig=ACfU3U2ndT733ELaxI6H52-o1_PmoIynPg%26hl=en%26sa=X%26ved=2ahUKEwi-kOa-66_lAhUB6qQKHYb5BoIQ6AEwDHoECAkQAQ#v=onepage%26q=vmaxchangeimmobili, accessed October 22, 2019 Search PubMed
- J. Hu and S. Li,
et al., Properties of immobilized pepsin on modified pmma microspheres, Biotechnol. J., 2006, 1, 75–79, DOI:10.1002/biot.200500022
- G. Zoldák and A. Zubrik,
et al., Irreversible thermal denaturation of glucose oxidase from aspergillus niger is the transition to the denatured state with residual structure, J. Biol. Chem., 2004, 279, 47601–47609, DOI:10.1074/jbc.m406883200
- J. Atalah and P. Cáceres-Moreno,
et al., Thermophiles and the applications of their enzymes as new biocatalysts, Bioresour. Technol., 2019, 280, 478–488, DOI:10.1016/j.biortech.2019.02.008
- S. A. Ahmed and S. A. A. Saleh,
et al., Catalytic, kinetic and thermodynamic properties of free and immobilized caseinase on mica glass-ceramics, Heliyon, 2019, 5, e01674, DOI:10.1016/j.heliyon.2019.e01674
- M. S. Crosley and W. T. Yip, Multienzyme, multistep biosensor produced through kinetic doping, J. Phys. Chem. B, 2019, 123, 3962–3967, DOI:10.1021/acs.jpcb.9b01907
- B. Viswanath and B. Rajesh,
et al., Fungal laccases and their applications in bioremediation, Enzyme Res., 2014, 2014 DOI:10.1155/2014/163242
-
D. Avnir and J. Blum, et al., Better Catalysis with Organically Modified Sol-Gel Materials, in Sol-Gel Handb., Wiley Blackwell, 2015: pp. 963–986, DOI:10.1002/9783527670819.ch31
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05419g |
| This journal is © The Royal Society of Chemistry 2020 |