
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of the enamine pyramidalization direction on the reactivity of secondary amine organocatalysts†
Tobias
Schnitzer‡
 ,
Jasper S.
Möhler‡
and
Helma
Wennemers
*
,
Jasper S.
Möhler‡
and
Helma
Wennemers
*
Laboratory of Organic Chemistry, ETH Zürich, Vladimir-Prelog-Weg 3, 8093 Zürich, Switzerland. E-mail: Helma.Wennemers@org.chem.ethz.ch
First published on 7th January 2020
Abstract
Chiral secondary amines are valuable catalysts for reactions that proceed through an enamine intermediate. Here, we explored the importance of the pyramidalization direction of the enamine-N on the reactivity of chiral enamines with a combination of computational, NMR spectroscopic, and kinetic experiments. Studies with peptidic catalysts that bear cyclic amines with different ring sizes revealed that endo-pyramidalized enamines are significantly more reactive compared to exo-pyramidalized analogs. The results show that the pyramidalization direction can have a greater effect than n→π* orbital overlap on the reactivity of chiral enamines. The data enabled the development of a catalyst with higher reactivity compared to the parent catalyst.
Introduction
Since the late 1990s, numerous chiral secondary amines have emerged as valuable catalysts for asymmetric reactions that proceed via an enamine intermediate (Scheme 1a).1 Proline and other α-substituted five-membered cyclic amines are the most commonly used catalysts for such aldol and related reactions.1,2 Seminal work by Stork, Eschenmoser, and Seebach on stoichiometric reactions showed that enamines are most reactive when the enamine nitrogen is planar:3–7 The higher the sp2 character of the nitrogen is, the better is the donation of electron density from the non-bonding orbital (n) of N into the π* orbital of the enamine C–C double bond and the higher is the nucleophilicity (Scheme 1b).3–10 The degree of sp2 hybridization correlates with the degree of pyramidalization (Δ) of the enamine-N. Thus, the more planar, the better is the n→π* overlap and the more reactive is the enamine.3–10 But is n→π* overlap really the only reason for reactivity differences of chiral enamines?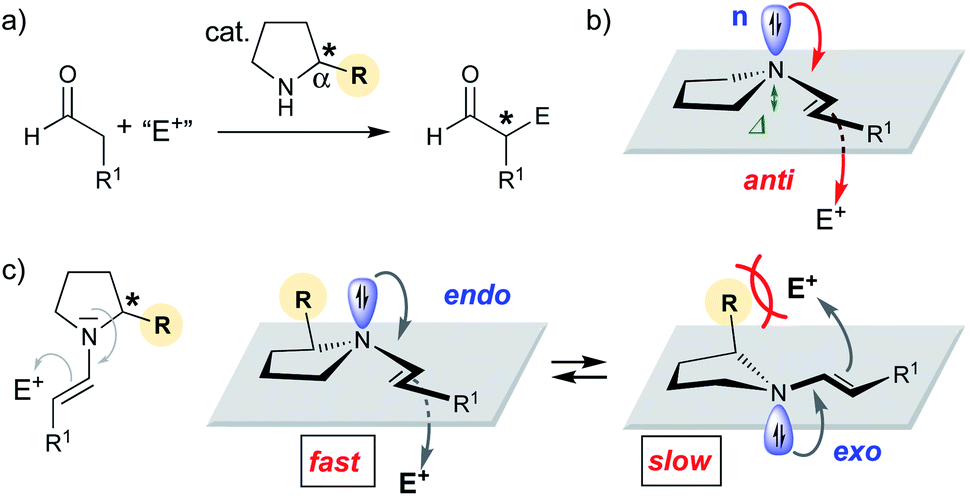 | ||
| Scheme 1 (a) Secondary amine catalyzed addition reaction of aldehydes to electrophiles. (b) Anti-attack of enamine. (c) Equilibrium between endo and exo pyramidalized enamines. | ||
Stereoelectronic considerations require an attack of the enamine onto the electrophile anti to the n orbital (Scheme 1b).11 We reasoned that the pyramidalization direction of the enamine-N should have a profound effect on the reactivity of enamines derived from chiral secondary amines. Enamines with an endo pyramidalization, where the n orbital is on the same side as the Cα-substituent should react faster than those with an exo pyramidalization due to the smaller steric hindrance for the incoming electrophile (Scheme 1c). Organocatalysts should be excellent for probing the reactivity of enamines – provided that the enamine intermediate is involved in the rate-determining step.
Herein we used peptidic catalysts bearing secondary amines with different ring sizes as tools to evaluate the effect of pyramidalization on the reactivity of enamines. We show that the pyramidalization direction can have a significant effect on the reactivity of Cα-substituted enamines. The results provided a guide that enabled the development of an organocatalyst with enhanced reactivity compared to the parent catalyst.
Results and discussion
The tripeptides H-DPro-Pro-Glu-NH2 (1) and H-DPro-Pip-Glu-NH2 (2, Pip = piperidine carboxylic acid) are highly reactive and stereoselective catalysts for conjugate addition reactions between aldehydes and nitroolefins.12,13 Mechanistic studies revealed that the enamine intermediate undergoes the rate and enantioselectivity determining step and off-cycle intermediates were not detected.14,15 Thus, this peptide-catalyzed reaction is a valid testing ground to probe for the effect of pyramidalization on enamine reactivity.We reasoned that the conformational properties of differently sized cyclic secondary amines should result in enamines with different extents of pyramidalization and different endo/exo pyramidalization ratios of the enamine-N. Derivatives of peptide catalysts 1 and 2 with four- and six-membered N-terminal cyclic amines should therefore allow for probing the effect of pyramidalization.
Computational analysis of enamines derived from cyclic amines with different ring sizes
We started by computationally evaluating whether the pyramidalization of enamines formed by four-, five- and six-membered amines differs. Enamines derived from α-methyl substituted azetidine (3a), pyrrolidine (3), and piperidine (3b) were used as model compounds that are sufficiently small to allow for calculations within a reasonable time (Fig. 1). Conformational searches with MacroModel (OPLS3e force field, GB/SA solvent models for DMSO and CHCl3)16,17 provided conformers in energy minima with geometries that were then optimized by DFT using the M06-2X-D3/6-31+G** level of theory.18 We used CHCl3 for the calculations to be as close as possible to the solvent mixture used for the reactions as well as DMSO, a solvent in which enamines are sufficiently stable to be studied by NMR spectroscopy (vide infra). These calculations resulted in two conformers of 3a, five of 3, and four of 3b with a population of >3% according to the Boltzmann distribution. s-Trans enamines are the lowest energy conformers for all three compounds (Fig. 1).§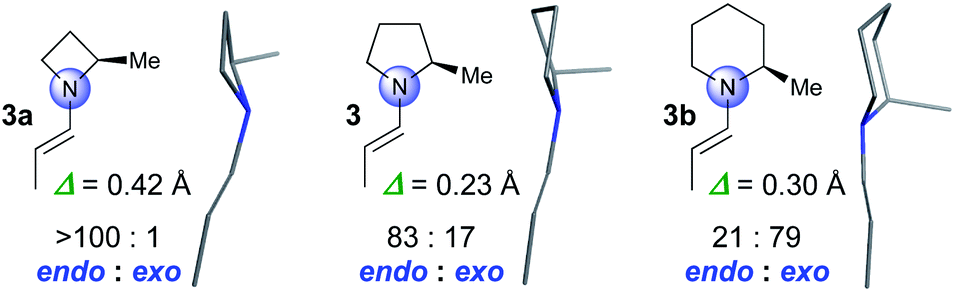 | ||
Fig. 1 Lowest energy structures of 3a, 3 and 3b. Endo/exo pyramidalization ratios and degree of pyramidalization (Δ) at N averaged over the absolute values of all calculated structures and weighted by their relative energies in CHCl3. Values in DMSO are comparable (3a: Δ = 0.43, >100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 endo:exo; 3: Δ = 0.24, 84:16 endo:exo; 3b: Δ = 0.34, 23:77 endo:exo). :1 endo:exo; 3: Δ = 0.24, 84:16 endo:exo; 3b: Δ = 0.34, 23:77 endo:exo). | ||
The calculations predict significant differences regarding the degree and the direction of pyramidalization of the enamine-N of 3a, 3, and 3b: (a) the enamine-N is most planar in the pyrrolidine-derived enamine 3 (Δ = 0.23 Å) followed by that of the piperidine 3b (Δ = 0.30 Å) and the azetidine 3a (Δ = 0.42 Å) derivatives (Fig. 1). (b) The enamine-N in four-membered 3a is exclusively endo-pyramidalized; for five-membered 3 a ratio of 83:17 in favour of endo is predicted and for six-membered 3b a ratio of 21:79 in favor of exo (Fig. 1).¶ The predictions for 3a and 3 are in good agreement with crystal structures by Dunitz, Eschenmoser, Seebach, and List of proline- and azetidine-derived enamines or enaminones in which the enamine-N is endo pyramidalized.4,19 In the structure of the piperidine derivative 3b, allylic strain enforces an axial position of the substituent at Cα20 and as a result the exo pyramidalization.
NMR spectroscopic analysis of enamines derived from model compounds and catalysts bearing amines with different ring sizes
To validate the computational findings we prepared enamines of butanal with the α-methylated cyclic amines (3a′, 3′, 3b′) and studied them by NMR spectroscopy (Fig. 2).|| In addition, we prepared and studied the enamines of analogs of peptidic catalyst 2 that bear four-, five-, and six-membered rings at the N-terminus (2a-En, 2-En, 2b-En; Fig. 2).** Specifically, we measured 13C NMR spectra and monitored the chemical shift of the enamine carbon C(2) (Fig. 2). This chemical shift is a good measure for the electron density at C(2) and has been used by Mayr to evaluate the nucleophilicity of enamines.9 The more upfield shifted the carbon signal is, the stronger is the n→π* donation and the more planar is the enamine. Since the enamines 2a-En and 2b-En proved too unstable to allow analysis in CDCl3, we analysed them in d6-DMSO, keeping in mind that the calculations had predicted comparable pyramidalization degrees and endo/exo ratios regardless of the solvent (Fig. 1). 13C NMR spectra showed as expected the highest upfield shift of C(2) for the five-membered enamine 2-En (98.6 ppm, Fig. 2). The C(2) of the piperidine-derived enamine 2b-En appears further upfield (100.9 ppm) than that of the azetidine-derived enamine 2a-En (104.7 ppm). The same trend was observed for the enamines 3a′ (101.9 ppm), 3′ (96.8 ppm), 3b′ (100.5 ppm) (Fig. 2) suggesting that the substituent at Cα does not affect the relative amount of n→π* overlap to a significant extent. These chemical shift differences indicate a higher electron density, and thus greater planarity and higher nucleophilicity, at C(2) of the enamine of piperidine compared to that of azetidine. Thus, the NMR spectroscopic data corroborate the higher planarity of the Pip-compared to the Aze-derived enamines predicted by the calculations.††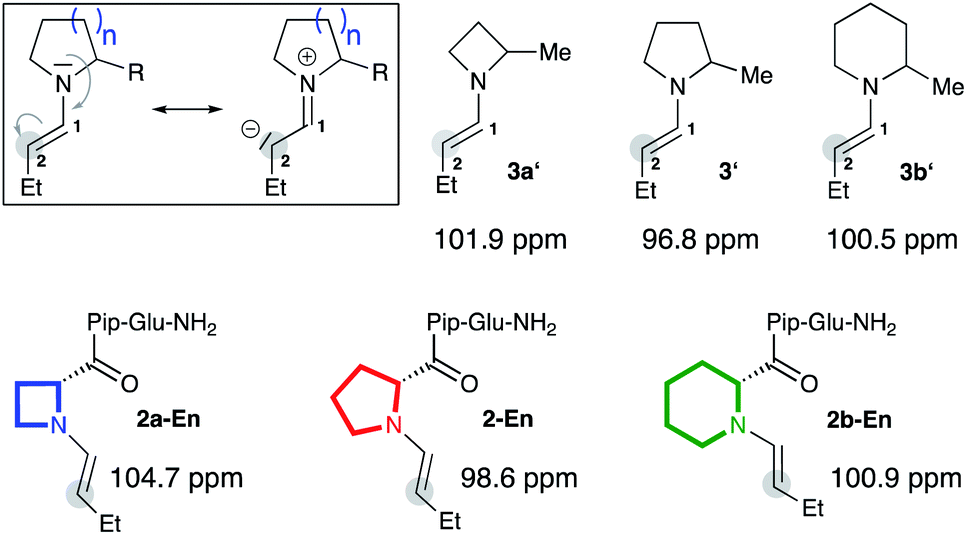 | ||
| Fig. 2 13C NMR chemical shifts of C(2) of enamines derived from α-methyl azetidine (3a′), pyrrolidine (3′), piperidine (3b′) and catalysts 2a, 2, and 2b in d6-DMSO. | ||
Catalysis with secondary amines of different ring sizes
Based on these findings enamines derived from pyrrolidines should react fastest. The comparison of the reactivity of enamines from azetidine versus piperidine will allow to elucidate whether the degree of pyramidalization (n→π* overlap and electron density at C(2)) or another factor such as the pyramidalization direction is more or equally as important for the reactivity of enamines of chiral amines: if the degree of pyramidalization (Δ) is key, piperidine derivatives should react faster than azetidine derivatives. In contrast, if enamines with an endo-pyramidalized N react faster than exo-pyramidalized enamines, azetidine derivatives should outcompete piperidine derivatives. To compare the importance of these two effects, we studied the catalytic properties of H-DPro-Pip-Glu-NH22 and its analogues with four- (Aze, 2a) and six-membered (Pip, 2b) cyclic amines at the N-terminal position. NMR spectra of these three peptides are similar, in particular with respect to the observed interresidue NOEs, suggesting that their secondary structures are comparable.† We used the conjugate addition of butanal to (E)-nitrostyrene to compare their reactivity (Scheme 2a).** The reaction was performed under conditions where the reaction rate is not influenced by the aldehyde (0 order), and the reaction of the enamine intermediate with the nitroolefin is rate-determining (CHCl3/iPrOH 9:1, 3 equiv. of butanal, 15 mM in catalyst). 3 mol% of 2a, 2, and 2b were used to ensure detectable product formation for all three catalysts within hours.‡‡ To ascertain that the enamine intermediate is involved in the rate-determining step (and not the formation of the enamine) in case of all three catalysts we determined the rate orders for the reactions catalysed by 2a and 2b. These studies showed that the reactions are, as previously found for 1,15 0 order in aldehyde at butanal concentrations >1.5 M, the concentration which was used to compare the reactivity of 2, 2a, and 2b.† The rate order of nitrostyrene is under these conditions ∼0.5 similarly to what was previously found for 114 and corroborating that the enamine is involved in the rate-determining step.§§
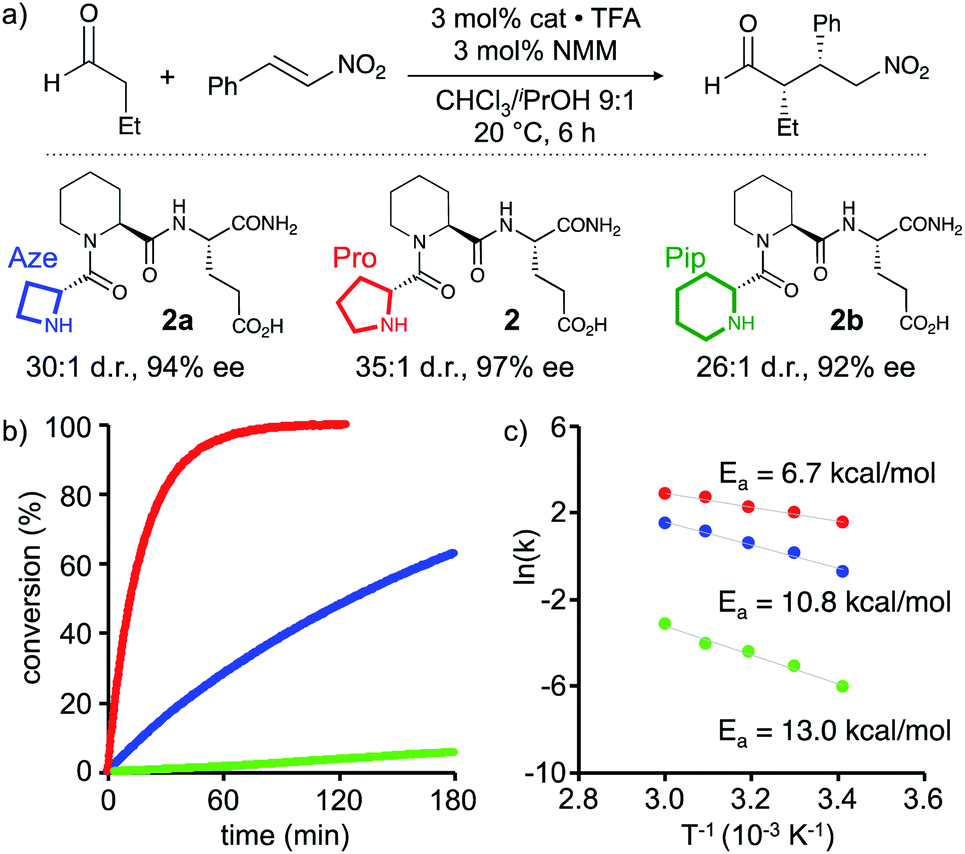 | ||
| Scheme 2 (a) Conjugate addition reaction of butanal to nitrostyrene catalyzed by 2a, 2 and 2b. (b) In situ IR monitoring of the formation of γ-nitroaldehyde, and (c) Arrhenius plot and activation energy of the peptide catalyzed reactions. | ||
Monitoring of the reactions in situ by infrared spectroscopy revealed significant reaction rate differences (Scheme 2b). After two hours, parent peptide 2 had converted the starting materials quantitatively into the conjugate addition product, which was obtained with high stereoselectivity (d.r. 35:1 (syn:anti), 97% ee, Scheme 2a and b, red). After the same time, only ∼50% conversion was observed in the presence of 2a with the 4-membered Aze as reactive centre (Scheme 2b, blue). The reactivity of 2b with the 6-membered Pip was even lower, with less than 5% conversion after two hours (Scheme 2b, green). Both catalysts provided the product with slightly lower but still good stereoselectivity (2a: d.r. 30:1, 94% ee and 2b: d.r. 26:1, 92% ee, Scheme 2a). Notably, in case of peptide 2b the same product enantiomer formed indicating that the minor endo N-pyramidalized enamine reacted predominantly. Reaction of the exo N-pyramidalized enamine with the nitroolefin via an anti-attack, would provide a different stereoisomer.
These relative reactivity differences of the three catalysts are also reflected in the activation energies (Ea) that were derived from Arrhenius plots of initial rates at different temperatures (20, 30, 40, 50, and 60 °C) under otherwise identical reaction conditions (Scheme 2c). The parent peptide 2 has the lowest activation energy (6.7 ± 0.5 kcal mol−1) followed by those of the four-membered N-terminal amine 2a (10.8 ± 1.2 kcal mol−1) and the six-membered amine 2b (13.0 ± 0.8 kcal mol−1).
These findings show that the five-membered Pro derivative 2, with the most planar and mainly endo pyramidalized enamine-N is most reactive. The four-membered Aze derivative 2a is significantly more reactive compared to the six-membered Pip derivative 2b. Combined with the computational and NMR spectroscopic data, these results are consistent with a greater importance of endo-pyramidalization than the degree of n→π* overlap for the reactivity of chiral enamines.
Enhancing the reactivity of secondary amine catalysts
The computational studies predicted an endo/exo pyramidalization ratio of 83:17 for the enamine formed by chiral 5-membered cyclic amines (Fig. 1, middle). The presented data suggests that pyrrolidine derivatives with a higher endo/exo ratio should be even more reactive. We reasoned that bicyclic all-cis-2,3-methanoproline should favor endo-pyramidalized enamines by “locking” the pseudorotation of the pyrrolidine ring of proline. Computational studies with MacroModel and DFT using the same procedure as for the calculations of 3a, 3, and 3b, indeed predict a >100:1 ratio of endo/exo pyramidalization for the methyl substituted 2,3-methanopyrrolidine 3c, with a degree of pyramidalization (Δ = 0.26 Å, Fig. 3) that is comparable to that of the proline derivative 3 (Δ = 0.23 Å, Fig. 3).21 Hence, a methanoproline derived catalyst should be more reactive than the corresponding Pro analogue. We therefore prepared peptide 2c with an N-terminal 4,5-methanoproline residue and compared its catalytic properties to those of parent peptide 2 (Fig. 3). Peptide 2c is indeed more reactive than 2 and converted butanal and nitrostyrene quantitatively within 30 min into the conjugate addition product, which was obtained with a d.r. of 30:1 and an enantioselectivity of 98% ee.
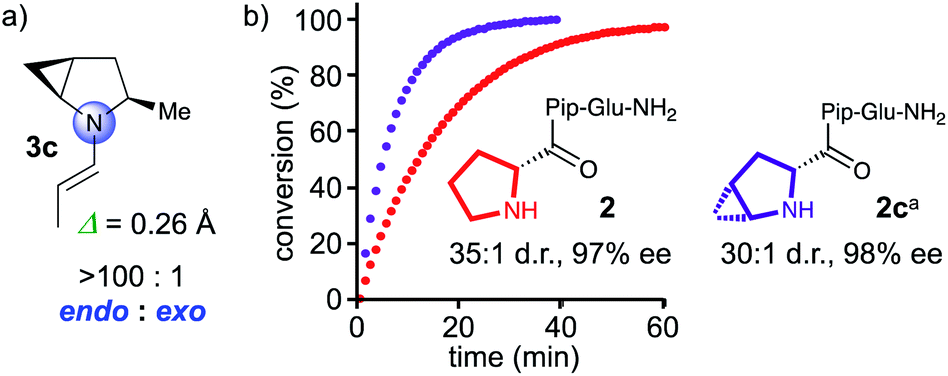 | ||
| Fig. 3 (a) Calculated endo/exo pyramidalization ratio of enamine formed by α-methyl 4,5-methanopyrrolidine. (b) In situ IR monitoring of the conjugate addition reaction between butanal and nitrostyrene, catalyzed by 2 and 2c. aNote, since D-methanoproline was not commercially available, the enantiomeric peptide was used and the enantiomeric product was obtained, see the ESI.† | ||
Conclusions
In summary, we have shown the importance of endo-pyramidalization for endowing enamines derived from α-substituted secondary amines with high reactivity. Exo-pyramidalized enamines react slower due to the steric hindrance between the incoming electrophile and the Cα-substituent. Thus, the pyramidalization direction of the enamine-N is, together with the extent of n→π* overlap, important for the reactivity of chiral enamines. These findings provide an additional design element for the development of catalytically active chiral secondary amines – one of the most utilized types of organocatalysts. The reactivity of chiral enamines is likely more complex and not only determined by n→π* overlap and the enamine-N pyramidalization direction. This study might therefore also inspire further research to unravel thus far overlooked contributors to enamine reactivity.Author contributions
T. S. and H. W. conceived the project and designed the experiments. T. S. and J. S. M. performed the experiments. T. S. developed the concept for the reactivity of enamines depending on their pyramidalization direction. J. S. M. developed catalyst 2c. T. S., J. S. M. and H. W. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Fonds der Chemischen Industrie (Germany) for a Kekulé Fellowship for T. S. and the Swiss National Science Foundation (Grant No. 200020_169423) for financial support. We thank Dr M.-O. Ebert for support with NMR spectroscopic experiments at low temperature and Prof. P. Chen for useful discussions.Notes and references
- For reviews see: (a) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed; (b) Y.-Q. Zou, F. M. Hörmann and T. Bach, Chem. Soc. Rev., 2018, 47, 278–290 RSC; (c) D. A. Alonso, A. Baeza, R. Chinchilla, C. Gómez, G. Guillena, I. M. Pastor and D. J. Ramón, Molecules, 2017, 22, 895–946 CrossRef PubMed.
- (a) Z. G. Hajos and D. R. Parrish, German Patent DE2102623, 1971; (b) U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed., 1971, 10, 496–497 CrossRef CAS; (c) B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- (a) G. Stork, R. Terrell and J. Szmuszkovicz, J. Am. Chem. Soc., 1954, 76, 2029–2030 CrossRef CAS; (b) G. Stork and H. K. Landesman, J. Am. Chem. Soc., 1956, 78, 5128–5129 CrossRef CAS; (c) G. Stork and H. K. Landesman, J. Am. Chem. Soc., 1956, 78, 5129–5130 CrossRef CAS; (d) G. Stork, A. Brizzolara, H. Landesman and J. Szmuszkovicz, J. Am. Chem. Soc., 1963, 85, 207–222 CrossRef CAS.
- K. L. Brown, L. Damm, J. Dunitz, A. Eschenmoser, R. Hobi and C. Kratky, Helv. Chim. Acta, 1978, 61, 3108–3135 CrossRef CAS.
- D. Seebach, A. K. Beck, D. M. Badine, M. Limbach, A. Eschenmoser, A. M. Treasurywala, R. Hobi, W. Prikoszovich and B. Linder, Helv. Chim. Acta, 2007, 90, 425–471 CrossRef CAS.
- D. Seebach and J. Golinski, Helv. Chim. Acta, 1981, 64, 1413–1423 CrossRef CAS.
- K. Müller, F. Previdoli and H. Desilvestro, Helv. Chim. Acta, 1981, 64, 2497–2507 CrossRef.
- (a) B. Kempf, N. Hampel, W. R. Ofial and H. Mayr, Chem.–Eur. J., 2003, 9, 2209–2218 CrossRef CAS PubMed; (b) R. C. Samanta, B. Maji, S. De Sarkar, K. Bergander, R. Fröhlich, C. Mück-Lichtenfeld, H. Mayr and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 5234–5238 CrossRef CAS PubMed; (c) D. S. Timofeeva, R. J. Mayer, P. Mayer, A. R. Ofial and H. Mayr, Chem.–Eur. J., 2018, 24, 5901–5910 CrossRef CAS PubMed; (d) H. Mayr, S. Lakhdar, B. Maji and A. R. Ofial, Beilstein J. Org. Chem., 2012, 8, 1458–1478 CrossRef CAS PubMed.
- (a) S. Lakhdar, B. Maji and H. Mayr, Angew. Chem., Int. Ed., 2012, 51, 5739–5742 CrossRef CAS PubMed; (b) H. Erdmann, F. An, P. Mayer, A. R. Ofial, S. Lakhdar and H. Mayr, J. Am. Chem. Soc., 2014, 136, 14263–14268 CrossRef CAS PubMed.
- For examples of mechanistic studies involving enamines, see: (a) T. Husch, D. Seebach, A. K. Beck and M. Reiher, Helv. Chim. Acta, 2017, 100, e1700182 CrossRef; (b) K. Patora-Komisarska, M. Benohoud, H. Ishikawa, D. Seebach and Y. Hayashi, Helv. Chim. Acta, 2011, 94, 719–745 CrossRef CAS; (c) D. Seebach, X. Sun, M.-O. Ebert, W. B. Schweizer, N. Purkayastha, A. K. Beck, J. Duschmalé, H. Wennemers, T. Mukaiyama, M. Benohoud, Y. Hayashi and M. Reiher, Helv. Chim. Acta, 2013, 96, 799–852 CrossRef CAS; (d) M. Schmid, K. Zeitler and R. M. Gschwind, J. Am. Chem. Soc., 2011, 133, 7065–7074 CrossRef CAS PubMed; (e) M. B. Schmid, K. Zeitler and R. M. Gschwind, Chem. Sci., 2011, 2, 1793–1803 RSC; (f) J. Bures, A. Armstrong and D. G. Blackmond, J. Am. Chem. Soc., 2011, 133, 8822–8825 CrossRef CAS PubMed; (g) J. Bures, A. Armstrong and D. G. Blackmond, Acc. Chem. Res., 2016, 49, 214–222 CrossRef CAS PubMed; (h) T. Földes, A. Madarasz, A. Revesz, Z. Dobi, S. Varga, A. Hamza, P. R. Nagy, P. M. Pihko and I. Papai, J. Am. Chem. Soc., 2017, 139, 17052–17063 CrossRef PubMed.
- (a) D. Seebach, A. K. Beck, J. Golinski, J. N. Hay and T. Laube, Helv. Chim. Acta, 1985, 68, 162–172 CrossRef CAS; (b) D. Seebach, R. Gilmour, U. Groselj, G. Deniau, C. Sparr, M.-O. Ebert, A. K. Beck, L. B. McCusker, D. Sisak and T. Uchimaru, Helv. Chim. Acta, 2010, 93, 603–634 CrossRef CAS.
- (a) M. Wiesner, J. D. Revell, S. Tonazzi and H. Wennemers, J. Am. Chem. Soc., 2008, 130, 5610–5611 CrossRef CAS PubMed; (b) M. Wiesner, J. D. Revell and H. Wennemers, Angew. Chem., Int. Ed., 2008, 47, 1871–1874 CrossRef CAS PubMed; (c) M. Wiesner, M. Neuburger and H. Wennemers, Chem.–Eur. J., 2009, 25, 10103–10109 CrossRef PubMed; (d) J. Duschmalé, S. Kohrt and H. Wennemers, Chem. Commun., 2014, 50, 8109–8112 RSC; (e) T. Schnitzer, M. Wiesner, P. Krattiger, J. D. Revell and H. Wennemers, Org. Biomol. Chem., 2017, 15, 5877–5881 RSC.
- T. Schnitzer and H. Wennemers, J. Am. Chem. Soc., 2017, 139, 15356–15362 CrossRef CAS PubMed.
- M. Wiesner, G. Upert, G. Angelici and H. Wennemers, J. Am. Chem. Soc., 2010, 132, 6–7 CrossRef CAS PubMed.
- (a) J. Duschmalé, J. Wiest, M. Wiesner and H. Wennemers, Chem. Sci., 2013, 4, 1312–1318 RSC; (b) F. Bächle, J. Duschmalé, C. Ebner, A. Pfaltz and H. Wennemers, Angew. Chem., Int. Ed., 2013, 52, 12619–12623 CrossRef PubMed; (c) T. Schnitzer and H. Wennemers, Helv. Chim. Acta, 2019, 102, e1900070 CrossRef; (d) C. Rigling, J. K. Kisunzu, J. Duschmalé, D. Häussinger, M. Wiesner, M.-O. Ebert and H. Wennemers, J. Am. Chem. Soc., 2018, 140, 10829–10838 CrossRef CAS PubMed.
- (a) W. C. Still, A. Tempczyk, R. C. Hawley and T. Hendrickson, J. Am. Chem. Soc., 1990, 112, 6127–6129 CrossRef CAS; (b) D. Qiu, P. S. Shenkin, F. P. Hollinger and W. C. Still, J. Phys. Chem. A, 1997, 101, 3005–3014 CrossRef CAS; (c) L. L. C. Schrödinger, Schrödinger Release 2018-3, New York, NY, 2018 Search PubMed.
- E. Harder, W. Damm, J. Maple, C. Wu, M. Reboul, J. Y. Xiang, L. Wang, D. Lupyan, M. K. Dahlgren, J. L. Knight, J. W. Kaus, D. S. Cerutti, G. Krilov, W. L. Jorgensen, R. Abel and R. A. Friesner, J. Chem. Theory Comput., 2016, 12, 281–296 CrossRef CAS PubMed.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. J. Goerigk, Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed; (c) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (d) R. Krishnan, J. S. Binkley, R. Seeger and J. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS; (e) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- (a) D. Seebach, U. Groselj, D. M. Badine, W. B. Schweizer and A. K. Beck, Helv. Chim. Acta, 2008, 91, 1999–2034 CrossRef CAS; (b) U. Groselj, D. Seebach, D. M. Badine, W. B. Schweizer and A. K. Beck, Helv. Chim. Acta, 2009, 92, 1225–1259 CrossRef CAS; (c) G. Mloston, M. Celeda, A. Linden and H. Heimgartner, Helv. Chim. Acta, 2009, 92, 1520–1537 CrossRef CAS; (d) D. A. Bock, C. W. Lehmann and B. List, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20636–20641 CrossRef CAS PubMed.
- (a) F. Johnson, Chem. Rev., 1968, 68, 375–413 CrossRef CAS; (b) T. C. Coombs, G. H. Lushington, J. Douglas and J. Aube, Angew. Chem., Int. Ed., 2011, 50, 2734–2737 CrossRef CAS PubMed.
- For the use of 4,5-methanopyrrolidine carboxylic acid as an organocatalyst, see: P. H.-Y. Cheong, K. N. Houk, J. S. Warrier and S. Hanessian, Adv. Synth. Catal., 2004, 346, 1111–1115 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on the synthesis and analytical data of all compounds, and additional spectroscopic, chromatographic, and computations data. See DOI: 10.1039/c9sc05410c |
| ‡ These authors contributed equally. |
| § Note, for 3 and 3b, conformers with the methyl group in equatorial and axial positions were found and conformers that differ in the ring pucker. For a complete listing of all structures and more details, see the ESI† (chapter 3). |
| ¶ Comparable degrees of pyramidalization at N and the same trend of endo/exo pyramidalization ratios were obtained in calculations with enamines derived from pyrrolidinyl amides of Aze, Pro, and Pip; see the ESI† (chapter 3, p. S28) for details. |
| || We used butanal since prior studies with peptidic catalysts also used this aldehyde as a model compound (see, e.g.ref. 12–15) and enamines derived from butanal are less reactive compared to those made from propanal. |
| ** Derivatives of peptide 2 were used since the trans/cis ratio of the DPro–Pip amide bond, which affects the reactivity of the tripeptidic catalysts, is higher compared to that of DPro–Pro of peptide 1, see ref. 13. The kinetic experiments were also performed with analogues of peptide 1 bearing Aze and Pip, respectively, at the N-terminus and yielded the same conclusions, see the ESI† (chapter 5.3) for details. |
| †† NMR spectroscopic studies at −80 °C did not allow for monitoring exo- and endo-pyramidalized conformers of 3′ and 3b′ suggesting that the activation energy between them is <20 kcal mol−1 and rendering direct experimental evidence for the calculated endo/exo pyramidalization ratios difficult. (Note, a N-inversion barrier of ∼8 kcal mol−1 has been determined for N-methylpyrrolidine: J. M. Lehn, J. Wagner, Tetrahedron, 1970, 26, 4227–4240.) |
| ‡‡ The TFA-salts of the peptides were used for ease in accessibility and since previous studies had shown, that the presence of TFA/NMM does not affect the performance of the peptidic catalysts 1 and 2, ref. 12b and c. Higher amounts of aldehyde led to the formation of homoaldol products, which hindered the analysis. For analogous experiments with 1 mol% of the catalysts, see the ESI† (chapter 5.2). |
| §§ Note, the rate order of the nitroolefin is known to depend on the amount of water consistent with the hydrolysis of the iminium ion to the product as an additional rate-limiting step of the reaction (ref. 14). |
| This journal is © The Royal Society of Chemistry 2020 |