
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluenza as a molecular walker
P. H. (Erik)
Hamming†
 ,
Nico J.
Overeem†
and
Jurriaan
Huskens
*
,
Nico J.
Overeem†
and
Jurriaan
Huskens
*
Molecular Nanofabrication Group, MESA + Institute for Nanotechnology, Faculty of Science and Technology, University of Twente, P.O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: j.huskens@utwente.nl
First published on 14th November 2019
Abstract
The surface of the influenza virus is decorated with the receptor-binding protein hemagglutinin (HA) and the receptor-cleaving enzyme neuraminidase (NA). HA is responsible for host cell recognition, while NA prevents aggregation and entrapment, but the intricate mechanism of how the functions of these glycoproteins cooperate and how they are regulated by mutational responses to environmental pressures remains unclear. Recently, several groups have described the motion of influenza over surfaces and reported that this motion is inhibited by NA inhibitors. We argue that the motion of influenza resembles the motility of artificial receptor-cleaving particles called “molecular spiders”. The cleaving of receptors by this type of molecular walkers leads to self-avoiding motion across a surface. When the binding and cleaving rates of molecular spiders are balanced, they move both rapidly and efficiently. The studies of molecular spiders offer new insights into the functional balance of HA and NA, but they do not address the asymmetric distribution of HA and NA on the surface of influenza. We propose that receptor-cleaving molecular walkers could play an important role in the further investigation of the motility of influenza viruses.
 P. H. (Erik) Hamming | P. H. (Erik) Hamming (1993) studied Chemical Engineering at the University of Twente, where he received his Bachelor's in 2014 and his Master's in 2017, working under supervision of Prof. Dr Ir. Jurriaan Huskens and studying weak multivalent binding at surfaces. During his Master's he did an internship in the group of Prof. Kimoon Kim working on Cucurbit[7]uril monolayers and kinetics of complexation. Since July 2017, he is a PhD candidate working in Prof. Dr Ir. Jurriaan Huskens' group. His project is focused on the dynamics of multivalent systems made from intrinsically weak interactions. |
 Nico J. Overeem | Nico Overeem (1993) studied Chemical Engineering at the University of Twente. During his Master's he worked in the group of Prof. Dr Jeroen Cornelissen on the controlled encapsulation of enzyme cascades in virus-like particles and did an internship in the group of Prof. Havazelet Bianco-Peled on the encapsulation of 5-fluorouracil in biodegradable polymersomes. Since October 2016, he is a PhD candidate in the Molecular Nanofabrication group under the supervision of Prof. Dr Ir. Jurriaan Huskens. His research is part of the interdisciplinary “FLAP-chips” project to develop a method to quantify the multivalent binding of influenza viruses using receptor gradients. |
 Jurriaan Huskens | Jurriaan Huskens (1968) obtained his PhD (1994) at the Delft University of Technology with Herman van Bekkum. After postdoctoral stays with Dean Sherry and Manfred Reetz, he became assistant professor with David Reinhoudt at the University of Twente in 1998, and full professor in 2005. He received the Unilever Research Award (1990), a Marie Curie fellowship (1997), the Gold Medal 2007 of the Royal Netherlands Chemical Society, and a Fellowship from the Institute of Advanced Study, Durham University, UK (2019). Present research interests encompass: supramolecular chemistry at interfaces, supramolecular materials, multivalency, nanofabrication, and solar fuels. He is (co)author of about 400 refereed research papers and five patents. |
Introduction
Influenza is among the most common diseases in the world. The disease and mortality of seasonal outbreaks and the deadliness of the rarer pandemic outbreaks have made influenza a prime target of virology. Its facile adaptation to a range of different hosts and its rapid evolution under antigenic pressure originate from two cooperating glycoproteins, the receptor-binding hemagglutinin (HA) and the receptor-cleaving enzyme neuraminidase (NA), and from the rapid mutations that occur in the genetic material that encodes for these proteins.1,2 HA and NA make up the characteristic ‘spikes’, and are jointly responsible for the surface interactions of the virus with a host cell and for its passage through the mucus layer that protects the host cell.1,2 These interactions are far more complex than an ordinary receptor–ligand equilibrium and are only partially understood.It is widely accepted that the receptor-binding function of HA and the receptor cleaving function of NA must be balanced for successful infection.3–5 This functional balance between HA and NA is reflected in the evolution of HA and NA.6–10 It has therefore been suggested that all changes in activity of HA or NA must be followed by an adjustment of the activity of the other to maintain a functional balance.6,7,11,12In vivo studies have shown that lower NA activity leads to less efficient virus replication, but stronger binding by HA can have the same effect.13 It is believed that the role of NA in this balance is to prevent aggregation of the virus and entrapment of progeny viruses on the surface of host cells.1,3,14
Recently, several groups have described a new function of NA in imparting motility of the virus on a surface.15–18 Sakai et al. were the first to report that the motion of influenza over a surface is NA-dependent.1 They also showed that this motility increased cellular uptake of the virus. De Haan et al. found that the receptor-cleaving activity of a few adsorbed viruses is enough to prevent adsorption of new viruses and proposed that the viruses roll over the surface while cleaving off the receptors across the path they follow.2 Vahey and Fletcher found that the organization of HA and NA on filamentous viruses imparts directionality to their motion, and these viruses crawl rather than roll.18 These new observations call for a model that can account for this motility and has predictive power.
Surface-confined motility is not new, neither in biological nor in synthetic systems. In biology, the most famous examples are the kinesin and myosin V motor enzymes, which transport cargo unidirectionally along microtubules and actin filaments.19–21 Vogel et al. showed that kinesin immobilized on a surface could impart motility onto microtubules in a synthetic environment as well.22 Synthetic systems which aim to achieve motion over a surface or track are called ‘molecular walkers’.23 In the simplest form a molecular walker is a biped with feet that can bind to and release from a surface sequentially, and it will act like a molecular walker for as long as at least one foot remains attached to the surface.24 Its movement depends on Brownian motion and will therefore be diffusive and non-directional, unless it can move over a gradient or is inhibited in one direction by ratcheting.25,26
Mimicking directional motion as shown by kinesin and myosin V requires the walker to overcome Brownian motion and requires energy input.27 The natural motor enzymes use ATP as fuel, whereas synthetic systems have a wider array of possible energy sources.28,29 The first molecular walker used DNA strands as fuel, quickly followed by an example that used ATP as fuel.23,30 Instead of using the consumption of a chemical fuel to impose a strict directionality in each individual step, overall directionality can be attained when the direction of the steps is biased, for example using an enzyme with a chiral preference to cleave the back leg.31 For a comprehensive review of different walker designs, energy sources and mechanisms, see: Leigh et al.29
Stojanovic et al. introduced a class of molecular walkers, called ‘molecular spiders’, that use their receptors as fuel.32 A molecular spider consists of a body with multiple catalytically active legs that cleave the receptors to which they bind. The cleaving of its receptors is the energy source for biased motion away from their starting position, leading to self-avoiding walking. Its essential design is simple enough to allow systematic study, both experimentally and in silico, while their receptor-cleaving dependent motion may be comparable to that of influenza, as will be explored below.
In this review we aim to show that the combined roles of HA and NA in the surface-bound motility of influenza can be understood by looking at receptor-cleaving molecular walkers. We first explain the mechanism of surface-bound motion in receptor-cleaving molecular walkers. Then we describe how this mechanism leads to faster-than-diffusive and self-avoiding motion and how these properties are influenced by the design of the walker. The roles of the HA and NA proteins are discussed, as well as their presentation on the surface of influenza. We comprehensively review the proposed mechanisms that have been published about influenza movement on cell surfaces. We argue that both the “rolling” and the “Brownian ratchet” mechanism requires collaborative interactions of HA and NA that make influenza a molecular walker and propose that both models can explain how influenza can efficiently cross the mucus layer. We then discuss how properties of molecular spiders may be favorable for influenza in avoiding clearance by the mucus and in finding a suitable site for host cell infection. After that, we discuss the similarities and differences in the motility of molecular spiders and influenza. Finally, we indicate research directions for molecular walkers that have high relevance for understanding influenza. We will not discuss the role of HA and NA inside the cell, as their role in the complete cycle of infection has been described elsewhere.33,34
Molecular spiders and superdiffusive walking
Molecular spiders interact with a surface through their legs. All legs are identical and cleave the sites to which they bind.32,35,36 Experimental setups of molecular spiders generally use DNA on the surface and DNAzymes as legs.32,35,37,38 The DNA on the surface is cleaved by the DNAzyme, resulting in a shorter DNA strand (Fig. 1a). The DNAzymes are still able to bind to these shorter strands but have a diminished affinity and residence time.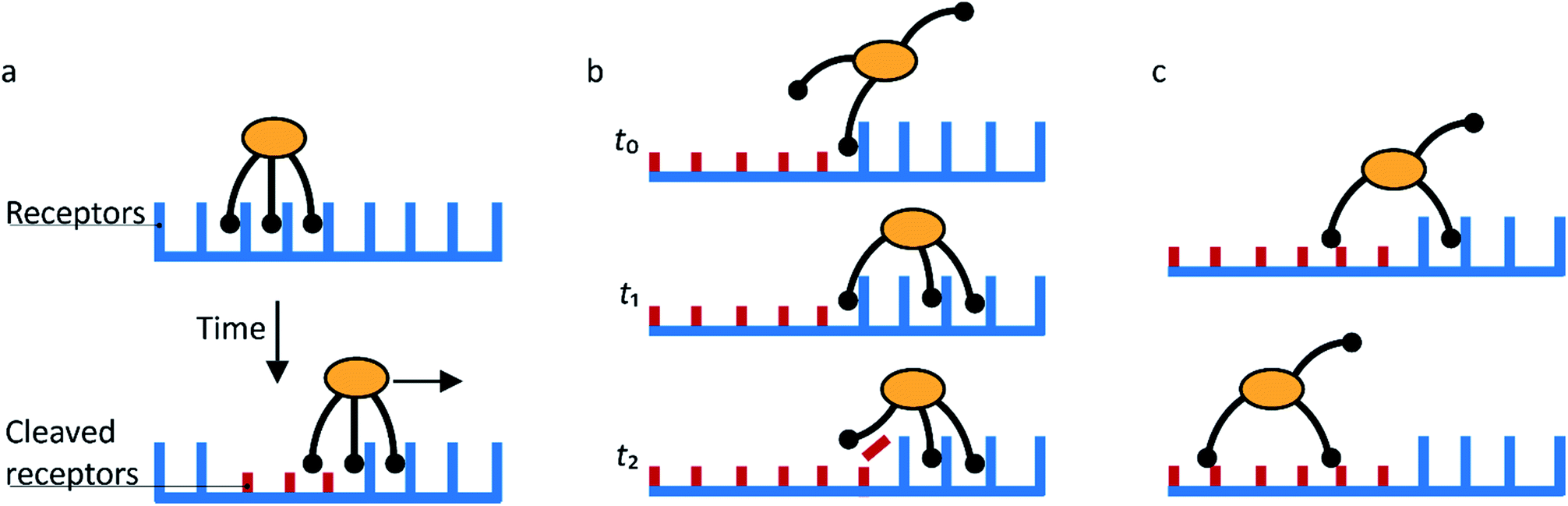 | ||
| Fig. 1 Mechanism for directional motility in molecular spiders.36,39 (a) A molecular spider consists of a rigid body with several legs. The legs bind to receptors on a surface, cleaving them as they go. The legs can bind to a cleaved receptor, but have a lower residence time. (b) The difference in residence time can lead to a bias in movement. At t0, the spider is attached at the boundary between fresh and cleaved receptors with one leg. At t1, the legs are more likely to be bound to fresh than to cleaved receptors due to the difference in residence times. At t2, the first leg detaches and leaves a cleaved receptor behind, shifting the boundary. (c) The spider is either at the boundary and moving with a bias towards fresh receptors, or the spider is in a patch of cleaved receptors where all legs have low residence time and diffusion is fast. | ||
When a molecular spider is on the boundary between uncleaved substrates and cleaved products, it experiences a gradient in affinity and residence times, leading to a directional bias towards the uncleaved substrates. The cleaving of receptors acts as a fuel to continually create the gradient. With each step of the spider, the boundary shifts one step as well (Fig. 1b). For as long as the spider remains at this self-propelling boundary, it moves away from the starting position faster than it would by normal diffusion. This mode of motion is called superdiffusivity.36 The directionality is temporarily lost if the spider moves a step away from the boundary (Fig. 1c). The overall bias depends therefore primarily on the chance that the spider remains at the moving boundary.
The superdiffusive motion of a molecular spider is weakly self-avoiding; it is possible for the spider to revisit a site, but it will spend most of its time visiting new sites. Some molecular spiders cannot revisit former sites at all and are therefore strictly self-avoiding; these are also called ‘burnt-bridges’ walkers.29 When a spider leaves the boundary between cleaved and uncleaved receptors or reaches a previously visited area, it undergoes non-directional diffusive motion over the surface. The low residence time of the legs on cleaved receptors results in fast diffusion of the spider. The spider thus spends only limited time in this diffusive state and quickly returns to a boundary where it again undergoes biased motion away from the cleaved receptors. This weakly self-avoiding walk is a more efficient way of probing an area than diffusion.36,40
The superdiffusivity of a molecular spider is determined by how well it recognizes the gradient at the boundary between cleaved and uncleaved receptors. The state of a spider on this boundary is defined by the length of its legs L and the number of legs n (Fig. 2a). The superdiffusive behavior of a spider emerges from its multivalent character, because one-legged spiders cannot recognize a gradient.41 Whether additional legs increase superdiffusivity depends on the length of the legs. The superdiffusive behavior of molecular spiders with short legs benefits from a high number of legs,40 whereas a high number of legs decreases the superdiffusivity of molecular spiders with long and flexible legs.42
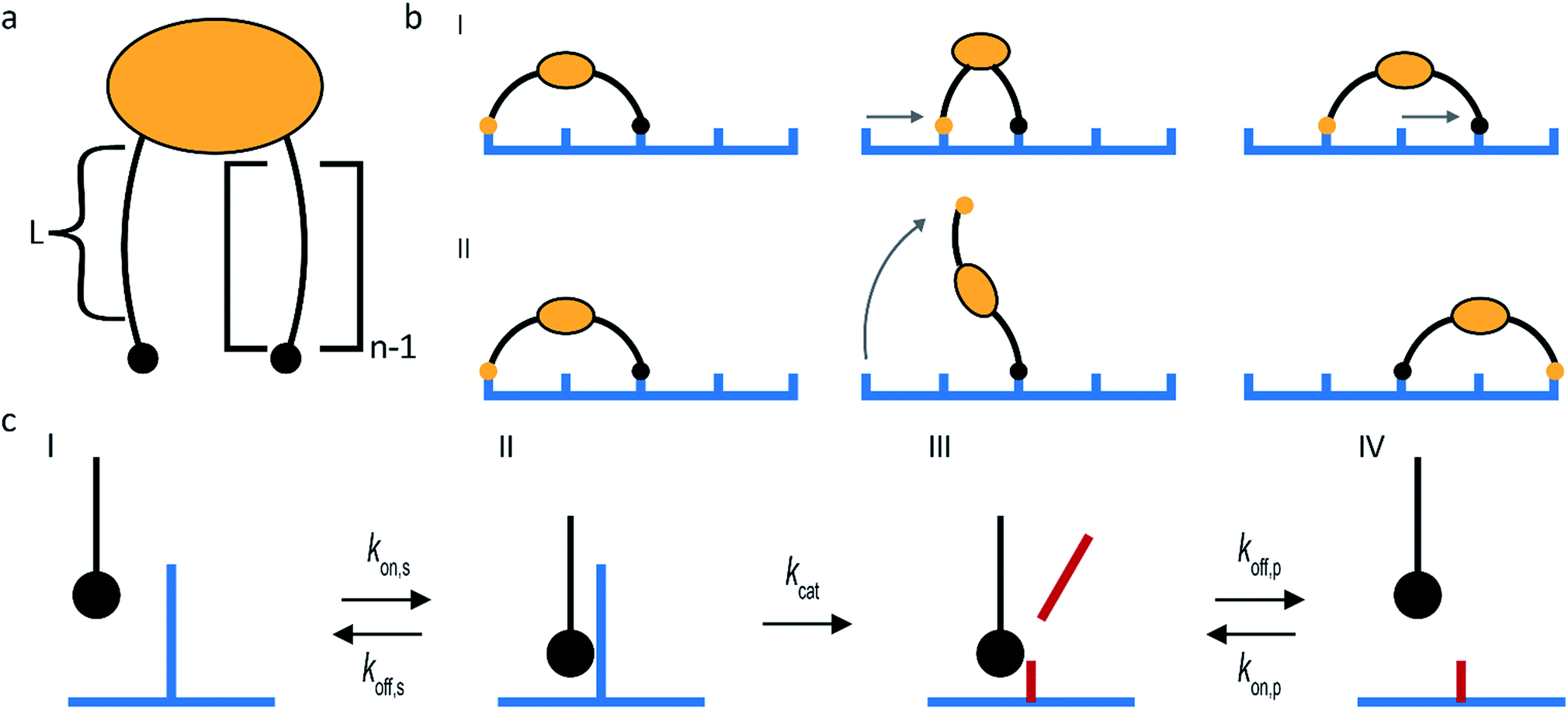 | ||
| Fig. 2 Main design parameters in molecular spiders. (a) The number of legs (n) of a spider and the length of each leg (L) define the boundary state of the spider. (b) Molecular spiders can walk using an inchworm (I) or hand-over-hand (II) motif. (c) The kinetics of interaction between the substrate and an individual leg. | ||
The maximum size of a step is determined by the length of the legs of a molecular spider and by its gait. Molecular spiders tend to move faster if they use a hand-over-hand or rolling motion than if they use inchworm motion.38 It is faster because a hand-over-hand gait limits the step size to how far the spider can stretch, whereas the inchworm gait limits the step size to the farthest point that is already attached (Fig. 2b).
The rate of each step is determined by the kinetics of the catalytically active legs and by the dissociation rate of a leg from a cleaved receptor (Fig. 2c). Stronger binding of a leg towards an uncleaved receptor means a lower Michaelis–Menten constant and can therefore increase the speed of walking. Less intuitive is that stronger binding to both the uncleaved and the cleaved receptors can increase the superdiffusivity of a spider, not only compensating the slower release of its legs but even increasing its velocity.35,38,43
To maintain a molecular walk over longer distances, a functional balance is needed between the cleaving activity and the binding activity of a molecular spider. A higher cleaving activity leads to faster motion, but if its cleaving activity exceeds its binding activity the spider dissociates.32,42
For the exploration of larger surfaces, dissociation of a molecular spider can even be favorable. If a spider finds itself in an area that was visited before, the low residence time of legs on cleaved receptors increases the probability that all legs are unbound at the same time, releasing the spider from the surface.36,38 After dissociation, the spider can diffuse through the solution for some distance and associate again with a higher association constant for unvisited areas. This type of motion could be regarded as a “self-avoiding hop”, so that molecular spiders are weakly self-avoiding over multiple length scales.
Motility of influenza virus
Of the two glycoproteins on the surface of influenza virus that are involved in host cell binding, HA is the most abundant. HA is a trimeric protein that can bind sialic acid-terminated glycans with millimolar affinities (Fig. 3a).5,44 The HA conveys host cell specificity to the virus by binding with greater affinity to a sialic acid that has either a 2,3-linkage to a neighboring galactose (2,3-SLN), which is more abundant in avian intestines, or a 2,6-linkage (2,6-SLN), which is more abundant in the human upper respiratory tract.1,34 This specificity is amplified by the multivalent presentation of HA on the virus so that small differences in the affinity of HA lead to major differences in affinity of the virus.44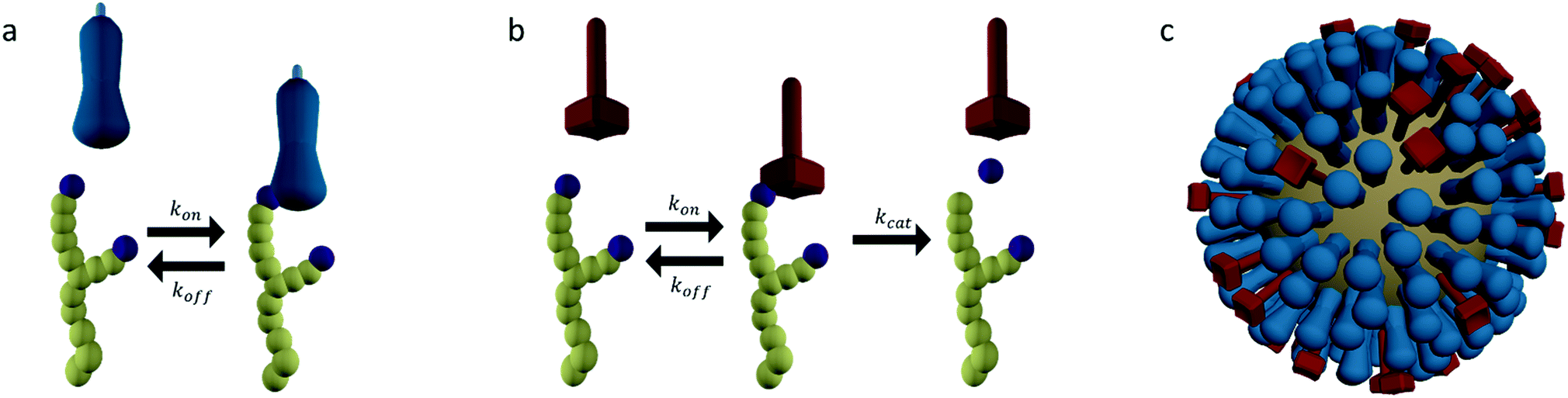 | ||
Fig. 3 (a) Hemagglutinin (HA) is a trimeric protein that binds to sialic acid-terminated glycans in a reversible manner. (b) Neuraminidase (NA) is a tetrameric protein that binds and cleaves sialic acid-terminated glycans. (c) HA and NA are present on the surface of influenza in high copy numbers, approximately in a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. :1 ratio. | ||
The secondary glycoprotein, NA, is a tetrameric enzyme that binds sialic acid-terminated glycans and then cleaves the bond between the sialic acid and galactose units (Fig. 3b). NA typically has a Michaelis–Menten constant Km in the low millimolar range and a turnover number kcat of several 10s per second.5,45 In some virus strains kcat is reduced to tenths per second.46 This magnitude of Km means that the initial contribution of NA to the binding of a virus is comparable to that of HA. The enzyme–substrate complex is, however, short lived as its lifetime is not greater than kcat−1. Cleaving off the terminal sialic acid by NA reduces the affinity of HA to the glycan by more than 10-fold.47,48
The HA and NA are presented on the surface of influenza in high copy numbers in an approximately 6:1 ratio (Fig. 3c).49 The virus is approximately 100 nm in diameter and can be almost spherical or filamentous, with lengths up to a micrometer. HA and NA are densely packed on the surface, but not entirely random. Patches or even complete separation of HA and NA into domains have been reported.18,49,50 This distribution likely originates from an interplay between entropic factors and differences in the preferred membrane curvature between HA and NA.50–53 It is likely that the organization of HA and NA has an influence on the way their functions cooperate.
Motility of influenza was first reported by Sakai et al.15 They tracked the motion of influenza viruses over fetuin-modified glass with total internal reflection fluorescence microscopy (TIRF). When adding an NA inhibitor, the movement of the viruses was completely blocked. They concluded that the virus moves over a cell surface not by lateral diffusion of the virus along receptor sites, but by NA-initiated exchange of receptors from one binding pocket to the next. They distinguished two modes of movement: a slower and more frequent “crawling”, and a faster and rarer “gliding” (Fig. 4a). Lower NA activity in mutant viruses led to slower motility and lower occurrence of gliding steps. The gliding steps were also shorter. Therefore, they concluded that NA is not only necessary to initiate movement, but also to sustain gliding steps. The NA-dependent motion of influenza was not only demonstrated on artificial surfaces, but also on live cells. Adding an NA inhibitor or using a mutant with less active NA both led to decreased endocytosis of the viruses.
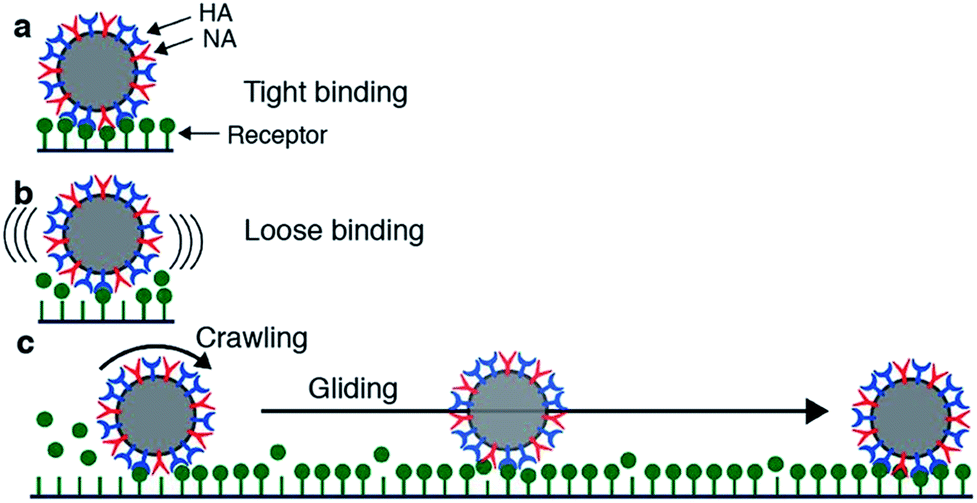 | ||
| Fig. 4 Mechanism for virus motion proposed by Sakai et al.15 (a) Influenza binds tightly to a surface with multiple HA-receptor interactions. (b) NA cleaves receptors, which decreases the number of interactions and initiates motility. (c) A loosely attached virus performs crawling and gliding motions by iterative association and dissociation of HA-receptor interactions, until it reaches a site where it can form multiple interactions and again bind tightly to the surface. Reprinted from ref. 15, with permission from Springer Nature, licensed under CC BY 4.0. | ||
In a kinetic analysis of the binding of influenza HA and NA with biolayer interferometry (BLI), De Haan et al. found that the cleaving of receptors by adsorbed viruses was far more complete than suggested by Fig. 4c.16 They allowed various concentrations of influenza to bind to a receptor-functionalized sensor in the presence of NA inhibitor, followed by transfer of the sensor with adhering viruses to a buffer solution. If the buffer also contained NA inhibitor, the viruses remained adhering, but without inhibitor, they dissociated completely. When a lower amount of virus was adsorbed, the viruses dissociated more slowly. Then they regenerated the sensors at pH 2, removing all viruses, but leaving the receptors on the sensors. The regenerated sensors were subsequently allowed to re-bind viruses at the maximum concentration. While the sensors that were placed in buffer with NA inhibitor were unaffected, the sensors where the virus was dissociated could bind significantly fewer new viruses.
To test whether the deactivation of the sensors was due to receptor cleavage by NA, De Haan et al. blocked two sensors partly with a virus with inactive NA (Fig. 5). Then they incubated the partly blocked sensors and a third sensor with a different virus that had active NA, with NA inhibitor present at one of the two blocked sensors. Then they regenerated the sensors and allowed the virus with inactive NA to bind on all three sensors. Binding was uninhibited at the sensor that was protected with NA inhibitor, partly inhibited at the sensor that was partly blocked, and fully inhibited at the exposed sensor. With these two experiments they showed that NA-dependent “rolling” of a low amount of virus could cleave receptors from a large surface area.
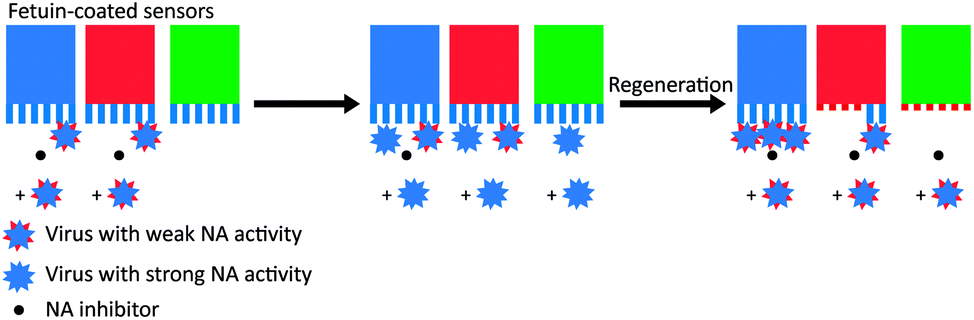 | ||
| Fig. 5 Motility of influenza is driven by NA activity. De Haan et al. partially blocked receptors on BLI sensors with viruses that had inactive NA and then exposed the sensors to a virus with active NA.16 One sensor (in blue) was protected with the NA inhibitor oseltamivir carboxylate (OC), one sensor (in red) was only locally blocked by the NA-inactive virus, and one sensor (in green) was left fully unprotected. After regeneration of the sensors, exposure to new virus showed that receptors were cleaved from all unprotected areas. Adapted from ref. 16 with permission from Public Library of Sciences, licensed under CC BY 4.0. | ||
Vahey and Fletcher showed that the asymmetric distribution of HA and NA on filamentous viruses imparted a directional bias in over several micrometers.18 Filamentous viruses are more common in clinical isolates, whereas laboratory-grown strains often produce more spherical viruses.52,54 Vahey and Fletcher hypothesized that the filamentous shape together with non-uniform distribution of HA and NA promote efficient penetration of viruses through mucus. They labelled the HA, NA and nucleoprotein (NP) of a filamentous virus and showed that HA was more or less homogenously distributed, NP was clustered at one pole and NA was more abundant at the same pole (Fig. 6). Tracking the viruses with TIRF and imaging with super-resolution microscopy, they showed that the virus moved directionally away from the NA-rich pole. By labeling cleaved receptors with the fluorescent lectin ECL, they showed that receptors were cleaved in the path of the virus. They posed that the cleaving of receptors functions as a Brownian ratchet by restricting backward motion. To predict the effect of the diffusion coefficient of receptors on the mean square displacement of viruses, they made a computational model. They found that the velocity of viruses is the highest when the diffusion coefficient of receptors is small, but not zero. The values for the diffusion coefficient where motility is fastest are consistent with the diffusion coefficients of sialic acid in mucus. They cultured mucus-secreting cells to test whether the virus could also move through a three-dimensional environment and observed tracks similar to those observed on two-dimensional surfaces.
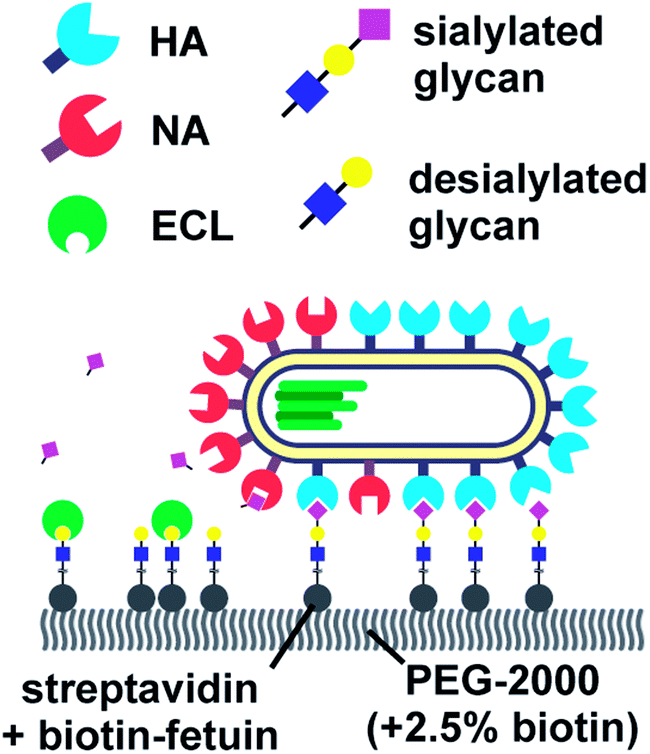 | ||
| Fig. 6 By labelling HA, NA and cleaved receptors, Vahey and Fletcher showed that the asymmetric organization of HA and NA imparts directional motility in filamentous viruses.18 Reprinted from ref. 18 with permission from eLife Sciences Publications, licensed under CC BY 4.0. | ||
Block et al. also tracked influenza viruses, but with the aim of using its mobility to quantify the number of receptor–ligand interactions.17 They tracked labelled influenza viruses with TIRF on a supported lipid bilayer (SLB) containing glycolipid receptors (Fig. 7). Because multivalent binding decreases the dissociation rate constant koff and the diffusion coefficient D, the average valency scales with 1/D. In a plot of koffversus 1/D, the effect of multivalent binding appears as an exponential decline, but is actually composed of subpopulations that share the same D values (Fig. 7). Interestingly, there is a peak structure visible with elevated koff values for 1/D ∼ 5 s μm−2. This peak decreases with added NA inhibitor and is therefore ascribed to receptor cleaving by NA. Block et al. claim that the lateral diffusion of the glycolipids to which influenza binds remains significantly faster than the NA-dependent motility that was reported by Sakai et al.15,17
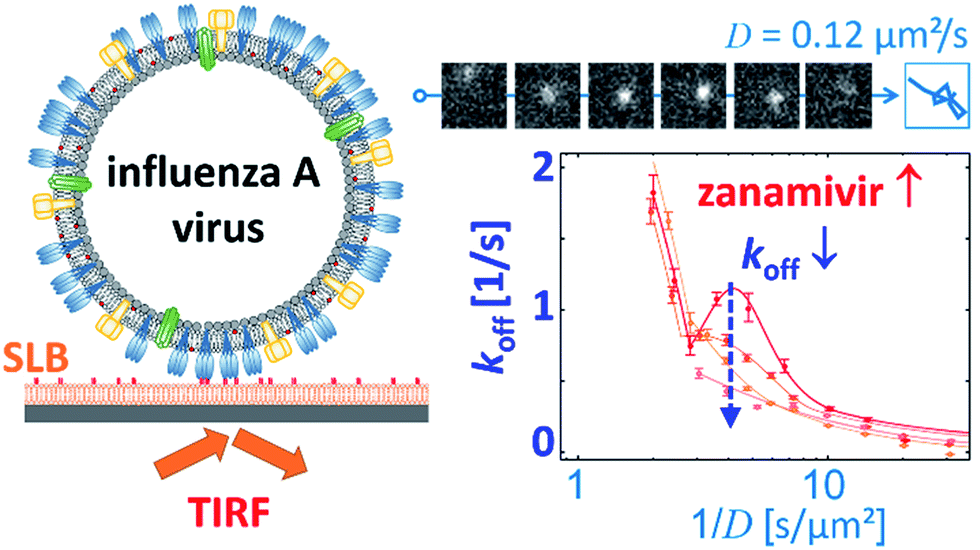 | ||
| Fig. 7 Block et al. tracked influenza viruses on supported lipid bilayers to quantify the number of interactions with glycolipids.17 They observed elevated koff values for 1/D ∼ 5 s μm−2, which decreased with added NA inhibitor. Reprinted with permission from ref. 17. Copyright 2019 American Chemical Society. | ||
It is interesting to note the differences in setup and observed mobility between these four publications. Both Block et al. and Sakai et al. used an egg-adapted influenza strain with HA and NA from the IAV strain Aichi/2/68 (H3N2).15,17 Vahey and Fletcher used HA and NA from A/WSN/33 (H1N1).18 De Haan et al. used the HA and NA from the same strain for its lower NA activity and used A/Puerto Rico/8/34 Mount Sinai (H1N1) to demonstrate the removal of receptors.16 In an assay of NA activity, Aichi/2 was approximately fivefold less active than WSN, which was fivefold less active than PR/8 Mt. Sinai.5,16 It is therefore remarkable that Block et al. report a motion that is up to two orders of magnitude faster than Sakai et al., whereas Vahey and Fletcher reported a motion that is an order of magnitude slower than Sakai et al.15,17,18 Unlike De Haan et al., who reported that NA contributes to the association of viruses,16 Block et al. report an increase of association rate with added NA inhibitor.17
We think it likely that the differences described above are primarily due to the different structures of the surfaces that were used in these studies. Block et al. used 1 mol% of glycolipid GDa1 in an SLB;17 Sakai et al. used 10% fetuin with albumin adsorbed on glass;15 De Haan et al. used BLI sensors with streptavidin and biotinylated fetuin;16 and Vahey and Fletcher used a PEG2000 brush on glass with streptavidin and biotinylated fetuin.18 Sakai et al. reported an optimum in the receptor density, while Vahey and Fletcher reported an optimum in the diffusion coefficient of sialic acid.15,18 We expect that there exists also an optimum in the conformational flexibility of the receptors. These differences show that the motility of a virus depends on a combination of many properties of both the virus and the surface.
The role of motility in host cell recognition
Sakai et al. found that the NA-dependent motility of influenza contributes to its endocytosis, but did not study the mechanism of this contribution.15 They propose that the motility of the virus allows lateral motion even if receptors are restricted from diffusion by membrane rafts or other rigid structures. They also argue that it must play an important role in intercellular migration.The studies of molecular spiders offer two additional possible advantages of molecular walking over random lateral diffusion. Firstly, the weakly self-avoiding walk provides a more efficient pattern to search the surface of a cell for a suitable location to bind to induce endocytosis (Fig. 8a). Secondly, the molecular walk provides a bias towards high receptor densities (Fig. 8b). Because endocytosis of influenza is mediated by clathrin-coated as well as noncoated pits, it is likely that influenza searches for these pits or for locations where they will form.34,55 The bias towards higher receptor densities follows from the mechanism of walking and may provide a means of recognition to the virus if such locations prove to have a higher density of receptors. Higher densities are also favored by the ability of influenza to bind multiple receptors through HA,56 but without motility the chance of binding to an area of high receptor density remains relatively low for an individual virus.
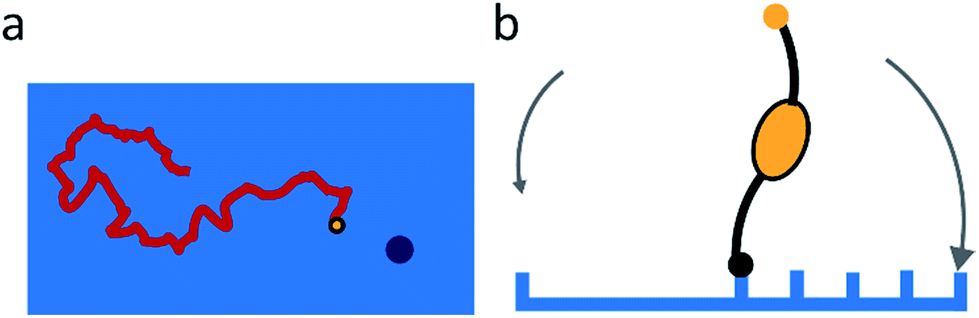 | ||
| Fig. 8 (a) Self-avoiding walking gives rise to an efficient search pattern to find clathrin-coated pits. (b) The motility of molecular spiders is biased towards a higher density of receptors. | ||
De Haan et al. and Vahey and Fletcher claim that the primary advantage of motility for the virus is to penetrate the mucus layer.16,18 This mucus layer consists of a dense gel of mucins that is propelled by a layer of cilia that extend from the epithelial cells in the respiratory tract (Fig. 9a).57,58 The mucins are glycoproteins with a sugar content of 50–80 wt% and rich in sialic acid.59 The mucus can therefore easily entrap sialic acid-binding viruses such as influenza and clear them from the respiratory tract.60
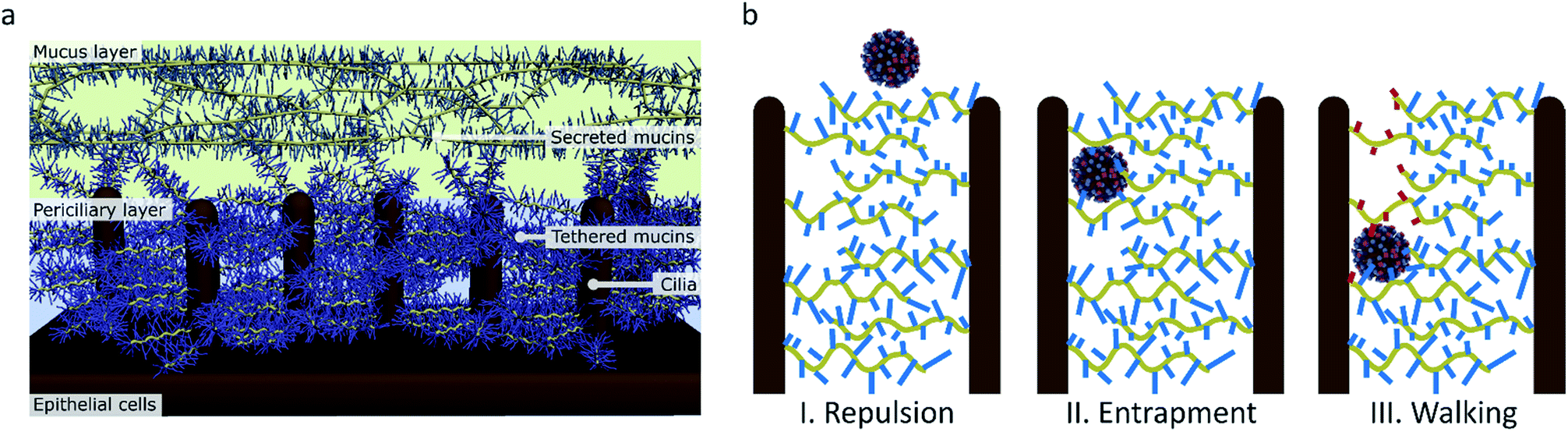 | ||
| Fig. 9 (a) The structure of the mucus in human airways as proposed by Rubinstein et al.57 (b) The function of molecular walking of influenza in crossing the mucus. (I) When there is no interaction, particles are repelled by the charged brush. (II) Particles that have an affinity for sialic acid are entrapped. (III) Influenza, which binds and cleaves sialic acid, can walk through the mucus. | ||
It has been shown that influenza uses NA to cleave sialic acid when the virus passes through the mucus and thus prevents entrapment.61,62 The sialic acid in the human mucus is mostly 2,3-linked, and human-adapted viruses usually have an HA specificity for 2,6-SLN, but an NA specificity for both 2,3- and 2,6-SLN.1,2,63,64 The effect of this cleaving on influenza was evidenced by an in vitro experiment where infection by a virus with weak NA activity was inhibited by human mucus, but for a virus with stronger NA activity it was not.65 It is believed that this prevention of entrapment of entering viruses in the mucus is essential for aerosol transmission between humans.4,66
Preventing entrapment and clearance of the viruses may, however, not be the only reason why this receptor cleavage is important for the virus in crossing the mucus. While the mucus layer allows diffusion of inert particles of the size of influenza virus, the periciliary layer underneath contains a mesh that is dense enough to exclude even 40 nm particles.57,60 This mesh is formed by mucins that are tethered in a bottle brush-like structure to the cilia.67 It is therefore likely that influenza, instead of relying on diffusion that is inhibited by interactions with sialic acid, must use an active motion to cross over the mucins and cilia towards the surface of the respiratory epithelial cells (Fig. 9b).
Lessons from molecular walkers for influenza
We have described how directional motion emerges both in molecular spiders and influenza from their receptor cleaving properties. In this section we first address the lessons from molecular spiders that may apply to influenza and how they can help understand the changes that influenza viruses undergo in their adaptation. Thereafter, we discuss the properties of the receptor–ligand interactions of influenza that are not addressed by molecular spiders and how they may be addressed by other molecular walkers.Probably the most interesting lesson from molecular spiders is that a higher cleaving activity leads to faster motion, but if the cleaving activity exceeds the binding activity the spider dissociates.32,42 This suggests that if there is a selective pressure on an influenza population to develop increasing motility, both the NA activity and the affinity of HA will increase. Conversely, if the affinity of HA decreases, there will be a selective pressure to decrease its NA activity as well. This co-optimization may offer an explanation why influenza acquired both stronger binding to, and faster cleaving of, 2,6-SLN during circulation in the human population.7
Another trend that was reported for molecular spiders is that the superdiffusivity of a spider with short legs increases with the number of legs.40 The HA and NA of influenza are too short and rigid to reach behind another trimer or tetramer, so it is likely that the superdiffusivity of influenza can benefit from the high number and density of glycoproteins on its surface. Because filamentous viruses have more interactions with the cell surface because of their larger contact area, their superdiffusivity may be increased over spherical viruses.
Different from molecular spiders is the gait of influenza. Molecular spiders move faster by rolling than by inchworm motion,38 but Vahey and Fletcher demonstrated that the motion of influenza is more inchworm-like.18 It remains, however, interesting to see if spherical influenza viruses would move like the filamentous viruses of Vahey and Fletcher or would shift to rolling.
The main reason why Vahey and Fletcher found an inchworm gait is that HA and NA are organized in trimers and tetramers that are asymmetrically distributed on the surface of the virus,18 unlike molecular spiders which have identical legs that perform both the binding and cleaving actions. It would be interesting to see how a molecular walker would behave where only a fraction of the legs is catalytically active and how an asymmetric distribution of these legs would affect its behavior.
In contrast with Vahey's and Fletcher's observations for influenza, molecular spiders do not need a separation of their binding and cleaving action for directional motion. This separation of actions may benefit influenza by allowing adjustment of its functional balance of binding and cleaving through independent variation of the cleaving activity and the binding activity of NA, the affinity of HA, and the ratio of HA to NA copy numbers. Indeed, some H3N2 strains have adapted in the human population by developing binding through their NA with almost undetectable HA binding.46,68
Large rolling motors developed by Salaita et al. share their walking mechanism with molecular spiders, although they are powered by enzymes in solution.69,70 These rolling motors demonstrate that receptor-cleaving molecular walkers of micrometer size can be propelled with high speed over long distances due to their polyvalency. Interestingly, if these particles form dimers, they roll over their shared longitudinal axis,69 in contrast with influenza's inchworm motion.18 This may indicate that the organization of HA and NA on influenza dictates its gait.
Perhaps the most important aspect of influenza motility that has neither been addressed with molecular spiders nor in the experimental studies of influenza motility, is the relation between receptor specificity and directionality of the virus. We briefly mentioned that the human mucus is rich in 2,3-SLN, whereas the epithelial cells that are most commonly infected by influenza are richer in 2,6-SLN. We hypothesize that the balance between cleaving and binding in human influenza for 2,3-SLN is different from 2,6-SLN. Moderate binding to 2,3-SLN combined with fast cleaving would result in strongly superdiffusive motion with a high chance of dissociation. This dissociation is, however, strongly inhibited by the 3D structure of the mucus. At the cell surface, where dissociation is more disadvantageous, a stronger binding to 2,6-SLN combined with moderate cleaving still results in superdiffusive motion to find a suitable spot for endocytosis, but with lower chance of dissociation. At the interface between the mucus and the cell, the binding to 2,6-SLN on the cell would then be strongly favored over 2,3-SLN on the mucus.
Conclusion
We have argued that the multivalent display of NA and HA on the surface of influenza makes the virus a receptor-cleaving molecular walker. In its molecular walking, the virus resembles the synthetic molecular spiders, but the virus may achieve additional directionality by the asymmetric organization of HA and NA on its surface. It is likely that the surface-bound motility of influenza plays a key role in the transmission and propagation of the virus.We expect that studies of artificial receptor-cleaving molecular walkers on heterogeneous surfaces can further our understanding of influenza as viruses that actively find access to their host cells, can help to explain and predict the evolutionary changes of influenza viruses, and can help identify possible new targets for antivirals in the transmission cycle of influenza.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Erhard van der Vries (UMC Utrecht), Erik de Vries and Cornelis A. M. de Haan (University Utrecht) for helpful discussions. We acknowledge the Volkswagen Stiftung (Flap-Chips project) and the Netherlands Organization for Scientific Research (NWO, TOP project 715.015.001) for funding.References
- M. de Graaf and R. A. M. Fouchier, EMBO J., 2014, 33, 823–841 CrossRef CAS PubMed.
- L. Byrd-Leotis, R. D. Cummings and D. A. Steinhauer, Int. J. Mol. Sci., 2017, 18, 1541 CrossRef PubMed.
- G. Neumann and Y. Kawaoka, Virology, 2015, 479–480, 234–246 CrossRef CAS PubMed.
- R. Xu, X. Zhu, R. McBride, C. M. Nycholat, W. Yu, J. C. Paulson and I. A. Wilson, J. Virol., 2012, 86, 9221–9232 CrossRef CAS.
- D. J. Benton, S. R. Martin, S. A. Wharton and J. W. McCauley, J. Biol. Chem., 2015, 290, 6516–6521 CrossRef CAS PubMed.
- A. S. Gambaryan and M. N. Matrosovich, Biochemistry, 2015, 80, 872–880 CAS.
- R. Wagner, M. Matrosovich and H.-D. Klenk, Rev. Med. Virol., 2002, 12, 159–166 CrossRef CAS PubMed.
- A. K. Behera, S. Basu and S. S. Cherian, Gene, 2015, 557, 19–27 CrossRef CAS PubMed.
- M. J. Ward, S. J. Lycett, D. Avila, J. P. Bollback and A. J. Leigh Brown, BMC Evol. Biol., 2013, 13, 222 CrossRef PubMed.
- S. Diederich, Y. Berhane, C. Embury-Hyatt, T. Hisanaga, K. Handel, C. Cottam-Birt, C. Ranadheera, D. Kobasa and J. Pasick, J. Virol., 2015, 89, 10724–10734 CrossRef CAS PubMed.
- U. Gulati, W. Wu, S. Gulati, K. Kumari, J. L. Waner and G. M. Air, Virology, 2005, 339, 12–20 CrossRef CAS PubMed.
- S. J. Baigent and J. W. McCauley, Virus Res., 2001, 79, 177–185 CrossRef CAS PubMed.
- F. Gen, S. Yamada, K. Kato, H. Akashi, Y. Kawaoka and T. Horimoto, Arch. Virol., 2013, 158, 1003–1011 CrossRef CAS PubMed.
- Q. Chen, S. Huang, J. Chen, S. Zhang and Z. Chen, PLoS One, 2013, 8, e54334 CrossRef CAS PubMed.
- T. Sakai, S. I. Nishimura, T. Naito and M. Saito, Sci. Rep., 2017, 7, 45043 CrossRef CAS PubMed.
- H. Guo, H. Rabouw, A. Slomp, M. Dai, F. van der Vegt, J. W. M. van Lent, R. McBride, J. C. Paulson, R. J. de Groot, F. J. M. van Kuppeveld, E. de Vries and C. A. M. de Haan, PLoS Pathog., 2018, 14, e1007233 CrossRef PubMed.
- M. Müller, D. Lauster, H. H. K. Wildenauer, A. Herrmann and S. Block, Nano Lett., 2019, 19, 1875–1882 CrossRef PubMed.
- M. D. Vahey and D. A. Fletcher, eLife, 2019, 8, 1–24 CrossRef PubMed.
- S. M. Block, Biophys. J., 2007, 92, 2986–2995 CrossRef CAS PubMed.
- W. O. Hancock, Nat. Rev. Mol. Cell Biol., 2014, 15, 615–628 CrossRef CAS PubMed.
- J. A. Hammer and J. R. Sellers, Nat. Rev. Mol. Cell Biol., 2012, 13, 13–26 CrossRef CAS PubMed.
- J. Clemmens, H. Hess, R. Lipscomb, Y. Hanein, K. F. Böhringer, C. M. Matzke, G. D. Bachand, B. C. Bunker and V. Vogel, Langmuir, 2003, 19, 10967–10974 CrossRef CAS.
- W. B. Sherman and N. C. Seeman, Nano Lett., 2004, 4, 1203–1207 CrossRef CAS.
- D. A. Leigh, U. Lewandowska, B. Lewandowski and M. R. Wilson, in Multistage Molecular Methods in Applied chemistry, 2014, pp. 111–138 Search PubMed.
- A. Perl, A. Gomez-Casado, D. Thompson, H. H. Dam, P. Jonkheijm, D. N. Reinhoudt and J. Huskens, Nat. Chem., 2011, 3, 317–322 CrossRef CAS PubMed.
- J. W. Fredy, A. Méndez-Ardoy, S. Kwangmettatam, D. Bochicchio, B. Matt, M. C. A. Stuart, J. Huskens, N. Katsonis, G. M. Pavan and T. Kudernac, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11850–11855 CrossRef CAS PubMed.
- T. R. Kelly, Angew. Chem., Int. Ed., 2005, 44, 4124–4127 CrossRef CAS PubMed.
- Z. Zhang, Y. Goldtzvik and D. Thirumalai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E9838–E9845 CrossRef CAS PubMed.
- M. von Delius and D. A. Leigh, Chem. Soc. Rev., 2011, 40, 3656 RSC.
- P. Yin, H. Yan, X. G. Daniell, A. J. Turberfield and J. H. Reif, Angew. Chem., Int. Ed., 2004, 43, 4906–4911 CrossRef CAS PubMed.
- C. J. Martin, A. T. L. Lee, R. W. Adams and D. A. Leigh, J. Am. Chem. Soc., 2017, 139, 11998–112002 CrossRef CAS PubMed.
- R. Pei, S. K. Taylor, D. Stefanovic, S. Rudchenko, T. E. Mitchell and M. N. Stojanovic, J. Am. Chem. Soc., 2006, 128, 12693–12699 CrossRef CAS PubMed.
- P. Chlanda and J. Zimmerberg, FEBS Lett., 2016, 590, 1940–1954 CrossRef CAS PubMed.
- X. Sun and G. R. Whittaker, in Advances in Experimental Medicine and Biology, 2006, vol. 790, pp. 72–82 Search PubMed.
- K. Lund, A. J. Manzo, N. Dabby, N. Michelotti, A. Johnson-Buck, J. Nangreave, S. Taylor, R. Pei, M. N. Stojanovic, N. G. Walter, E. Winfree and H. Yan, Nature, 2010, 465, 206–209 CrossRef CAS PubMed.
- O. Semenov, M. J. Olah and D. Stefanovic, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 021117 CrossRef PubMed.
- Y. Tian, Y. He, Y. Chen, P. Yin and C. Mao, Angew. Chem., Int. Ed., 2005, 44, 4355–4358 CrossRef CAS PubMed.
- L. Samii, H. Linke, M. J. Zuckermann and N. R. Forde, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 81, 1–11 CrossRef PubMed.
- M. J. Olah and D. Stefanovic, in Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics), 2011, vol. 6937 LNCS, pp. 160–174 Search PubMed.
- C. S. Korosec, M. J. Zuckermann and N. R. Forde, Phys. Rev. E, 2018, 98, 1–11 CrossRef.
- D. Stefanovic, in Proceedings of the 5th ACM International Conference on Nanoscale Computing and Communication - NANOCOM'18, ACM Press, New York, USA, 2018, pp. 1–2 Search PubMed.
- L. Samii, G. A. Blab, E. H. C. Bromley, H. Linke, P. M. G. Curmi, M. J. Zuckermann and N. R. Forde, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 84, 1–11 CrossRef PubMed.
- T. Antal and P. L. Krapivsky, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 1–9 Search PubMed.
- X. Xiong, P. J. Coombs, S. R. Martin, J. Liu, H. Xiao, J. W. McCauley, K. Locher, P. a. Walker, P. J. Collins, Y. Kawaoka, J. J. Skehel and S. J. Gamblin, Nature, 2013, 497, 392–396 CrossRef CAS PubMed.
- M. A. Rameix-Welti, S. Munier, S. Le Gal, F. Cuvelier, F. Agou, V. Enouf, N. Naffakh and S. Van Der Werf, Antiviral Ther., 2011, 16, 597–603 CrossRef CAS PubMed.
- X. Zhu, R. McBride, C. M. Nycholat, W. Yu, J. C. Paulson and I. A. Wilson, J. Virol., 2012, 86, 13371–13383 CrossRef CAS PubMed.
- O. Blixt, S. Head, T. Mondala, C. Scanlan, M. E. Huflejt, R. Alvarez, M. C. Bryan, F. Fazio, D. Calarese, J. Stevens, N. Razi, D. J. Stevens, J. J. Skehel, I. van Die, D. R. Burton, I. a. Wilson, R. Cummings, N. Bovin, C.-H. Wong and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17033–17038 CrossRef CAS PubMed.
- J. Stevens, O. Blixt, L. Glaser, J. K. Taubenberger, P. Palese, J. C. Paulson and I. A. Wilson, J. Mol. Biol., 2006, 355, 1143–1155 CrossRef CAS PubMed.
- A. Harris, G. Cardone, D. C. Winkler, J. B. Heymann, M. Brecher, J. M. White and A. C. Steven, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19123–19127 CrossRef CAS PubMed.
- P. Chlanda, O. Schraidt, S. Kummer, J. Riches, H. Oberwinkler, S. Prinz, H.-G. Kräusslich and J. A. G. Briggs, J. Virol., 2015, 89, 8957–8966 CrossRef CAS PubMed.
- T. Ohkura, F. Momose, R. Ichikawa, K. Takeuchi and Y. Morikawa, J. Virol., 2014, 88, 10039–10055 CrossRef PubMed.
- J. S. Rossman and R. A. Lamb, Virology, 2011, 411, 229–236 CrossRef CAS PubMed.
- J. J. Madsen, J. M. A. Grime, J. S. Rossman and G. A. Voth, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E8595–E8603 CrossRef CAS PubMed.
- B. Dadonaite, S. Vijayakrishnan, E. Fodor, D. Bhella and E. C. Hutchinson, J. Gen. Virol., 2016, 97, 1755–1764 CrossRef CAS PubMed.
- M. J. Rust, M. Lakadamyali, F. Zhang and X. Zhuang, Nat. Struct. Mol. Biol., 2004, 11, 567–573 CrossRef CAS PubMed.
- F. J. Martinez-Veracoechea and D. Frenkel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10963–10968 CrossRef CAS PubMed.
- B. Button, L.-H. Cai, C. Ehre, M. Kesimer, D. B. Hill, J. K. Sheehan, R. C. Boucher and M. Rubinstein, Science, 2012, 337, 937–941 CrossRef CAS PubMed.
- F. Taherali, F. Varum and A. W. Basit, Adv. Drug Delivery Rev., 2018, 124, 16–33 CrossRef CAS PubMed.
- R. Bansil and B. S. Turner, Adv. Drug Delivery Rev., 2018, 124, 3–15 CrossRef CAS PubMed.
- B. S. Schuster, J. S. Suk, G. F. Woodworth and J. Hanes, Biomaterials, 2013, 34, 3439–3446 CrossRef CAS PubMed.
- M. Cohen, X.-Q. Zhang, H. P. Senaati, H.-W. Chen, N. M. Varki, R. T. Schooley and P. Gagneux, Virol. J., 2013, 10, 321 CrossRef PubMed.
- X. Yang, L. Steukers, K. Forier, R. Xiong, K. Braeckmans, K. Van Reeth and H. Nauwynck, PLoS One, 2014, 9, e110026 CrossRef PubMed.
- P. Gagneux, M. Cheriyan, N. Hurtado-Ziola, E. C. M. B. van der Linden, D. Anderson, H. McClure, A. Varki and N. M. Varki, J. Biol. Chem., 2003, 278, 48245–48250 CrossRef CAS PubMed.
- J. N. S. S. Couceiro, J. C. Paulson and L. G. Baum, Virus Res., 1993, 29, 155–165 CrossRef CAS PubMed.
- M. Zanin, B. Marathe, S.-S. Wong, S.-W. Yoon, E. Collin, C. Oshansky, J. Jones, B. Hause and R. Webby, J. Virol., 2015, 89, 5935–5948 CrossRef CAS PubMed.
- M. Zanin, P. Baviskar, R. Webster and R. Webby, Cell Host Microbe, 2016, 19, 159–168 CrossRef CAS PubMed.
- R. J. Pickles, J. K. Sheehan, M. Kesimer, C. W. Davis, K. A. Burns and C. Ehre, Mucosal Immunol., 2012, 6, 379–392 Search PubMed.
- Y. P. Lin, X. Xiong, S. A. Wharton, S. R. Martin, P. J. Coombs, S. G. Vachieri, E. Christodoulou, P. A. Walker, J. Liu, J. J. Skehel, S. J. Gamblin, A. J. Hay, R. S. Daniels and J. W. McCauley, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 21474–21479 CrossRef CAS PubMed.
- K. Yehl, A. Mugler, S. Vivek, Y. Liu, Y. Zhang, M. Fan, E. R. Weeks and K. Salaita, Nat. Nanotechnol., 2016, 11, 184–190 CrossRef CAS PubMed.
- P. K. Dutta, Y. Zhang, A. T. Blanchard, C. Ge, M. Rushdi, K. Weiss, C. Zhu, Y. Ke and K. Salaita, Nano Lett., 2018, 18, 4803–4811 CrossRef CAS PubMed.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |