
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtocells programmed through artificial reaction networks
Yifan
Lyu
ab,
Ruizi
Peng
b,
Hui
Liu
b,
Hailan
Kuai
b,
Liuting
Mo
b,
Da
Han
a,
Juan
Li
*acd and
Weihong
Tan
 *abd
*abd
aInstitute of Molecular Medicine (IMM), State Key Laboratory of Oncogenes and Related Genes Renji Hospital, Shanghai Jiao Tong University School of Medicine, College of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China
bMolecular Science and Biomedicine Laboratory, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology, Aptamer Engineering Center of Hunan Province, Hunan University, Changsha, Hunan 410082, China
cMOE Key Laboratory for Analytical Science of Food Safety and Biology, Fujian Provincial Key Laboratory of Analysis and Detection Technology for Food Safety, State Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, China
dInstitute of Cancer and Basic Medicine (IBMC), Chinese Academy of Sciences, The Cancer Hospital of the University of Chinese Academy of Sciences, Hangzhou, Zhejiang 310022, China. E-mail: tan@hnu.edu.cn; lijuan@fzu.edu.cn
First published on 19th December 2019
Abstract
As the smallest unit of life, cells attract interest due to their structural complexity and functional reliability. Protocells assembled by inanimate components are created as an artificial entity to mimic the structure and some essential properties of a natural cell, and artificial reaction networks are used to program the functions of protocells. Although the bottom-up construction of a protocell that can be considered truly ‘alive’ is still an ambitious goal, these man-made constructs with a certain degree of ‘liveness’ can offer effective tools to understand fundamental processes of cellular life, and have paved the new way for bionic applications. In this review, we highlight both the milestones and recent progress of protocells programmed by artificial reaction networks, including genetic circuits, enzyme-assisted non-genetic circuits, prebiotic mimicking reaction networks, and DNA dynamic circuits. Challenges and opportunities have also been discussed.
1. Introduction
According to the Chemoton model propounded by Tibor Gánti, the primitive form of life should be as simple as possible, with only three fundamental features: metabolism, self-replication, and a bilipid membrane.1 At the molecular level, metabolism, self-replication processes, and even membrane structures are organized and modulated by a series of spatiotemporally ordered chemical reactions termed chemical reaction networks.2 The single cell is the basic structural and functional unit of living organisms, and its genetic and metabolic processes have been significantly studied since 1839.3 One plausible way of understanding the mechanism of cellular life involves the assembly of inanimate components into artificial cells by creating artificial reaction networks. Since synthetic biology and chemistry are limited, these artificial reaction networks can program artificial cells4,5 or protocells,6,7 which mimic some natural cellular features.8 However, if we consider protocells as cell-sized automatons with autonomous computing ability, then artificial reaction networks become the computational core or software of protocells, similarly to how natural reaction networks control the behavior, operation and communication of natural cells. Therefore, benefiting from the cell-like characteristics, protocell constructs can perhaps act as feasible candidates to work in biological circumstances with biologically and artificially combined algorithms and should find wide applications in some emerging fields such as biomedicine and bioengineering in the future.Some previous reviews have focused on the bionic features of protocells.3,5 However, herein, we prefer to expand our review to include the various types of artificial reaction networks9–11 used to build protocells. Accordingly, we will discuss four types of protocells in the following sections: protocells programmed by genetic circuitry, protocells programmed by enzyme-assisted non-genetic circuitry, protocells propelled by prebiotically mimicked reaction networks and protocells equipped with DNA dynamic circuitry (Fig. 1). Specifically, protocells programmed by genetic circuitry and enzyme-assisted non-genetic circuitry are mainly constructed to study and understand the genetic and metabolic processes of modern cells through mimicry, while protocells propelled by prebiotically mimicked reaction networks are built to study the origin of the cellular system on primitive Earth. The purpose of constructing protocells equipped with DNA dynamic circuitry is different from the others because this field is focused on building cell-like automations. Because here we mainly focus on artificial reaction networks, top-down approaches12 for manipulating genes in living cells to achieve different phenotypes are not discussed and some similar nomenclatures such as artificial cells, minimal cells, protocells and semi-synthetic cells are not differentiated in this review. For those who want a comprehensive understanding of these nomenclatures, the review by Caschera et al.13 is recommended.
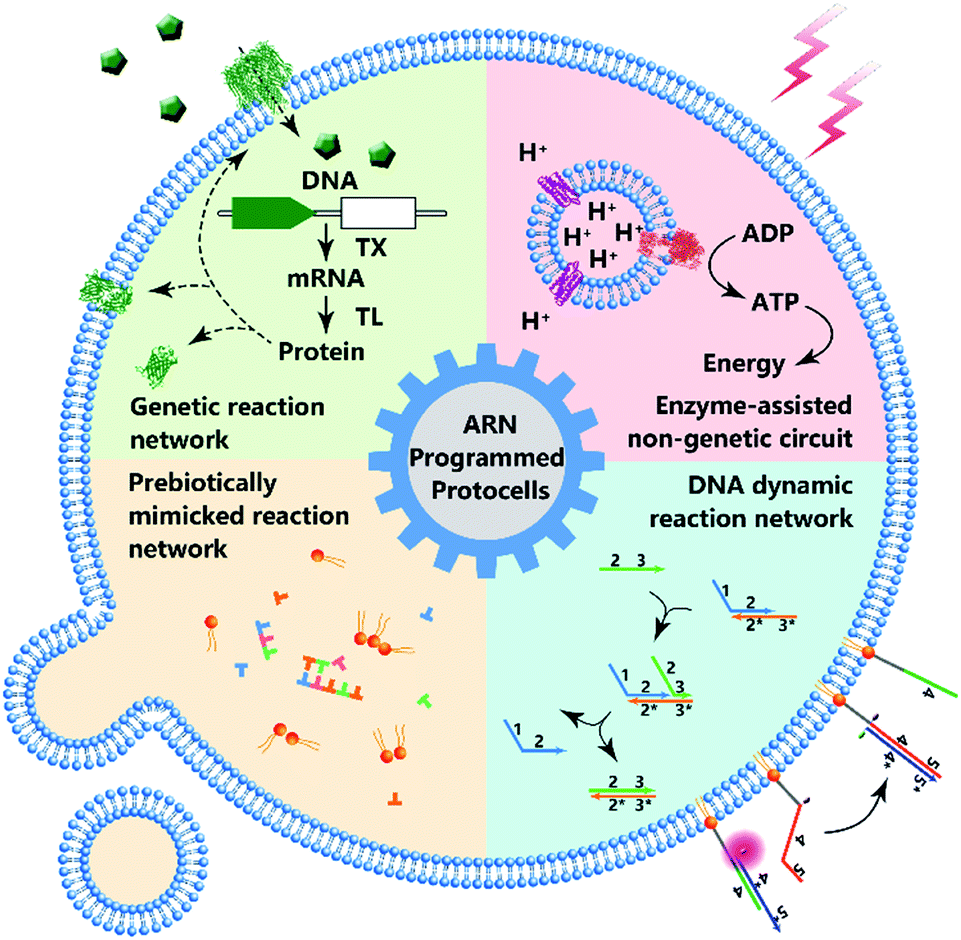 | ||
| Fig. 1 Schematic diagram of protocell programmed by various types of artificial reaction networks (ARN), including genetic reaction network, enzyme-assisted non-genetic circuit, prebiotically mimicked reaction network and DNA dynamic reaction network. | ||
2. Protocells programmed by genetic circuitry
The most straightforward way to construct a protocell from scratch is to directly encapsulate naturally existing genetic circuits such as transcription/translation (TX/TL) into an artificially prepared vesicle with a continuous membrane structure. Then further modification of the TX/TL system to achieve more controllable computational capability can be expected. The final goal of this semi-synthetic approach is the maximum utilization of naturally existing systems or the recovery of metabolic mechanisms. With rational genetic engineering, we show that a natural TX/TL system can be reprogrammed into complex genetic circuits with a customized operational mechanism and readout strategy.2.1 Brief timeline of protocells programmed for gene expression
Pioneering attempts in this field were initiated in 1994 when Walde et al. and Chakrabarti et al. independently used RNA polymerase to polymerize ADP to poly A in oleic acid/oleate or phospholipid vesicles.14,15 Inspired by these works, templated RNA replication by Qβ replicase,16 polymerase chain reaction (PCR)-based DNA replication,17 and T7 polymerase-based transcription using a DNA template18 were subsequently achieved in phospholipids or fatty acid vesicles within a few years (Fig. 2a). In 1999, Oberholzer et al. reported the first successful trial of ribosomal synthesis of a peptide (poly(phenylalanine)) inside vesicles.19 Two years later, in a seminal report, Yu et al.20 synthesized GFP with detectable fluorescent emission inside liposomes, showing that proteins synthesized inside an artificial vesicle could retain their function. Reconstituting a complete gene expression system inside an artificial vesicle was first achieved in 2003 when Nomura et al. used a plasmid to express the red-shifted green fluorescent protein (rsGFP) in cell-sized giant vesicles.21 However, owing to the impermeability of the phospholipid membrane, this system could not work for long time, and the protein expression inside stopped after only a few hours as a result of the exhaustion of energy and nutrients. To solve this problem, Noireaux et al. constructed cell-like bilayer vesicles with combined internal expression of α-hemolysin (αHL) pore protein and enhanced GFP.22 The expressed αHL pore protein, after inserting into the membrane, could create selective permeability for nutrients and assist vesicle exchange with the environment so that the protocell system could then sustain expression for up to 4 days. Another remarkable work was proposed by Ishikawa et al.23 who created a two-stage cascading genetic network inside a liposome. Specifically, a T7 RNA polymerase was produced in the first stage and then utilized for GFP synthesis in the second stage. These pioneering works, although containing only limited components and simple in function, demonstrated the potential of using cell-mimicking bilayer vesicles as artificial containers to isolate encapsulated genetic reactions, including transcription and translation, from the external environment. Unlike the transcription procedure, which can be easily performed in vitro by using purified components, a translation procedure is much more complicated. In these early studies, natural protein synthesis systems from cells (e.g., Escherichia coli (E. coli), wheat germ, rabbit reticulocytes, insect cells, and human cells) were extracted (S30 fraction, named from the supernatant of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g centrifugation24) and encapsulated into artificial vesicles so that all components needed in a translation procedure were automatically functional. However, an obvious limitation of this strategy is that crude cell extract has unknown compositions and cannot be customized for diverse purposes. Besides, proteases or nucleases extant in cell extracts may damage nucleic acid templates and protein products. Thus, to solve these problems, an artificially recombinant cell-free translation system, now known as PURE (Protein Synthesis Using Recombinant Elements) (Fig. 2a), was first revealed by Shimizu et al.25 in 2001. Based on an understanding of the natural translation procedure, PURE is composed of 36 individually purified components, including His-tagged proteins, ribosomes, and tRNAs mixture, and is highly customized and suitable for the cell-free production of natural and unnatural proteins. However, compared to cheap cell extracts, PURE, as an alternative method, is very expensive. As a result, PURE-encapsulated protocells for protein expression were not widely used in cell-like vesicles until Sunami et al.26 constructed PURE-encapsulated liposomes for in vitro GPF expression, and Murtas et al.27 tried to synthesize membrane proteins in liposomes with a minimal set of enzymes. Since then, it has been gradually accepted that PURE has tremendous advantage to customize artificial genetic circuits compared with cell extract. In the case of the bottom-up reconstitution of protocells, a more flexible and controllable artificial genetic system with well-defined composition and full functions of cell-free TX/TL is more attractive and useful.28 Such artificially reconstituted cell-free genetic systems paved the way for the construction of more sophisticated protocells with customized genetic circuit, as follows.
000g centrifugation24) and encapsulated into artificial vesicles so that all components needed in a translation procedure were automatically functional. However, an obvious limitation of this strategy is that crude cell extract has unknown compositions and cannot be customized for diverse purposes. Besides, proteases or nucleases extant in cell extracts may damage nucleic acid templates and protein products. Thus, to solve these problems, an artificially recombinant cell-free translation system, now known as PURE (Protein Synthesis Using Recombinant Elements) (Fig. 2a), was first revealed by Shimizu et al.25 in 2001. Based on an understanding of the natural translation procedure, PURE is composed of 36 individually purified components, including His-tagged proteins, ribosomes, and tRNAs mixture, and is highly customized and suitable for the cell-free production of natural and unnatural proteins. However, compared to cheap cell extracts, PURE, as an alternative method, is very expensive. As a result, PURE-encapsulated protocells for protein expression were not widely used in cell-like vesicles until Sunami et al.26 constructed PURE-encapsulated liposomes for in vitro GPF expression, and Murtas et al.27 tried to synthesize membrane proteins in liposomes with a minimal set of enzymes. Since then, it has been gradually accepted that PURE has tremendous advantage to customize artificial genetic circuits compared with cell extract. In the case of the bottom-up reconstitution of protocells, a more flexible and controllable artificial genetic system with well-defined composition and full functions of cell-free TX/TL is more attractive and useful.28 Such artificially reconstituted cell-free genetic systems paved the way for the construction of more sophisticated protocells with customized genetic circuit, as follows.
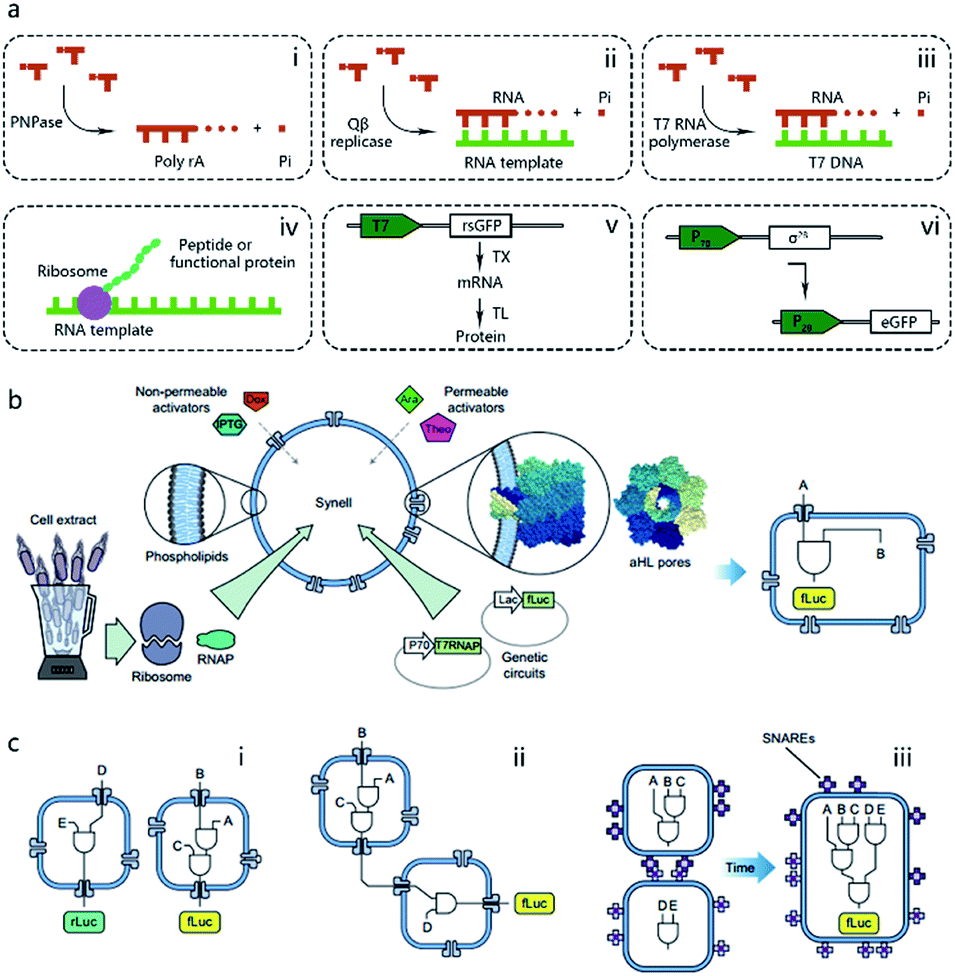 | ||
| Fig. 2 (a) Schematic illustration of pioneering attempts to construct genetic circuits in artificial vesicles (dashed lines). (i) ADP is polymerized into poly(A) by polynucleotide phosphorylase (PNPase). (ii) Templated RNA replication by Qβ replicase. (iii) T7 polymerase-based transcription. (iv) Peptide/protein translation. (v) Hybrid bacteriophage-E. coli system by coupling the transcription procedure of bacteriophage and the translation procedure of E. coli. rsGFP, red-shifted green fluorescent protein. (vi) All-E. coli cell-free TX/TL system. eGFP, enhanced green fluorescent protein. (b) Schematic representation of Synells with a phospholipid bilayer membrane and its components. Synells membranes are permeable to theophylline (Theo) and arabinose (Ara), and are impermeable to IPTG and doxycycline (Dox). But when channel-forming proteins aHL (grey membrane pores) are present, IPTG and Dox can traverse the membrane through aHL channels. Synells contain programmed genetic circuits that can be triggered by these molecules. fLuc, firefly luciferase. rLuc, Renilla luciferase. (c) Different genetic circuits running within and between Synells. (i) Two genetic circuits working in independent protocells without crosstalk. (ii) Genetic circuits in two different protocells interacting in a cascading way. (iii) Genetic circuits running in parallel in separate protocells can be joined hierarchically after protocell fusion. Reproduced from ref. 40 with permission. Copyright 2016 Springer Nature. | ||
2.2 Protocells programmed by customized genetic circuitry
The construction of biochemical systems with programmed genetic information in vitro requires the development of novel experimental platforms that offer broad capabilities and enough flexibility to allow for change, as well as the investigation of biochemical and biophysical parameters otherwise inaccessible in vivo. Thus, protocells programmed by genetic circuitry are increasingly employed as a steppingstone for constructing, understanding, and interrogating complex biochemical systems, such as cell signaling pathways and protein integration.As the first type of popular cell-free TX/TL system, a hybrid bacteriophage-E. coli system29 was invented in 1991 by coupling the transcription procedure of bacteriophage and the translation procedure of E. coli. The transcription procedure of a hybrid bacteriophage-E. coli system is performed by a bacteriophage RNA polymerase with its promoter, usually T7, in simplicity, affinity, and specificity, while the translation procedure is carried out by a cytoplasmic extract or reconstituted components (PURE system, as mentioned in the previous section) from E. coli (Fig. 2a). While hybrid T7 TX/TL systems are powerful tools for a vast array of applications, such as recombinant protein expression and minimal cell construction,30 sometimes obvious disadvantages are shown in using them to build more complex biological systems. This results from their limited bacteriophagic transcription machinery consisting of only a few promoters, which are not enough to construct DNA-programmed synthetic gene circuits. Thus, to solve this problem, an all-E. coli cell-free TX/TL system with all components derived solely from E. coli was developed by Shin et al. in 2010.31 As shown in Fig. 2a, this new system used the endogenous E. coli RNA polymerase and the entire sigma factor 70 for transcription, substantially expanding the programmability of cell-free expression technology for use in building artificial cells with multistage genetic circuits, simple Boolean logic gates and feedback loops. Recently, an updated version of the all-E. coli system with improved ATP regeneration pathway was reported and proven to be compatible with liposomes.32
With the powerful genetic circuit tools described above, protocells can be implemented in a programmable way by purposely choosing the encapsulated compounds and reaction networks. An impressive example of this is the protocell-based in vitro evolution or selection system. Inspired by the protein evolution strategy in a water-in-oil emulsion proposed by Ghadessy et al.,33 Ichihashi et al. constructed a protocell containing artificial genomic RNA that could be replicated by a replicase translated from itself. Introduced replication error allows this protocell to mimic a Darwinian evolutionary process.34 Through evolution in protocells, genomic RNA with improved interaction toward translated replicase dominates the population, regardless of parasitic replicators. Artificial genetic circuits also offer an entirely in vitro membrane protein evolutionary process, termed liposome display, which was reported by Fujii et al., who used this method to evolve the pore-forming activity of αHL from Staphylococcus aureus. Compared to the wild type, the evolved αHL mutant possessed only two point mutations, but with a 30-fold increase in pore-forming tendency.35 As a physical channel, artificially expressed αHL can also be utilized to send a chemical message from protocells to E. coli. In a work reported by Lentini et al.,36 a protocell was constructed with built-in TX/TL machinery for αHL synthesis. The translation process was modulated by a DNA coded riboswitch responding to the presence of theophylline. In the presence of theophylline, an αHL pore forms on the membrane so that the entrapped signal molecules, β-D-1-thiogalactopyranoside (IPTG), are released from the protocell and recognized by E. coli carrying a plasmid encoding an IPTG-responsive lac operator sequence, followed by a fluorescent protein sequence. Another membrane protein integration strategy was accomplished by Matsubayashi et al. In their design, in vitro synthesized E. coli Sec translocon was first assembled onto the artificial cell membrane then induced the membrane translocation of single- and multi-span membrane proteins.37
One of the dominant features of biological cells is their spatial organization of contents and functions with different biochemical processes confined to specific cellular regions such as organelles. Thus, in contrast to performing a sophisticated genetic reaction network in a single chamber, another strategy of artificial genetic circuit construction is using a multicompartment vesicle to spatially control each stage of a reaction network.38 Elani et al., for the first time, demonstrated protein expression in separate compartments of a protocell to achieve segregation of functions.39 A further modular and controllable compartmentalization system was reported by Adamala et al., who engineered genetic circuit cascade reactions within and between liposome-based protocells termed genetic circuit-containing synthetic minimal cells (Synells) (Fig. 2b).40 Some membrane permeable (theophylline and arabinose), or impermeable (IPTG and doxycycline) small molecules were used as triggers to module genetic circuits within Synells and genetically expressed luciferases were used as output molecules of various circuits. These circuits include three parts. First, two genetic circuits were designed to work in independent protocells without crosstalk. Second, genetic circuits equipped in two different protocells interacted in a cascading way. Third, genetic circuits ran in parallel in separate protocells. If the reaction-encapsulated protocells carried fusogenic peptides, such as the SNARE protein, an acronym derived from SNAP (Soluble NSF Attachment Protein) REceptor, as used in this work, the genetic circuits from different protocell populations could be joined hierarchically (Fig. 2c). This example represents the most extensive artificial genetic circuit currently realized in synthetic compartments, or protocells.
3. Protocells programmed by enzyme-assisted non-genetic circuitry
Almost all metabolic processes in a cell need enzymes to accelerate reactions so that organisms can survive. Compared to genetic systems in which various components interact with each other in a naturally defined way, enzyme-assisted non-genetic circuits have a higher degree of freedom when applied to construct artificial reaction networks.In the mid-20th century, Oparin et al. published a series of papers describing the enzymatic reactions in prebiotic cell-like coacervates.41 However, owing to the instability and morphological difference of coacervates compared to natural cells, coacervates were gradually replaced by vesicles with continuous membrane structures, such as liposomes, when constructing protocells. Starting with investigations of enzymatic activities in vesicles, enzyme-reconstituted protocells have been studied for decades.42 Interestingly, one such attempt involved the enzymatic production of lecithin molecules inside lecithin liposomes to induce protocell self-reproduction from inside.43 Although lecithin molecules were synthesized, no vesicle growth could be observed in this work. Actually, the first experiment showing vesicle growth by enzymatic catalysis that produced a vesicle boundary from inside was reported by Murtas et al. via the biochemical synthesis of palmitate based on FAS type I enzyme catalysis.44,45 In a recent study, Exterkate et al. reported a cascading phospholipid biosynthesis pathway containing eight enzymes to synthesize phospholipid using fatty acid and glycerol 3-phosphate, and observed membrane expansion via this system.46 All these works used integral membrane proteins to synthesize the vesicle boundary, as inspired by the enzymatical synthesis of phospholipids in nature. However, considering the early evolution process of complex membrane structures, using a soluble enzyme seems to be more rational because in prebiotic Earth there was no pre-existing membrane. Based on this hypothesis, Bhattacharya et al. designed a plausible lipid synthesizing system using a soluble mycobacterial ligase, FadD10, for phospholipid formation.47 In their design, FadD10 first catalyzed the generation of fatty acyl adenylates (FAAs) from dodecanoic acid (DDA), Mg2+, and ATP. Then FAA spontaneously reacted with an amine-functionalized lipid fragment to produce a membrane-forming phospholipid.
The energy consumed in many cellular processes is provided by ATP. The typical ATP synthase F0F1 needs an electrochemical proton gradient to synthesize ATP from ADP and inorganic phosphate (Pi). Studies in the last century have succeeded in gaining an understanding of the mechanism behind ATP generation, purification of ATP synthesis-related proteins and preliminary construction of ATP synthesis devices using liposomes.48–50 In 2005, Choi et al. reconstituted both bacteriorhodopsin (BR) and F0F1-ATP synthase onto membrane of proteopolymersomes and realized light-driven ATP generation.51 BR was used to generate a photo-induced proton gradient across the membrane, which could be utilized by F0F1-ATP synthase. A similar strategy was reported by Feng et al. as a mimic of chloroplast.52 In their work, photosystem II (PSII), the only protein complex with the capability of catalyzing water into protons, electrons, and oxygen, was used to generate the proton gradient for ATP synthesis. F0F1-ATPases were assembled on the surface of proteoliposomes coated on a PSII-based microsphere. Based on these highly feasible ATP-generating artificial organelles, artificial cells with a self-sustaining energy system were consequently constructed. Lee et al. engineered switchable photosynthetic organelles (∼100 nm in diameter, as shown in Fig. 3a) as energy generators and encapsulated them within a giant phospholipid vesicle embedded with ionophores to form a protocell for carbon fixation and actin polymerization (Fig. 3b).53 Berhanu et al. demonstrated that photosynthesized ATP could be further consumed as: (i) substrates for messenger RNA (mRNA) synthesis, (ii) energy for phosphorylation of guanosine diphosphate (GDP), and (iii) energy for aminoacylation of transfer RNA (tRNA) (Fig. 3c).54 Interestingly, the aminoacylated transfer tRNA was finally used to synthesize two types of proteins (bR and Fo), which were initially used for ATP photosynthesis inside a protocell. This positive feedback loop demonstrated that this protocell can use light as an energy source to synthesize its own part in an autotrophic way, just like primordial cells. Artificial organelle-containing protocells can also be achieved by using multi-compartmentalized vesicles structurally resembling biological cells with encapsulated organelles. For example, Lecommandoux et al. constructed a polymersome-based protocell with encapsulated polymersome-based artificial organelles.55 A cofactor-dependent cascade reaction involving two different enzyme-containing artificial organelles, as well as a ‘cytosolic’ enzyme, was performed in this protocell. Elani et al. carried out multi-step enzymatic pathways in multi-compartment protocells to mimic biological boundary-combined reactions. The products of each step could traverse into adjacent compartments with the aid of transmembrane protein pores.56
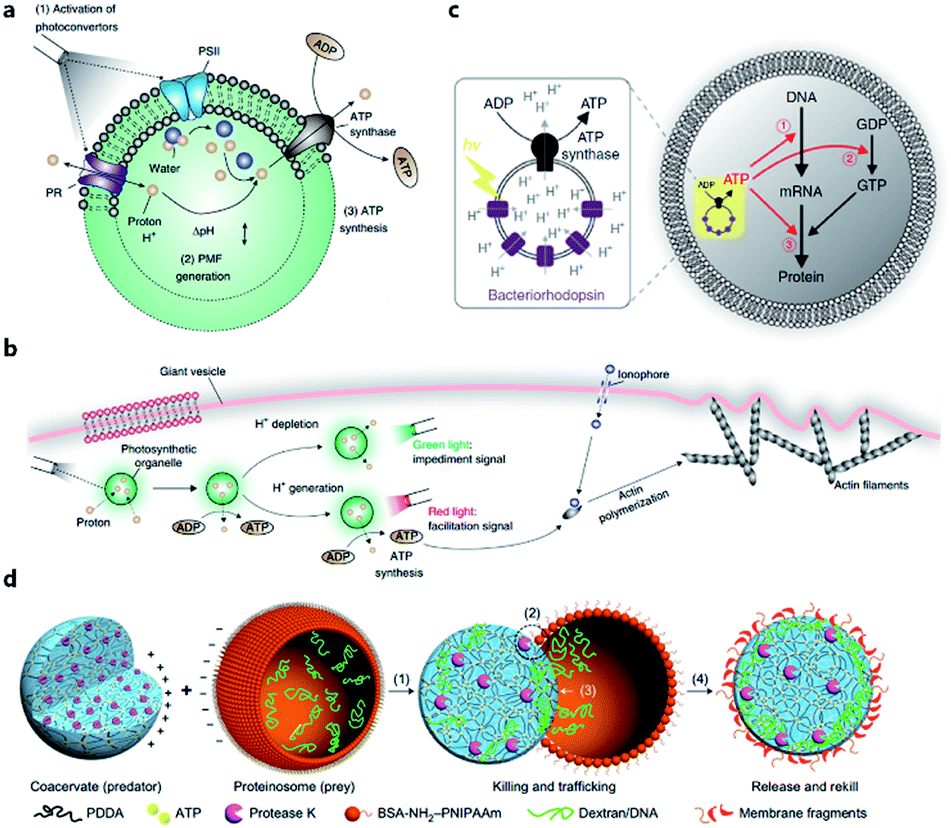 | ||
| Fig. 3 (a) Structure and function of an ATP-generating artificial organelle with two photoconverters, plant-derived photosystem II (PSII) and bacteria-derived proteorhodopsin (PR), and an ATP synthase integrated into the membrane. PSII can be activated by red light to generate protons inside the organelle and PR can be activated by green light to deplete protons. The proton gradient across the organelle membrane drives the conversion of ADP to ATP by ATP synthase. PMF, proton motive force. (b) ATP-generating artificial organelles are encapsulated in a protocell. The synthesized ATP fuels ATP-dependent actin polymerization, thus inducing a morphological change in the protocell. Reproduced from ref. 53 with permission. Copyright 2018 Springer Nature. (c) Schematic of a protocell encapsulating an artificial photosynthetic organelle equipped with bacteriorhodopsin (bR) and F0F1-ATP synthase. Synthesized ATP is consumed for (1) mRNA transcription, (2) phosphorylation of guanosine diphosphate (GDP), and (3) aminoacylation of tRNA. Reproduced from ref. 54 with permission. Copyright 2019 Springer Nature. (d) Schematic representation of a synthetic protocell community including predator and prey. A protease K-containing coacervate microdroplet acts as an artificial predator protocell, which can capture the proteinosome-based prey through four steps: (1) electrostatic attachment; (2) protease-induced disassembly; (3) payload transfer; and (4) release of the compositionally modified predator protocell. Reproduced from ref. 57 with permission. Copyright 2017 Springer Nature. | ||
Apart from cell-like structures or functions, some inter-cellular behaviors such as predation can also be mimicked by enzyme-driven reactions. Qiao et al. developed two types of protocells acting as predator and prey, respectively, and studied the predatory behavior in interacting artificial protocell communities (Fig. 3d).57 The created protocell was a protease-containing coacervate with no continuous membrane structure, and thus was not regarded as a plausible structure of primitive cells. Still, the study on collective behavior illustrated an approach to design of synthetic protocell communities. Kumar et al. used an organoclay/DNA hybrid to construct protocells with buoyancy-derived motility powered by gas bubbles generated by encapsulated catalase in the presence of hydrogen peroxide.58 This strategy was more like a bioengineering design than a bionic approach, demonstrating the feasibility of cellular motion driven by artificial reaction network.
4. Protocells propelled by prebiotically mimicked reaction networks
In the Earth's prebiotic oceans, prelife chemical reaction networks gave rise to life. It is widely accepted that primitive cells living on Earth did not have complex structures or sophisticated metabolic processes such as protein enzyme-dependent reactions. Thus, to explore the minimal condition that a protocell needs to realize some basic features, such as growth, division, replication, or even chemotaxis, researchers try to use prebiotically mimicked reaction networks, mainly composed of small molecules, to propel protocells.4.1 Membrane growth and division-associated circuits
One of the pioneering studies reporting the plausible mechanisms of membrane growth and division was conducted by P. L. Luisi et al.59 who fed vesicles in buffered solution with alkaline fatty acid micelles and observed their growth. A previous hypothesis60 was that vesicle division occurred by extrusion through small pores in prebiotic Earth. However, this was difficult to replicate owing to insufficient pressure gradients and porous rock without larger channels. Furthermore, the extrusion process could lead to significant loss of contents in the protocell during each division cycle. Zhu et al.61 and Budin et al.62 observed that with the addition of fatty acid micelles or solvent evaporation, the initially spherical fatty acid vesicles transformed into long thread-like vesicles. This transformation process is accompanied by the division of the vesicles into multiple daughter vesicles without loss of internal contents under modest shear forces (Fig. 4a). An investigation on division without external mechanical forces was reported in 2012 by Zhu et al.63 They built filamentous fatty acid vesicles containing either fluorescent molecules in aqueous solution in the chamber or hydroxypyrene molecules in the membrane. In the presence of thiols, illuminating the vesicles rapidly induced pearling and subsequent division. This is perhaps because the reactive oxygen species generated oxidized thiols into disulfide-containing compounds, which subsequently associated with fatty acid membranes and induced a change in surface tension (Fig. 4b).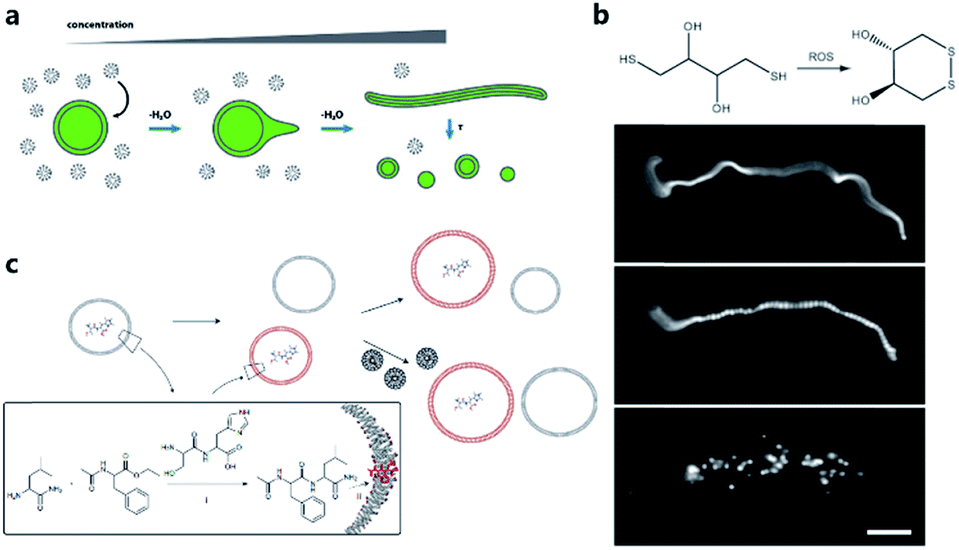 | ||
| Fig. 4 (a) Schematic illustration of the concentration-driven growth and division of model protocell membranes. This model indicates that spherical protocells can grow into long, filamentous vesicles resulting from evaporative concentration, followed by shear force or photochemically induced division. Reproduced from ref. 62 with permission. Copyright 2012 American Chemical Society. (b) Oleate vesicle pearling and division caused by radical oxidation of DTT. After the addition of oleate micelles, an oleate vesicle grew into a long thread-like vesicle, which then pealed and divide under illumination. Reproduced from ref. 63 with permission. Copyright 2012 National Academy of Sciences. (c) Dipeptide catalyst Ser-His encapsulated in fatty acid vesicles catalyzes the condensation between LeuNH2 and AcPheOEt to afford AcPheLeuNH2. The dipeptide AcPheLeuNH2 binds to the bilayer membrane of protocells (red), induces the growth of the protocells by drawing fatty acids from protocells without dipeptide (grey). Upon addition of micelles, protocells with AcPheLeuNH2 in the membrane grow larger than protocells without the dipeptide. Reproduced from ref. 65 with permission. Copyright 2013 Macmillan Publishers Limited. | ||
Combining membrane behavior with encapsulated small molecules or reactions in protocells is another way to understand the growth or division process of primitive cells. Chen et al. found that protocells encapsulating RNA would exert an osmotic pressure on the vesicle membrane so that protocells could grow by taking additional membrane components from, for example, empty vesicles or micelles.64 Adamala et al. constructed a protocell containing a dipeptide catalyst that catalyzed the synthesis of a hydrophobic dipeptide. The hydrophobic dipeptide product could bind with the fatty acid membrane of the protocell and further promote vesicle growth (Fig. 4c).65 Protocells encapsulated with PCR were also studied.17,66 Research interest in encapsulating a PCR reaction inside a protocell is rooted in the hypothesis that primitive cells in prebiotic Earth may have grown and evolved in a PCR-like manner because the thermal cycling process could be fulfilled by prebiotic cells around a hydrothermal vent in the deep prebiotic sea. Based on this hypothesis, Kurihara et al. employed a PCR-based DNA replication process to induce the growth and division of protocells.67 With the addition of vesicular membrane precursors, the division process was observed to be accelerated by the PCR process of encapsulated DNA, which was finally distributed into divided daughter protocells.
4.2 Oligomerization-associated circuits
Considering the key functions of nucleic acids and peptides in modern cells, the polymerization processes of nucleotides and amino acids have been regarded as a portal to gain more understanding of the origin of cellular life on prebiotic Earth. Some origin-of-life scenarios hold that nucleotides or amino acids present on prebiotic Earth underwent oligomerization to form catalytically active RNA (ribozymes) or peptides, which then were incorporated into protocells. Although numerous efforts have been made to investigate plausible pathways for the synthesis and degradation of biopolymers in different buffer solutions that mimic the prebiotic ocean,68 studies about nonenzymatic polymerization reactions in protocells do not have a long history. Early studies tended to directly encapsulate all reagents inside an artificial vesicle to observe the polymerized oligomers or just simply incubate monomers with liposomes to study liposome-assisted selectivity or catalytic ability.69,70 Studying the polymerization inside a protocell in mimicked prebiotic environment raises a pertinent question about how building blocks or nutrients could traverse the membrane, which quite probably consisted of amphiphiles. In 2007, Zepik et al.71 combined nonenzymatic polymerization with permeation of amino acids into a lipid bilayer membrane and demonstrated the possibility of nonenzymatic oligomerization of thio-glutamic acid in lipid vesicles. A more comprehensive work was carried out by Mansy et al.72 who examined the effect of different membrane compositions on solute permeability and concluded that solute permeability could be enhanced by using short, unsaturated, or branched acyl chain, or amphiphiles with larger head groups to form fatty acid vesicles. Using a mixture of the most plausible prebiotic amphiphiles, they further constructed artificial cells with high permeability to simple sugars and nucleotide nutrients, finally achieving the template-directed polymerization of imidazole-activated 2′-amino-guanosine in the fatty acid-based vesicles. The membrane structure proved to be extremely thermostable and could retain internal RNA and DNA oligonucleotides at temperatures ranging from 0 °C to 100 °C.73 In 2013, Adamala et al.74 reported that the presence of citrate could protect fatty acid membranes from the disruptive effects of high Mg2+ concentrations, while allowing RNA copying to proceed and also protecting single-stranded RNA from Mg2+-catalyzed degradation. However, despite these advances, only homopolymers have been efficiently replicated inside vesicles. Thus, to replicate and evolve functional RNA sequences, in 2018, O'Flaherty et al.75 identified a way to replicate RNAs with mixed nucleotides within vesicles, with a strand length limitation of 5 nucleotides (Fig. 5a). Recently, the limitation was increased up to a length of 25 nucleotides with an average stepwise yield of 96–97%.76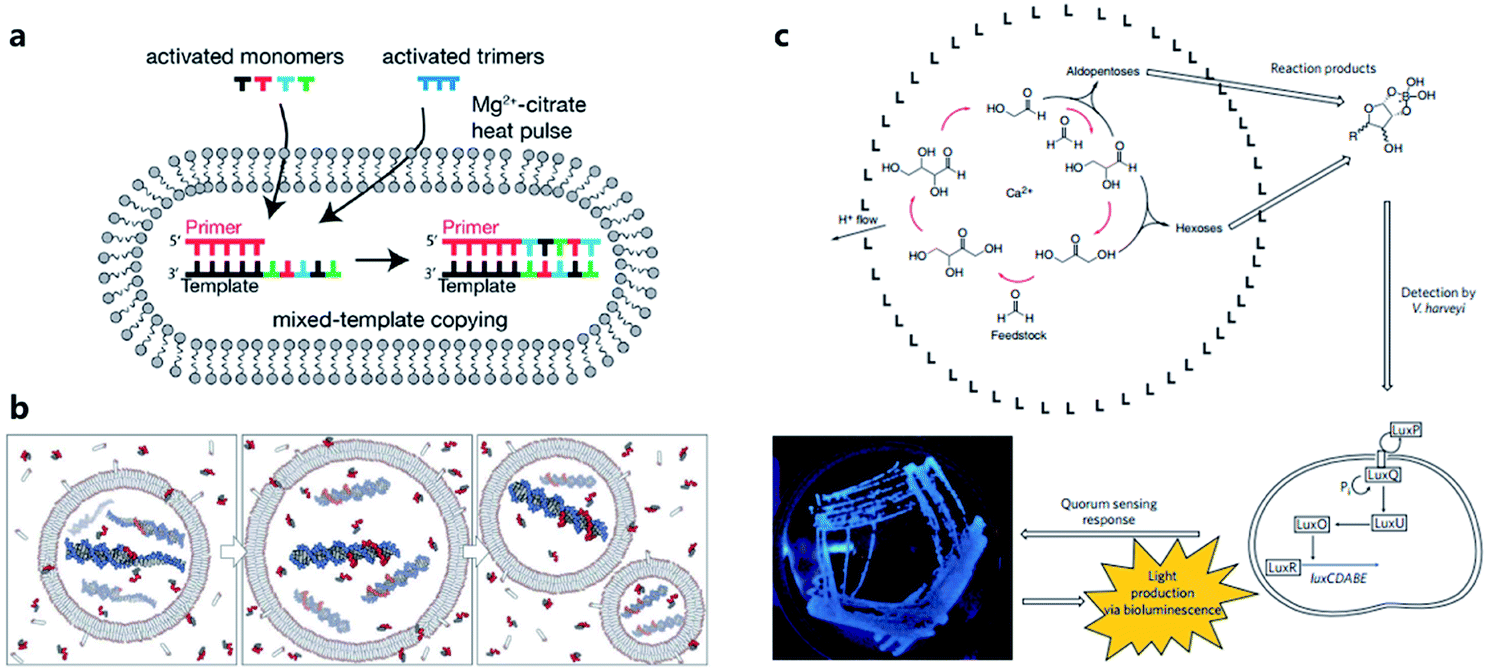 | ||
| Fig. 5 (a) Schematic representation of a recently developed method for nonenzymatic RNAs replication with mixed nucleotides within vesicles. Reproduced from ref. 75 with permission. Copyright 2018 American Chemical Society. (b) Conceptual model of a heterotrophic protocell. The protocells grow by taking amphiphiles from the environment and dividing by plausible intrinsic or extrinsic physical forces. Nonenzymatic copying of internal templates is implemented by externally supplied activated nucleotides that can permeate across the protocell membrane. Reproduced from ref. 72 with permission. Copyright 2008 Springer Nature. (c) Protocell that can produce carbohydrates by an autocatalytic formose reaction. The carbohydrates generated then form complexes with borates, which are input signals that induce Vibrio harveyi bacteria to emit a detectable bioluminescent output. Reproduced from ref. 81 with permission. Copyright 2009 Macmillan Publishers Limited. | ||
4.3 Metabolism-associated circuits
According to a widely recognized model of primitive cells, a self-replicating protocell requires two minimum but essential components, a membrane compartment that can grow and divide and an encapsulated, chemically replicating nucleic acid, or catalytic ribozyme, for synthesis or metabolism (Fig. 5b).77 However, integrating ribozymes into a protocell is challenging because the high concentrations of divalent cations (typically in the order of 10−3 to 10−2 M)78 needed for ribozyme catalysis will disrupt fatty acid-based protocell membrane. A potential solution to this problem is to construct Mg2+-tolerant protocells using a combination of fatty acids and their glycerol ester.79 However, this strategy is limited due to the hydrolytic tendency of esters. Recently, Adamala et al.80 used an amide analogue of a monoacylglycerol (lipid amide) instead of monoacylglycerol as a membrane component to construct protocells and found that the protocells obtained could tolerate millimolar concentrations of magnesium, under which ribozymes could work within. In addition, by encapsulating a lipid amide precursor and a catalyst mimicking primitive acyltransferase ribozyme, lipid amide could be synthesized inside the protocell so that these protocells showed increased tolerance to magnesium with the expectation that they would survive during evolution.However, as the third major class of biomolecules, sugars, have been rarely studied in a protocell-based evolution process. In fact, owing to their simplicity of elementary composition, carbohydrates can be synthesized using a primitive form of metabolism consisting only of small molecules, albeit absent from the genetic central dogma. Gardner et al.81 built a chemical protocell containing an autocatalytic formose reaction that could produce sugars from formaldehyde in the interior (Fig. 5c). Then the synthesized carbohydrates exited the protocell and formed a complex with borate in the medium. The carbohydrate–borate complexes diffused through the medium to interact with the bacterium Vibrio harveyi and activated a signaling pathway, which finally gave a detectable bioluminescent output. This work realized a simplified communication process between chemical cells and natural cells (bacteria). The protometabolism encapsulated inside the protocell is fueled by small-molecule precursors, which is a notable achievement in the field of protocell construction.
5. DNA dynamic circuit-equipped protocells
With the development of DNA dynamic technology, the computational capability and scaling up ability of DNA logic circuits have been demonstrated.82 Because all basic theories and operative strategies of DNA dynamic circuits have been established within the last several decades, the mechanism of DNA dynamic circuit-based artificial reaction networks is more like a man-made engineering process. The goal of constructing protocells programmed by DNA dynamic circuits is never simple mimicry of some key characterizations of natural cells, but rather to build cell-like automatons with controllable behavior. As the most widely used material possessing both programmability and biomolecular property, DNA has huge potential in constructing powerful artificial reaction networks to propel protocells. From the aspect of programmability, DNA circuits have been proven to be quite efficient in parallel computation.83 As a directional and linear molecule, for a certain length of nucleic acid, there is an exponentially large number of different possible combinations of nucleotides, which indicates a very high information density and numerous potential reaction pathways.82 Benefiting from biomolecular properties, e.g., catalytic activity and recognition capability,84 DNA circuits can function seamlessly with biological inputs and outputs, just like other nucleic acids, proteins, small molecules, and even cellular states, allowing DNA dynamic circuit-equipped protocells to work in electrolyte- and biomolecule-rich biological environments such as vasculatures. In this section, we will review the recent progress in constructing DNA circuits running on protocell membrane and building DNA circuits as a computational core in a protocell chamber.5.1 DNA circuits on artificial membranes
Similarly to silicon-based electronic circuits running on chips, DNA logic circuits also need a carrier so that species can be concentrated, and reactions can be constrained in a limited region instead of diffusing away. For a natural cell, numerous reactions occur on the surface such as dimerization and phosphorylation of membrane protein and cause different downstream signaling pathways, which inspired researchers to build DNA reaction networks on the surface of a protocell to fulfill some membrane functions by mimicking these naturally existing reaction networks. Besides, compared to encapsulating DNA species in the protocell chamber, constructing DNA reaction networks on the surface is much more straightforward. As in known, lipid bilayer-based compartmentalization is of great significance for the spatiotemporally ordered metabolism process of a natural cell. However, the low permeability and negative charge of this lipid bilayer, conversely, acts as a barrier for encapsulating negatively charged DNA strands into the chamber when constructing protocells. In contrast, the lipid membrane itself, especially the outer leaflet, is much more accessible. Thanks to mature DNA solid-phase synthesis and DNA modification technology, DNA strands can be easily conjugated with different hydrophobic moieties (Fig. 6a) such as phospholipid,85 cholesterol,86 tocopherol,87 porphyrin88 and even multiple ethyl groups,89 which will act as molecular anchors for on-membrane insertion with various microdomain preferences. While chemical synthesis grants facile access to specific sequences and a variety of lipid anchors, Watson–Crick-based recognition can be used to control the proximity of the appended entities, as demonstrated by pioneering research involving the construction of a DNA reaction network on a cell-like giant unilamellar vesicle surface reported by Schade et al.90 Another notable work was designed by Czogalla et al.91 who engineered amphipathic DNA origami structures as membrane-scaffolding tools to mimic the oligomerization steps of biological membrane scaffolding proteins that drive membrane deformation. The amphipathic DNA origami structure has a flat membrane-binding interface decorated with cholesterol-derived anchors and sticky oligonucleotide overhangs on its side facet. Only DNA origami monomers that laterally bind to neighboring monomers were shown to form ordered arrays capable of deforming free-standing lipid membranes.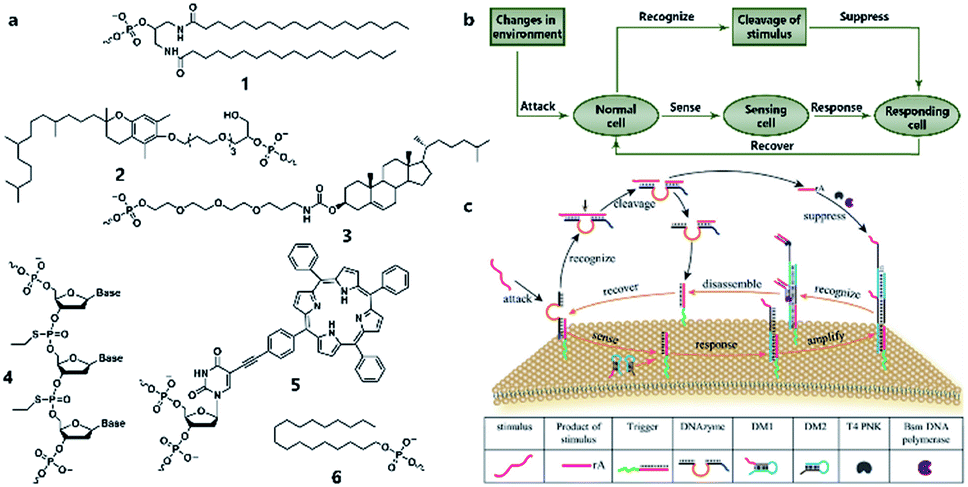 | ||
| Fig. 6 (a) Different lipophilic moieties can be conjugated with DNA strands to anchor the reaction network on the protocell membrane. (1) Phospholipid, (2) tocopherol, (3) cholesterol, (4) ethyl group, (5) porphyrin, and (6) stearyl. (b) Design concept of DASsys-based protocell model responding to environmental stimuli. (c) Recyclable DASsys anchored on protocell membrane surface was composed of three stages: external stimulus, cell sensing and self-protection, and stimulus elimination and cell recovery. When attacked by the external stimulus (attacker strand), the system was activated to trigger two signal pathways including a feedforward loop. As a result, the attacker strand was eliminated, and the membrane surface was fully restored to the initial state and able to respond to the next incoming stimulus. Reproduced from ref. 94 with permission. Copyright 2018 American Chemical Society. | ||
All the above research indicated the feasibility of constructing artificial DNA reaction networks on the surface of artificial vesicles. These synthetic lipid membranes were widely used in synthetic biology approaches when constructing protocells, but they lacked the inherent complexity of natural membranes. To solve this problem, Peng et al.92 used living mammalian cells to generate cell-mimicking micrometer-scale giant unilamellar vesicles93 and reported the assembly and disassembly of DNA nanoprisms on the membrane of these vesicles controlled by DNA strand hybridization and toehold-mediated strand displacement, which is the dynamic basis for constructing scaled-up DNA reaction networks on the protocell surface. Recently, a sophisticated artificial DNA reaction network was successfully fitted to the surface of giant vesicles to create a protocell, which could both sense incoming stimuli and emit a feedback response to eliminate the stimuli (Fig. 6b)94 as a mimic of natural cell adaptation. As shown in Fig. 6c, this protocell system, termed DNA-based artificial signal system (DASsys), was composed of three stages, external stimulus, cell sensing and self-protection, and stimulus elimination and cell recovery. The initial state of the artificial cell was defined by a cholesterol-labeled DNA trigger that was anchored on the artificial cell membrane and the hybridized DNAzyme strand. When the system was attacked by the external stimulus, acting by a piece of ssDNA, the initial strand was released after a strand-displacement reaction, and the DNAzyme strand was hybridized with the added attacker strand. Afterwards, two signal pathways were activated. The released initial strand triggered a hybridization chain reaction by recruiting two DNA monomers (DMs), generating a DNA polymer on the membrane. Meanwhile, the attacker strand was cut into two pieces of smaller strands by DNAzyme in the presence of magnesium ions. Consequently, the digested attacker strand was then captured by the DNA polymer mentioned above and elongated by DNA polymerase as a primer, leading to the disassembly of the DNA polymer and release of the DNA trigger strand. Finally, the DNA trigger strand recaptured the DNAzyme strand, and the membrane surface was fully restored to the initial state and able to respond to the next incoming stimulus. This process has a feedforward loop that can respond to environmental stimuli, thus providing an engineered approach to introduce interaction between protocells and the environment. With the help of a DNA reaction network built on the surface, protocells can both sense and respond to stimuli, as well as self-renew by returning to the pre-stimulus state, thereby permitting continuous sensing and responding in the micromilieu.
5.2 DNA circuits in an artificial chamber
Although the lipid membrane of an artificial cell can be used as a perfect carrier or biological disk to run the built-in DNA reaction network, a direct interface with the external environment may easily interrupt a signal pathway consisting of DNA hybridization and strand displacement. In addition, without the compartmentalization of the lipid bilayer, which is impermeable to many electrolytes and biological macromolecules, the diversity of a DNA reaction network may be restricted by surrounding buffer conditions, which are unfavorable for the design of independent computing systems. As discussed before, two goals generally propel studies on artificial cells, originology and bionics. From both aspects, it is anticipated that the created protocell can become an independent thinker, just like its natural counterpart, the independence of which is guaranteed by different membranous structures. For this reason, Lyu et al.95 encapsulated a DNA reaction network, termed artificial immune response simulator (AIRS),96 inside an artificial cell as the computing core (Fig. 7a). In AIRS, key species existing in a vertebrate adaptive immune response are mimicked by DNA strands to show the computing ability of DNA circuits. By constructing an artificial pathogen, the artificial cell becomes infected through a strand migration-induced membrane fusion process. Then, the mimicked host immune response happens in a concise way, as defined by AIRS, but within an artificially constructed intracellular microenvironment to eliminate the artificial exogenous pathogenic challenge (Fig. 7b). The encapsulated DNA reaction network consists of three steps, recognition and tolerance, immune response, killing and memory, as shown in Fig. 7a. After pathogenic DNA (P) is injected into the artificial cell via a mimicked infection process, the first step of built-in AIRS, recognition and tolerance, is triggered. Pathogenic DNA is recognized by antigen-presenting cell mimicry (AM) and T cell mimicry (TM) is activated via a strand displacement process. When the amount of infected pathogen DNA is below the threshold of immune tolerance, the next steps (step 2 and step 3) will not be triggered. However, if the amount of pathogenic DNA is beyond the threshold, the released TM acts as a catalyst to accelerate the immune response against excessive pathogenic DNA by activating B cell mimicry (BM) to release the antibody initiator (AI). Then a rolling circle amplification (RCA)-based artificial immune response is activated, involving the production of antibody mimicry (RCA product) (step 2), immunological memory, and the specific destruction of the foreign DNA by a restriction enzyme (step 3).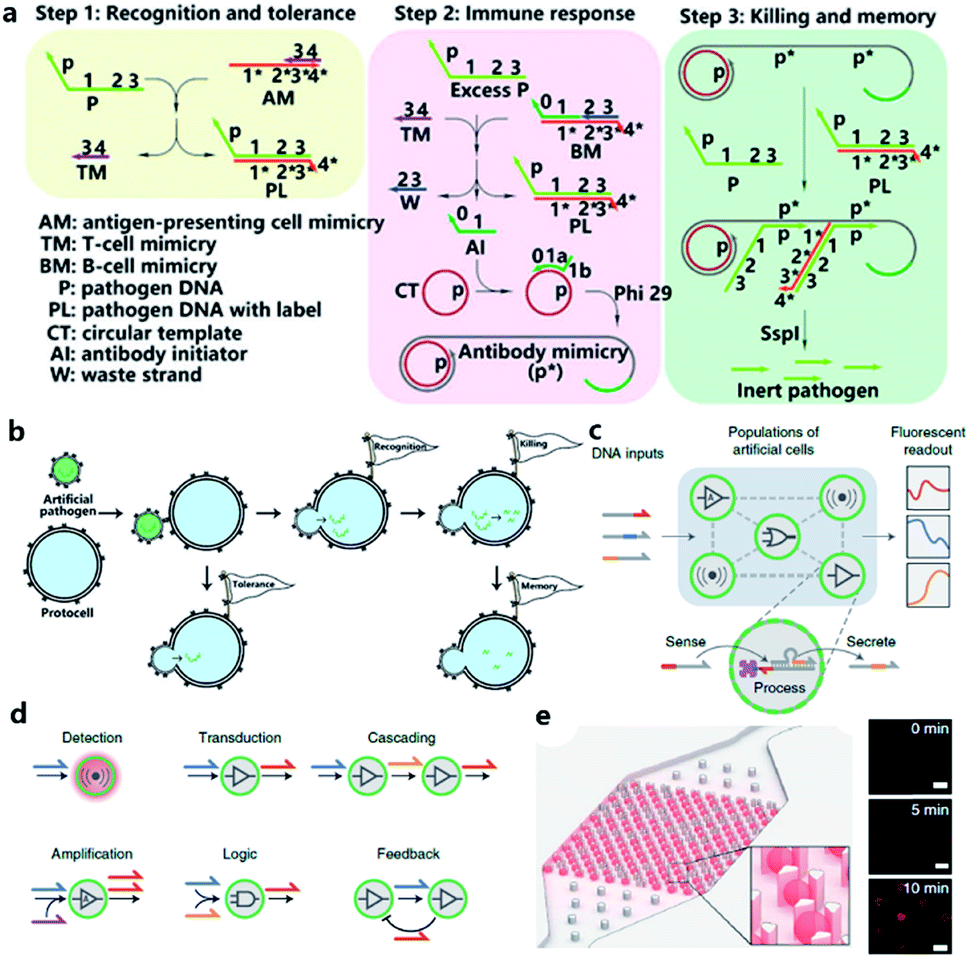 | ||
| Fig. 7 (a and b) Working principle of protocell with built-in AIRS. (a) Mechanism of AIRS as a DNA computational core built inside the protocell. Step 1: recognition and tolerance. Pathogen DNA is recognized by AIRS and if the amount of pathogen DNA is below the threshold of immune tolerance, further immune response will not be triggered. Step 2: immune response. If the amount of pathogen is excessive, an RCA-based immune response will be accelerated and antibody-mimicry (RCA product) will be generated. Step 3: killing and memory. Pathogen DNA is specifically captured by generated antibody-mimicry via hybridization and subsequently digested by a restriction enzyme. (b) Pathogen DNA is injected from an artificial pathogen into a protocell via a mimicked infection process. The delivered pathogen DNA triggers AIRS inside the protocell and a mimicked host immune response is activated to eliminate the infected pathogen DNA via a DNA reaction network-based computation. Reproduced from ref. 95 with permission. Copyright 2018 American Chemical Society. (c–f) Design of biomolecular implementation of protocellular communication (BIO-PC). (c) Basic principle of a BIO-PC platform that can sense, process and secrete short ssDNA-based signals. The semipermeable membrane allows an input strand to diffuse into a protocell followed by activation of a DNA gate complex via toehold-mediated strand displacement reaction. (d) Individual protocells can be designed as functional modules and combined to implement more complex population behaviors such as detection, transduction, cascading, amplification, logic operation, and feedback circuit. (e) Protocells can be captured and imaged on a microfluidic protocell trap array. Right panel: confocal imaging of eight protocells showing time-dependent signal increase after activation. Scale bar, 50 μm. Reproduced from ref. 97 with permission. Copyright 2019 Springer Nature. | ||
The scalability and cascading reaction of DNA reaction networks can also be built between artificial cells to engineer an artificial multicellular system. Joesaar et al.97 engineered a series of DNA logic circuit-based protocells (Fig. 7c) capable of cascading amplification, bidirectional communication and distributed computational operations, and further constructed a highly programmable protocellular messaging system, termed ‘Biomolecular Implementation Of Protocellular Communication’ (BIO-PC) (Fig. 7d). The mechanism to achieve signal transduction between each protocell is quite ingenious. Protocells in this system were based on protein-polymer crosslinked microcapsules called proteinosomes, which have a porous membrane permeable to input and output strands that are shorter than 100 bases. In contrast, biotinylated DNA gate strands were conjugated with streptavidin in order to remain in the interior instead of diffusing out of the membrane. Such a confinement-based strategy offers a concise solution to combine both permeability and compartmentalization into a single protocell. The DNA reaction network encapsulated in protocells was programmed by classical toehold-mediated strand displacement reactions, which were proved to be capable of constructing powerful algorithms.98,99 To observe the reaction kinetics, a microfluidic protocell trap array was prepared to capture and image a single protocell (Fig. 7e). Using recovered fluorescence as the output signal, they achieved a signaling cascade, negative feedback network between two proteinosome populations, and protocell-based three-population AND network in the BIO-PC system. Another achievement in their work is that by encapsulating DNA circuits in proteinosomes, the BIO-PC system can operate in 50% serum, which indicates a potential application of constructing programmable protocells in mammalian vasculatures.
6. Conclusions and perspectives
Protocells were initially created to study the origin and early evolution of life and to understand the mechanism of modern cells. Although both goals have not yet been achieved, numerous model protocells with cell-like structures and at least some of the essential properties of a natural cell have been developed and described in the present review. Rationally designed artificial reaction networks based on genetic systems were employed to reconstitute cellular functions in protocells. Complex behaviors, such as cell growth and division, metabolism, host immune response, and intercellular communication, were realized via enzyme-assisted or small molecule-based artificial reaction networks. DNA dynamic circuits were built on the surface or in the chamber of protocells as the computing core to construct cell-like automatons with a “logical mind”. As synthetic equivalents of natural cells, these properties or behaviors of protocells are modulated and controlled by equipping them with various artificial reaction networks so that future researchers can endow protocells with different functions in a customized way using programmed language and a bio-compiler.Although great achievements have been made, there are still many important challenges to be answered. One obvious limitation of the protocells reported is that almost all complex functions need external supplies of energy molecules or substrates and cannot sustain for a long time. Although the advancement of synthetic biology and technology allows the construction of more sophisticated bio-circuits, building more independent artificial reaction network propelled protocells is more profound and rewarding. Besides, it is the key to answer the question of the origin of life, since on primitive Earth no modern biomolecules existed. Although our attempts to generate an artificial entity able to evolve and be considered as a living cell have, thus far, failed, more rational hypotheses about the growth, division and replication of protocells on early Earth will be proposed. Another challenge is to segregate different artificial reaction networks effectively to afford protocells with organelles that operate spontaneously and even synergistically through the communication between these organelles. In addition, DNA reaction network-encapsulated artificial cells will attract increasing interest in the future because this strategy provides new principles of protocell construction and a biocompatible carrier or platform to build smart bionic automatons. A possible future direction is to integrate DNA circuits encapsulated inside a protocell with circuits built onto the protocell membrane so that DNA computation can be performed over the whole cell with enhanced programmability.
Although this review has focused on the remarkable advances in the construction of protocells programmed from sketches of artificial reaction networks or a bottom-up approach to construct protocells, a top-down approach100 that includes genetic manipulation or theoretical analysis of minimal genomes also shows potential in understanding the rules of cellular life. Perhaps developing a strategy that combines both top-down and bottom-up approaches will accelerate the studies of protocell construction and protocell-based biotics and bioengineering. Prospectively, prototissues101 formed by interacting protocells that can sense and adapt to their surroundings will be the next stage of exploration and understanding of life.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by Renji Hospital, Shanghai Jiao Tong University, by National Natural Science Foundation Major Instrument Development Project (21827811), China Postdoctoral Science Foundation Funded Project (Project No. 2018M640398) and Shanghai Rising-Star Program (Grant No. 19QA1405400 to J. L.).Notes and references
- S. Van Segbroeck, A. Nowé and T. Lenaerts, Artif. Life, 2009, 15, 213–226 CrossRef PubMed.
- B. N. Kholodenko, Nat. Rev. Mol. Cell Biol., 2006, 7, 165–176 CrossRef CAS PubMed.
- C. Xu, S. Hu and X. Chen, Mater. Today, 2016, 19, 516–532 CrossRef CAS PubMed.
- V. Noireaux, Y. T. Maeda and A. Libchaber, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3473–3480 CrossRef CAS PubMed.
- B. C. Buddingh' and J. C. van Hest, Acc. Chem. Res., 2017, 50, 769–777 CrossRef PubMed.
- A. Lopez and M. Fiore, Life, 2019, 9, 49 CrossRef PubMed.
- G. F. Joyce and J. W. Szostak, Cold Spring Harbor Perspect. Biol., 2018, 10, a034801 CrossRef PubMed.
- P. Stano and P. L. Luisi, Curr. Opin. Biotechnol., 2013, 24, 633–638 CrossRef CAS PubMed.
- D. Han, H. Kang, T. Zhang, C. Wu, C. Zhou, M. You, Z. Chen, X. Zhang and W. Tan, Chem.–Eur. J., 2014, 20, 5866–5873 CrossRef CAS PubMed.
- H. W. van Roekel, B. J. Rosier, L. H. Meijer, P. A. Hilbers, A. J. Markvoort, W. T. Huck and T. F. de Greef, Chem. Soc. Rev., 2015, 44, 7465–7483 RSC.
- O. Sinanoglu, J. Am. Chem. Soc., 1975, 97, 2309–2320 CrossRef CAS.
- J. I. Glass, N. Assad-Garcia, N. Alperovich, S. Yooseph, M. R. Lewis, M. Maruf, C. A. Hutchison, H. O. Smith and J. C. Venter, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 425–430 CrossRef CAS PubMed.
- F. Caschera and V. Noireaux, Curr. Opin. Chem. Biol., 2014, 22, 85–91 CrossRef CAS PubMed.
- P. Walde, A. Goto, P.-A. Monnard, M. Wessicken and P. L. Luisi, J. Am. Chem. Soc., 1994, 116, 7541–7547 CrossRef CAS.
- A. C. Chakrabarti, R. R. Breaker, G. F. Joyce and D. W. Deamer, J. Mol. Evol., 1994, 39, 555–559 CrossRef CAS PubMed.
- T. Oberholzer, R. Wick, P. L. Luisi and C. K. Biebricher, Biochem. Biophys. Res. Commun., 1995, 207, 250–257 CrossRef CAS PubMed.
- T. Oberholzer, M. Albrizio and P. L. Luisi, Chem. Biol., 1995, 2, 677–682 CrossRef CAS PubMed.
- K. Tsumoto, S.-i. M. Nomura, Y. Nakatani and K. Yoshikawa, Langmuir, 2001, 17, 7225–7228 CrossRef CAS.
- T. Oberholzer, K. H. Nierhaus and P. L. Luisi, Biochem. Biophys. Res. Commun., 1999, 261, 238–241 CrossRef CAS PubMed.
- W. Yu, K. Sato, M. Wakabayashi, T. Nakaishi, E. P. Ko-Mitamura, Y. Shima, I. Urabe and T. Yomo, J. Biosci. Bioeng., 2001, 92, 590–593 CrossRef CAS PubMed.
- S.-i. M. Nomura, K. Tsumoto, T. Hamada, K. Akiyoshi, Y. Nakatani and K. Yoshikawa, ChemBioChem, 2003, 4, 1172–1175 CrossRef CAS PubMed.
- V. Noireaux and A. Libchaber, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17669–17674 CrossRef CAS PubMed.
- K. Ishikawa, K. Sato, Y. Shima, I. Urabe and T. Yomo, FEBS Lett., 2004, 576, 387–390 CrossRef CAS PubMed.
- Y. Shimizu, T. Kanamori and T. Ueda, Methods, 2005, 36, 299–304 CrossRef CAS PubMed.
- Y. Shimizu, A. Inoue, Y. Tomari, T. Suzuki, T. Yokogawa, K. Nishikawa and T. Ueda, Nat. Biotechnol., 2001, 19, 751 CrossRef CAS PubMed.
- T. Sunami, K. Sato, T. Matsuura, K. Tsukada, I. Urabe and T. Yomo, Anal. Biochem., 2006, 357, 128–136 CrossRef CAS PubMed.
- G. Murtas, Y. Kuruma, P. Bianchini, A. Diaspro and P. L. Luisi, Biochem. Biophys. Res. Commun., 2007, 363, 12–17 CrossRef CAS PubMed.
- J. W. Whittaker, Biotechnol. Lett., 2013, 35, 143–152 CrossRef CAS PubMed.
- D. E. Nevin and J. M. Pratt, FEBS Lett., 1991, 291, 259–263 CrossRef CAS PubMed.
- Y. Kuruma, P. Stano, T. Ueda and P. L. Luisi, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 567–574 CrossRef CAS PubMed.
- J. Shin and V. Noireaux, J. Biol. Eng., 2010, 4, 8 CrossRef PubMed.
- J. Garamella, R. Marshall, M. Rustad and V. Noireaux, ACS Synth. Biol., 2016, 5, 344–355 CrossRef CAS PubMed.
- F. J. Ghadessy, J. L. Ong and P. Holliger, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4552–4557 CrossRef CAS PubMed.
- N. Ichihashi, K. Usui, Y. Kazuta, T. Sunami, T. Matsuura and T. Yomo, Nat. Commun., 2013, 4, 2494 CrossRef PubMed.
- S. Fujii, T. Matsuura, T. Sunami, Y. Kazuta and T. Yomo, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16796–16801 CrossRef CAS PubMed.
- R. Lentini, S. P. Santero, F. Chizzolini, D. Cecchi, J. Fontana, M. Marchioretto, C. Del Bianco, J. L. Terrell, A. C. Spencer and L. Martini, Nat. Commun., 2014, 5, 4012 CrossRef CAS PubMed.
- H. Matsubayashi, Y. Kuruma and T. Ueda, Angew. Chem., Int. Ed., 2014, 53, 7535–7538 CrossRef CAS PubMed.
- Y. Elani, A. Gee, R. V. Law and O. Ces, Chem. Sci., 2013, 4, 3332–3338 RSC.
- Y. Elani, R. V. Law and O. Ces, Phys. Chem. Chem. Phys., 2015, 17, 15534–15537 RSC.
- K. P. Adamala, D. A. Martin-Alarcon, K. R. Guthrie-Honea and E. S. Boyden, Nat. Chem., 2016, 9, 431 CrossRef PubMed.
- A. I. Oparin, A. F. Orlovskiĭ, V. Bukhlaeva and K. L. Gladilin, Dokl. Akad. Nauk SSSR, 1976, 226, 972–974 CAS.
- P. R. Green and R. M. Bell, J. Biol. Chem., 1984, 259, 14688–14694 CAS.
- P. K. Schmidli, P. Schurtenberger and P. L. Luisi, J. Am. Chem. Soc., 1991, 113, 8127–8130 CrossRef CAS.
- G. Murtas, Syst. Synth. Biol., 2010, 4, 85–93 CrossRef PubMed.
- E. Schweizer and J. Hofmann, Microbiol. Mol. Biol. Rev., 2004, 68, 501–517 CrossRef CAS PubMed.
- M. Exterkate, A. Caforio, M. C. Stuart and A. J. Driessen, ACS Synth. Biol., 2017, 7, 153–165 CrossRef PubMed.
- A. Bhattacharya, R. J. Brea, H. Niederholtmeyer and N. K. Devaraj, Nat. Commun., 2019, 10, 300 CrossRef PubMed.
- B. Deisinger, T. Nawroth, K. Zwicker, S. Matuschka, G. John, G. Zimmer and H. J. Freisleben, Eur. J. Biochem., 1993, 218, 377–383 CrossRef CAS PubMed.
- P. Richard, B. Pitard and J.-L. Rigaud, J. Biol. Chem., 1995, 270, 21571–21578 CrossRef CAS PubMed.
- B. Pitard, P. Richard, M. Duñach and J. L. Rigaud, Eur. J. Biochem., 1996, 235, 779–788 CrossRef CAS PubMed.
- H.-J. Choi and C. D. Montemagno, Nano Lett., 2005, 5, 2538–2542 CrossRef CAS PubMed.
- X. Feng, Y. Jia, P. Cai, J. Fei and J. Li, ACS Nano, 2015, 10, 556–561 CrossRef PubMed.
- K. Y. Lee, S.-J. Park, K. A. Lee, S.-H. Kim, H. Kim, Y. Meroz, L. Mahadevan, K.-H. Jung, T. K. Ahn and K. K. Parker, Nat. Biotechnol., 2018, 36, 530 CrossRef CAS PubMed.
- S. Berhanu, T. Ueda and Y. Kuruma, Nat. Commun., 2019, 10, 1325 CrossRef PubMed.
- R. J. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. Van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS PubMed.
- Y. Elani, R. V. Law and O. Ces, Nat. Commun., 2014, 5, 5305 CrossRef CAS PubMed.
- Y. Qiao, M. Li, R. Booth and S. Mann, Nat. Chem., 2017, 9, 110–119 CrossRef CAS PubMed.
- B. P. Kumar, A. J. Patil and S. Mann, Nat. Chem., 2018, 10, 1154–1163 CrossRef CAS PubMed.
- E. Blöchliger, M. Blocher, P. Walde and P. L. Luisi, J. Phys. Chem. B, 1998, 102, 10383–10390 CrossRef.
- M. M. Hanczyc, S. M. Fujikawa and J. W. Szostak, Science, 2003, 302, 618–622 CrossRef CAS PubMed.
- T. F. Zhu and J. W. Szostak, J. Am. Chem. Soc., 2009, 131, 5705–5713 CrossRef CAS PubMed.
- I. Budin, A. Debnath and J. W. Szostak, J. Am. Chem. Soc., 2012, 134, 20812–20819 CrossRef CAS PubMed.
- T. F. Zhu, K. Adamala, N. Zhang and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9828–9832 CrossRef CAS PubMed.
- I. A. Chen, R. W. Roberts and J. W. Szostak, Science, 2004, 305, 1474–1476 CrossRef CAS PubMed.
- K. Adamala and J. W. Szostak, Nat. Chem., 2013, 5, 495 CrossRef CAS PubMed.
- K.-i. Shohda, M. Tamura, Y. Kageyama, K. Suzuki, A. Suyama and T. Sugawara, Soft Matter, 2011, 7, 3750–3753 RSC.
- K. Kurihara, M. Tamura, K.-i. Shohda, T. Toyota, K. Suzuki and T. Sugawara, Nat. Chem., 2011, 3, 775–781 CrossRef CAS PubMed.
- H. Kaddour and N. Sahai, Life, 2014, 4, 598–620 CrossRef CAS PubMed.
- M. Blocher, D. Liu, P. Walde and P. L. Luisi, Macromolecules, 1999, 32, 7332–7334 CrossRef CAS.
- T. Hitz, M. Blocher, P. Walde and P. L. Luisi, Macromolecules, 2001, 34, 2443–2449 CrossRef CAS.
- H. Zepik, S. Rajamani, M.-C. Maurel and D. Deamer, Origins Life Evol. Biospheres, 2007, 37, 495–505 CrossRef CAS PubMed.
- S. S. Mansy, J. P. Schrum, M. Krishnamurthy, S. Tobé, D. A. Treco and J. W. Szostak, Nature, 2008, 454, 122–125 CrossRef CAS PubMed.
- S. S. Mansy and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13351–13355 CrossRef CAS PubMed.
- K. Adamala and J. W. Szostak, Science, 2013, 342, 1098–1100 CrossRef CAS PubMed.
- D. K. O'Flaherty, N. P. Kamat, F. N. Mirza, L. Li, N. Prywes and J. W. Szostak, J. Am. Chem. Soc., 2018, 140, 5171–5178 CrossRef PubMed.
- D. K. O'Flaherty, L. Zhou and J. W. Szostak, J. Am. Chem. Soc., 2019, 141, 10481–10488 CrossRef PubMed.
- J. W. Szostak, D. P. Bartel and P. L. Luisi, Nature, 2001, 409, 387–390 CrossRef CAS PubMed.
- A. S. Petrov, J. C. Bowman, S. C. Harvey and L. D. Williams, RNA, 2011, 17, 291–297 CrossRef CAS PubMed.
- I. A. Chen, K. Salehi-Ashtiani and J. W. Szostak, J. Am. Chem. Soc., 2005, 127, 13213–13219 CrossRef CAS PubMed.
- K. P. Adamala, A. E. Engelhart and J. W. Szostak, Nat. Commun., 2016, 7, 11041 CrossRef CAS PubMed.
- P. M. Gardner, K. Winzer and B. G. Davis, Nat. Chem., 2009, 1, 377–383 CrossRef CAS PubMed.
- T. Fu, Y. Lyu, H. Liu, R. Peng, X. Zhang, M. Ye and W. Tan, Trends Biochem. Sci., 2018, 43, 547–560 CrossRef CAS PubMed.
- L. M. Adleman, Science, 1994, 1021–1024 CrossRef CAS PubMed.
- J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS PubMed.
- L. Qiu, T. Zhang, J. Jiang, C. Wu, G. Zhu, M. You, X. Chen, L. Zhang, C. Cui and R. Yu, J. Am. Chem. Soc., 2014, 136, 13090–13093 CrossRef CAS PubMed.
- J. R. Burns, A. Seifert, N. Fertig and S. Howorka, Nat. Nanotechnol., 2016, 11, 152–156 CrossRef CAS PubMed.
- M. You, Y. Lyu, D. Han, L. Qiu, Q. Liu, T. Chen, C. S. Wu, L. Peng, L. Zhang and G. Bao, Nat. Nanotechnol., 2017, 12, 453–459 CrossRef CAS PubMed.
- J. R. Burns, K. Göpfrich, J. W. Wood, V. V. Thacker, E. Stulz, U. F. Keyser and S. Howorka, Angew. Chem., Int. Ed., 2013, 52, 12069–12072 CrossRef CAS PubMed.
- J. R. Burns, E. Stulz and S. Howorka, Nano Lett., 2013, 13, 2351–2356 CrossRef CAS PubMed.
- M. Schade, A. Knoll, A. Vogel, O. Seitz, J. r. Liebscher, D. Huster, A. Herrmann and A. Arbuzova, J. Am. Chem. Soc., 2012, 134, 20490–20497 CrossRef CAS PubMed.
- A. Czogalla, D. J. Kauert, H. G. Franquelim, V. Uzunova, Y. Zhang, R. Seidel and P. Schwille, Angew. Chem., Int. Ed., 2015, 54, 6501–6505 CrossRef CAS PubMed.
- R. Peng, H. Wang, Y. Lyu, L. Xu, H. Liu, H. Kuai, Q. Liu and W. Tan, J. Am. Chem. Soc., 2017, 139, 12410–12413 CrossRef CAS PubMed.
- Q. Liu, C. Bi, J. Li, X. Liu, R. Peng, C. Jin, Y. Sun, Y. Lyu, H. Liu and H. Wang, Research, 2019, 2019, 6523970 Search PubMed.
- H. Liu, Q. Yang, R. Peng, H. Kuai, Y. Lyu, X. Pan, Q. Liu and W. Tan, J. Am. Chem. Soc., 2019, 141, 6458–6461 CrossRef CAS PubMed.
- Y. Lyu, C. Wu, C. Heinke, D. Han, R. Cai, I.-T. Teng, Y. Liu, H. Liu, X. Zhang and Q. Liu, J. Am. Chem. Soc., 2018, 140, 6912–6920 CrossRef CAS PubMed.
- D. Han, C. Wu, M. You, T. Zhang, S. Wan, T. Chen, L. Qiu, Z. Zheng, H. Liang and W. Tan, Nat. Chem., 2015, 7, 835–841 CrossRef CAS PubMed.
- A. Joesaar, S. Yang, B. Bögels, A. van der Linden, P. Pieters, B. P. Kumar, N. Dalchau, A. Phillips, S. Mann and T. F. de Greef, Nat. Nanotechnol., 2019, 14, 369–378 CrossRef CAS PubMed.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- G. Seelig, D. Soloveichik, D. Y. Zhang and E. Winfree, Science, 2006, 314, 1585–1588 CrossRef CAS PubMed.
- C. A. Hutchison, R.-Y. Chuang, V. N. Noskov, N. Assad-Garcia, T. J. Deerinck, M. H. Ellisman, J. Gill, K. Kannan, B. J. Karas, L. Ma, J. F. Pelletier, Z.-Q. Qi, R. A. Richter, E. A. Strychalski, L. Sun, Y. Suzuki, B. Tsvetanova, K. S. Wise, H. O. Smith, J. I. Glass, C. Merryman, D. G. Gibson and J. C. Venter, Science, 2016, 351, aad6253 CrossRef PubMed.
- P. Gobbo, A. J. Patil, M. Li, R. Harniman, W. H. Briscoe and S. Mann, Nat. Mater., 2018, 17, 1145–1153 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |