
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOpen-circuit-voltage shift of over 0.5 V in organic photovoltaic cells induced by a minor structural difference in alkyl substituents†‡
Mitsuharu
Suzuki
 *a,
Kengo
Terai
b,
Cassandre
Quinton§
b,
Hironobu
Hayashi
b,
Naoki
Aratani
b and
Hiroko
Yamada
*b
*a,
Kengo
Terai
b,
Cassandre
Quinton§
b,
Hironobu
Hayashi
b,
Naoki
Aratani
b and
Hiroko
Yamada
*b
aDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: msuzuki@mls.eng.osaka-u.ac.jp
bDivision of Materials Science, Nara Institute of Science and Technology (NAIST), 8916-5 Takayama-cho, Ikoma, Nara 630-0192, Japan. E-mail: hyamada@ms.naist.jp
First published on 13th January 2020
Abstract
The recent surge in the efficiency of organic photovoltaic devices (OPVs) largely hinges on the reduction of energy loss (Eloss) that leads to improvements in open-circuit voltage (VOC). However, there are still many unclarified factors regarding the relationship between the molecular structure and VOC, hampering the establishment of widely applicable, effective principles for the design of active-layer materials. In this contribution, we examine the origin of the large VOC shifts induced by minor structural differences in end-alkyl substituents on a series of anthracene-based p-type compounds. The examined p-type compounds are all highly crystalline, thereby enabling detailed investigation of the molecular packing with X-ray diffraction analysis. At the same time, they are strongly aggregating and hardly soluble; therefore, they are deposited with the aid of a photoprecursor approach which we have been employing for controlled deposition of insoluble acene-based organic semiconductors. The resultant OPVs afford the highest VOC of 0.966 V when the end-alkyl groups are 2-ethylbutyl, and the lowest of 0.419 V when n-butyl is used in replacement of 2-ethylbutyl. X-ray diffraction analyses and density-functional-theory calculations indicate a critical impact of the non-slipped herringbone arrangement on the observed large loss in VOC. This type of molecular arrangement is prohibited when branched alkyl chains are introduced at the ends of linear π-systems, which we consider an important factor contributing to the relatively high VOC obtained with the 2-ethylbutyl derivative. These results may serve as a basis of useful molecular-design rules to avoid unnecessary losses in VOC.
Introduction
The open-circuit voltage (VOC) is currently a main limiting factor in the development of organic photovoltaic cells (OPVs). The energy loss (Eloss), which is the difference between the optical band-gap energy (Eoptg) of an active layer and the VOC multiplied by the elementary charge (eVOC), is rarely smaller than 0.6 eV in OPVs, while it is only 0.3–0.5 eV in higher-efficiency options such as crystalline silicon and perovskite PVs. Accordingly, considerable effort has been devoted to the elucidation of the origin of VOC and to the reduction of Eloss.1–5 For example, the extensive comparison of a number of different OPVs has offered empirical and semiempirical formulae that correlate VOC with the frontier-orbital energies of active-layer compounds.6–8 These formulae have been serving as practical guidelines for molecular design toward minimizing Eloss from the view point of orbital-energy matching. Indeed, studies in this context have led to the finding that the apparent loss from Eoptg to charge-transfer-state energy (i.e., the driving force for formation of the charge-transfer state) can be reduced to near zero for certain p- and n-type material combinations.9 Another aspect revealed by previous studies is the critical impact of the microstructure at interfaces such as p/n and active layer/electrode.10–18 Among others, Izawa et al. has recently reported that the crystallinity at the donor/acceptor interface affects VOC, demonstrating a shift by about 0.25 V upon changing the length of alkyl substituents on perylene diimide, a typical n-type material employed in OPVs.10 In another example, Ran et al. showed that a difference in molecular orientation at the p/n interface led to a VOC shift of about 0.15 V in planar heterojunction (PHJ) OPVs.12However, there are still open questions regarding the relationship between the molecular structure/packing and VOC. One question that is seemingly simple, yet remains to be answered is about the shift of frontier-orbital energies induced by the minor modification of alkyl substituents.19–26 A typical example can be found in the paper by Seo, wherein the ionization energy (IE) was decreased from 4.92 to 4.61 eV upon changing the N-alkyl groups of a diketopyrrolopyrrole (DPP)-based p-type compound from 2-ethylhexyl to n-hexyl.25 The VOC in the corresponding OPVs also decreased from 0.75 to 0.47 V. The author assumed that the shift in IE was caused by the difference in packing motif, but detailed analysis was left to be reported. The difference of about 0.3 V in VOC is significant, being comparable to the non-radiative voltage loss: one of the largest factors unfavourably affecting VOC in OPVs.6 Therefore, it is valuable to clarify how such a minor structural difference in substituents causes a major alteration in electronic structure of organic semiconductors.
Meanwhile, we are working on controlled deposition of small-molecule organic semiconductors through a photoprecursor approach.27–33 In this approach, an α-diketone (DK)-type precursor of acene derivative is deposited by a wet process, and then converted to a target compound through the visible-light-induced decarbonylative aromatization in the thin-film state (Fig. 1).33 As DK derivatives are generally more soluble than corresponding acenes, this technique makes highly crystalline acene derivatives compatible with solution processes without relying on bulky solubilizing groups. Thus, substituents can be designed purely for packing control, which is advantageous in gaining a clear understanding of the relationship between the molecular structure and packing in thin films.
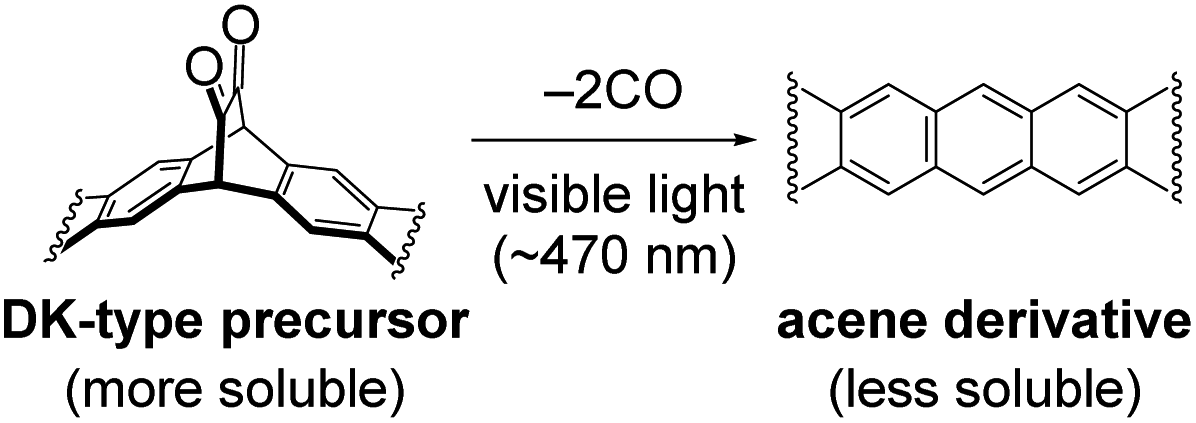 | ||
| Fig. 1 Formation of acene derivatives via light-induced decarbonylative aromatization of DK-type photoprecursors. | ||
By taking advantage of the photoprecursor approach, this study compares four alkylated derivatives of 2,6-di(2,2′-bithien-5-yl)anthracene (RDBTA; R = C4 (n-butyl), C6 (n-hexyl), C8 (n-octyl), or EB (2-ethylbutyl)) and 2,6-di(2-thienyl)anthracene (DTA) as p-type materials in OPVs (Fig. 2). Each of these crystalline compounds is deposited from a solution of the corresponding DK-type precursor (RDBTADK) and [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM) to form a p:n-intermixed sublayer (i-sublayer) in p–i–n-type devices. Among the resultant OPVs, the highest VOC of 0.966 V is obtained with EBDBTA, while the lowest is only 0.419 V with C4DBTA. The origin of this large VOC shift is investigated by using single-crystal and thin-film X-ray diffraction (XRD) analyses in combination with density-functional-theory (DFT) calculations. The results indicate a critical impact of non-slipped π–π stacking on the increase in IE, providing a basis of substituent design to avoid unnecessary decreases in VOC.
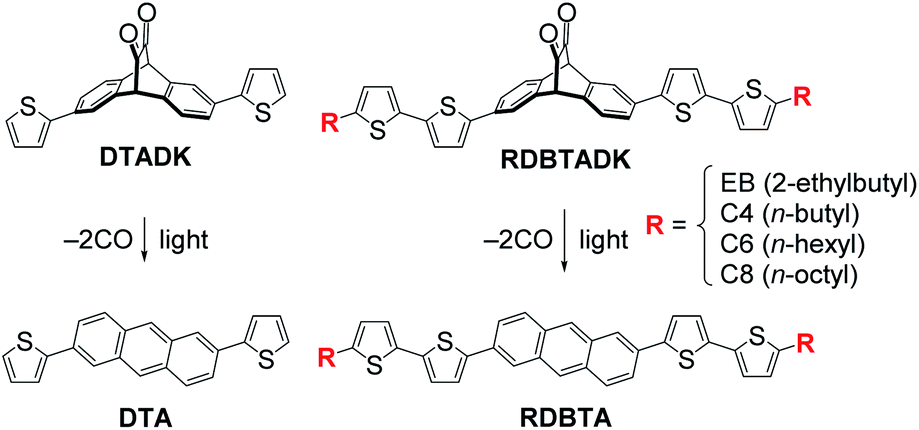 | ||
| Fig. 2 Chemical structures of the p-type materials examined in this work and the corresponding DK-type photoprecursors. | ||
Results and discussion
Molecular design
This study originated from the observation in our previous work, wherein 2,6-di[5′-(2-ethylhexyl)-2,2′-bithien-5-yl]anthracene (EHDBTA, Fig. S1‡) gave a slightly higher VOC as compared to DTA (0.86–0.93 V vs. 0.82–0.89 V) when used as a p-type material in p–i–n-type OPVs.32 This result was totally unexpected: EHDBTA should have a higher energy level for the highest occupied molecular orbital (HOMO) and thus give a lower VOC than DTA, because of the extension of the π-system with electron-rich thienylene units, and the substitution with electron-donating alkyl groups.In search of plausible explanations for this previous observation, we decided to examine another alkylated DBTA, EBDBTA. While EHDBTA was used as a mixture of stereoisomers regarding the alkyl groups, EBDBTA does not involve such isomerism, and therefore is more suitable for detailed investigation on solid-state molecular packing. We also designed the derivatives with linear alkyl chains, C4, C6, and C8DBTA, for systematic evaluation of the effect of alkyl groups.
Photovoltaic performance
DTA and RDBTA were evaluated as p-type materials in the i-sublayer of p–i–n-type OPVs. The organic active layers were prepared through the photoprecursor approach according to our previous papers.27,32 Specifically, the p-sublayer composed of 2,8-diphenylanthra[1,2-b:5,6-b′]dithiophene (PhBADT) was prepared by spin-coating of the corresponding DK-type photoprecursor (PhBADTDK) followed by photoirradiation (Fig. S1‡).27 The i-sublayer was then deposited in the same manner using a DTA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC71BM or RDBTADK:PC71BM (2:1, w/w) blend solution instead of PhBADTDK. The resulting i-sublayers do not fully dissolve due to the insoluble DTA or RDBTA, and thus the n-sublayer could be deposited by simply spin-coating PC71BM on the i-sublayer. Details of device preparation are provided in ESI.‡
:PC71BM or RDBTADK:PC71BM (2:1, w/w) blend solution instead of PhBADTDK. The resulting i-sublayers do not fully dissolve due to the insoluble DTA or RDBTA, and thus the n-sublayer could be deposited by simply spin-coating PC71BM on the i-sublayer. Details of device preparation are provided in ESI.‡
Photovoltaic parameters of the p–i–n OPVs are summarized in Table 1 and the corresponding current-density–voltage (J–V) curves and external quantum efficiency (EQE) spectra are plotted in Fig. 3. As reported in the previous paper,32 DTA afforded a relatively high VOC of 0.900 V. Note that the p-sublayer material and cathode buffer in the present work are different from those employed in the previous paper (ESI‡), which may be the cause of the minor deviation from the previous data. EBDBTA showed a higher VOC of 0.966 V, in good agreement with the case of EHDBTA mentioned above. This result indicates that the structural difference between 2-ethylhexyl and 2-ethylbutyl makes minimal impact on the electronic structure of the DBTA system in the thin-film state. Apart from the VOC, EBDBTA gave a considerably higher short-circuit current density (JSC, 6.71 mA cm−2) and fill factor (FF, 62.0%) than DTA (2.94 mA cm−2 and 55.2%). These differences can be ascribed to the higher light-absorption capability and better film morphology of EBDBTA, as previously reported for EHDBTA.
| p-Type material | J SC/cm2 V−1 s−1 | V OC/V | FF/% | PCE/% |
|---|---|---|---|---|
| a Data of best-PCE cells followed by average of six cells in parentheses. b Measured under simulated AM1.5G illumination at 100 mW cm−2. c Active-layer structure: [PhBADT/(DTA or RDBTA):PC71BM/PC71BM]. | ||||
| DTA | 2.94 (3.08 ± 0.39) | 0.900 (0.854 ± 0.037) | 55.2 (47.2 ± 5.5) | 1.46 (1.23 ± 0.15) |
| EBDBTA | 6.71 (6.61 ± 0.08) | 0.966 (0.960 ± 0.004) | 62.0 (60.4 ± 1.8) | 4.02 (3.84 ± 0.14) |
| C4DBTA | 7.20 (7.02 ± 0.12) | 0.419 (0.421 ± 0.002) | 48.3 (48.2 ± 0.5) | 1.46 (1.42 ± 0.03) |
| C6DBTA | 7.19 (7.09 ± 0.27) | 0.499 (0.501 ± 0.009) | 56.2 (53.2 ± 2.4) | 2.02 (1.89 ± 0.12) |
| C8DBTA | 6.83 (6.89 ± 0.09) | 0.602 (0.595 ± 0.004) | 54.1 (52.4 ± 1.0) | 2.22 (2.15 ± 0.05) |
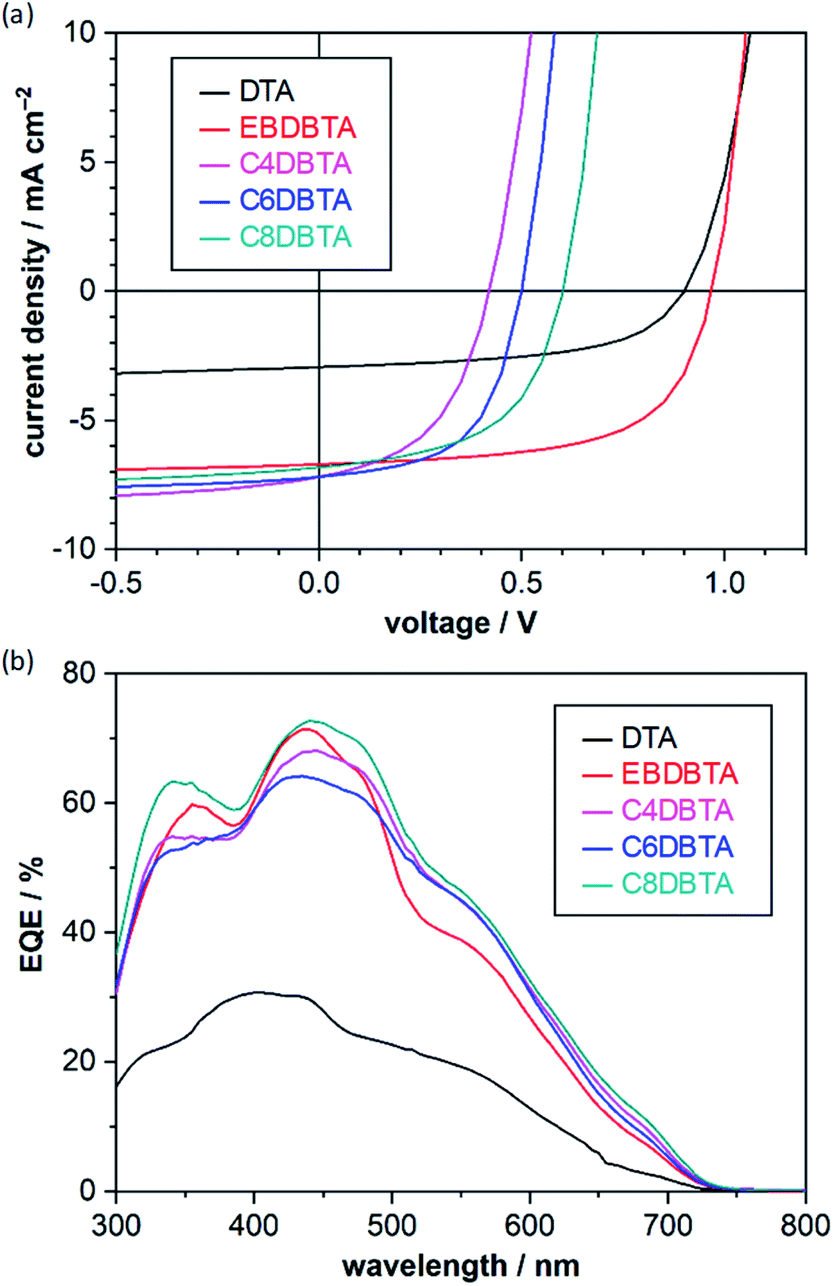 | ||
| Fig. 3 Photovoltaic response of the p–i–n OPVs. (a) J–V curves; (b) EQE spectra. Data are of the best-performing cells in each entry shown in Table 1. The J–V measurements were performed under simulated AM1.5G illumination at 100 mW cm−2. Active-layer structure: [PhBADT/(DTA or RDBTA):PC71BM/PC71BM]. | ||
The DBTA derivatives with linear alkyl substituents gave considerably lower VOCs: 0.419, 0.499, and 0.602 V for C4, C6, and C8DBTA, respectively. Indeed, the VOC of 0.419 V is less than half of the highest VOC obtained with EBDBTA (0.966 V). Such a large difference in VOC (>0.5 V) has been rarely observed for bulk-heterojunction (BHJ)-type blend films in which molecules are less organized and the effect of molecular structure is rather obscured compared to neat films. Note that the analysis of surface topology with atomic force microscopy has shown that the RDBTA:PC71BM blends generally form a smooth surface without large crystallites (root-mean-square roughness < 1 nm, Fig. S2b–e‡), indicating relatively well-mixed nature of these blend films. On the other hand, DTA forms large grains in the blend film with PC71BM indicating a significant degree of phase separation as reported in previous papers (Fig. S2a‡).29,32 The PCE obtained with EBDBTA reaches 4.02%, an efficiency which compares well with that of the conventional benchmark bulk-heterojunction (BHJ) system comprising poly(3-hexylthiophene) (P3HT) and PC71BM.34 This is respectable if one considers the simple, small structure of EBDBTA. By contrast, C4DBTA afforded a PCE of only 1.46% at best. Comparison of C4DBTA with DTA shows that the gain in JSC due to the enhanced light-absorption capability (2.94 → 7.20 mA cm−2) was mostly cancelled out by the loss in VOC (0.900 → 0.419 V).
The above results confirm that the high VOC for EHDBTA can be reproduced with the more crystalline derivative EBDBTA. In addition, it has turned out that the DBTA derivatives having linear end-alkyl groups afford significantly lower VOCs. It should be also noted that VOC increases with the elongation of the linear end-alkyl chains: 0.419, 0.499, and 0.602 V for C4, C6, and C8DBTA. We will investigate the origin of these observations in the following sections.
Electronic properties
As the first step of the investigation, we measured the HOMO energies of the DBTA derivatives. Note here that the term “HOMO” is expanded to describe electronic properties of molecular thin films or molecular aggregates, and the HOMO energy in the thin-film state (HOMOfilm) is defined as negated IE (i.e., HOMOfilm = –IE). The IE is measured with photoelectron spectroscopy in air (PESA) (Fig. S3‡). As summarized in Table 2, EBDBTA showed the deepest HOMOfilm of −5.39 eV, and DTA gave a slightly higher value of −5.36 eV. On the other hand, C4, C6, and C8DBTA showed considerably higher values of −4.91, −5.00, and −5.02 eV, respectively. The dependency of HOMOfilm on the structure of alkyl groups matches very well with that of VOC in the p–i–n OPVs.| Compound | HOMOfilma/eV | E oxp /V | E optg /eV |
|---|---|---|---|
| a HOMO in the thin-film state determined from the IE measured by PESA. b Potential of the first oxidation peak measured by DPV. Values are vs. the Fc/Fc+ redox couple. c Absorption-onset energy of thin films. | |||
| DTA | −5.36 | 0.62 | 2.66 |
| EBDBTA | −5.39 | 0.44 | 2.42 |
| C4DBTA | −4.91 | 0.43 | 2.39 |
| C6DBTA | −5.00 | 0.43 | 2.39 |
| C8DBTA | −5.02 | 0.43 | 2.39 |
We also measured the oxidation potentials of the five p-type compounds in solution by differential pulse voltammetry (DPV). Although the compounds are hardly soluble in common solvents, they dissolve in o-dichlorobenzene/benzonitrile (9:1, v/v) barely enough for detecting oxidation peaks (Fig. S4‡). As listed in Table 2, the oxidation peak potential (Eoxp) of DTA was determined as 0.62 V vs. the ferrocene/ferrocenium (Fc/Fc+) reference, while all the DBTA derivatives showed Eoxp of 0.43–0.44 V. This observation is reasonable in terms of the relationship between molecular and electronic structures; namely, the extension of π-system from DTA to DBTA causes decrease in oxidation potential (i.e., increase in HOMO level), and the difference of alkyl substituents poses little effects on the easiness of oxidation.
The PESA and DPV measurements have revealed that the linear and branched alkyl substituents induce significantly different degrees of perturbation in the electronic structure of DBTA upon condensation from a solution to a solid. This effect can be seen in the photoabsorption spectra of thin films as well: the shapes of spectra are different between EBDBTA and the other DBTA derivatives especially at shorter wavelengths below 400 nm, although the absorption onsets are similar regardless of alkyl groups (Fig. 4 and Table 2). Importantly, the strong correlation between the VOC and HOMOfilm among all the five p-type compounds suggests that the shifts in VOC are mostly due to the interactions between molecules of the same kind rather than those at the p/n and active layer/electrode interfaces.
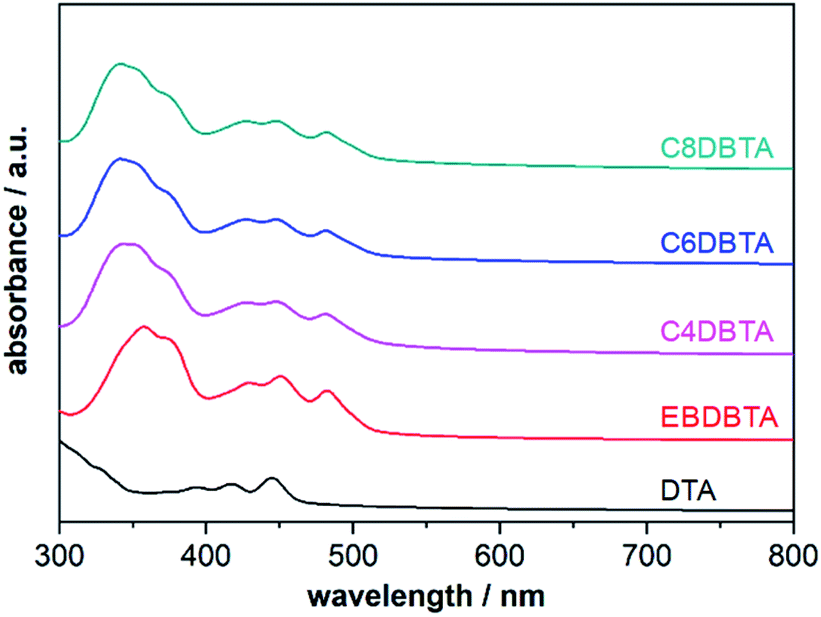 | ||
| Fig. 4 UV-vis absorption spectra of thin films of DTA and the DBTA derivatives. | ||
Molecular packing in single crystals
We then investigated molecular packing by single-crystal X-ray diffraction (XRD) analysis. Single crystals of EB, C4, and C6DBTA suitable for the measurement were obtained by exposing the DK-type precursors in o-dichlorobenzene to weak light such that the target compounds were generated and crystallized out slowly. Unfortunately, single crystals of C8DBTA could not be obtained despite multiple trials under different conditions. The single-crystal structure of DTA is available in literature.35 In the following discussion, we concentrate on DTA, EBDBTA, and C6DBTA for simplicity. C4DBTA packs similarly to the other DBTA derivatives as depicted in Fig. S5.‡ Single-crystal X-ray diffraction parameters and thermal ellipsoid plots are shown in Table S1 and Fig. S6.‡Comparison between DTA and EBDBTA revealed a clear difference in packing of π-frameworks: while the two compounds both crystallized in the herringbone motif, DTA molecules packed straight without a longitudinal slip, but EBDBTA molecules formed a slipped stacking motif (Fig. 5a and b). Calculations at the PBEPBE/6-31G(d) level of theory suggested that this difference might cause an appreciable impact on the electronic structure of molecules in the solid state (Fig. 6). When calculations were performed on isolated molecules in the single-crystal X-ray structures, DTA gave a lower HOMO level (−4.39 eV) than EBDBTA (−4.30 eV) as expected. However, for extended packing structures along the herringbone stack, the HOMO energy of DTA quickly increased with the number of molecules than that of EBDBTA. As a result, DTA showed a higher HOMO energy (−4.11 eV) than EBDBTA (−4.14 eV) already at the stage of 7 molecules. In addition, the difference between these two compounds kept increasing at least up to the point of 13 molecules. Thus, it seemed that the high VOC observed with EBDBTA could be explained by the packing motif in the single-crystalline state.
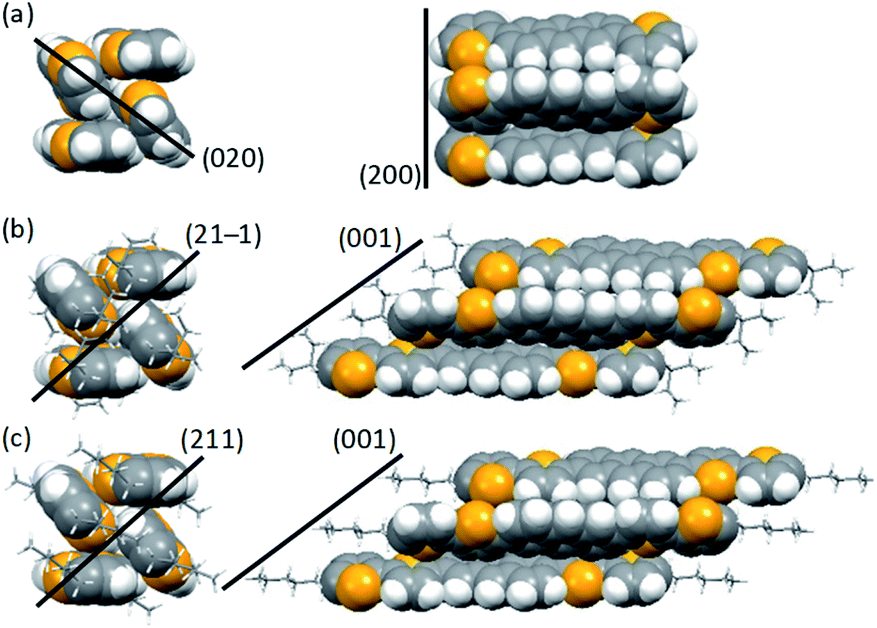 | ||
| Fig. 5 Molecular packing in single crystals of (a) DTA,25 (b) EBDBTA, and (c) C6DBTA. Left and right columns are views along and normal to the long molecular axis, respectively. Black lines indicate approximate positions of the crystal planes relevant to the slipped or non-slipped herringbone motifs. | ||
 | ||
| Fig. 6 Calculated HOMO energies for single and sets of molecules in the single-crystal X-ray structures of DTA,25 EBDBTA, and C6DBTA. Calculations were performed at the PBEPBE/6-31G(d) level of theory. | ||
It turned out, however, that the same argument was not applicable to the case of C6DBTA. Single-crystal XRD analysis revealed that C6DBTA packed in a slipped-herringbone motif similarly to EBDBTA. Further, DFT calculations suggested a similar degree of electronic perturbation for C6DBTA in the single-crystalline state as compared to EBDBTA. Specifically, the HOMO levels of C6DBTA were calculated as −4.27, −4.12, and −4.08 eV for 1, 4, and 7 molecules, and only negligible increase was indicated after the 7-molecule point. Here, the small deviation between the calculated HOMO energies of the two compounds can be attributed to minor differences in molecular conformation and packing motif, which in turn confirms that the large difference in VOC cannot be explained by the single-crystal X-ray structures.
Molecular packing in thin films
We performed out-of-plane XRD analysis also on neat films of DTA and RDBTA to see if there are any differences in molecular packing between single crystals and thin films. The resultant diffraction profiles of DTA, EBDBTA, and C6DBTA are plotted in Fig. 7, while those of C4 and C8DBTA are shown in Fig. S7.‡ For DTA, relatively strong X00 diffractions are apparent, and thus molecules of this compound tend to stack in an end-on mode with clear repeating order in the out-of-plane direction of the film (see Fig. 5 for the corresponding crystal plane). In addition, a closer look reveals a small peak that can be assigned as the 020 diffraction, which indicates that the ordered herringbone motif exists as well in the film. Therefore, it can be assumed that DTA molecules maintain the single-crystal packing motif both the longitudinal and lateral molecular axes in the thin-film state, although the orientation of crystallites seems random.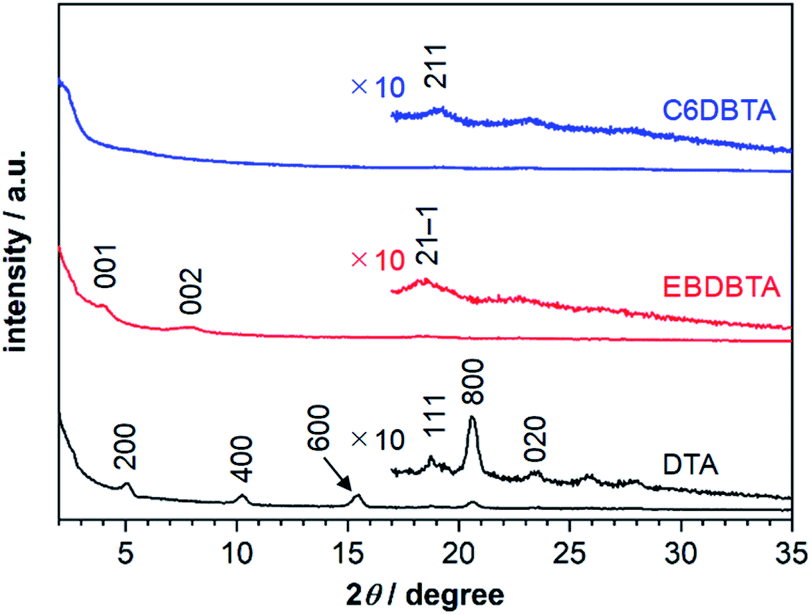 | ||
| Fig. 7 Out-of-plane thin-film XRD patterns of DTA, EBDBTA, and C6DBTA. | ||
The situation is similar for EBDBTA; namely, the 00X and 21−1 diffractions indicate the ordered arrangements along the longitudinal and lateral molecular axes are both maintained in the film. In contrast, the C6DBTA film does not show the 00X peaks, while the 211 diffraction is still observable. This indicates that the herringbone motif exists, but the longitudinal molecular arrangement is random. Based on this observation and other results described in the earlier sections, we postulate that the randomness in longitudinal slip has led to the high HOMO level of C6DBTA. This randomness can certainly include the non-slipped arrangement or similar that induces significant increase of the HOMO level as in the case of DTA. On the other hand, EBDBTA cannot adopt such an arrangement because of the steric demand associated with the branched alkyl groups, and thus its HOMO level stays relatively low in the thin film state.
The differences in HOMO level among C4, C6, C8DBTA may be explained by the degree of randomness along the longitudinal molecular axis, too. It is expected that longer alkyl chains would bring about stronger intermolecular interactions that facilitate the formation of a thermodynamically stable, ordered arrangement (i.e., the slipped-herringbone packing for C4 and C6DBTA) during the preparation of thin films. Therefore, C6DBTA must have lower randomness and thus less chances to form non-slipped herringbone arrangement than C4DBTA, leading to a somewhat attenuated increase of the HOMO energy. Although the single-crystal X-ray structure is unknown, the same discussion seems valid for C8DBTA as well, considering that the length of end-alkyl groups does not cause critical changes in overall packing motif for many small molecules.36 Accordingly, the HOMO level is expected to decrease slightly in the order of C4 > C6 > C8DBTA, which is in accordance with the experimental observation.
Conclusions
This contribution has demonstrated that a minor structural modification of alkyl groups in an active-layer compound can cause a significant change in VOC. Spectroscopic analyses have indicated that the change in VOC originates from the difference in intermolecular interactions between the same kind of molecules, rather than the hetero-interactions at p/n or active-layer/electrode interfaces. The single crystal XRD data show that DTA and EB, C4, C6DBTA all adopt the herringbone packing; however, DTA is different from the other compounds in terms of the packing motif along the longitudinal molecular axis (i.e., slipped vs. non-slipped packing). This aspect can explain the unexpected similarity in VOC between DTA and EBDBTA. On the other hand, the out-of-plane thin-film XRD analysis has revealed that the degree of ordering along the longitudinal molecular axis is different between EBDBTA and the other DBTA derivatives. This latter aspect can explain the exceptionally high VOC of EBDBTA among the four DBTA derivatives.All these observations point toward the importance of avoiding the herringbone packing with non-slipped stacking along the π-extended framework. This molecular arrangement leads to significant increase of the HOMO level and thus decrease in VOC. Having bulky substituents such as branched alkyl chains is effective for avoiding the non-slipped herringbone packing. Note here that similar observations can be found in previous papers, but to our knowledge detailed investigation has not been reported, probably because of the low crystallinity of employed materials that obscures molecular packing. In contrast, the compound employed in the present study are crystalline such that single-crystal and thin-film XRD analyses can be conducted without difficulties. The knowledge obtained here will serve as a useful guideline in the molecular design of organic semiconductors for OPVs or other optoelectronic applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST CREST and MEXT/JSPS Grants-in-Aid for Scientific Research (KAKENHI) (Nos. JP26105004, JP16H02286, JP17H03042, JP18K14190, JP18K14298, JP19K22112). We thank Ms Y. Nishikawa, Mr S. Katao, and Ms M. Furukawa at NAIST for help in mass spectrometry, X-ray diffraction analysis, and chemical synthesis, respectively. C. Q. thanks the JSPS for a post-doctoral fellowship.Notes and references
- X. He, L. Yin and Y. Li, J. Mater. Chem. C, 2019, 7, 2487–2521 RSC.
- M. Azzouzi, T. Kirchartz and J. Nelson, Trends Chem., 2019, 1, 49–62 CrossRef.
- S. M. Menke, N. A. Ran, G. C. Bazan and R. H. Friend, Joule, 2018, 2, 25–35 CrossRef CAS.
- T. Linderl, T. Zechel, M. Brendel, D. M. González, P. Müller-Buschbaum, J. Pflaum and W. Brütting, Adv. Energy Mater., 2017, 7, 1700237 CrossRef.
- N. K. Elumalai and A. Uddin, Energy Environ. Sci., 2016, 9, 391–410 RSC.
- J. Benduhn, K. Tvingstedt, F. Piersimoni, S. Ullbrich, Y. Fan, M. Tropiano, K. A. McGarry, O. Zeika, M. K. Riede, C. J. Douglas, S. Barlow, S. R. Marder, D. Neher, D. Spoltore and K. Vandewal, Nat. Energy, 2017, 2, 1–6 Search PubMed.
- A. Maurano, R. Hamilton, C. G. Shuttle, A. M. Ballantyne, J. Nelson, B. O'Regan, W. Zhang, I. McCulloch, H. Azimi, M. Morana, C. J. Brabec and J. R. Durrant, Adv. Mater., 2010, 22, 4987–4992 CrossRef CAS.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- J. Yuan, N. A. Ran, M. J. Ford, M. Wang, M. K. Ravva, C.-K. Mai, X. Liu, J.-L. Brédas, T.-Q. Nguyen, W. Ma and G. C. Bazan, J. Mater. Chem. A, 2017, 5, 18618–18626 RSC.
- S. Izawa, N. Shintaku, M. Kikuchi and M. Hiramoto, Appl. Phys. Lett., 2019, 115, 153301 CrossRef.
- F. Wang, K. Nakano, H. Yoshida, K. Hashimoto, H. Segawa, C.-S. Hsu and K. Tajima, J. Mater. Chem. A, 2018, 6, 22889–22898 RSC.
- N. A. Ran, S. Roland, J. A. Love, V. Savikhin, C. J. Takacs, Y.-T. Fu, H. Li, V. Coropceanu, X. Liu, J.-L. Brédas, G. C. Bazan, M. F. Toney, D. Neher and T.-Q. Nguyen, Nat. Commun., 2017, 8, 79 CrossRef PubMed.
- S. M. Ryno, Y.-T. Fu, C. Risko and J.-L. Brédas, ACS Appl. Mater. Interfaces, 2016, 8, 15524–15534 CrossRef CAS PubMed.
- B. Kitchen, O. Awartani, R. J. Kline, T. McAfee, H. Ade and B. T. O'Connor, ACS Appl. Mater. Interfaces, 2015, 7, 13208–13216 CrossRef CAS PubMed.
- S. Izawa, K. Nakano, K. Suzuki, K. Hashimoto and K. Tajima, Adv. Mater., 2015, 27, 3025–3031 CrossRef CAS PubMed.
- Y. Zhong, A. Tada, S. Izawa, K. Hashimoto and K. Tajima, Adv. Energy Mater., 2014, 4, 1301332 CrossRef.
- U. Hörmann, C. Lorch, A. Hinderhofer, A. Gerlach, M. Gruber, J. Kraus, B. Sykora, S. Grob, T. Linderl, A. Wilke, A. Opitz, R. Hansson, A. S. Anselmo, Y. Ozawa, Y. Nakayama, H. Ishii, N. Koch, E. Moons, F. Schreiber and W. Brütting, J. Phys. Chem. C, 2014, 118, 26462–26470 CrossRef.
- A. Tada, Y. Geng, Q. Wei, K. Hashimoto and K. Tajima, Nat. Mater., 2011, 10, 450–455 CrossRef CAS PubMed.
- J. Min, Y. N. Luponosov, N. Gasparini, L. Xue, F. V. Drozdov, S. M. Peregudova, P. V. Dmitryakov, K. L. Gerasimov, D. V. Anokhin, Z.-G. Zhang, T. Ameri, S. N. Chvalun, D. A. Ivanov, Y. Li, S. A. Ponomarenko and C. J. Brabec, J. Mater. Chem. A, 2015, 3, 22695–22707 RSC.
- R. B. Zerdan, N. T. Shewmon, Y. Zhu, J. P. Mudrick, K. J. Chesney, J. Xue and R. K. Castellano, Adv. Funct. Mater., 2014, 24, 5993–6004 CrossRef CAS.
- J. Min, Y. N. Luponosov, A. Gerl, M. S. Polinskaya, S. M. Peregudova, P. V. Dmitryakov, A. V. Bakirov, M. A. Shcherbina, S. N. Chvalun, S. Grigorian, N. Kaush-Busies, S. A. Ponomarenko, T. Ameri and C. J. Brabec, Adv. Energy Mater., 2014, 4, 1301234 CrossRef.
- Y. J. Kim, K. H. Park, J. Ha, D. S. Chung, Y.-H. Kim and C. E. Park, Phys. Chem. Chem. Phys., 2014, 16, 19874–19883 RSC.
- D. Deng, Y. Zhang, L. Yuan, C. He, K. Lu and Z. Wei, Adv. Energy Mater., 2014, 4, 1400538 CrossRef.
- J. Liu, Y. Sun, P. Moonsin, M. Kuik, C. M. Proctor, J. Lin, B. B. Hsu, V. Promarak, A. J. Heeger and T.-Q. Nguyen, Adv. Mater., 2013, 25, 5898–5903 CrossRef CAS PubMed.
- J. H. Seo, Synth. Met., 2012, 162, 748–752 CrossRef CAS.
- D. Baran, A. Balan, T. Stubhan, T. Ameri, L. Toppare and C. J. Brabec, Synth. Met., 2012, 162, 2047–2051 CrossRef CAS.
- M. Suzuki, Y. Yamaguchi, K. Uchinaga, K. Takahira, C. Quinton, S. Yamamoto, N. Nagami, M. Furukawa, K. Nakayama and H. Yamada, Chem. Sci., 2018, 9, 6614–6621 RSC.
- K. Kawajiri, T. Kawanoue, M. Yamato, K. Terai, M. Yamashita, M. Furukawa, N. Aratani, M. Suzuki, K. Nakayama and H. Yamada, ECS J. Solid State Sci. Technol., 2017, 6, M3068–M3074 CrossRef CAS.
- M. Suzuki, Y. Yamaguchi, K. Takahashi, K. Takahira, T. Koganezawa, S. Masuo, K. Nakayama and H. Yamada, ACS Appl. Mater. Interfaces, 2016, 8, 8644–8651 CrossRef CAS PubMed.
- C. Quinton, M. Suzuki, Y. Kaneshige, Y. Tatenaka, C. Katagiri, Y. Yamaguchi, D. Kuzuhara, N. Aratani, K. Nakayama and H. Yamada, J. Mater. Chem. C, 2015, 3, 5995–6005 RSC.
- S. Masuo, W. Sato, Y. Yamaguchi, M. Suzuki, K. Nakayama and H. Yamada, Photochem. Photobiol. Sci., 2015, 14, 883–890 RSC.
- Y. Yamaguchi, M. Suzuki, T. Motoyama, S. Sugii, C. Katagiri, K. Takahira, S. Ikeda, H. Yamada and K. Nakayama, Sci. Rep., 2014, 4, 7151 CrossRef CAS PubMed.
- M. Suzuki, T. Aotake, Y. Yamaguchi, N. Noguchi, H. Nakano, K. Nakayama and H. Yamada, J. Photochem. Photobiol., C, 2014, 18, 50–70 CrossRef CAS.
- P. A. Troshin, H. Hoppe, J. Renz, M. Egginger, J. Y. Mayorova, A. E. Goryachev, A. S. Peregudov, R. N. Lyubovskaya, G. Gobsch, N. S. Sariciftci and V. F. Razumov, Adv. Funct. Mater., 2009, 19, 779–788 CrossRef CAS.
- H. Meng, F. Sun, M. B. Goldfinger, G. D. Jaycox, Z. Li, W. J. Marshall and G. S. Blackman, J. Am. Chem. Soc., 2005, 127, 2406–2407 CrossRef CAS PubMed.
- H. Minemawari, M. Tanaka, S. Tsuzuki, S. Inoue, T. Yamada, R. Kumai, Y. Shimoi and T. Hasegawa, Chem. Mater., 2017, 29, 1245–1254 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Michael R. Wasielewski on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Supporting figures and a table, experimental details including spectroscopic measurements, X-ray diffraction analysis, computation, and chemical synthesis. CCDC 1956921–1956923. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04956h |
| § Current address: Université de Rennes, CNRS, ISCR-UMR 6226, F-35000 Rennes, France. |
| This journal is © The Royal Society of Chemistry 2020 |