
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn intramolecular catalytic hairpin assembly on a DNA tetrahedron for mRNA imaging in living cells: improving reaction kinetics and signal stability†
Zhihe
Qing
 *a,
Jinlei
Hu
a,
Jingyuan
Xu
a,
Zhen
Zou
a,
Yanli
Lei
a,
Taiping
Qing
b and
Ronghua
Yang
*a
*a,
Jinlei
Hu
a,
Jingyuan
Xu
a,
Zhen
Zou
a,
Yanli
Lei
a,
Taiping
Qing
b and
Ronghua
Yang
*a
aHunan Provincial Key Laboratory of Materials Protection for Electric Power and Transportation, Hunan Provincial Engineering Research Center for Food Processing of Aquatic Biotic Resources, School of Chemistry and Food Engineering, Changsha University of Science and Technology, Changsha, 410114, P. R. China. E-mail: qingzhihe@hnu.edu.cn; Yangrh@pku.edu.cn
bCollege of Environment and Resources, Xiangtan University, Xiangtan, 411105, P. R. China
First published on 18th December 2019
Abstract
Enzyme-free amplification techniques based on dynamic DNA self-assembly (DDSA) have recently been developed for the in situ detection of mRNA in living cells. However, signal generation in traditional DDSA amplifiers is mainly dependent on the random diffusion of dissociative probes in a bulk solution, which is generally accompanied by poor kinetics and interference from complex biological systems. In this work, a new amplifier based on the design of an intramolecular catalytic hairpin assembly (intra-CHA) is proposed for the FRET imaging of mRNA in living cells. Compared with that in the free catalytic hairpin assembly (free-CHA), probes H1 and H2 in intra-CHA were simultaneously fixed on a DNA tetrahedron. The distance between them was closer, the local concentration of H1 and H2 in intra-CHA was theoretically approximately 808-times higher than that in free-CHA, and the initial reaction rate was enhanced 15.6 fold. Due to the spatial confinement effect, the reaction kinetics for target-catalyzed signal generation were significantly improved. By virtue of the three-dimensional nanostructure, H1 and H2 in the intra-CHA amplifier entered cells without any transfection or nanocarrier, and the probes and their products were free from biological interference, providing much higher signal stability for the reliable imaging of mRNA in living cells.
Introduction
Messenger RNA (mRNA) can be translated into functional proteins in the ribosomes, and differential expression of mRNA in cells has been shown to be correlated with some diseases.1,2 For example, the imbalanced expression of manganese superoxide dismutase (MnSOD) mRNA is closely related to tumor proliferation.3,4 Thus, mRNA has been a significant biomarker in disease diagnosis, and the development of highly efficient strategies for detecting mRNA is required, especially in situ imaging to analyze it in living cells.5–8Reverse transcription-polymerase chain reaction (RT-PCR) is a traditional amplification method that has been developed to detect RNA at low concentration levels, but it only operates in cell lysate or buffer, and cannot reflect the difference in the expression level between cells.9,10 Isothermal enzymatic signal amplification-based fluorescence in situ hybridization (FISH) has been used to achieve in situ imaging of mRNA in single cells, but because the fixation of cells is required for inward transportation of enzymes, it is inapplicable for living cell imaging.11,12
In recent years, enzyme-free amplification techniques based on dynamic DNA self-assembly (DDSA) have been developed for amplification detection of targets of interest and also imaging of RNA in living cells, such as the hybridization chain reaction (HCR)13–15 and catalyzed hairpin assembly (CHA).16–18 However, some inescapable issues still remain to be addressed for DDSA amplifiers. First, with traditional DDSA amplifiers, reactions generally occur through random diffusion of dissociative probes in a bulk solution, which is difficult for collision of probes, and then leads to low efficiency and low kinetics of signal generation.19–21 Second, dissociative DNA probes cannot independently enter cells. They are dependent on transfection or nanocarriers,22–24 and the complex biological environment (especially the presence of nuclease) would degrade dissociative DNA probes and their reaction products, resulting in signal instability.25,26 Therefore, it is desirable to develop a highly efficient DDSA amplifier that not only improves the kinetics, but also the signal stability, and ensures good cell permeability and nuclease resistance.
Herein, inspired by this demand, we propose an intramolecular catalytic hairpin assembly (intra-CHA) on a DNA tetrahedron for fluorescence resonance energy transfer (FRET) imaging of mRNA in living cells. Two hairpin probes H1 and H2 labeled with fluorophores Cy3 and Cy5, respectively, were linked to two vertices of a DNA tetrahedron via base complementary pairing. Compared with the distance between dissociative H1 and H2 in the free catalytic hairpin assembly (free-CHA), there is less distance between reactive H1 and H2 in intra-CHA, and the distance is controllable. By virtue of the spatial confinement effect and three-dimensional nanostructure, the intra-CHA system can remarkably accelerate the speed of target-triggered signal generation, with excellent cell permeability and FRET signal stability.
Experimental
Materials
All high-performance liquid chromatography (HPLC)-purified DNA oligonucleotides (Table S1†) were obtained from Sangon Biotech Company, Ltd. (Shanghai, China). The modification of fluorophores Cy3 and Cy5 on nucleotides and their molecular structures are shown in Fig. S1.† DNase I enzyme was purchased from Beyotime Biotechnology Company, Ltd. (Shanghai, China). Lipofectamine 3000 was purchased from Invitrogen Company, Ltd. (USA). Other reagents were of analytical grade and purchased from Sinopharm Chemical Reagent Company, Ltd. (Shanghai, China). L0-2, MCF-7, and MDA-MB-231 cells were obtained from the cell bank of the Central Laboratory of Xiangya Hospital (Changsha, China). Cell culture medium was obtained from Thermo Scientific HyClone (USA). Hoechst nuclear dye, Mito Tracker Deep Red FM, and Lyso Tracker Red DND-99 were purchased from Aokang Biological Technology Co., Ltd. (Shanghai, China).Apparatus
Fluorescence spectra were measured using a PTI QM8000 fluorescence spectrophotometer system (Photo Technology International, USA). The excitation slit of the spectrophotometer was set to 4.00 nm, and the emission slit was set to 5.00 nm; the response time was 0.2 s, and fluorescence data was then recorded using a 1.0 cm × 1.0 cm quartz cuvette containing 100 μL of solution. Biocompatibility of the amplifiers was tested by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assay using a VersaMax Microplate reader (USA). An FV3000 confocal laser-scanning microscope (Olympus, Tokyo, Japan) was used for the fluorescence imaging studies.Preparation of intra-CHA amplifiers
A DNA tetrahedron was self-assembled according to procedures from a previous report.21 In brief, equimolar amounts of S1, S2, S3, and S4 were combined in phosphate-buffered saline (PBS) buffer (pH 7.2). Samples were annealed with the following procedure: 95 °C for 5 minutes, room temperature for 15 minutes, and 4 °C for 1 h; samples were stored at 4 °C for the next experiment. Then, equimolar amounts of DNA tetrahedron, H1, and H2 were combined in PBS buffer and incubated at 25 °C for 1 h at 600 r h−1.Electrophoretic characterization of the amplifiers
To characterize the self-assembly formation of the DNA tetrahedron and intra-CHA amplifiers, samples were analyzed using agarose gel electrophoresis. Samples were prepared by mixing 7 μL DNA, 1 μL 6× loading buffer, and 2 μL SYBR Gold dye so that the final concentration of amplifiers was 500 nM. The samples were electrophoresed on a 2.5% agarose gel in 1× Tris–borate–EDTA (TBE) buffer at 120 V and analyzed using an image scanner (ChemiDoc XRS+, Bio-Rad, USA).Reaction kinetics analysis
First, the real-time fluorescence signal of the CHA system was measured under 25 nM target. The fluorescence spectra were measured every 3 minutes for the first 12 minutes, and then, every 6 minutes for a total of 3 hours. Subsequently, FRET signals (FCy5/FCy3) were calculated by normalizing the starting value of each system to 1. Finally, the relationship between the normalized FRET signal and time was fitted and derived, and the relationship between the reaction rate and time was obtained.Detection performance
To investigate the sensitivity and selectivity in buffer solution, different concentrations of target (0, 1, 3, 5, 8, 10, 15, 20, 25, 50, and 100 nM) were added to 100 μL PBS buffer containing 50 nM CHA amplifiers. Then, after incubation at 25 °C for 3 h, the fluorescence spectra of samples were measured in a 200 μL quartz cuvette; the excitation wavelength was set at 540 nm, and the emission wavelength was collected from 555 to 720 nm. In addition, several other substances (c-myc mRNA, miRNA 21, miRNA 221, miRNA 205, random sequence) were introduced for a selectivity study. All substances were diluted to a final concentration of 25 nM and were then incubated with the intra-CHA system, and the fluorescence responses were measured after 3 h.Nuclease resistance of CHA amplifiers and products
To demonstrate the stability of intra-CHA amplifiers, 0.5 U mL−1 DNase I was added to the intra-CHA system, which was incubated for different amounts of time (0, 10, 20, 30, 40, 50, and 60 min) at 37 °C. Next, 10 μL solution containing 500 nM amplifier was analyzed by 15% polyacrylamide gel electrophoresis (PAGE). The fluorescence spectra of the above-treated intra-CHA amplifiers were measured over increasing time. In addition, to investigate the signal stability of intra-CHA and free-CHA products, CHA amplifiers (1 μM) were reacted with 500 nM target for 2 h under the same conditions. Then, 0.5 U mL−1 DNase I was added for cleavage at different times, and 10 μL enzyme-treated solutions were separated via 15% PAGE. Fluorescence signals were measured every 10 min for 1 h.Cell culture
Human normal liver cells (L0-2) and two human breast adenocarcinoma cell lines (MCF-7 and MDA-MB-231) were obtained from the cell bank of the Central Laboratory of Xiangya Hospital (Changsha, China). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 1% penicillin–streptomycin (PS, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 IU penicillin and 10000 μg mL−1 streptomycin, multi-cell) and 10% fetal bovine serum (FBS) at 37 °C in a humidified incubator containing 5% CO2 and 95% air.
000 IU penicillin and 10000 μg mL−1 streptomycin, multi-cell) and 10% fetal bovine serum (FBS) at 37 °C in a humidified incubator containing 5% CO2 and 95% air.
Real-time monitoring of FRET signal stability in living cells
To investigate the FRET signal stability under the same conditions, we further used Lipofectamine 3000 transfection reagent to transfect CHA products into MDA-MB-231 cells. Briefly, cells were cultured at 37 °C with 5% CO2 for 24 h. Next, the cells were washed three times with PBS buffer and incubated with Lipofectamine 3000 to transfect amplifiers at 37 °C for different amounts of time (2, 3, 4, 5, 6, and 7 h). After washing the cells three times with PBS, confocal fluorescence imaging of MDA-MB-231 cells was carried out using a FV3000 confocal laser-scanning microscope with an 80× objective lens.Fluorescence co-localization in living cells
MDA-MB-231 cells were cultured at 37 °C with 5% CO2 for 24 h. Next, the cells were washed three times with PBS buffer and incubated with Lipofectamine 3000 to transfect free-CHA products at 37 °C for 8 h. Then, nuclear, lysosomal, and mitochondrial dyes were introduced, and the cells were incubated for another 15 min. After washing the cells three times with PBS, confocal fluorescence imaging of MDA-MB-231 cells was carried out.Cytotoxicity
The cytotoxicity of the intra-CHA system was assessed with a standard MTT assay. L0-2, MDA-MB-231, and MCF-7 cells were cultured in 96-well plates, followed by incubation at 37 °C in 5% CO2 for 24 h. Subsequently, 250 nM intra-CHA amplifier was added, and the cells were incubated for different amounts of time (12, 24, and 36 h). Then, after washing three times with PBS, the cells were mixed with 10 μL of MTT and incubated at 37 °C for 4 h. Finally, a multimode microplate reader was used to measure the absorbance of samples at 490 nm.Investigation on self-delivery of the intra-CHA amplifiers into cells
L0-2 cells were cultured in 96-well plates, followed by incubation at 37 °C in 5% CO2 for 24 h. Next, the cells were washed three times with PBS buffer and incubated with CHA amplifiers at 37 °C for another 4 h. After washing the cells three times with PBS, confocal fluorescence imaging of L0-2 cells was carried out.In situ imaging of MnSOD mRNA in different cells
L0-2, MDA-MB-231, and MCF-7 cells were cultured at 37 °C in 5% CO2 for 24 h. Next, the cells were washed three times with PBS buffer and incubated with intra-CHA amplifiers at 37 °C for another 4 h. After washing the cells three times with PBS, confocal fluorescence imaging of different types of cells was carried out using an FV3000 confocal laser-scanning microscope with an 80× objective lens.Results and discussion
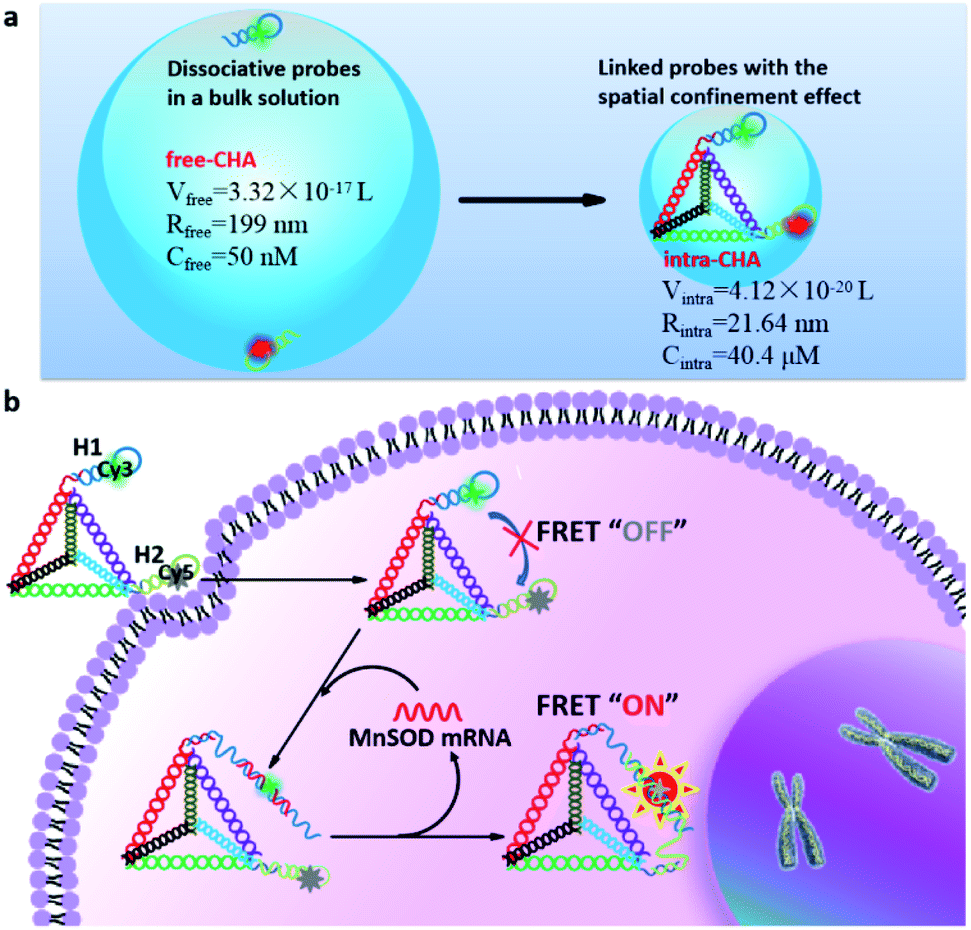
To explain the mechanism of distance-dependent improvement of reaction kinetics and efficiency (Scheme 1a), some physicochemical parameters were theoretically analyzed using collision frequency theory (V = 1/CN),27,28 in which the local sphere volume (V) and the concentration (C) of probes are connected with the Avogadro constant (N). For free-CHA, the concentrations of H1 and H2 are both 50 nM, and the volume of a local sphere for the collision of H1 and H2 is calculated to 3.32 × 10−17 L with a radius of 199 nm. For intra-CHA, the distance between H1 and H2 is calculated to be 21.64 nm, including the section linking H1 and H2 and the edge length of the tetrahedron, approximately 63 bp in total. Linking structures and detailed sequence information are shown in Fig. S2, ESI.† The local concentration of H1 and H2 in intra-CHA is calculated to 40.4 μM, which is 808-fold higher than that in free-CHA. Because the molecular collision frequency is proportional to the local concentration of reactants, the reaction kinetics in intra-CHA will be much faster than that in free-CHA. As known, the step with the largest difficulty in a reaction sequence is the rate-limiting step.29 In the free-CHA system, the reaction sequence is as follows:| target + H1 → target–H1 | (1) |
| target–H1 + H2 → H1–H2 + target | (2) |
 | ||
| Scheme 1 (a) A schematic illustration of the collision model for improving the reaction kinetics in intra-CHA. (b) A schematic illustration of the mechanism for the FRET imaging of mRNA in living cells using the intra-CHA system. | ||
After reaction (1), a larger complex target–H1 is formed, and thus, the reaction between target–H1 and H2 becomes more different. In the intra-CHA system, the distance between target–H1 and H2 is shortened, and their local concentration is increased by virtue of the spatial confinement effect. Thus, the difficulty of the rate-limiting step is reduced, and the reaction kinetics will be rationally improved.
Due to the cell permeability of the DNA tetrahedron through a caveolin-dependent pathway,30,31 the intra-CHA system can independently enter cells, and the intramolecular catalytic hairpin assembly between H1 and H2 will be triggered by the target MnSOD mRNA in cells (Scheme 1b). With the process of circular cascade reactions, the distance between Cy3 and Cy5 changes from far to close, resulting in a fluorescence resonance energy transfer (FRET) signal that will be free from the interference of environmental fluctuations.
First, the preparation of the intra-CHA system and the detection feasibility were verified. As shown by agarose gel electrophoresis (Fig. S3, ESI†), with the gradual hybridization of DNA strands, the DNA tetrahedron was successfully prepared, as evidenced by the gradual decrease in electrophoretic mobility, due to the increase in molecular weight of the hybridization complexes. The linking of H1 and H2 on the tetrahedron was also demonstrated by electrophoresis (Fig. 1a), as evidenced by the disappearance of the H1 and H2 bands and clear band shifts after conjugation with the tetrahedron. The feasibility of MnSOD mRNA detection was demonstrated by the fluorescence spectra change as a function of the addition of the target (Fig. 1b), as evidenced by the FRET signal from Cy3 (Em = 565 nm) to Cy5 (Em = 670 nm). Considering the easy synthesis of DNA, corresponding DNA sequences were used instead of RNA targets in in vitro experiments, because of their same base-pairing recognition and similar fluorescence response from the intra-CHA system (Fig. S4† and 1b).
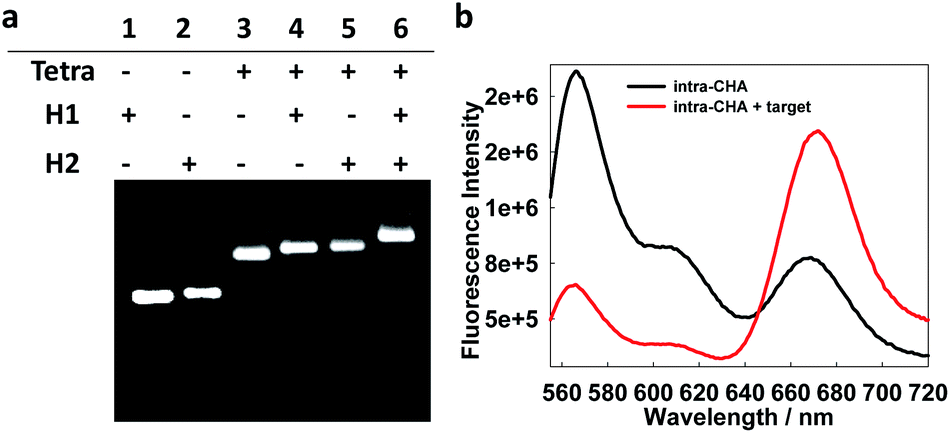 | ||
| Fig. 1 (a) Electrophoresis characterization of the construction of the intra-CHA system via the conjugation of H1 and H2 with the tetrahedron. (b) Fluorescence spectra of the intra-CHA system in the absence (black curve) and presence (red curve) of the target MnSOD mRNA. | ||
Then, real-time monitoring of fluorescence signal (FCy5/FCy3) was performed to investigate the reaction kinetics. As shown in Fig. 2a, in the absence of the target, no FRET signal could be detected in both the intra-CHA and free-CHA systems. As a function of introduction of the target, the intra-CHA system displayed a much faster response to the target than the free-CHA system, indicating the distinct increase in the reaction speed. The intra-CHA system reached a detectable signal-to-background (S/B) ratio in only 3 min, while the free-CHA system required approximately 48 min to reach the analogous signal level (Fig. 2b and S5, ESI†), indicating an approximately 16-fold improvement of the reaction kinetics in the intra-CHA. In addition, quantitative analysis of the reaction rates of intra-CHA and free-CHA was carried out. As shown in Fig. S6,† there was a much faster initial rate in intra-CHA than that in free-CHA, with an enhancement of 15.6 fold. This occurred because the rate-limiting step of the target–H1 and H2 interaction was significantly accelerated by reducing the distance and increasing the local concentration.
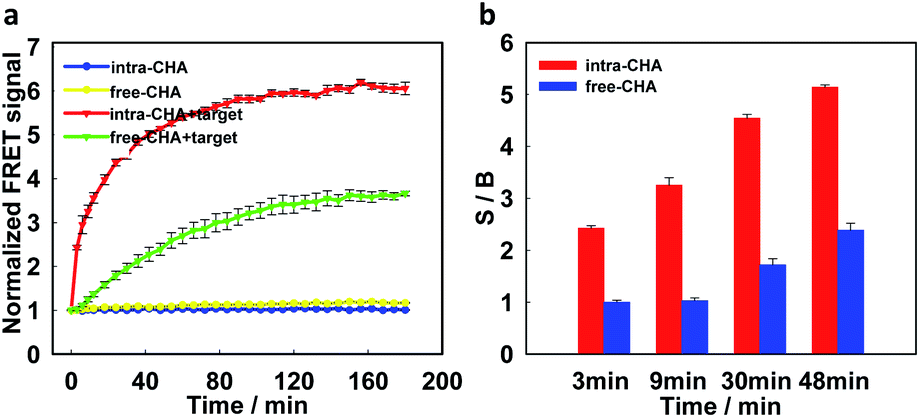 | ||
| Fig. 2 (a) The real-time monitoring of the FRET signal (FCy5/FCy3) of different systems. The starting value of each system is normalized to 1. (b) A comparison of the signal-to-background ratio (S/B) at different reaction time points. | ||
Subsequently, the capability of intra-CHA to detect the target MnSOD mRNA was investigated. To increase the detection performance, the concentration ratio of H1 to H2 and the detection temperature were optimized. As shown in Fig. S7 and S8, ESI,† a probe ratio of 1:1 was selected, and the intra-CHA system worked well under a wide temperature range from 25 to 37 °C. As shown in Fig. S9, ESI,† from the spectrum response of the intra-CHA system to different concentrations of the target, the Cy3 fluorescence emission gradually decreased with the increase in the target concentration, and the Cy5 fluorescence emission increased, indicating a ratiometric signal output based on FRET. Compared with the free-CHA system, the intra-CHA system held a higher detection efficiency toward the target, which should result from the improved reaction kinetics. Based on the three-time standard deviation of the blank signal (3σ), the limit of detection (LOD) was calculated to be 0.15 nM, and compared with previous studies, the intra-CHA system exhibited a comparable performance.14,32,33 Different sequences were also detected by the intra-CHA system to investigate the selectivity for MnSOD mRNA (Fig. S10, ESI†), and only the target could induce a high signal-to-background ratio. Little FRET signal was detected for other sequences, indicating good selectivity of the intra-CHA system.
A well-known difficulty during the application of nucleic acid probes in complex biological samples is degradation by nuclease. From the electrophoretic characterization (Fig. S11a, ESI†), the proposed intra-CHA system was stable under treatment with 0.5 U mL−1 of DNase I, which was higher than its intracellular level.19,34 Additionally, the FRET signal (FCy5/FCy3) of the blank was not affected by nuclease (Fig. S11b, ESI†), which prevented any false-positive signal. More attractively, the intra-CHA products exhibited high nuclease resistance and signal stability, and their electrophoresis bands did not change with the increase in the treatment time by DNase I, while the free-CHA products were digested and gradually disappeared (Fig. 3a). Simultaneously, the FRET signal of the intra-CHA products remained high and unchanged, while that of the free-CHA products decreased with lengthening degradation time (Fig. 3b and S12, ESI†).
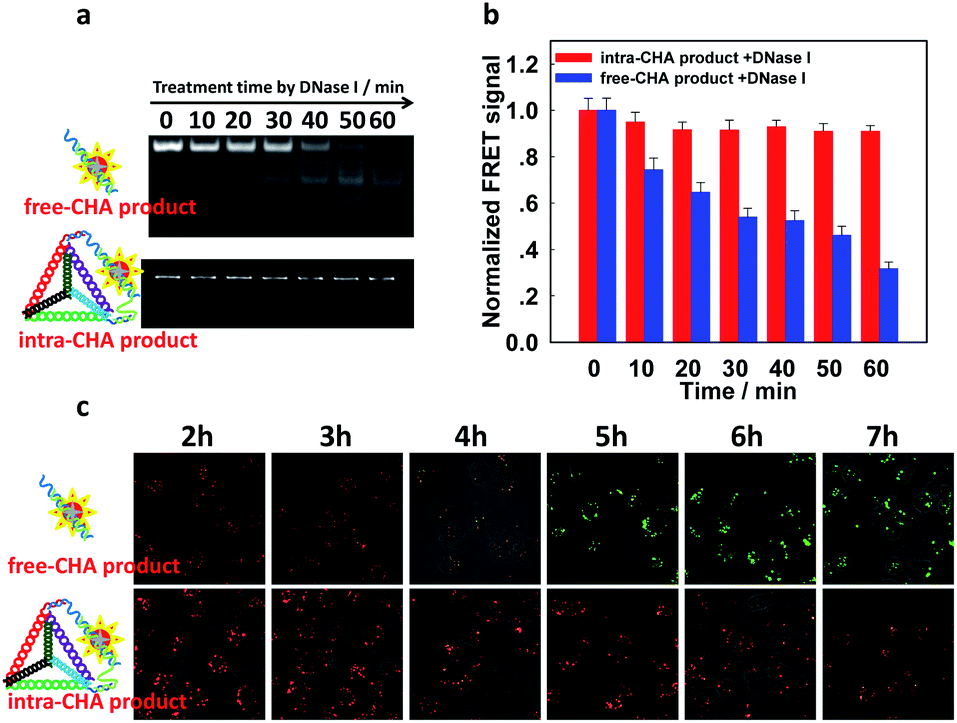 | ||
| Fig. 3 The stability of the FRET signal after intra-CHA or free-CHA. (a) A gel electrophoresis demonstration and (b) the FRET signal (FCy5/FCy3) of CHA products after increasing the treatment time with DNase I (0.5 U mL−1); the starting value of each group is normalized to 1. (c) Merged fluorescence images of Cy3 and Cy5 in MDA-MB-231 human breast cancer cells transfected with CHA products, with increasing incubation time. The excitation wavelength for imaging was set at 560 nm. | ||
In order to further demonstrate the signal stability under the same conditions, CHA products were transfected into MDA-MB-231 cells by Lipofectamine 3000, and real-time imaging of intracellular fluorescence was carried out. It was observed that for traditional free-CHA products, Cy3 fluorescence recovered after incubation for 2 h in cells, while Cy5 fluorescence gradually decreased, indicating instability of the free-CHA signal in a complex biologic system (upper Fig. 3c and S13, ESI†). Co-localization with tracking dyes for organelles showed that the degraded free-CHA products still remained in the cytoplasm without change in the cellular distribution (Fig. S14, ESI†). For intra-CHA products, the FRET signal was maintained for 6 h with little change, indicating enhanced stability of the intra-CHA signal (lower Fig. 3c and S15, ESI†). These results fully demonstrate that the proposed intra-CHA system maintained a higher signal stability than the traditional free-CHA system, which is significant for intracellular application, especially for tracking over long periods of time.
The biocompatibility of the intra-CHA system was investigated by standard MTT assay. After incubation with 250 nM intra-CHA amplifiers for different amounts of time (12, 24, and 36 h), three types of cells, including human normal liver cells (L0-2) and human breast adenocarcinoma cells (MCF-7 and MDA-MB-231), all exhibited good viability, with percentages over 90% (Fig. S16, ESI†). This indicated that the proposed intra-CHA amplifiers were safe for cells and suitable for subsequent intracellular experiments. Then, the investigation on the self-delivery of the intra-CHA amplifiers into cells was performed on L0-2 cells through Cy3 (Fig. S17, ESI†) and Cy5 (Fig. S18, ESI†) fluorescence imaging. For dissociative H1 and H2, negligible signal was measured in cells. However, obvious fluorescence indicated the successful L0-2 cellular uptake of the intra-CHA amplifiers. The intracellular localization of the intra-CHA amplifier was further visualized by z-stack images of L0-2 cells (Fig. S19 and S20, ESI†), and the results demonstrated the excellent cell permeability of the intra-CHA amplifiers, without any transfection or nanocarrier.
By virtue of its fast kinetics, signal stability, and self-delivery into cells, the intra-CHA system was finally used to visualize intracellular target MnSOD mRNA. The same concentrations of amplifiers (250 nM) were incubated with the three types of cells for 4 hours, and their signals were subsequently recorded by confocal fluorescence imaging. As shown in Fig. 4, for L0-2 cells, bright Cy3 fluorescence was recorded, with negligible Cy5 fluorescence, suggesting no FRET signal. However, Cy3 fluorescence decreased and Cy5 fluorescence increased in MCF-7 and MDA-MB-231 cells, indicating the generation of FRET signal and the presence of overexpressed target mRNA. In addition, the difference in MnSOD mRNA expression was verified by quantitative RT-PCR, and there was much higher expression in cancer cells than in normal cells (Fig. S21, ESI†), which is consistent with the intra-CHA-based imaging. Thus, these results successfully demonstrated that the intra-CHA system is capable of visually distinguishing cancer cells from normal cells, based on the different expression of biomarker mRNA.
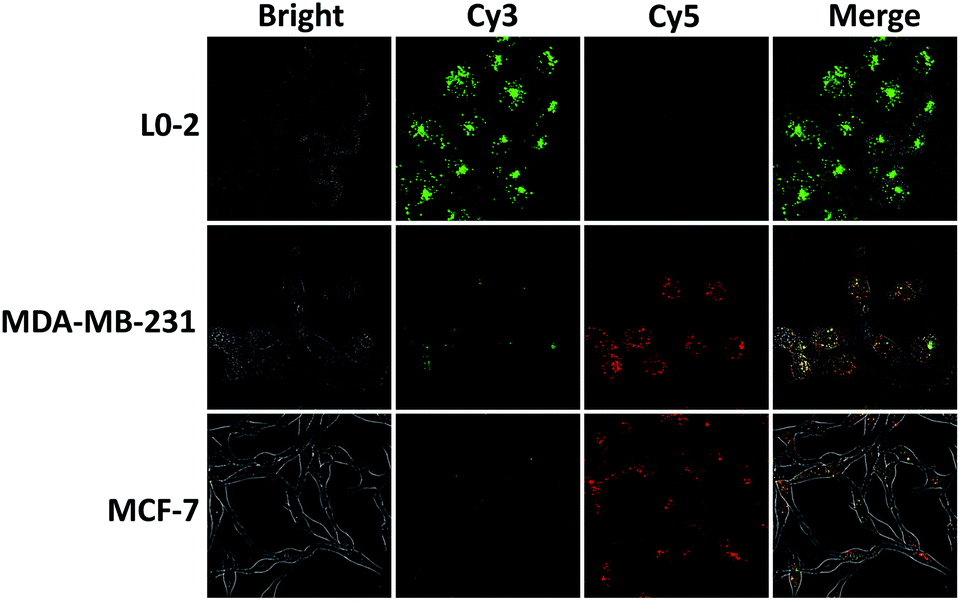 | ||
| Fig. 4 The in situ imaging of MnSOD mRNA in living cells: normal cells (L0-2) and cancer cells (MDA-MB-231, MCF-7). All cells were incubated with intra-CHA amplifiers for 4 h. | ||
Conclusions
Inspired by the demand to develop a highly efficient DDSA amplifier that improves not only the kinetics, but also the signal stability with good cell permeability, we proposed an intramolecular catalyzed hairpin assembly (intra-CHA) on a three-dimensional DNA tetrahedron for mRNA imaging in living cells. As a result, multiple advantages were demonstrated: (1) by virtue of reducing the distance between reactive probes and significantly increasing the local concentration, the target-triggered signal generation speed of the intra-CHA system was obviously increased by comparison with traditional free-CHA; (2) the good biocompatibility and signal stability of the intra-CHA system were also demonstrated, which are significant for intracellular application, especially for tracking over long periods of time; (3) the intra-CHA system could enter cells via self-delivery without any transfection or nanocarrier, for the selective and amplification imaging of the target mRNA in living cells. The proposed strategy can be applied to a wide range of targets of interest in living cells, through only adjustments of the recognition segments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by financial support from the National Natural Science Foundation of China (21605008, 21575018, 21735001, 21705010), the Natural Science Foundation of Hunan Province (2019JJ30025), and the Hunan Graduate Research and Innovation Project (CX2018B569).Notes and references
- D. G. Spiller, C. D. Wood, D. A. Rand and M. R. H. White, Nature, 2010, 465, 736–745 CrossRef CAS PubMed.
- Y. L. Liu, X. Gao, Y. Jiang, G. Zhang, Z. C. Sun, B. B. Cui and Y. M. Yan, J. Cancer Res. Clin. Oncol., 2015, 141, 661–669 CrossRef CAS PubMed.
- S. L. Church, J. W. Grant, L. A. Ridnour, L. W. Oberley, P. E. Swanson, P. S. Meltzer and J. M. Trent, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3113–3117 CrossRef CAS PubMed.
- F. J. Huang, M. H. Lin, R. L. Duan, X. D. Lou, F. Xia and I. Willner, Nano Lett., 2018, 18, 5116–5123 CrossRef CAS PubMed.
- N. L. Xie, J. Huang, X. H. Yang, Y. J. Yang, K. Quan, H. Wang, L. Ying, M. Ou and K. M. Wang, Chem. Commun., 2016, 52, 2346–2349 RSC.
- T. Xie, M. Li and Y. T. Long, Chem. Commun., 2017, 53, 7768–7771 RSC.
- Z. Qing, J. Xu, J. Hu, J. Zheng, L. He, Z. Zou, S. Yang, W. Tan and R. Yang, Angew. Chem., Int. Ed., 2019, 58, 11574–11585 CrossRef CAS.
- Z. Qing, A. Bai, S. Xing, Z. Zou, X. He, K. Wang and R. Yang, Biosens. Bioelectron., 2019, 137, 96–109 CrossRef CAS.
- R. H. Sedlak, T. Nguyen, I. Palileo, K. R. Jerome and J. Kuypers, J. Clin. Microbiol., 2017, 55, 442–449 CrossRef CAS PubMed.
- W. Ali, M. Habib, S. Sajid, A. R. S. Khan, M. U. Mazhar, I. U. Khan, U. Saliha, M. Farooq, M. S. U. D. Shah and H. M. Muzammil, Matrix Sci. Med., 2017, 1, 27–29 CrossRef.
- J. R. Moffitt, J. J. Hao, D. B. Mukku, T. Lu, C. Dulac and X. W. Zhuang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14456–14461 CrossRef CAS PubMed.
- C. H. Cui, W. Shu and P. N. Li, Front. Cell Dev. Biol., 2016, 4, 89 Search PubMed.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS PubMed.
- Z. Cheglakov, T. M. Cronin, C. He and Y. Weizmann, J. Am. Chem. Soc., 2015, 137, 6116–6119 CrossRef CAS PubMed.
- J. Zheng, G. Z. Zhu, Y. H. Li, C. M. Li, M. X. You, T. Chen, E. Song, R. H. Yang and W. H. Tan, ACS Nano, 2013, 7, 6545–6554 CrossRef CAS PubMed.
- J. T. Liu, P. Du, J. Zhang, H. Shen and J. P. Lei, Chem. Commun., 2018, 54, 2550–2553 RSC.
- A. P. K. K. Karunanayake Mudiyanselage, Q. K. Yu, M. A. L. Duque, B. Zhao, R. Wu and M. X. You, J. Am. Chem. Soc., 2018, 140, 8739–8745 CrossRef CAS PubMed.
- Z. H. Qing, X. X. He, J. Huang, K. M. Wang, Z. Zou, T. P. Qing, Z. G. Mao, H. Shi and D. G. He, Anal. Chem., 2014, 86, 4934–4939 CrossRef CAS PubMed.
- L. Liu, Q. M. Rong, G. L. Ke, M. Zhang, J. Li, Y. Q. Li, Y. C. Liu, M. Chen and X. B. Zhang, Anal. Chem., 2019, 91, 3675–3680 CrossRef CAS PubMed.
- Q. M. Wei, J. Huang, J. Li, J. L. Wang, X. H. Yang, J. B. Liu and K. M. Wang, Chem. Sci., 2018, 9, 7802–7808 RSC.
- K. W. Ren, Y. F. Xu, Y. Liu, M. Yang and H. X. Ju, ACS Nano, 2018, 12, 263–271 CrossRef CAS PubMed.
- C. C. Wu, S. Cansiz, L. Zhang, T. Teng, L. Qiu, J. Li, Y. Liu, C. Zhou, R. Hu, T. Zhang, C. Cui, L. Cui and W. H. Tan, J. Am. Chem. Soc., 2015, 137, 4900–4903 CrossRef CAS PubMed.
- W. H. Dai, H. F. Dong, K. K. Guo and X. J. Zhang, Chem. Sci., 2018, 9, 1753–1759 RSC.
- L. He, D. Q. Lu, H. Liang, S. T. Xie, X. B. Zhang, Q. L. Liu, Q. Yuan and W. H. Tan, J. Am. Chem. Soc., 2018, 140, 258–263 CrossRef CAS PubMed.
- Y. J. Yang, J. Huang, X. H. Yang, K. Quan, H. Wang, L. Ying, N. L. Xie, M. Ou and K. M. Wang, J. Am. Chem. Soc., 2015, 137, 8340–8343 CrossRef CAS PubMed.
- L. M. Xian, H. Y. Ge, F. Xu, N. Xu, J. L. Fan, K. Shao and X. J. Peng, Chem. Sci., 2019, 10, 7111–7118 RSC.
- L. P. Xu, Y. X. Chen, G. Yang, W. X. Shi, B. Dai, G. N. Li, Y. H. Cao, Y. Q. Wen, X. J. Zhang and S. T. Wang, Adv. Mater., 2015, 27, 6878–6884 CrossRef CAS PubMed.
- H. Q. Zhang, F. Li, B. Dever, C. Wang, X. F. Li and X. C. Le, Angew. Chem., Int. Ed., 2013, 52, 10698–10705 CrossRef CAS PubMed.
- W. J. Ray Jr, Biochemisty, 1983, 22, 4625–4637 CrossRef CAS PubMed.
- J. Li, H. Pei, B. Zhu, L. Liang, M. Wei, Y. He, N. Chen, D. Li, Q. Huang and C. H. Fan, ACS Nano, 2011, 5, 8783–8789 CrossRef CAS PubMed.
- L. Liang, J. Li, Q. Li, Q. Huang, J. Y. Shi, H. Yan and C. H. Fan, Angew. Chem., Int. Ed., 2014, 53, 7745–7750 CrossRef CAS PubMed.
- Z. Y. Xu, X. Y. Song, W. L. Song and S. Bi, Chem. Sci., 2019, 10, 1651–1658 RSC.
- Z. M. Ying, Z. Wu, B. Tu, W. H. Tan and J. H. Jiang, J. Am. Chem. Soc., 2017, 139, 9779–9782 CrossRef CAS PubMed.
- C. Y. Tay, L. Yuan and D. T. Leong, ACS Nano, 2015, 9, 5609–5617 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional documentation (1 table and 21 figures). See DOI: 10.1039/c9sc04916a |
| This journal is © The Royal Society of Chemistry 2020 |