
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detection of key transient Cu intermediates in SSZ-13 during NH3-SCR deNOx by modulation excitation IR spectroscopy†
Alex G.
Greenaway
ab,
Adrian
Marberger
c,
Adam
Thetford
ab,
Inés
Lezcano-González
ab,
Miren
Agote-Arán
ab,
Maarten
Nachtegaal
c,
Davide
Ferri
 c,
Oliver
Kröcher
c,
C. Richard A.
Catlow
abd and
Andrew M.
Beale
*ab
c,
Oliver
Kröcher
c,
C. Richard A.
Catlow
abd and
Andrew M.
Beale
*ab
aUK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Laboratory, Didcot OX11 0FA, UK. E-mail: andrew.beale@ucl.ac.uk
bDepartment of Chemistry, 20 Gordon Street, London, WC1H 0AJ, UK
cPaul Scherrer Institut, 5232 Villigen, Switzerland
dCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
First published on 18th November 2019
Abstract
The small pore zeolite Cu-SSZ-13 is an efficient material for the standard selective catalytic reduction of nitrogen oxides (NOx) by ammonia (NH3). In this work, Cu-SSZ-13 has been studied at 250 °C under high conversion using a modulation excitation approach and analysed with phase sensitive detection (PSD). While the complementary X-ray absorption near edge structure (XANES) spectroscopy measurements showed that the experiments were performed under cyclic Cu+/Cu2+ redox, Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS) experiments provide spectroscopic evidence for previously postulated intermediates Cu–N(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–NH2 and Cu–NO3 in the NH3-SCR deNOx mechanism and for the role of [Cu2+(OH−)]+. These results therefore help in building towards a more comprehensive understanding of the reaction mechanism which to date has only been postulated in silico.
O)–NH2 and Cu–NO3 in the NH3-SCR deNOx mechanism and for the role of [Cu2+(OH−)]+. These results therefore help in building towards a more comprehensive understanding of the reaction mechanism which to date has only been postulated in silico.
Introduction
Ammonia selective catalytic reduction (NH3-SCR) is an efficient way to prevent nitrogen oxide (NO, NO2) emissions from heavy duty diesel powered vehicles.1–5 Cu-exchanged chabazite (in its synthetic form SSZ-13), a small pore zeolite (maximum ring size 8 T-sites), is the subject of a considerable amount of research as it exhibits both a higher activity and selectivity and is less susceptible to (hydro)thermal degradation than other zeolite-based systems (e.g. ZSM-5 and beta).5–7 Mechanistically, the study of NH3-SCR is complicated and as of yet, not fully rationalised in terms of reaction kinetics, temperature regimes, and perhaps more relevant here, reaction intermediates. A number of plausible mechanisms have been proposed on the basis of spectroscopic and theoretical data (including works by Paolucci et al., Kwak et al., Janssens et al., Gao et al.),8–11 leading to several points of disagreement regarding a consistent mechanism linking the nature of the catalytically active site to the reaction pathway that enables NOx species to be converted to N2. Operando spectroscopic studies have proven to be successful at providing insight into both active sites and intermediate species for NH3-SCR.12,13 Cu-SSZ-13 has been studied extensively under operando conditions with many of the pertinent results outlined in recent reviews.4,12 For each of the various spectroscopic methods employed, there are limitations when building mechanistic cycles, an example of which is diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), which has been exploited for several in situ NH3-SCR studies.14–17 In this technique, the spectra are dominated by species with large molar extinction coefficients (NH3, NH4+, NO3−) which renders the detection of short-lived, transient, species difficult. Similarly, for X-ray absorption near edge structure (XANES) spectroscopy, which provides a method to determine the oxidation and coordination state of copper within the sample (e.g. Cu2+/Cu+), it can be challenging to deconvolute signals relevant to the active site from those that originate from other sites in the catalyst that may not be pertinent to the reaction; the large majority of spectroscopic studies are equilibrium-based experiments and many phenomena such as adsorption of reactants, desorption of products and the formation of spectator species occur within similar time frames.4 It is thus difficult to rationalise the features that are part of the catalytic process against those that do not correspond to the main reaction pathway. From a mechanistic viewpoint, spectroscopic transient experiments are highly advantageous in studies of NH3-SCR catalysts, as demonstrated for V-based and Cu-SSZ-13 catalysts using DRIFTS/UV-Vis and XANES, respectively.18,19Concentration modulation excitation (ME) represents an approach to enable the detection of dynamic species directly involved in a reaction,18–20 which is achieved by removing all parts of the spectroscopic signal from species which are not changing with the same frequency as the modulation excitation frequency. The procedure used in a typical concentration ME experiment is briefly explained here; the theory and data processing required are covered in detail in the ESI† and elsewhere.20–25
In this study, all ME experiments utilise a pulse sequence which repeatedly stops/starts the flow of NO through the catalyst bed mimicking the standard NH3-SCR reaction (see Fig. 1a). Spectra are recorded continuously throughout this work using DRIFTS or XANES in a time-resolved manner in order to follow structural changes. To the eye, these spectra look similar, as the changes between each spectrum are barely discernible (Fig. 1b). After a few cycles of the perturbation, a quasi-equilibrium state is attained, in which the state of the catalyst is considered the same at the start of each cycle (Fig. 1c); these spectra, which correspond to the same point (time) in each cycle, can then be averaged to produce a series of spectra with improved signal-to-noise ratio which are subsequently processed using phase sensitive detection (PSD) (see ESI† for more detail). For this study in particular, features due to strongly adsorbed species (particularly ‘stored’ ammonia), which are not involved in the reaction, are removed.17 This also applies to background noise that does not respond to the same frequency as the pulse sequence. The result is a series of spectra with enhanced signal-to-noise ratio providing enhanced sensitivity to species present in the catalytic cycle.
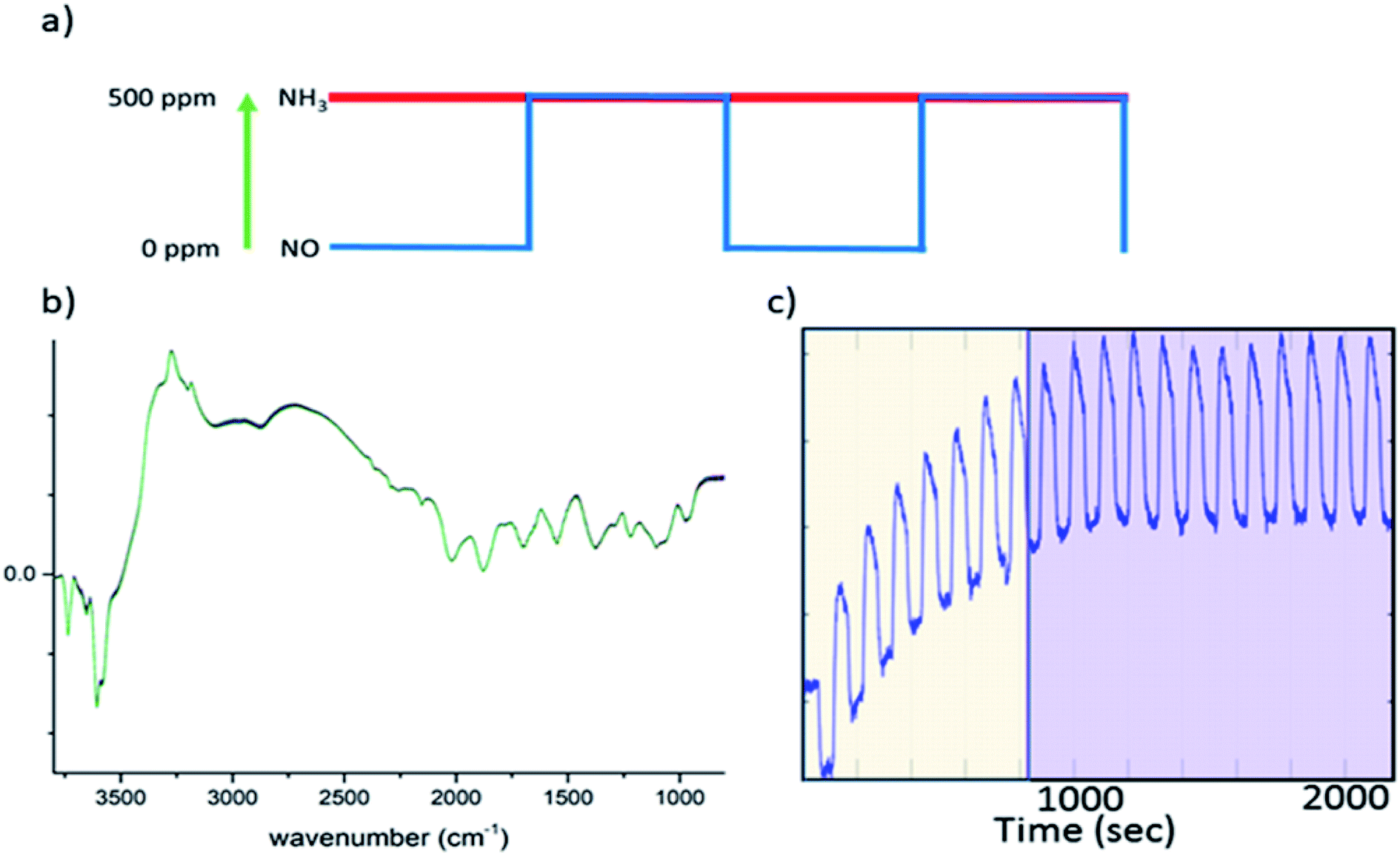 | ||
| Fig. 1 (a) A schematic of a concentration pulse sequence used for the ME experiment, in which NO is turned “on/off” repeatedly between 0 and 500 ppm while the concentration of other reactive components, NH3 and O2, remains constant. (b) An example of the 2400 DRIFTS spectra collected during this ME experiment (note that there are no significant changes). (c) Time dependent signal at 3655 cm−1, demonstrating how the catalyst achieves a quasi-equilibrium state during experiment; the yellow area indicates the cycles where the quasi-equilibrium is not achieved while the purple area highlights the quasi equilibrium region. It is from this latter region that the time-resolved information is derived. | ||
Importantly the concentration modulation excitation DRIFTS data augmented with the application of Density Functional Theory (DFT) allowed us to observe the formation of key intermediates including a copper nitrosamine (Cu–N(O)–NH2) and copper bidentate nitrate species (Cu–NO3), while highlighting the regeneration and importance of [Cu2+(OH−)]+ sites in the catalytic process. Ancillary ME XANES experiments confirmed that the measurements were conducted in the presence of the Cu2+/+ redox behaviour in the cycle.
Experimental
Operando ME experiments
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1) and the temperature at 250 °C. All DRIFT spectra were obtained by accumulating 10 interferograms at 4 cm−1 resolution and 80 kHz scanner velocity (0.9 s per spectrum). During a concentration modulation excitation experiment, solenoid valves were used to automatically switch between gases and were operated by the OPUS software (Bruker). The following pulse sequences were used; 20 cycles of 120 s with 60 s of 500 ppm NO flow on followed by 60 s of NO flow off, into a constant stream of 500 ppm NH3, 10000 ppm O2 make up N2 (during the “NO off” section of the pulse cycle additional N2 was used to keep the total flow constant). The sets of time-resolved DRIFTS spectra obtained from the modulation experiments at equilibrium conditions (Fig. 1c) were time-averaged and subsequently converted into phase-resolved spectra using eqn (S1) (see ESI†). A concentration ME DRIFTS experiment was conducted on an activated sample of Cu-SSZ-13 at 250 °C. The concentration sequence followed 20 cycles in which a 500 ppm flow of NO was added into a stream containing 500 ppm NH3, 10 vol% O2 and made up with N2 for 60 s, followed by 60 s without NO flow (see Fig. 3a and ESI Fig. S17† for out gas analysis).
000 h−1) and the temperature at 250 °C. All DRIFT spectra were obtained by accumulating 10 interferograms at 4 cm−1 resolution and 80 kHz scanner velocity (0.9 s per spectrum). During a concentration modulation excitation experiment, solenoid valves were used to automatically switch between gases and were operated by the OPUS software (Bruker). The following pulse sequences were used; 20 cycles of 120 s with 60 s of 500 ppm NO flow on followed by 60 s of NO flow off, into a constant stream of 500 ppm NH3, 10000 ppm O2 make up N2 (during the “NO off” section of the pulse cycle additional N2 was used to keep the total flow constant). The sets of time-resolved DRIFTS spectra obtained from the modulation experiments at equilibrium conditions (Fig. 1c) were time-averaged and subsequently converted into phase-resolved spectra using eqn (S1) (see ESI†). A concentration ME DRIFTS experiment was conducted on an activated sample of Cu-SSZ-13 at 250 °C. The concentration sequence followed 20 cycles in which a 500 ppm flow of NO was added into a stream containing 500 ppm NH3, 10 vol% O2 and made up with N2 for 60 s, followed by 60 s without NO flow (see Fig. 3a and ESI Fig. S17† for out gas analysis).
Density function theory (DFT) calculations
The VASP code using the Perdew–Burke–Ernzerhof (PBE) functional was employed for the Density Functional Theory simulations. The projector augmented wave (PAW) method with a plane-wave cut-off of 450 eV was used with single k-point at the gamma point.29–33 The structures were optimised with a convergence criterion of 0.02 eV Å−1. The converged bulk energies are within 10−4 eV. The vibration frequencies were calculated using finite differences to determine the second derivatives. A cube of 8 unit cells of the CHA structure was constructed, which has dimensions of 18.69 Å × 18.69 Å × 18.69 Å containing 94 Si atoms, 2 Al atoms and 192 O atoms. The Cu is placed in the 8 ring as a Cu2+ with the framework acting as counter charge due to the acid site hydrogen atoms being removed. In calculations on structures where the Cu2+ has been reduced to Cu+, an additional NH4+ cation has been included to maintain charge neutrality for the model.Results and discussion
Operando ME DRIFTS experiments
The 2400 spectra collected during the ME DRIFTS experiments (Fig. 2, top panel), on visual inspection, appear to be the same, with no clear changes being detected. The spectra display several prominent bands in both the stretching and bending regions, previously identified as vibrations associated with ammonium ions, formed on the zeolite Brønsted acid sites, and NH3 coordinated to Cu2+ ions (see Table 1 for further assignments). In addition, intense negative bands are seen in the hydroxyl region, corresponding to both Brønsted acidic bridging hydroxyls and silanol groups, along with a weaker feature due to [Cu2+(OH−)]+ centres, showing the formation of adsorbed complexes on these particular sites. The region below 2000 cm−1 is, in contrast, dominated by alternating positive–negative signals, attributable to the interaction of NH3 molecules, rendering the identification of changes around other features challenging.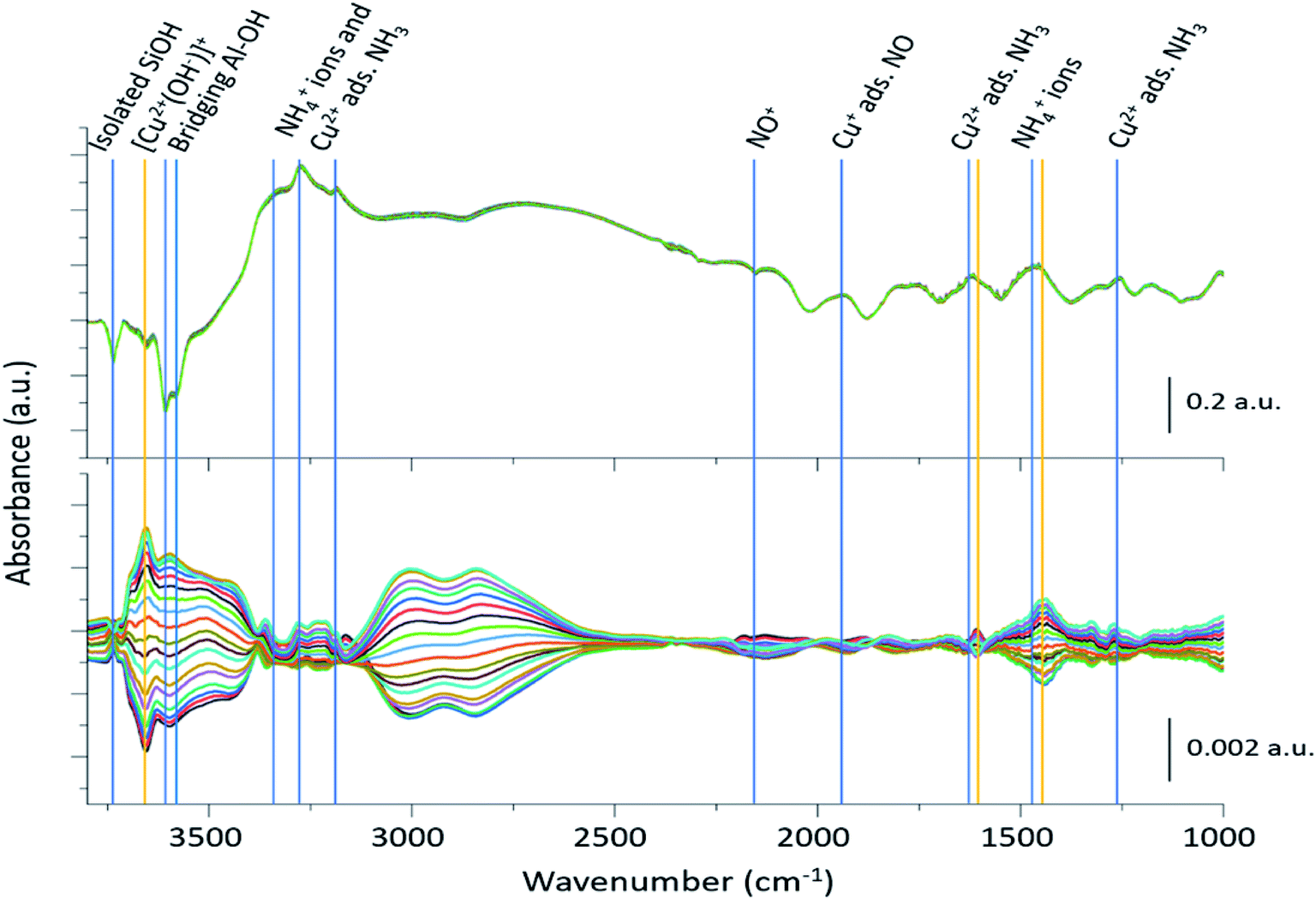 | ||
| Fig. 2 Time-resolved spectra collected during the NO concentration ME DRIFTS experiment (top), and corresponding phase-resolved spectra (bottom). Blue vertical lines indicate features previously reported in the literature (Table 1), whereas orange vertical lines represent features that are enhanced in the phase-resolved spectra, corresponding to newly observed species. The bottom traces indicate how the spectral features are changing with time/gas composition. Note the difference in scale bar units. | ||
| Wavenumber (cm−1) | Assignment | Ref. |
|---|---|---|
| 3737 | Isolated silanol groups (strong) | 17 and 34 |
| 3655 | [Cu2+(OH−)]+ (very weak) | 17 and 34 |
| 3602–3588 | Brønsted acidic bridging hydroxyls (very strong) | 17 and 34 |
| 3332, 3182, 1620 | Cu2+ adsorbed NH3 (medium) | 15 and 17 |
| 3272, 1454 | NH4+ (Brønsted acid site adsorbed NH3) (strong) | 15 and 17 |
| 2158 | NO+ (very weak) | 14 |
| 1812 | Cu+ adsorbed NO (strong) | 14 |
| 1460 | NH4+/solvating NH3 | 34 |
| 1327 | Extra framework Al (EFAl) adsorbed NH3 (weak) | 17 |
| 1270 | [Cu2+(NH3)4]2+ (weak) | 17 |
The corresponding phase-resolved spectra (Fig. 2, bottom panel) reveal how species that are actively involved in the catalytic process evolve over the duration of one complete modulation cycle. These spectra clearly show that most of the contribution of the zeolite framework is removed because it is unresponsive, and that the intense features in the time-resolved spectra do not necessarily correlate to large features in the phase-resolved one. For instance, the relative intensity of the absorption bands of the bridging hydroxyl groups and the [Cu2+(OH−)]+ species is completely reversed. For clarity, the time-resolved response of several of the relevant features and regions of interest in the ME data have been expanded in Fig. 3. Importantly, for completeness, we note that the varying signal of gas-phase NH3 measured online to the DRIFTS cell confirms that the ME experiment was performed under actual SCR conditions (Fig. 3b).
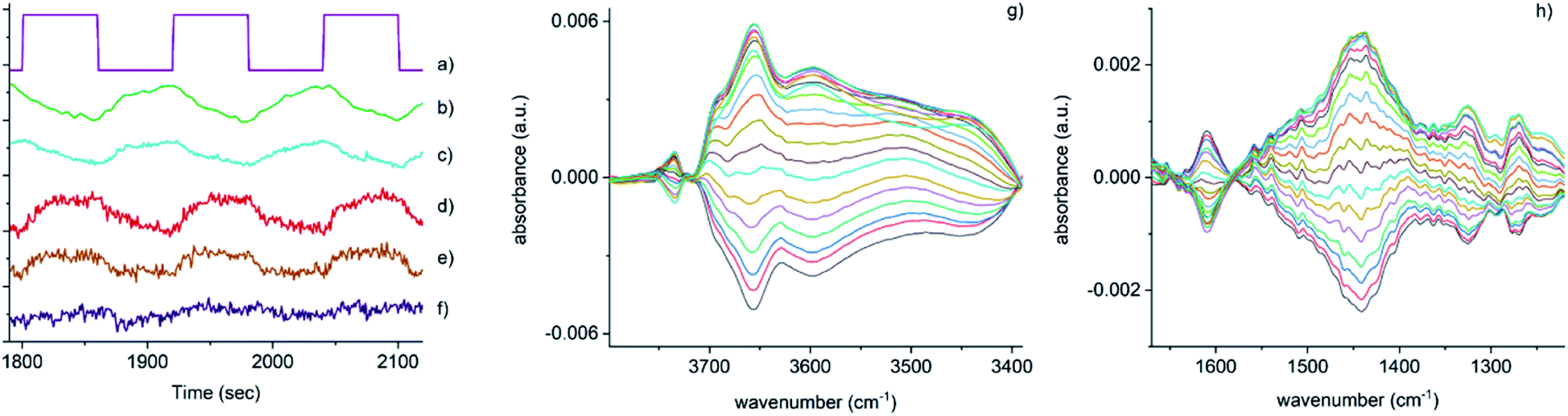 | ||
| Fig. 3 Time-resolved response of several features present in Fig. 2 during the 15–18 cycles of the NO concentration modulation experiment. (a) NO pulse sequence, (b) out-gas NH3 integral response (online IR detected), (c) [Cu2+(OH−)]+ integral, (d) 1436 cm−1 and (e) 1258 cm−1 bands due to the presence of Cu–N(O)–NH2 species, (f) response of the band at 3605 cm−1 (Brønsted acid site). Close-up of regions in the phase-resolved spectra of Fig. 2, (g) 3800–3300 cm−1 region containing the Brønsted acid site (zeolite OH) and the [Cu2+(OH−)]+ stretches, (h) 1650–1200 cm−1 region relevant for NH3 coordinated Cu and zeolite. | ||
Fig. 3 shows that the set of phase-resolved spectra exhibits several features (particularly between 1250 and 1700 cm−1) that appear in the time-resolved data, such as NH4+ ions formed on the Brønsted acid sites and NH3 adsorbed on Cu2+. In addition, bands at 1812 and 2158 cm−1 are also observed that were previously attributed to Cu+-NO and NO+ (i.e. formed by either NO2 disproportionation or NO oxidation on Cu2+ sites), respectively. It is important to note, however, that the most intense feature in the phase-resolved spectra is that at 3655 cm−1, characteristic of [Cu2+(OH−)]+ sites, presenting a larger variation than those corresponding to either silanol groups and bridging hydroxyls. This observation can be attributed to a response of this species to the variation of gas composition, pointing to an active role of this species in the catalytic mechanism, as is clearly illustrated in Fig. 3c, which shows the temporal response of [Cu2+(OH−)]+ species to be affected during the entirety of the NO pulse, which is clear evidence for its consumption and subsequent regeneration under SCR conditions.
A second interesting feature observed in the phase-resolved data concerns a band centred at 1436 cm−1, which on first sight appears to be due to NH4+ species (i.e. typically, a combination of both symmetric and asymmetric bending vibrations). Upon closer inspection, this band is clearly red-shifted from the centre of the band due to adsorbed ammonia species, observed in the time-resolved spectra, by approximately 20 cm−1. There have been previous studies into the nature and evolution of the broad feature associated with the NH4+ species, notably by Giordanino et al. and Lezcano-Gonzalez et al.17,34 The former study identified the presence of a combination of bands in this region at low temperatures, caused by solvated NH4+ ions (i.e. NH4+·nNH3 associations) and the two bending vibrations of NH4+ ions. Desorption of solvating NH3 molecules with increasing temperatures was seen to lead to a gradual intensity decrease and shift to lower wavenumbers of the component at 1463 cm−1, and eventually to the appearance of a single broad band at 1430 cm−1 due to un-solvated NH4+ ions attached to the zeolite framework. Lezcano-Gonzalez et al. showed that at 250 °C, these NH4+ ions slowly react under a flow of NO and O2, concluding that these species are not an intricate part of the NH3-SCR mechanism and more likely remain as a ‘reservoir’ of NH3.17 Given the time scale over which the (NO pulse) modulation experiment is occurring and the relative intensity of the 1436 cm−1 band, it seems unlikely that this is due to the consumption and regeneration of NH4+ ions. Nevertheless, to further probe the origin of the band at 1436 cm−1, a series of additional time-resolved pulse experiments were conducted.
In order to visualize differences in the time response of the bands in the 1400–1530 cm−1 region (Fig. S7–S13†) the following pulse sequences were used: (1) adsorption of NH3 into a pre-equilibrated catalyst bed of NO and O2, and (2) the consequent desorption of NH3 (after pre-equilibration of NO, NH3 and O2). While the features at 1460 and 1436 cm−1 share the same response when NO is omitted from the catalyst bed (NH3 adsorption under O2 flow, Fig. S7 and S8†), clear differences are detected when the catalyst is previously equilibrated with NH3, NO and O2 (Fig. S9 and S10†), with the intensity of the band at 1436 cm−1 increasing more rapidly than the feature at 1460 cm−1 upon NH3 adsorption. Similarly, a different evolution of these signals is detected after switching off the stream of NH3 (Fig. S11 and S12†), with again the band at 1436 cm−1 showing a different response from those of the bands more typically observed. This difference in evolutions confirms the 1436 cm−1 band to be a separate species to the adsorbed ammonium species typically seen in this part of the spectrum.
To understand the possible origin of the 1436 cm−1 band, DFT simulations were performed, focusing on transient species postulated in various reaction mechanisms, but for which there is currently no definitive spectroscopic evidence. The vibrational frequencies of different Cu complexes were calculated, where the active Cu site is placed in the 8 ring as a Cu2+ in a cube of 8 unit cells of the CHA structure zeolite (see ESI section† on DFT IR band prediction for further information of all structures calculated and vibrational modes assigned). Of the structures calculated, the closest assignment to such a signal was identified as a Cu nitrosamine (Cu–N(O)–NH2), with an NO stretching (with some Cu–N component) frequency determined to 1431.2 cm−1 (Fig. 4a). In addition to this band, the DFT calculation also predicted a weak band originating from the N–N stretch of the Cu–N(O)–NH2 species at 1258 cm−1. Upon (re)inspection of the PSD data there is a weak band in the spectra that evolves and is consumed at exactly the same time (see Fig. 3d and e) and phase angle as the band at 1436 cm−1, providing additional support for the presence of a Cu nitrosamine intermediate species. Importantly, Cu–N(O)–NH2 is a key intermediate postulated (on the basis of DFT calculations) in the recent works of Janssens et al. and Paolucci et al.,10,35 although this is the first spectroscopic confirmation of its formation and role in the NH3-SCR reaction mechanism.
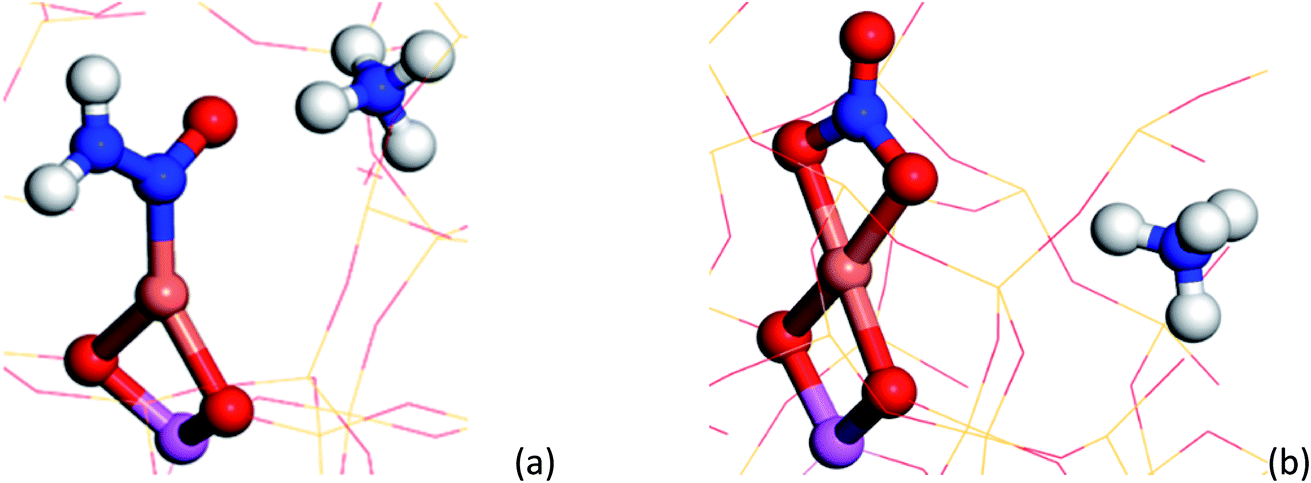 | ||
| Fig. 4 (a) Cu–N(O)–NH2 structure yielded from DFT calculations, containing a NO and N–N stretches observable at ∼1431 and 1258 cm−1 respectively. (b) Bidentate Cu–NO3 structure yielded from DFT calculations, containing a NO stretch at ∼1609 cm−1 respectively. The following colour key has been used: Cu (orange), Al (pink), O (red), N (blue), H (white). | ||
To validate further the findings of the ME DRIFTS experiment, additional modulation experiments were conducted in the absence of NH3 (see Fig. S13†). Significantly, these data contain two large features that modulate with the NO pulses; species representative of adsorbed NO (1896 cm−1) and NO+ (2160 cm−1).16 The phase-resolved data show neither the consumption and regeneration of [Cu2+(OH−)]+ or any feature close to the signal observed at 1436 cm−1 in the experiments described above, thereby supporting the proposal that Cu–N(O)–NH2 intermediate species is part of the SCR catalytic cycle. In addition, the results confirm that the various species observed in the NO pulse ME experiment are consumed and regenerated during the SCR reaction and that they are not spectator species or by-products caused by unselective NO oxidation.
Another feature that shows a strong response to the ME concentration stimulation (NO) is a band centred at ca. 1606 cm−1. Although the exact origin of this band is unclear, its position falls in the range characteristic of nitrate/nitrite and Cu-amine species, also postulated as relevant intermediates in the SCR mechanism.10 We exclude the presence of diamine/tetra-amine Cu species on the basis that although this band could be assigned to an asymmetric bending mode, as can be seen in Fig. 3h, the behaviour of this part of the spectrum does not match at all with that of the 1278 ‘region’ corresponding to the symmetric bending mode of these species.17
Examination of possible stable species predicted using DFT suggests a few candidate mono, bidentate nitrate and nitrite species with typical NO stretching frequencies ca. 1595 cm−1 (±30 cm−1). Of the three structures, the bidentate Cu–NO3 structure A (see ESI†) possesses a NO stretching frequency (νNO, ca. 1609 cm−1) closest to that observed in the phase-resolved IR spectrum. The observation of only one IR active stretch mode (E mode) also implies that the D3h symmetry of the NO3− molecule is preserved on coordination with the Cu ion; the absence of two bands with similar stretching frequencies (νas/νs modes) suggests the absence of a nitrate species of monodentate geometry (C2v symmetry). We note, however, that there have been many studies performed on Cu-containing zeolites and that a number of possible nitrates/nitrites have been proposed, including some similar to the structures shown here (particularly Cu–NO2, surface adsorbed NO2), further suggesting that the accurate assignment of nitrates/nitrites is not trivial.16,36–38 These results also contrast with some observations in which the appearance of ‘Cu-NOx’ only occurs after the system has moved away from SCR conditions (i.e. after NH3 flow is switched off).19 We note, however, that the Cu nitrate species observed by XAS probably possess a higher Cu–(N)O coordination number or higher number of nitrate ligands than those postulated or observed here. This difference in the nature of the nitrate species may be a consequence of the operating conditions, i.e. different temperatures and conversion regimes, but also of their fraction being too short lived for XAS to be detected compared to all other species and thus of the different sensitivity of the two spectroscopic techniques. This observation allows us to conclude that there is probably a difference between the nitrates that play a role in the catalytic cycle as seen here, compared with those that form in the absence of NH3 (i.e. those that form slowly during NO oxidation).
Additional weak bands are seen in the phase-resolved data of Fig. 2 at 3272 and 3182 cm−1, which can be assigned to the stretching modes of both NH4+ ions and adsorbed NH3 on Cu2+, respectively, together with a broad feature at 2156 cm−1 due to NO+.14 Finally, two very broad features between 2600 and 3100 cm−1 are also seen to be present, which have previously been assigned to hydrogen-bonded coordination spheres around NH4+ (2800 cm−1) and H3O+ ions (3025 cm−1).39,40 These changes are not unexpected since the Cu ions and zeolite are fully saturated with NH3 before the SCR reaction begins with the loss of at least one NH3 ligand around the Cu ions necessary for the SCR reaction to take place. The absence of the more commonly observed, yet weaker NH3/NH4+ bending modes in the phase-resolved data suggests that these species undergo only minor modulation during the SCR reaction, which is largely due to their playing an indirect role in the reaction; solvating NH3 and NH4+ species are proposed to provide a reservoir of ammonia for the reaction although as has been shown previously for this catalyst, NH4+ ions are not particularly reactive.17
The intermediate species seen in this study have previously been proposed as protagonists in the reaction mechanism proposed by Janssens et al. (see Fig. 5).10 However, analysis of the phase-resolved data, specifically of the in-phase data (φPSD value where signal intensity is highest; Fig. 6) can provide additional insight on the mechanism. Table S2† lists the φPSD values obtained for the signals of the species discussed above. Similar angular values indicate that the species behave kinetically very similarly, while large differences suggest that species are temporally well separated in the chemical process. In the previous work by Janssens et al., it was proposed that the reaction begins with [Cu2+(OH−)]+ and is followed by the formation of Cu–N(O)–NH2.10 From Table S2† it appears that both components have a similar phase angle suggesting similar rates and time of formation. Furthermore, the subsequent formation of Cu–NO3 followed by NO+ (2160 cm−1) from the disproportionation of NO2, is observed at its maximum intensity with roughly the same phase angle as those associated with Cu–NH3 stretch (3182 cm−1) which suggests a complex is formed containing both Cu–NH3 and NO2 towards the end of the cycle. Indeed, if we normalise the phase angle to the start of the catalytic cycle (i.e. presence of [Cu2+(OH−)]+ as shown in Fig. 6) it can be seen that there is excellent correlation concerning the order in the phase angle that a species appears and the catalytic cycle shown in Fig. 5.
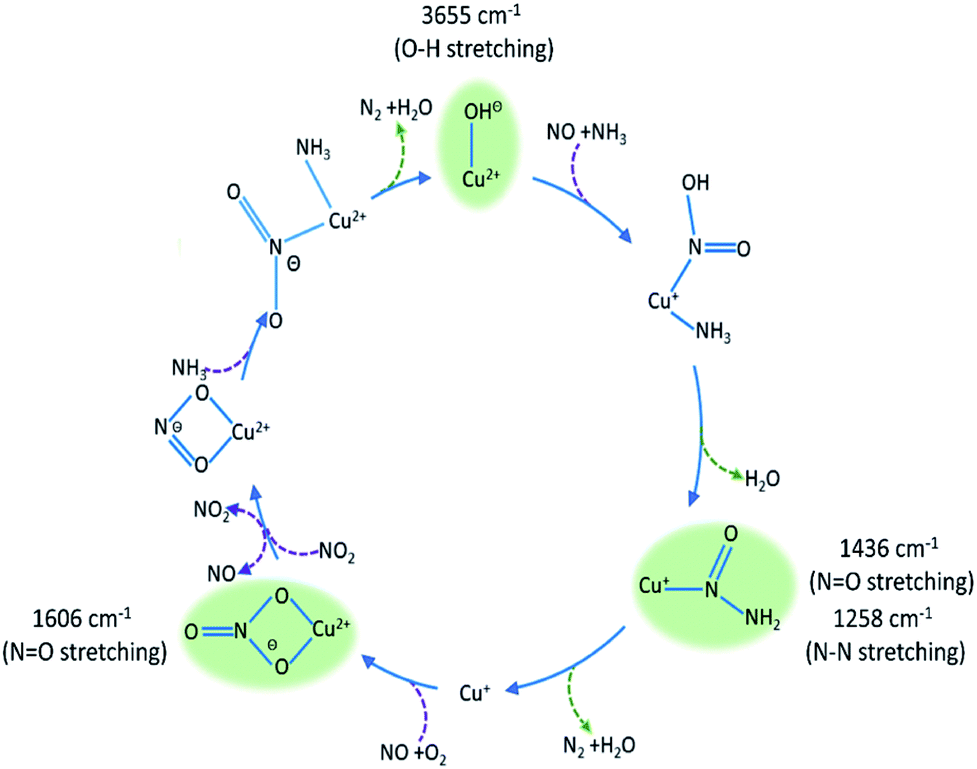 | ||
| Fig. 5 Summary of species evolution during the catalytic cycle, species shaded in green observed in the ME drifts experiments. [Cu2+(OH−)]+ is consumed to make Cu–N(O)–NH2, followed by bidentate nitrate and finally a Cu complex with both NO type and NH3 ligands (as anticipated in the mechanism proposed by Janssens et al.10). | ||
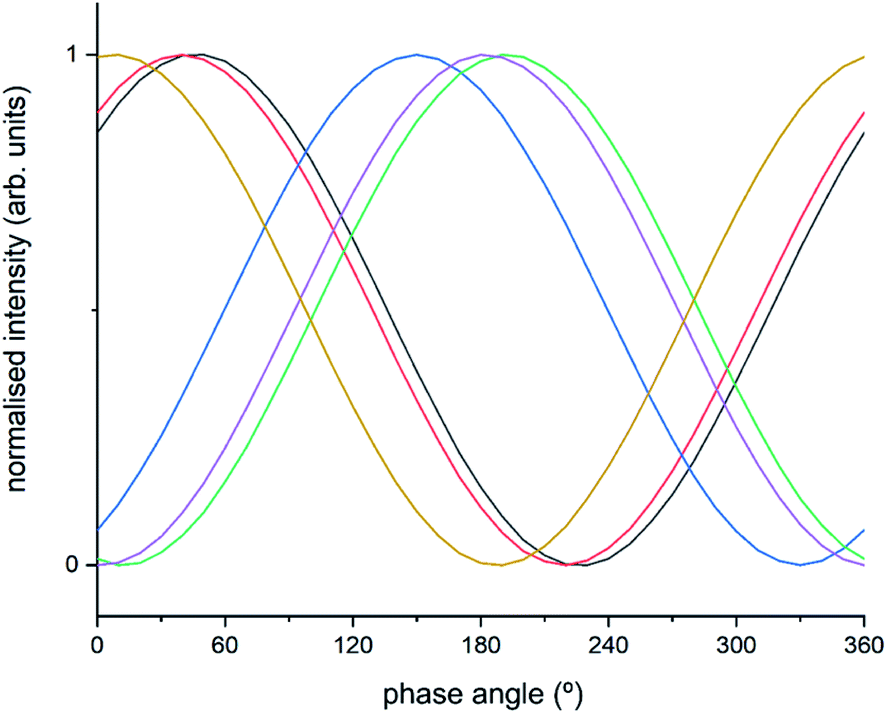 | ||
| Fig. 6 Normalised sinusoidal curves for the respective protagonist IR bands listed in Table S2.† All signals are normalised between 0 and 1, with the response at 3655 cm−1 ([Cu2+(OH−)]+) set to 0° phase angle and all other species adjusted accordingly. These species are listed in Table S2† and correspond to band intensity responses due to black = 1258 cm−1, red = 1436 cm−1, blue = 1606 cm−1, green = 2158 cm−1, purple = 3182 cm−1, orange = 3655 cm−1. | ||
Operando ME XANES experiments
To probe the oxidation state of the Cu present in the sample under SCR conditions a concentration stimulation ME XANES experiment was conducted on an activated sample of Cu-SSZ-13. The concentration stimulation XANES experiment at the Cu K-edge shows a strong rising absorption edge at 9000 eV due to a 1s–4p dipole transition typical of Cu2+, followed by an evolving feature with time attributed to a 1s–4p dipole transition at 8982 eV due Cu+.4 The phase-resolved data show these two significant features relating to both the Cu2+ and Cu+ species in the time-resolved data much more clearly. Significantly, the maximum amplitude of the Cu+ feature correlates exactly with the minimum amplitude for the Cu2+ feature. This experimental observation is a clear indication of the accompanying Cu2+/Cu+ redox process occurring during the SCR catalytic cycle in these experiments (further details available in ESI Fig. S14†).Mechanistic implications of the results
The concentration ME experiments have provided key mechanistic details from both DRIFTS and XANES experiments. The DRIFTS data suggest that the catalytic cycle follows a reaction pathway whereby [Cu2+(OH−)]+ is consumed/reduced to form the transient Cu–N(O)–NH2 species. We note that the NO stretch of this particular species is red-shifted by ∼400 cm−1 with respect to where stretches are typically found for heteronuclear NOx species and which, we argue, is caused by electron density removal from the NO double bond brought about by the formation of an L-type ligand interaction between Cu and N(O) leading to a distribution of 7 electrons over 4 bonds. It is interesting to note that we do not see the initial Cu–NH3 or Cu–NO(OH) interactions suggesting that although these must take place they are simply too fast to be observed in this experiment. Indeed, it is well known that Cu+/2+–NH3 and Cu+–NO interactions occur readily, which for NH3 even leads to reaction inhibition at low temperatures.4 The observation of a sharp feature at 1606 cm−1, which could be tentatively attributed to nitrate-type species, and subsequently the appearance of NO+ (which has been proposed to be formed through the disproportionation of NO2) suggests an important role in the catalytic cycle also for nitrates and nitrites, particularly for completing the Cu redox cycle as has been previously postulated for both NH3-SCR in Cu zeolites and observed in hydrogen-promoted hydrocarbon SCR on silver–alumina catalysts.38,41,42 The exact determination of these features is challenging for conventional operando DRIFTS experiments as the spectra are dominated by species originating from NH3. Interestingly the identification of these intermediates indicates a pathway through which gas phase NO2 can be produced in the cycle to circumvent the need to co-feed it to effect fast SCR as is typical in Fe-based systems.1 The ME XANES data clearly indicate a Cu2+/Cu+ redox cycle operating at the same frequency as the concentration pulse. It is interesting to note that previous experiments under steady-state, high-temperature (i.e. >250 °C) operational conditions contain no evidence for Cu reduction although it is to be expected that this must occur.4 Overall, we find that the observed intermediates have previously been postulated as key species in the catalytic cycle proposed by Janssens et al., although we do note that such species appear in a number of proposed cycles to date.10,43,44
Summary and conclusions
Cu-SSZ-13 has been studied by concentration ME using both DRIFTS and XANES. The data obtained have provided mechanistic insight into the NH3-SCR process on copper containing zeolites. The ME DRIFTS and XANES data reveal the start of the reaction to involve [Cu2+(OH−)]+, highlighting two key intermediates, Cu–N(O)–NH2 and Cu–NO3, that would be very difficult to observe using standard operando DRIFTS measurements. Using the mechanism in Fig. 5 as a framework to put these observations into a catalytic context, it is possible to argue that these intermediates are perhaps the most significant species in the standard SCR cycle since they lead respectively to the desired product (N2), and with the latter species, show how the Cu ion reoxidation occurs. Furthermore, within this mechanism, these species also help to rationalise how NO2 is produced circumventing the necessity to utilise additional NO2 for effective NH3-SCR behaviour. These results, moreover, are in line with predictions from theory, which helps benchmark the ME approach for obtaining potential mechanistic insight importantly obtained under more relevant conditions.10
Despite the new insight obtained, we observe that the ME technique is not able to allow us to rationalise fully all the steps in the catalytic mechanism at this temperature or to discriminate definitively between active and spectator species that may evolve at the same rate as the dose response. It may be that better resolution regarding these steps could be realised by performing experiments at lower conversions and/or at lower temperatures. We note, furthermore, that the temperature employed for this study is also unable to discriminate between the low and high temperature mechanism because of the Cu loading, but as it was performed at 250 °C, these results are likely to be more pertinent to a high temperature mechanism; indeed some of the intermediates observed here are different from those recently reported at much lower temperatures (i.e. Cu2+(NH3)3(NO3)).4,45,46 It is clear, however, that the identification of intermediate states as achieved here can be very useful in guiding the design of the next generation of new and related technologies for deNOx applications, perhaps using cationic components that could be more active or at least more environmentally benign or at risk than copper.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thank the Swiss Light Source at the Paul Scherrer Institute in Switzerland for beamtime (Proposal ID: 20160380). The UK Catalysis Hub is kindly thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC (portfolio grants EP/K014706/1, EP/K014668/1, EP/K014854/1, EP/K014714/1 and EP/I019693/1). All calculations were performed using the computational facilities provided by University College London.References
- S. Brandenberger, O. Kröcher, A. Tissler and R. Althoff, Catal. Rev.: Sci. Eng., 2008, 50, 492–531 CrossRef CAS.
- L. A. Chen, J. H. Li, M. F. Ge and R. H. Zhu, Catal. Today, 2010, 153, 77–83 CrossRef CAS.
- J. H. Li, H. Z. Chang, L. Ma, J. M. Hao and R. T. Yang, Catal. Today, 2011, 175, 147–156 CrossRef CAS.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- F. Gao, J. H. Kwak, J. Szanyi and C. H. F. Peden, Top. Catal., 2013, 56, 1441–1459 CrossRef CAS.
- R. Q. Long and R. T. Yang, J. Catal., 2002, 207, 224–231 CrossRef CAS.
- J. H. Kwak, R. G. Tonkyn, D. H. Kim, J. Szanyi and C. H. F. Peden, J. Catal., 2010, 275, 187–190 CrossRef CAS.
- C. Paolucci, A. A. Verma, S. A. Bates, V. F. Kispersky, J. T. Miller, R. Gounder, W. N. Delgass, F. H. Ribeiro and W. F. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11828–11833 CrossRef CAS.
- J. H. Kwak, J. H. Lee, S. D. Burton, A. S. Lipton, C. H. F. Peden and J. Szanyi, Angew. Chem., Int. Ed., 2013, 52, 9985–9989 CrossRef CAS.
- T. V. W. Janssens, H. Falsig, L. F. Lundegaard, P. N. R. Vennestrom, S. B. Rasmussen, P. G. Moses, F. Giordanino, E. Borfecchia, K. A. Lomachenko, C. Lamberti, S. Bordiga, A. Godiksen, S. Mossin and P. Beato, ACS Catal., 2015, 5, 2832–2845 CrossRef CAS.
- F. Gao, E. D. Walter, M. Kollar, Y. L. Wang, J. Szanyi and C. H. F. Peden, J. Catal., 2014, 319, 1–14 CrossRef CAS.
- E. Borfecchia, P. Beato, S. Svelle, U. Olsbye, C. Lamberti and S. Bordiga, Chem. Soc. Rev., 2018, 47, 8097–8133 RSC.
- A. G. Greenaway, I. Lezcano-Gonzalez, M. Agote-Aran, E. K. Gibson, Y. Odarchenko and A. M. Beale, Top. Catal., 2018, 61, 175–182 CrossRef CAS.
- J. Szanyi, J. H. Kwak, H. Y. Zhu and C. H. F. Peden, Phys. Chem. Chem. Phys., 2013, 15, 2368–2380 RSC.
- H. Y. Zhu, J. H. Kwak, C. H. F. Peden and J. Szanyi, Catal. Today, 2013, 205, 16–23 CrossRef CAS.
- L. Ma, Y. S. Cheng, G. Cavataio, R. W. McCabe, L. X. Fu and J. H. Li, Appl. Catal., B, 2014, 156, 428–437 CrossRef.
- I. Lezcano-Gonzalez, U. Deka, B. Arstad, A. Van Yperen-De Deyne, K. Hemelsoet, M. Waroquier, V. Van Speybroeck, B. M. Weckhuysen and A. M. Beale, Phys. Chem. Chem. Phys., 2014, 16, 1639–1650 RSC.
- A. Marberger, D. Ferri, M. Elsener and O. Kröcher, Angew. Chem., Int. Ed., 2016, 55, 11989–11994 CrossRef CAS.
- A. Marberger, A. W. Petrov, P. Steiger, M. Elsener, O. Kröcher, M. Nachtegaal and D. Ferri, Nat. Catal., 2018, 1, 221–227 CrossRef CAS.
- P. Müller and I. Hermans, Ind. Eng. Chem. Res., 2017, 56, 1123–1136 CrossRef.
- V. Marchionni, D. Ferri, O. Kröcher and A. Wokaun, Anal. Chem., 2017, 89, 5802–5810 CrossRef.
- A. Urakawa, T. Bürgi and A. Baiker, Chem. Eng. Sci., 2008, 63, 4902–4909 CrossRef CAS.
- D. Baurecht and U. P. Fringeli, Rev. Sci. Instrum., 2001, 72, 3782–3792 CrossRef CAS.
- C. F. J. König, T. J. Schildhauer and M. Nachtegaal, J. Catal., 2013, 305, 92–100 CrossRef.
- C. F. J. König, J. A. van Bokhoven, T. J. Schildhauer and M. Nachtegaal, J. Phys. Chem. C, 2012, 116, 19857–19866 CrossRef.
- P. M. Abdala, O. V. Safonova, G. Wiker, W. van Beek, H. Emerich, J. A. van Bokhoven, J. Sa, J. Szlachetko and M. Nachtegaal, Chimia, 2012, 66, 699–705 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS.
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. A. Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS PubMed.
- W. P. Shan, F. D. Liu, H. He, X. Y. Shi and C. B. Zhang, Chem. Commun., 2011, 47, 8046–8048 RSC.
- R. R. Zhang, Y. H. Li and T. L. Zhen, RSC Adv., 2014, 4, 52130–52139 RSC.
- M. Moreno-González, R. Millán, P. Concepción, T. Blasco and M. Boronat, ACS Catal., 2019, 9, 2725–2738 CrossRef.
- S. Bordiga, L. Regli, C. Lamberti, A. Zecchina, M. Bjorgen and K. P. Lillerud, J. Phys. Chem. B, 2005, 109, 7724–7732 CrossRef CAS.
- A. Zecchina, L. Marchese, S. Bordiga, C. Paze and E. Gianotti, J. Phys. Chem. B, 1997, 101, 10128–10135 CrossRef CAS.
- S. S. Lai, D. M. Meng, W. C. Zhan, Y. Guo, Y. L. Guo, Z. G. Zhang and G. Z. Lu, RSC Adv., 2015, 5, 90235–90244 RSC.
- K. Shimizu, J. Shibata, H. Yoshida, A. Satsuma and T. Hattori, Appl. Catal., B, 2001, 30, 151–162 CrossRef CAS.
- M. Bendrich, A. Scheuer, R. E. Hayes and M. Votsmeier, Appl. Catal., B, 2018, 222, 76–87 CrossRef CAS.
- J. Rudolph and C. R. Jacob, ACS Omega, 2019, 4, 7987–7993 CrossRef CAS.
- C. Paolucci, I. Khurana, A. A. Parekh, S. C. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-Caballero, A. Yezerets, J. T. Miller, W. N. Delgass, F. H. Ribeiro, W. F. Schneider and R. Gounder, Science, 2017, 357, 898–903 CrossRef CAS.
- C. Negri, E. Borfecchia, M. Cutini, K. A. Lomachenko, T. V. W. Janssens, G. Berlier and S. Bordiga, ChemCatChem, 2019, 11, 3828–3838 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04905c |
| This journal is © The Royal Society of Chemistry 2020 |