
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegio- and diastereoselective dearomatizations of N-alkyl activated azaarenes: the maximization of the reactive sites†
Hong-Jie
Miao
a,
Le-Le
Wang
a,
Hua-Bin
Han
a,
Yong-De
Zhao
ab,
Qi-Lin
Wang
 *a and
Zhan-Wei
Bu
*a
*a and
Zhan-Wei
Bu
*a
aCollege of Chemistry and Chemical Engineering, Henan University, Kaifeng 475004, China. E-mail: wangqilin@henu.edu.cn; buzhanwei@henu.edu.cn
bInstitute of Chemistry, Henan Academy of Sciences, Zhengzhou 450002, P. R. China
First published on 17th December 2019
Abstract
An unprecedented base-promoted multi-component one-pot dearomatization of N-alkyl activated azaarenes was developed, which enabled the synthesis of complex and diverse bridged cyclic polycycles with multiple stereocenters in a highly regio- and diastereoselective manner. Besides, we realized the step-controlled dearomative bi- and trifunctionalization of quinolinium salts. These transformations not only achieved the maximization of the reaction sites of pyridinium, quinolinium and isoquinolinium salts to enhance structural complexity and diversity, but also opened up a new reaction mode of these N-activated azaarenes. A unique feature of this strategy is the use of easily accessible and bench-stable N-alkyl activated azaarenes to provide maximum reactive sites for dearomative cascade cyclizations. In addition, the salient characteristics including high synthetic efficiency, short reaction time, mild conditions and simple operation made this strategy particularly attractive.
Introduction
The dearomatization reaction has emerged as an expedient and versatile synthetic strategy to construct three-dimensional complex molecular skeletons from easily available flat aromatic compounds.1 Within this realm, significant attention has been directed toward the dearomatization of arenes and heteroarenes to assemble cyclic and heterocyclic compounds. Despite the high synthetic value of this strategy, the inherent challenge lies in the low reactivity of the aromatic substrates due to their reluctance to lose aromaticity as well as the difficulty in controlling the regio- and stereoselectivity in the course of generating the target three-dimensional products. In view of the salient features of pyridine, quinoline and isoquinoline including ready availability, stability and ease of handling, the straightforward dearomatization reaction with electrophilically improved pyridinium, quinolinium and isoquinolinium salts represents one of the most efficient and robust approaches for the synthesis of hydrogenated azaarenes, which are common molecular architectures of a number of biologically active natural products and pharmaceuticals.2A survey of literature revealed that the reaction modes of pyridinium, quinolinium and isoquinolinium salts were mainly divided into two categories (Scheme 1a). The first one was the extensively studied monofunctionalization of pyridinium,3 quinolinium4 and isoquinolinium salts5 by taking advantage of their electrophilic activity via nucleophilic attack at the more electron-deficient carbons. However, for pyridinium and quinolinium salts, both the C-2 and C-4 positions are active sites. Thus, the regioselectivity of these additions was a main issue of concern. Generally, mixtures of regioisomers were observed in many cases. Introduction of an electron-withdrawing group at the C-3 position could regioselectively favor the C-4 nucleophilic attack.6 The second one was the sparsely reported bifunctionalization of these N-activated azaarenes, where the C-2 and C-4 positions of pyridinium and quinolinium salts and the C-1 and C-3 positions of isoquinolinium salts were skillfully employed to construct interesting bridged cyclic azaheterocycles (Scheme 1b).7 Although these elegant strategies have proved to be quite successful to assemble hydrogenated azaarenes via dearomative nucleophilic additions, only one or two reactive sites were exploited, which largely restricted their application for the construction of complex compounds. Therefore, it is highly important, but an extremely challenging task, to develop new reaction systems that facilitate the exploitation of all of the potential reactive sites of pyridinium, quinolinium and isoquinolinium salts to achieve structural complexity and diversity.
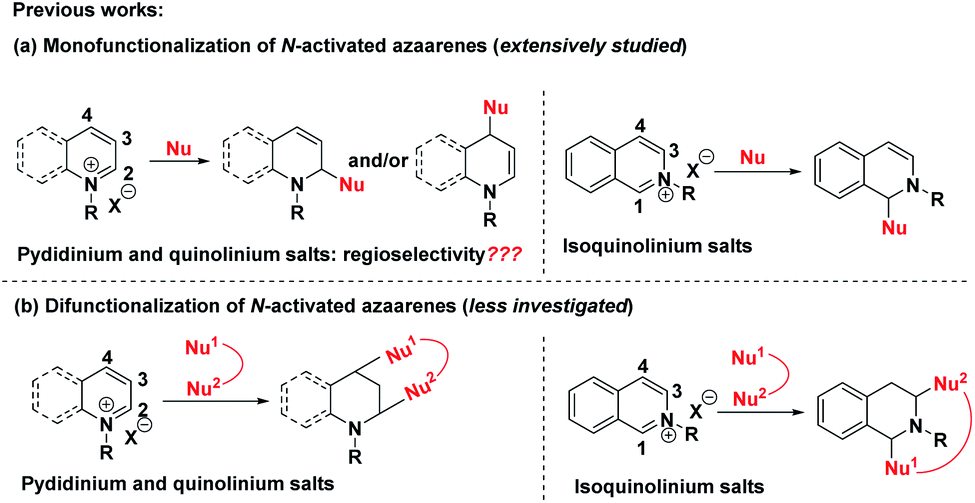 | ||
| Scheme 1 (a) Previous dearomative monofunctionalization of N-activated azaarenes. (b) Sparsely reported bifunctionalization of N-activated azaarenes. | ||
Theoretically, pyridinium salts possess three electrophilic sites at the C-2, C-4 and C-6 positions and two nucleophilic sites at the C-3 and C-5 positions once the first attack by a nucleophile at the C-4 position was initiated (Scheme 2a). Similarly, quinolinium and isoquinolinium salts also own two electrophilic sites and one nucleophilic site, respectively. Based on this knowledge, we envisioned that the maximization of the reactive sites of N-alkyl activated azaarenes could be achieved to enable the construction of complex and diverse bridged cyclic heterocycles by choosing the appropriate binucleophile and electrophile via a one-pot three-component dearomative cascade reaction (Scheme 2b). Actually, N-alkyl activated azaarenes themselves could be capable of behaving as electrophiles.
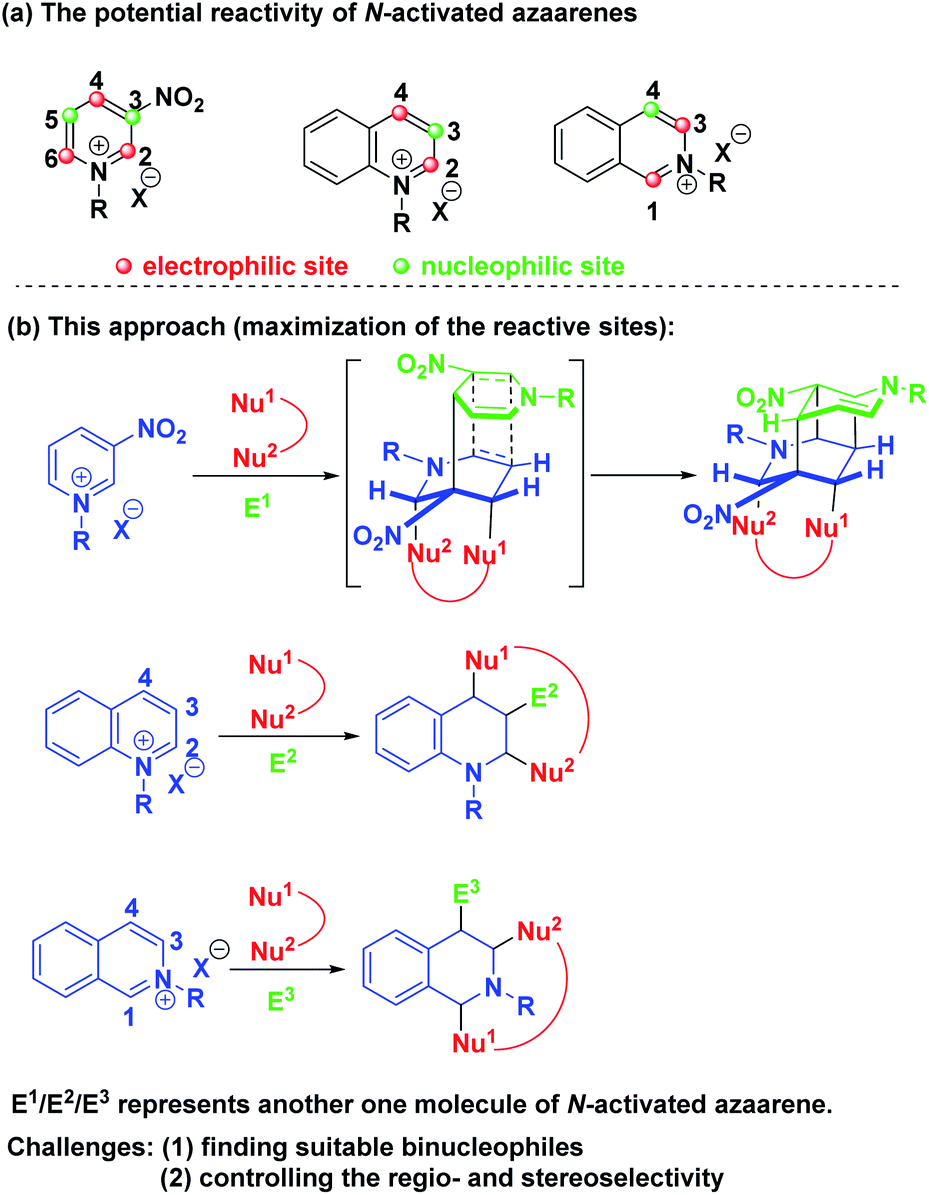 | ||
| Scheme 2 (a) The potential reactivity of N-activated azaarenes. (b) Our synthetic strategy to maximize the reactive sites of N-alkyl activated azaarenes. | ||
In this scenario, there were three inherent synthetic challenges: (1) the dearomative multifunctionalization of pyridinium, quinolinium and isoquinolinium salts has never been studied; (2) the appropriate binucleophile and suitable reaction conditions had to be found to improve the synthetic efficiency and control the regioselectivity and stereoselectivity; (3) the generation of multiple stereocenters embedded into bridged ring systems was a big challenge. As a continuation of our program aiming at the exploration of new cascade reactions for the construction of bridged rings with high synthetic efficiency and excellent stereocontrol,8 herein, we wish to present a base-promoted diastereoselective one-pot multicomponent dearomative cascade reaction of pyridinium, quinolinium and isoquinolinium salts, which realized the maximization of the active reactive sites of these N-alkyl activated azaarenes for dearomatization to construct diverse and complex bridged ring systems with multiple stereocenters. Such skeletons are privileged structural units of numerous natural products and biologically active pharmaceuticals.9
Results and discussion
As an initial study, enaminone 1a and N-benzyl-3-nitropyridinium salt 2a were chosen as the model substrates to test the feasibility of our synthetic design. When the reaction was conducted in CH3CN at 60 °C with 2.0 equivalents of Cs2CO3 as the base, gratifyingly, the desired bridged polycycle product 3a was delivered in 12% yield with complete regio- and diastereomeric control, whose structure was unambiguously verified by a comprehensive NMR, HRMS and X-ray analysis (Table 1, entry 1). This preliminary result definitely indicated that the dearomative cascade annulation of pyridinium salts was feasible. The formation of 3a was very appealing from the perspective of the reaction process and product structure. This transformation opened up an unprecedented reaction mode of pyridinium salt, where all of the five active sites were tactfully utilized to simultaneously construct five new bonds, two bridged rings as well as a challenging fused cyclobutane10 and eight stereocenters in a highly regio- and diastereoselective one-pot fashion. Moreover, the product contained some useful functional groups, such as nitro, carbonyl and a cyclic enamine, which offered versatile platforms for further architectural modifications.|
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Time | Yieldb (%) |
| a Unless otherwise noted, the reactions were conducted with 0.15 mmol 1a with 2.0 equivalents of 2a in the presence of 2.0 equivalents of base in 0.8 mL of specified solvent at 60 °C. b Isolated yields obtained by column chromatography. c Isolated yields obtained by filtration of the precipitate. d 2.2 equivalents of 2a were used. e 2.4 equivalents of 2a were used. DABCO = 1,4-diazabicyclo[2.2.2]octane; DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene; TMG = 1,1,3,3-tetramethylguanidine. | ||||
| 1 | Cs2CO3 | CH3CN | 58 h | 12 |
| 2 | K2CO3 | CH3CN | 18 h | 15 |
| 3 | Na2CO3 | CH3CN | 18 h | 12 |
| 4 | NaOH | CH3CN | 18 h | 30 |
| 5 | DABCO·6H2O | CH3CN | 27 h | 16 |
| 6 | DBU | CH3CN | 33 h | 45 |
| 7 | NEt3 | CH3CN | 3 h | 32 |
| 8 | TMG | CH3CN | 5 min | 87c |
| 9 | TMG | CHCl3 | 4 h | 82 |
| 10 | TMG | Toluene | 4 h | 74c |
| 11 | TMG | EtOAc | 20 min | 58 |
| 12d | TMG | CH3CN | 5 min | 95c |
| 13e | TMG | CH3CN | 5 min | 80c |
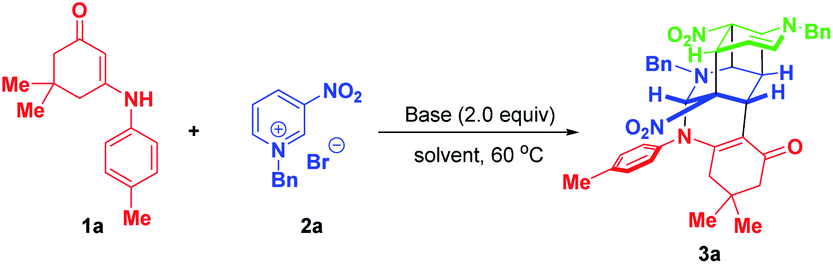
To improve the synthetic efficiency, some inorganic and organic bases were evaluated. Among them, 1,1,3,3-tetramethylguanidine (TMG) was identified to be optimal in terms of yield and reaction time, in which the reaction went to full conversion within 5 min, affording 3a in 87% yield (Table 1, entry 8). Remarkably, in this condition, the product was precipitated from the homogenous reaction mixture. So only a filtration process was needed to purify it, which largely simplified the purification procedure and met the requirement of green chemistry. Then, the reaction media were examined and the results suggested that they all gave inferior yields compared with CH3CN (Table 1, entries 9–11 vs. 8). Finally, the loadings of N-benzyl-3-nitropyridinium salt 2a were investigated. When 2.2 equivalents of 2a were used, product 3a was precipitated out in a yield of 95%. No increment in yield was observed on further raising the amount of 3a to 2.4 equivalents. To achieve the asymmetric synthesis of 3a, we also examined some chiral bases, including cinchona alkaloids and their derivatives and chiral guanidine (for details, see Table S1 on page S19 of the ESI†). However, the results with respect to yields and enantioselectivity were quite disappointing. Consequently, the optimal conditions for the formation of complex bridged cyclic polycycle 3a were identified to be: 0.15 mmol of 1a and 2.2 equivalents of 2a with 0.30 mmol of TMG in CH3CN at 60 °C for 5 min (Table 1, entry 12).
With the optimized reaction conditions in hand, the substrate scope with respect to different enaminones 1 was first explored and the results are highlighted in Scheme 3. These transformations had a wide tolerance of arylamine-derived enaminones with different substitution patterns and electron properties, affording the corresponding products 3a–p in 51–97% yields within 5 minutes. Notably, when R1 was located at the ortho-position of arylamine, new kinds of atropisomers 3m–p were delivered in 51–88% yields, albeit with low diastereoselectivities. This promising finding not only provides a robust and facile method to access new atropisomers, but also represents a valuable complement to the existing appealing protocols.11 Other than arylamine-derived enaminones, aliphatic amine-derived ones were also compatible, affording 3q–s in 24–41% yields with complete diastereomeric control. Particularly, furfuryl amine-derived enaminone also turned out to be a suitable reaction partner. Besides, acetylacetone and ethyl benzoylacetate derived acyclic enaminones could participate in this reaction successfully, thus facilitating the synthesis of 3u and 3v in 42% and 43% yields, respectively. An evaluation of the substituent effect of R2 on 3-nitropyridinium salts 2 revealed that not only were the methyl and ethyl groups accommodated well in this dearomative cascade cyclization, more hindered allyl, n-propyl and n-butyl groups were also amenable, enabling the construction of 3w–za in good to excellent yields with complete diastereomeric control.
 | ||
| Scheme 3 Scope of substrates. aIsolated yields obtained by filtration of the precipitate. bIsolated yields obtained by column chromatography. | ||
Later on, we investigated the effect of the substituents on the C3-position of pyridinium salts (Scheme 4). In sharp contrast to 3-nitropyridinium salt 2a, replacement of the nitro group with cyano and trifluoromethyl resulted in different reactivities, in which only bifunctionalization occurred for 2g and 2h by regioselective attack at C-2 and C-6 positions, affording bridged polycyclic N,N-ketals 3zb and 3zc in 50% and 41% yields, respectively.
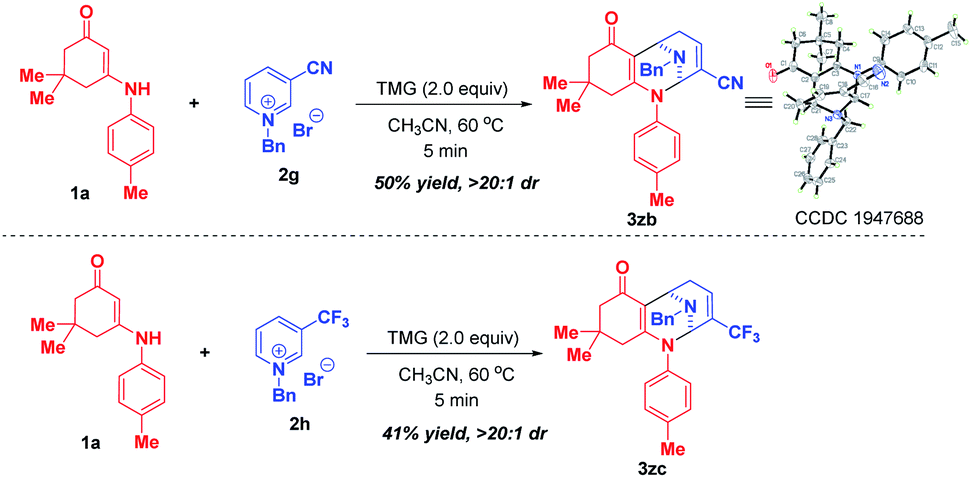 | ||
| Scheme 4 Scope of substrates with respect to pyridinium salts 2 with different substituents at the C-3 position. | ||
After successfully maximizing the active sites of pyridinium salts by the dearomative cascade strategy, we further extend this reaction system to quinolinium salts. With the first attempt of the reaction between enaminone 1a and N-benzyl quinolinium salt 4a in CH3CN at 60 °C, the trifunctionalized dearomatization product 5a, featured by an interesting fusion of bioactive bridged N,O-ketal and 1,2-dihydroquinoline, was precipitated out from the reaction mixture, and was obtained in 98% yield in a highly diastereoselective manner (Scheme 5). Although the product 5a contained four contiguous tertiary stereocenters including two bridgehead stereocenters, only one diastereoisomer was obtained. After flash silica gel column chromatography, 5a was partially decomposed into bifunctionalized product 6a through a ring-opening/closure sequence. This result indicated that 5a was not very stable and it might be sensitive to acid. So, we conducted a one-pot two-step reaction of 1a and 4a. After the first TMG-promoted three-component dearomative trifunctionalization went to completion, one equivalent of trifluoroacetic acid was added to the reaction mixture, and the desired bifunctionalized bridged N,N-ketal product 6a was generated smoothly in 99% yield with excellent diastereoselectivity.
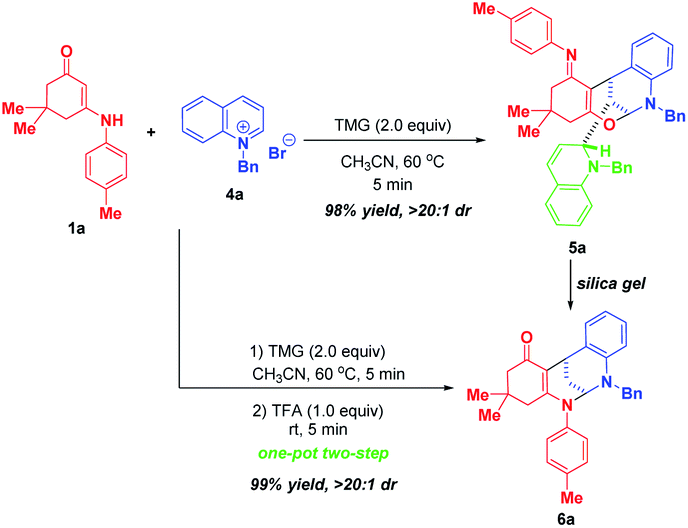 | ||
| Scheme 5 Step-controlled dearomative cascade cyclization of quinolinium salt 4a. | ||
This appealing step-controlled diverse synthesis prompted us to further investigate the substrate scope for the diastereoselective dearomative bi- and trifunctionalization of quinolinium salts. As demonstrated in Scheme 6, all of the reactions went to full conversion within 5 minutes to afford a series of trifunctionalized products 5a–s in 61–99% yields. Moreover, the workup procedure was extremely simple. All of the products were precipitated out from the reaction mixture and only an easy filtration process was needed to purify them. These prominent merits including high synthetic efficiency, short reaction time and simple operation make this transformation very attractive.
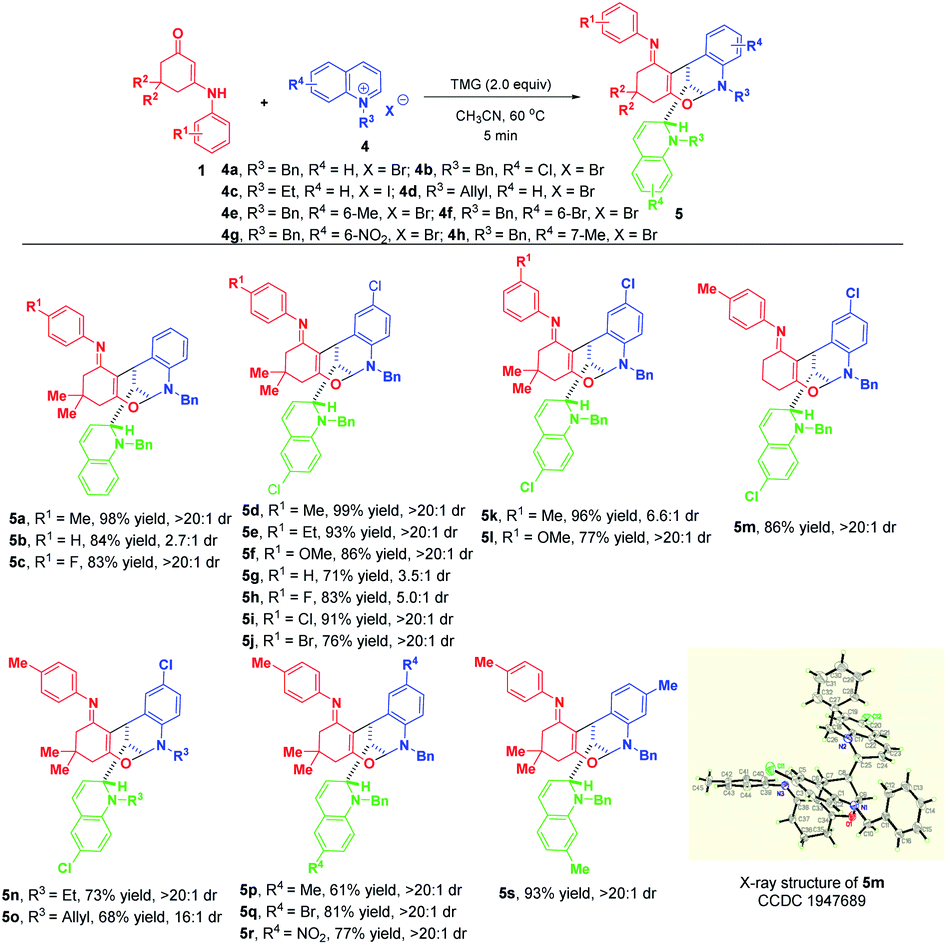 | ||
| Scheme 6 Diastereoselective dearomative trifunctionalization of quinolinium salts 4. | ||
When phenanthrolinium salt 4k and 3-methyl quinolinium salt 4j were used as substrates, only dearomative bifunctionalizations took place to afford 7 and 8 in 87% and 51% yields, respectively (Scheme 7). Next, we turned our attention to the one-pot two-step dearomative bifunctionalization of quinolinium salts. As summarized in Scheme 8, both enaminones 1 and quinolinium salts 4, irrespective of their substitution patterns and electron character, were tolerable, and participated in these transformations efficiently in a highly diastereomeric fashion, thus furnishing the corresponding products 6a–t in 79–99% yields.
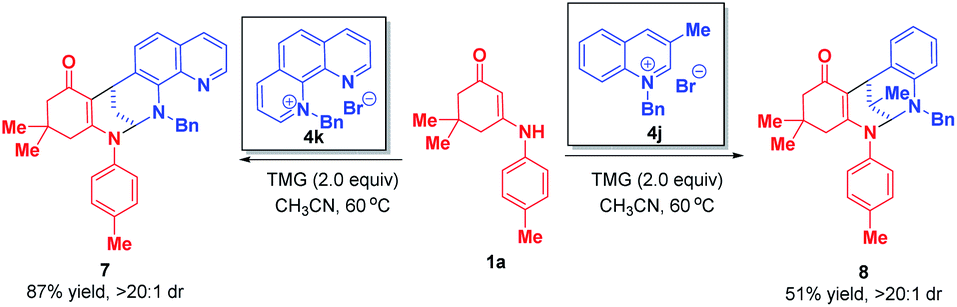 | ||
| Scheme 7 Diastereoselective dearomative bifunctionalization of phenanthrolinium salt 4k and 3-methyl quinolinium salt 4j. | ||
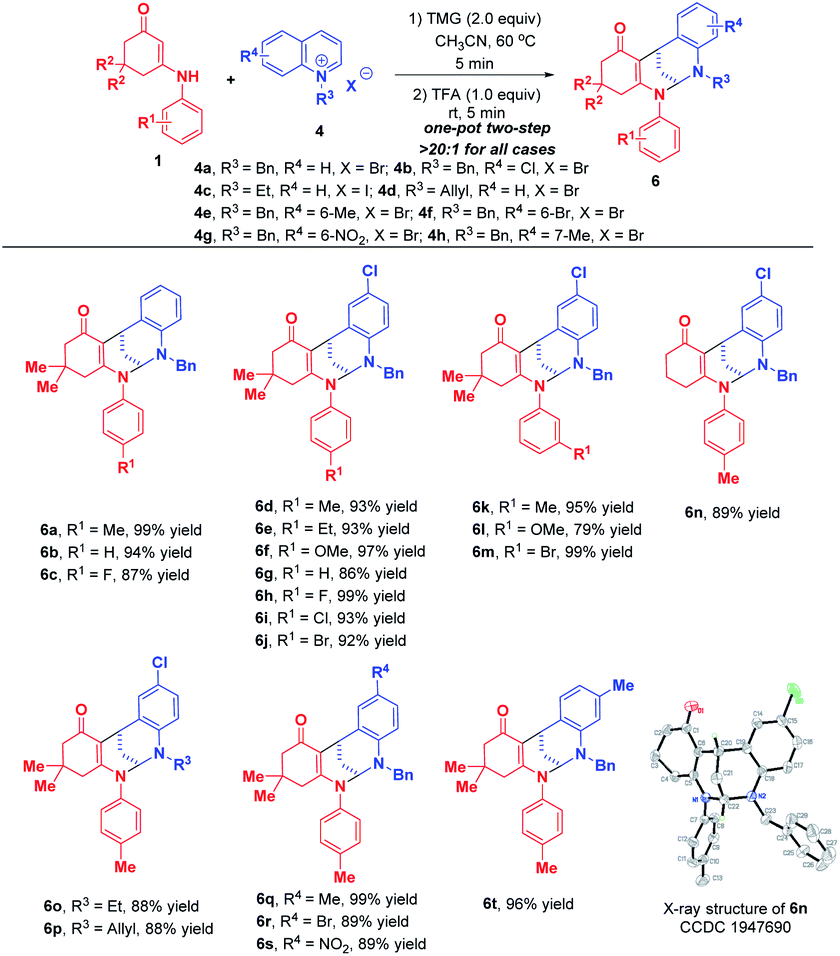 | ||
| Scheme 8 Diastereoselective dearomative bifunctionalization of quinolinium salts 4. | ||
Finally, we moved on to examine the reactivity of isoquinolinium salt 9a. Likewise, the highly regio- and diastereomeric trifunctionalized product 10a was generated in 36% yield when 2.0 equivalents of TMG were used as the base (Table S2,† entry 1). To benefit the yield, we investigated the effect of various bases and their loadings (for detailed condition optimization, see Table S2† on the page S48 of the ESI†), and we identified that 3.6 equivalents of DBU were the best of choice. Subsequently, the substrate scope was explored by using a variety of enaminones 1 and isoquinolinium salts 9 (Scheme 9). Gratifyingly, all of them could participate in this transformation successfully to afford 10a–o in 82–97% yields. Remarkably, use of ortho-fluoro-substituted aniline-derived enaminone as the substrate led to the synthesis of atropisomer 10l in 90% yield with 1.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. When 4-bromoisoquinolinium salt 9d was employed, the highly diastereoselective bifunctionalization occurred instead of the trifunctionalization to deliver 10p as a single diastereomer in 54% yield (Scheme 10). The difference in reactivities of 9d and 9a–c may be caused by the steric hindrance, which disfavored the subsequent addition with another isoquinolinium salt.
:1 dr. When 4-bromoisoquinolinium salt 9d was employed, the highly diastereoselective bifunctionalization occurred instead of the trifunctionalization to deliver 10p as a single diastereomer in 54% yield (Scheme 10). The difference in reactivities of 9d and 9a–c may be caused by the steric hindrance, which disfavored the subsequent addition with another isoquinolinium salt.
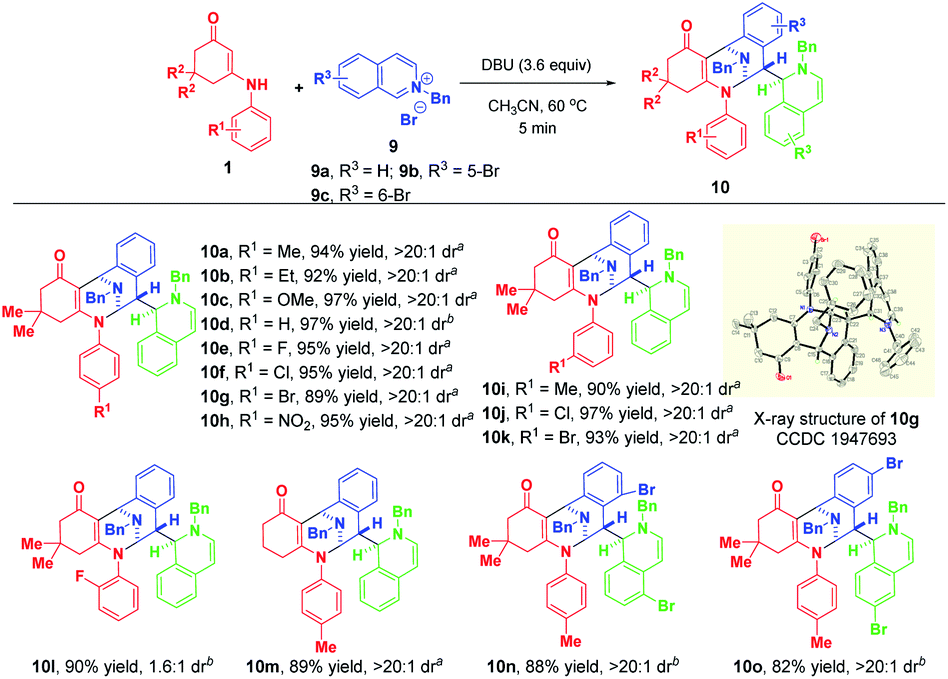 | ||
| Scheme 9 Diastereoselective dearomative trifunctionalization of isoquinolinium salts 9. aIsolated yields obtained by filtration of the precipitate. bIsolated yields obtained by column chromatography. | ||
 | ||
| Scheme 10 Diastereoselective dearomative bifunctionalization of 4-bromoisoquinolinium salt 9d. | ||
To demonstrate the synthetic robustness, a preparative-scale experiment was conducted with 2.0 mmol of enaminone 1d and 4.4 mmol of N-benzyl-3-nitropyridinium salt 2a (Scheme 11). Delightfully, the reaction proceeded smoothly to give 3d in diminished but acceptable yield within 5 minutes (0.89 g, 69% yield), thus suggesting that this protocol has the potential for scale-up preparation.
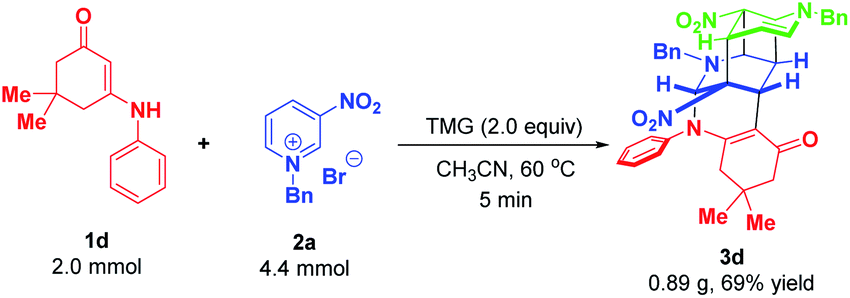 | ||
| Scheme 11 Scalable preparation of 3d. | ||
To further highlight the synthetic applicability, some chemical transformations of 3g were carried out (Scheme 12). By subjecting 3g to the NaBH4–NiCl2 reductive system, the two nitro groups in 3g were reduced into hydroxylamine and oxime respectively, accompanied by four-membered all-carbon ring opening. This transformation went to completion within 10 min with the formation of the corresponding functionalized product 11 in 62% yield with a ratio of 10:1 Z/E. In addition, the presence of a bromine atom in 3g offers a convenient platform for further elaboration to generate molecules with more diverse and complex structures. Thus, two palladium-catalyzed Suzuki couplings of 3g with 4-chlorophenzyl boronic acid and 2-indolyl boronic acid were performed, which afforded 12 and 13 in 99% and 73% yields, respectively.
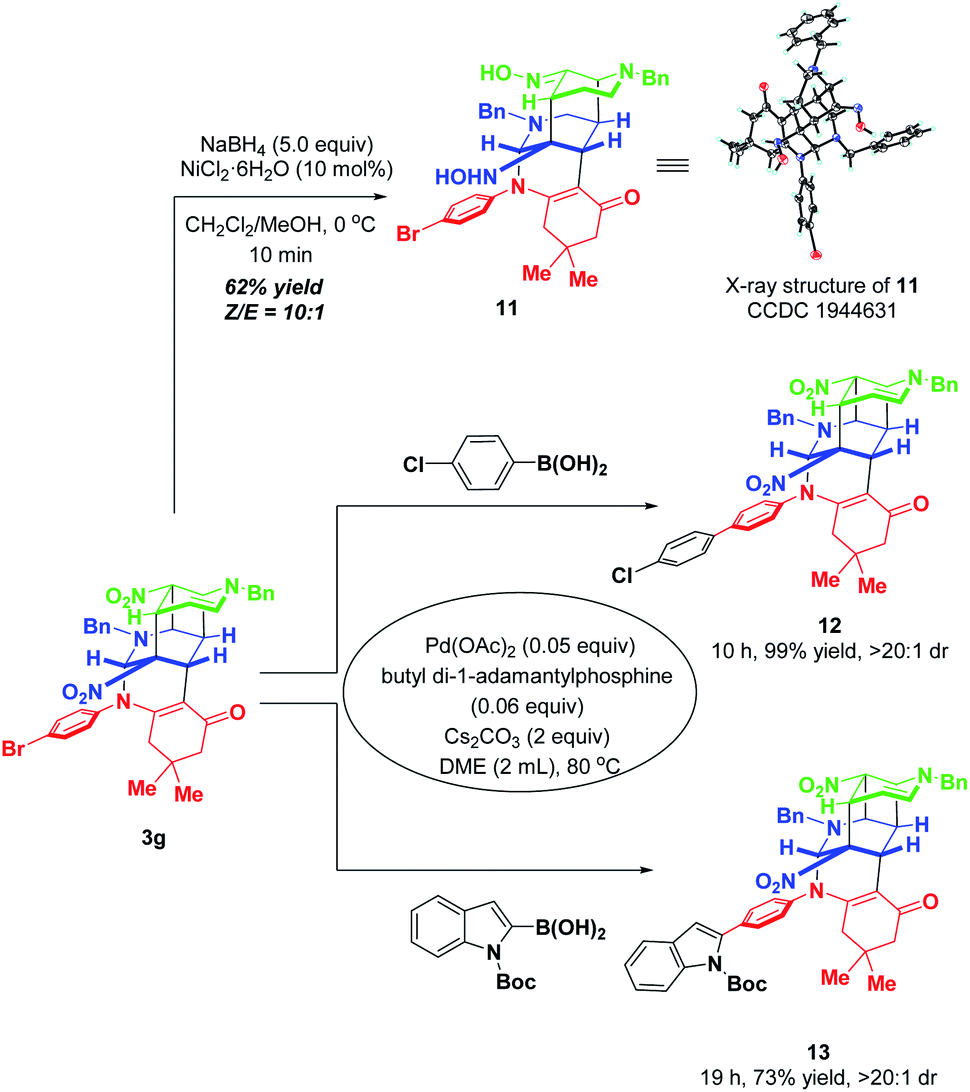 | ||
| Scheme 12 Chemical conversions of 3g. | ||
The structures and the relative configurations of 3a, 3zb, 5m, 6n, 10g, 10p and 11 were unequivocally verified by X-ray analysis.12 The relative configurations of other products 3, 5, 6 and 10 were assigned by analogy.
Based on the experimental results, plausible mechanisms were proposed to rationalize the reaction pathways. As shown in Scheme 13a, a Michael addition between enaminone 1a and 3-nitropyridinium salt 2a occurred first to generate the bienamine intermediate A. Nucleophilic Michael attack at the C-4 position of another 3-nitropyridinium salt molecule 2a resulted in the formation of active iminium ion intermediate B containing a trienamine moiety, followed by an intramolecular Mannich reaction to yield the intermediate C. A final [2 + 2] cycloaddition led to four-membered all-carbon cyclobutane formation. As for the regio- and diastereoselective dearomative tri- and bifunctionalization of quinolinium salt 4a (Scheme 13b), similarly, a 1,4-addition took place first to deliver intermediate D, which contained an enamine motif. Then, a Mannich reaction proceeded by regioselective nucleophilic attack at the C-2 position of another quinolinium salt 4a to generate intermediate E with an imine ion skeleton. Through electron transfer, the oxygen atom served as an active nucleophilic site to attack the imine ion via an intramolecular oxa-Mannich reaction, thus forming the trifunctionalized bridged cyclic product 5a. In the presence of TFA, 5a was susceptible to transform into bifunctionalized product 8a through an acid-catalyzed sequential ring-opening/enamine–imine tautomerization/aza-Mannich process. For the construction of trifunctionalized tetrahydroisoquinoline product 10a, a triple Mannich reaction of enaminone 1a and N-benzyl isoquinolinium salt 9a proceeded, which is shown in Scheme 13c.
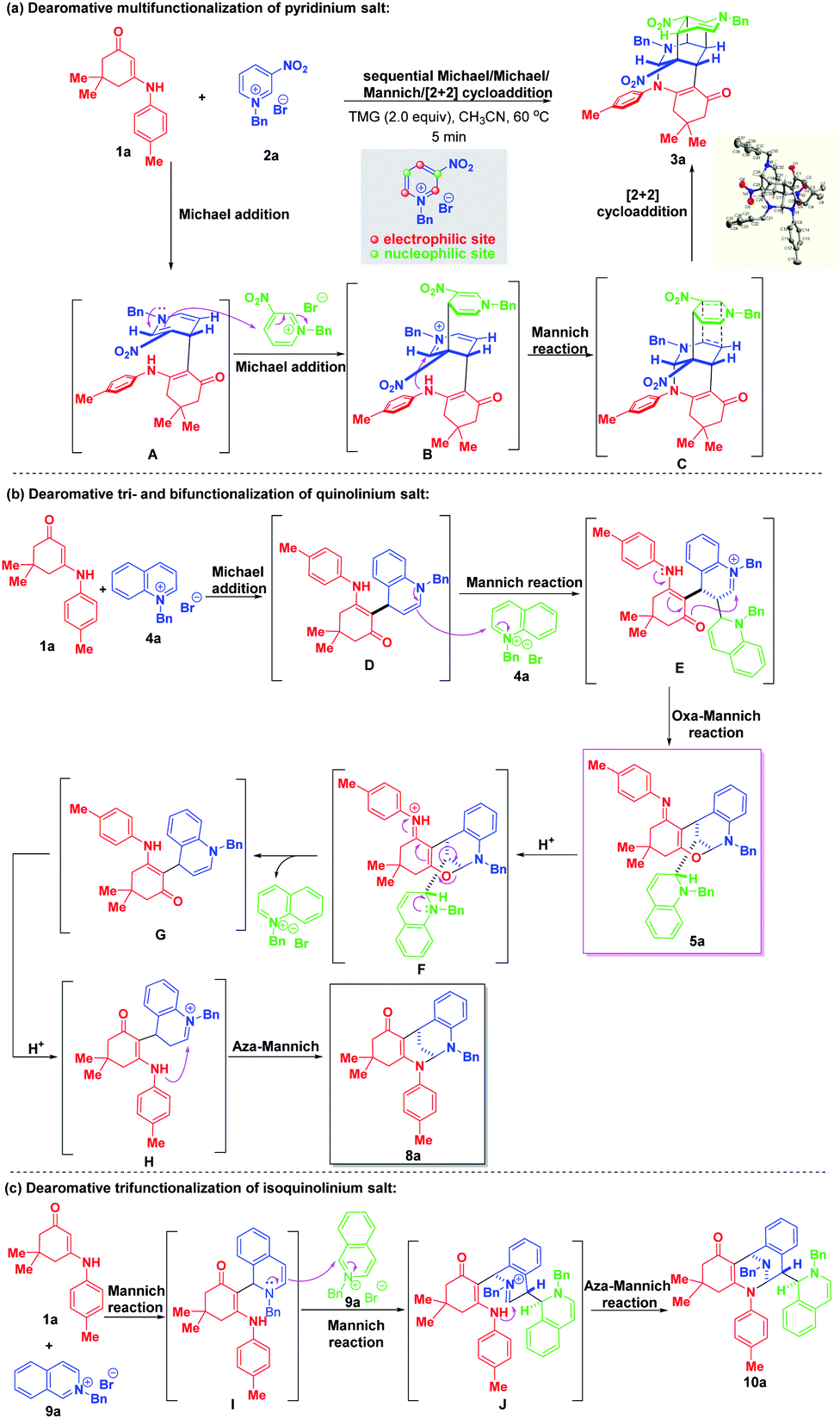 | ||
| Scheme 13 (a) Plausible reaction pathway for dearomative multifunctionalization of pyridinium salt 2a. (b) Possible mechanism for dearomative tri- and bifunctionalization of quinolinium salt 4a. (c) Proposed mechanism for the trifunctionalization of isoquinolinium salt 9a. | ||
Conclusions
In summary, we have successfully developed an unprecedented base-promoted multi-component one-pot dearomatization of pyridinium, quinolinium and isoquinolinium salts, which enabled the synthesis of complex and diverse bridged cyclic polycycles with multiple stereocenters in a highly regio- and diastereoselective manner with high synthetic efficiency. Besides, we realized the step-controlled dearomative bi- and trifunctionalization of quinolinium salts. These transformations not only achieved the maximization of the reactive sites of pyridinium, quinolinium and isoquinolinium salts to enhance structural complexity and diversity, but also opened up a new reaction mode of these N-activated azaarenes, which may largely benefit the prospective reaction designs. A unique feature of this strategy is the use of easily accessible and bench-stable N-alkyl activated azaarenes to provide maximum reactive sites for dearomative multi-component one-pot cascade cyclizations. In addition, the salient characteristics including high synthetic efficiency, short reaction time, mild conditions and simple operation made this strategy particularly attractive. We expect that this fascinating strategy will stimulate the design of new related reactions for the facile construction of natural products and biologically interesting compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (U1504206), the Foundation of He'nan Educational Committee (18A150002), and Henan University. We also thank Dr Zhenhua Wang and Dr Lei Yang from Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences for chiral HPLC analysis.Notes and references
- For selected reviews: (a) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed; (b) Q.-P. Ding, X.-L. Zhou and R.-H. Fan, Org. Biomol. Chem., 2014, 12, 4807 RSC; (c) W.-T. Wu, L.-M. Zhang and S.-L. You, Chem. Soc. Rev., 2016, 45, 1570 RSC; (d) W.-S. Sun, G.-F. Li, L. Hong and R. Wang, Org. Biomol. Chem., 2016, 14, 2164 RSC.
- For selected reviews: (a) V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157 CrossRef CAS PubMed; (b) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642 CrossRef CAS PubMed; (c) S. Sowmiah, J. M. S. S. Esperança, L. P. N. Rebelo and C. A. M. Afonoso, Org. Chem. Front., 2018, 5, 453 RSC.
- For selected examples on the mono-functionalization of pyridinium salts: (a) C. Nadeau, S. Aly and K. Belyk, J. Am. Chem. Soc., 2011, 133, 2878 CrossRef PubMed; (b) M. Mohiti, C. Rampalakos, K. Feeney, D. Leonori and V. K. Aggarwal, Chem. Sci., 2014, 5, 602 RSC; (c) O. G. Mancheño, S. Asmus, M. Zurro and T. Fischer, Angew. Chem., Int. Ed., 2015, 54, 8823 CrossRef PubMed.
- For selected examples on the mono-functionalization of quinolinium salts: (a) M. Pappoppula, F. S. P. Cardoso, B. O. Garrett and A. Aponick, Angew. Chem., Int. Ed., 2015, 54, 15202 CrossRef CAS PubMed; (b) S. Shirakawa, S. Liu, S. Kaneko, Y. Kumatabara, A. Fukuda, Y. Omagari and K. Maruoka, Angew. Chem., Int. Ed., 2015, 54, 15767 CrossRef CAS PubMed; (c) Y. Wang, Y.-L. Liu, D.-D. Zhang, H. Wei, M. S. Hi and F.-J. Wang, Angew. Chem., Int. Ed., 2016, 55, 3776 CrossRef CAS PubMed; (d) W. Jo, J. Kim, S. Choi and S. H. Cho, Angew. Chem., Int. Ed., 2016, 55, 9690 CrossRef CAS PubMed; (e) K. Kubota, Y. Watanabe, K. Hayama and H. Ito, J. Am. Chem. Soc., 2016, 138, 4338 CrossRef CAS PubMed; (f) T. Fischer, Q.-N. Duong and O. G. Mancheño, Chem.–Eur. J., 2017, 23, 5983 CrossRef CAS PubMed; (g) A. Grozavu, H. B. Hepburn, P. J. Smith, H. K. Potukuchi, P. J. Lindsay-Scott and T. J. Donohoe, Nat. Chem., 2019, 11, 242 CrossRef CAS PubMed.
- For selected examples on the mono-functionalization of isoquinolinium salts: (a) A. G. Schafer, J. M. Wieting, T. J. Fisher and A. E. Mattson, Angew. Chem., Int. Ed., 2013, 52, 11321 CrossRef CAS PubMed; (b) L. Mengozzi, A. Gualand and P. G. Cozzi, Chem. Sci., 2014, 5, 3915 RSC; (c) C. Guo, M. Fleige, D. Janssen-Müller, C. G. Daniliuc and F. Glorius, Nat. Chem., 2015, 7, 842 CrossRef CAS PubMed; (d) A. R. Choudhury and S. Mukherjee, Chem. Sci., 2016, 7, 6940 RSC; (e) M. Zurro, S. Asmus, J. Bamberger, S. Beckendorf and O. G. Mancheño, Chem.–Eur. J., 2016, 22, 3785 CrossRef CAS PubMed; (f) M. Zhang, W.-S. Sun, G.-M. Zhu, G.-J. Bao, B.-Z. Zhang, L. Hong, M. Li and R. Wang, ACS Catal., 2016, 6, 5290 CrossRef CAS.
- (a) G. Bertuzzi, A. Sinisi, L. Caruana, A. Mazzanti, M. Fochi and L. Bernardi, ACS Catal., 2016, 6, 6473 CrossRef CAS; (b) D. Zhang, Z.-H. Kang and W.-H. Hu, J. Org. Chem., 2017, 82, 5952 CrossRef CAS PubMed; (c) G. Bertuzzi, A. Sinisi, D. Pecorari, L. Caruana, A. Mazzanti, L. Bernardi and M. Fochi, Org. Lett., 2017, 19, 834 CrossRef CAS PubMed; (d) D. M. Flanigan and T. Rovis, Chem. Sci., 2017, 8, 6566 RSC; (e) R.-J. Yan, B.-X. Xiao, Q. Ouyang, H.-P. Liang, W. Du and Y.-C. Chen, Org. Lett., 2018, 20, 8000 CrossRef CAS PubMed; (f) G. D. Carmine, D. Ragno, O. Bortolini, P. P. Giovannini, A. Mazzanti, A. Massi and M. Fogagnolo, J. Org. Chem., 2018, 83, 2050 CrossRef PubMed.
- (a) M. Kiamehr, F. M. Moghaddam, P. V. Gormay, V. Semeniuchenko, A. Villinger, P. Langer and V. O. Iaroshenko, Tetrahedron, 2012, 68, 9685 CrossRef CAS; (b) Z.-H. Kang, D. Zhang and W.-H. Hu, Org. Lett., 2017, 19, 3783 CrossRef CAS PubMed; (c) M. A. Yawer, I. Hussain, J. P. Gütlein, A. Schmidt, H.-J. Jiao, H. Reinke, A. Spannenberg, C. Fischer and P. Langer, Eur. J. Org. Chem., 2008, 4193 CAS; (d) F. M. Moghaddam, S. Taheri, Z. Mirjafary and H. Saeidian, Synlett, 2010, 123 CrossRef CAS; (e) J.-H. Xu, S.-C. Zheng, J.-W. Zhang, X.-Y. Liu and B. Tan, Angew. Chem., Int. Ed., 2016, 55, 11834 CrossRef CAS PubMed.
- (a) Y.-S. Zhu, J. Zhou, S.-J. Jin, H.-H. Dong, J.-M. Guo, X.-G. Bai, Q.-L. Wang and Z.-W. Bu, Chem. Commun., 2017, 53, 11201 RSC; (b) J.-M. Guo, X.-G. Bai, Q.-L. Wang and Z.-W. Bu, J. Org. Chem., 2018, 83, 3679 CrossRef CAS PubMed; (c) W.-B. Wang, X.-G. Bai, S.-J. Jin, J.-M. Guo, Y. Zhao, H.-J. Miao, Y.-S. Zhu, Q.-L. Wang and Z.-W. Bu, Org. Lett., 2018, 20, 3451 CrossRef CAS PubMed; (d) S.-J. Jin, J. Guo, D.-M. Fang, Y.-W. Huang, Q.-L. Wang and Z.-W. Bu, Adv. Synth. Catal., 2019, 361, 456 CrossRef CAS.
- For a review: (a) V. H. Le, M. Inai, R. M. Williams and T. Kan, Nat. Prod. Rep., 2015, 32, 328 RSC. For selected examples: (b) H. Ueda, H. Satoh, K. Matsumoto, K. Sugimoto, T. Fukuyama and H. Tokuyama, Angew. Chem., Int. Ed., 2009, 48, 7600 CrossRef CAS PubMed; (c) M. Ding, K.-J. Liang, R. Pan, H.-B. Zhang and C.-F. Xia, J. Org. Chem., 2015, 80, 10309 CrossRef CAS PubMed; (d) X.-B. Wang, D.-L. Xia, L. Tan, H. Chen, H.-X. Huang, H. Song and Y. Qin, Chem.–Eur. J., 2015, 21, 14602 CrossRef CAS PubMed; (e) A. Takada, H. Fujiwara, K. Sugimoto, H. Ueda and H. Tokuyama, Chem.–Eur. J., 2015, 21, 16400 CrossRef CAS PubMed; (f) P. Lindovska and M. Movassaghi, J. Am. Chem. Soc., 2017, 139, 17590 CrossRef CAS PubMed; (g) J. D. Mason and S. M. Weinreb, Angew. Chem., Int. Ed., 2017, 56, 16674 CrossRef CAS PubMed; (h) K. N. Babu, A. Roy, M. Singh and A. Bisai, Org. Lett., 2018, 20, 6327 CrossRef CAS PubMed; (i) Y. Rebets, S. Nadmid, C. Paulus, C. Dahlem, J. Herrmann, H. Hübner, C. Rückert, A. K. Kiemer, P. Gmeiner, J. Kalinowski, R. Müller and A. Luzhetskyy, Angew. Chem., Int. Ed., 2019, 58, 12930 CrossRef CAS PubMed.
- For selected examples: (a) P.-L. Chen, P. J. Carroll and S. M. Sieburth, Org. Lett., 2009, 11, 4540 CrossRef CAS PubMed; (b) K. A. B. Austin, E. Herdtweck and T. Bach, Angew. Chem., Int. Ed., 2011, 50, 8416 CrossRef CAS PubMed; (c) S. C. Coote and T. Bach, J. Am. Chem. Soc., 2013, 135, 14948 CrossRef CAS PubMed; (d) S. C. Coote, A. Pöthig and T. Bach, Chem.–Eur. J., 2015, 21, 6906 CrossRef CAS PubMed; (e) A. Tröster, R. Alonso, A. Bauer and T. Bach, J. Am. Chem. Soc., 2016, 138, 7808 CrossRef PubMed; (f) C.-F. Wang and Z. Lu, Org. Lett., 2017, 19, 5888 CrossRef CAS PubMed; (g) C. Lorton, T. Castanheiro and A. Voituriez, J. Am. Chem. Soc., 2019, 141, 10142 CrossRef CAS PubMed.
- For selected examples: (a) H.-H. Zhang, C.-S. Wang, C. Li, G.-J. Mei, Y.-X. Li and F. Shi, Angew. Chem., Int. Ed., 2017, 56, 116 CrossRef CAS PubMed; (b) S.-Q. Jia, Z.-L. Chen, N. Zhang, Y. Tan, Y.-D. Liu, J. Deng and H.-L. Yan, J. Am. Chem. Soc., 2018, 140, 7056 CrossRef CAS PubMed; (c) L.-W. Qi, J.-H. Mao, J. Zhang and B. Tan, Nat. Chem., 2018, 10, 58 CrossRef CAS PubMed; (d) C. Ma, F. Jiang, F.-T. Sheng, Y.-C. Jiao, G.-J. Mei and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 3014 CrossRef CAS PubMed; (e) S.-C. Zheng, Q. Wang and J.-P. Zhu, Angew. Chem., Int. Ed., 2019, 58, 1494 CrossRef CAS PubMed; (f) H.-H. Li, X.-Q. Yan, J.-T. Zhang, W.-C. Guo, J.-J. Jiang and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 6732 CrossRef CAS PubMed; (g) J. Luo, T. Zhang, L. Wang, G. Liao, Q.-J. Yao, Y.-J. Wu, B.-B. Zhan, Y. Lan, X.-F. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2019, 58, 6708 CrossRef CAS PubMed; (h) S. Zhang, Q.-J. Yao, G. Liao, X. Li, H. Li, H.-M. Chen, X. Hong and B.-F. Shi, ACS Catal., 2019, 9, 1956 CrossRef CAS; (i) L.-W. Qi, S.-Y. Li, S.-H. Xiang, J. Wang and B. Tan, Nat. Catal., 2019, 2, 314 CrossRef CAS; (j) Y. Kwon, J.-Q. Li, J. P. Reid, J. M. Jacob, R. Crawford, M. S. Sigman, F. D. Toste and S. J. Miller, J. Am. Chem. Soc., 2019, 141, 6698 CrossRef CAS PubMed; (k) F. Jiang, K.-W. Chen, P. Wu, Y.-C. Zhang, Y.-C. Jiao and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 15104 CrossRef CAS PubMed.
- CCDC 1944660 (3a), CCDC 1947688 (3zb), CCDC 1947689 (5m), CCDC 1947690 (6n), CCDC 1947693 (10g), CCDC 1947691 (10p) and CCDC 1944631 (11) contain the supplementary crystallographic data for this paper.†.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures; characterization data of all the new products; copies of 1H NMR and 13C NMR. CCDC 1944660 (3a), 1947688 (3zb), 1947689 (5m), 1947690 (6n), 1947693 (10g), 1947691 (10p) and 1944631 (11). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04880d |
| This journal is © The Royal Society of Chemistry 2020 |