
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterization and C–H amination reactivity of nickel iminyl complexes†
Yuyang
Dong
 a,
James T.
Lukens
b,
Ryan M.
Clarke
a,
Shao-Liang
Zheng
a,
Kyle M.
Lancaster
b and
Theodore A.
Betley
*a
a,
James T.
Lukens
b,
Ryan M.
Clarke
a,
Shao-Liang
Zheng
a,
Kyle M.
Lancaster
b and
Theodore A.
Betley
*a
aDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, Massachusetts 02138, USA. E-mail: betley@chemistry.harvard.edu
bDepartment of Chemistry and Chemical Biology, Baker Laboratory, Cornell University, Ithaca, New York 14853, USA
First published on 11th December 2019
Abstract
Metalation of the deprotonated dipyrrin (AdFL)Li with NiCl2(py)2 afforded the divalent Ni product (AdFL)NiCl(py)2 (1) (AdFL: 1,9-di(1-adamantyl)-5-perfluorophenyldipyrrin; py: pyridine). To generate a reactive synthon on which to explore oxidative group transfer, we used potassium graphite to reduce 1, affording the monovalent Ni synthon (AdFL)Ni(py) (2) and concomitant production of a stoichiometric equivalent of KCl and pyridine. Slow addition of mesityl- or 1-adamantylazide in benzene to 2 afforded the oxidized Ni complexes (AdFL)Ni(NMes) (3) and (AdFL)Ni(NAd) (4), respectively. Both 3 and 4 were characterized by multinuclear NMR, EPR, magnetometry, single-crystal X-ray crystallography, theoretical calculations, and X-ray absorption spectroscopies to provide a detailed electronic structure picture of the nitrenoid adducts. X-ray absorption near edge spectroscopy (XANES) on the Ni reveals higher energy Ni 1s → 3d transitions (3: 8333.2 eV; 4: 8333.4 eV) than NiI or unambiguous NiII analogues. N K-edge X-ray absorption spectroscopy performed on 3 and 4 reveals a common low-energy absorption present only for 3 and 4 (395.4 eV) that was assigned via TDDFT as an N 1s promotion into a predominantly N-localized, singly occupied orbital, akin to metal-supported iminyl complexes reported for iron. On the continuum of imido (i.e., NR2−) to iminyl (i.e., 2NR−) formulations, the complexes are best described as NiII-bound iminyl species given the N K-edge and TDDFT results. Given the open-shell configuration (S = 1/2) of the iminyl adducts, we then examined their propensity to undergo nitrenoid-group transfer to organic substrates. The adamantyl complex 4 readily consumes 1,4-cyclohexadiene (CHD) via H-atom abstraction to afford the amide (AdFL)Ni(NHAd) (5), whereas no reaction was observed upon treatment of the mesityl variant 3 with excess amount of CHD over 3 hours. Toluene can be functionalized by 4 at room temperature, exclusively affording the N-1-adamantyl-benzylidene (6). Slow addition of the organoazide substrate (4-azidobutyl)benzene (7) with 2 exclusively forms 4-phenylbutanenitrile (8) as opposed to an intramolecular cyclized pyrrolidine, resulting from facile β-H elimination outcompeting H-atom abstraction from the benzylic position, followed by rapid H2-elimination from the intermediate Ni hydride ketimide intermediate.
1. Introduction
Late 3d transition metal complexes bearing metal–ligand multiple bonds (MLMBs) have been intensely scrutinized as potential reagents to deliver the multiply-bonded functionalities to organic substrates.1,2,4,5,8–43 One method to endow the metal–ligand multiply bonded species with the capacity to undergo functional group transfer is to attenuate the metal–ligand bond order. We and others have pursued the synthesis of late transition metal, low-coordinate MLMBs to investigate their resulting group transfer reactivity.1–6,8,9,14–18,35–43 Our group has focused on the generation of highly electrophilic MLMBs by employing weak-field dipyrrin ancillary ligands.37,39,40,42,44 Ferric imido complexes bearing the dipyrrin ligand feature high-spin (S = 5/2) electronic structures,37–40 while the CoIII analogues thermally access open-shell excited states.41,42 The open-shell electronic structures for the Fe37–40 and Co41,42 complexes provide access to reactivity along the MLMB vector, where both inter- and intramolecular amination transfer to C–H bond substrates were observed. In both cases, however, the MLMBs were best formulated as imido functionalities. Oxidation of the FeIII imidos elicits the formation of an open-shell iminyl, where oxidation is borne out at the NR functionality, not the metal center, leading to faster oxidative group transfer than their high spin imido analogues.39,40Formation of later transition metal MLMBs can also diminish MLMB bond order in two ways: (1) higher d-electron counts necessarily populate more M–L antibonding orbitals; (2) progression through the 3d transition metal series should lead to diminished energetic overlap between the transition metal and N valence orbitals, potentially impacting the electronic structure and reactivity of the resultant MLMB.17 Indeed, four Ni imido systems have been thus far reported by Hillhouse1,3–6,10–12 and Warren.2,13 The seminal work by Hillhouse and coworkers first described the synthesis and reactivity of phosphine-supported NiII imidos (Fig. 1a and b),10–12 later establishing both higher valent imido cations (Fig. 1a and b)4 and lower-coordinate, carbene supported imido complexes (Fig. 1c and d).3,5,6 Warren and coworkers reported β-diketiminate supported neutral, nominally NiIII imides (Fig. 1e).2,13 Of note, Hillhouse's two-coordinate, carbene supported NiII imido5 and Warren's NiIII imido complexes2,13 are competent for nitrene transfer into vinylic and benzylic C–H bonds, respectively. Each example represents a highly electrophilic imido complex capable of functional group transfer. Given the promising capability Ni imido species exhibit towards C–H bond functionalization, we were interested in investigating the synthesis of dipyrrin-supported Ni analogues. We report herein the synthesis, characterization, and reactivity of two Ni iminyl complexes bearing aryl and alkyl N-substituents.
 | ||
| Fig. 1 All previously reported Ni imides.1–6 | ||
2. Results and discussion
The synthesis of a modified adamantyl-substituted dipyrrin scaffold proceeded cleanly via substitution of pentafluorobenzaldehyde for 2-(dimethoxymethyl)-1,3,5-trimethylbenzene in the original procedure.37 The fluorinated aryl meso-substituent on the dipyrrin anodically shifts the bound transition metal and provides a useful 19F handle for 19F NMR characterization.45 Metalation of (AdFL)H with Ni followed previously reported protocols for preparing Fe and Co congeners.37,42,44 Lithiation of (AdFL)H is carried out by adding PhLi as a solid to a frozen benzene solution of (AdFL)H.37,44 The addition of resultant (AdFL)Li in THF to a frozen solution of NiCl2(py)2 in THF afforded a dark-red, paramagnetic product. Crystals of the product were obtained by storing a concentrated solution of the material in hexanes at −35 °C. Single-crystal X-ray diffraction analysis revealed the product as the trigonal bipyramidal, bis-pyridine complex (AdFL)NiCl(py)2 (1, Fig. 2a), which exhibits a paramagnetically-shifted 1H NMR spectrum, suggesting a triplet ground state at room temperature (Scheme 1).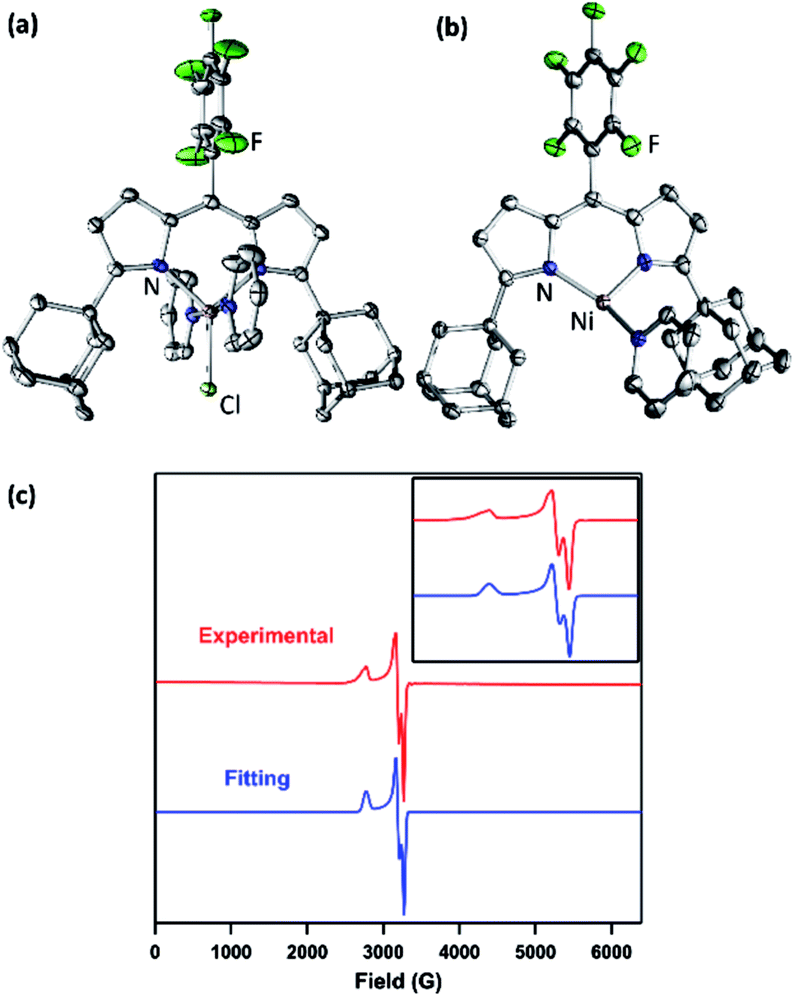 | ||
| Fig. 2 Solid-state molecular structure for (a) (AdFL)NiCl(py)2 (1) and (b) (AdFL)Ni(py) (2) with thermal ellipsoids at 50% probability level. Color scheme: Ni, pink; N, blue; C, gray; Cl, yellowgreen; F, green. H atoms omitted for clarity. (c) Frozen solution EPR spectrum of (AdFL)Ni(py) (2) collected at 77 K in toluene (red). Blue line represents a fit of the data using the program EasySpin.7 Fitting parameters: S = 1/2, g1 = 2.438, g2 = 2.121, g3 = 2.064. | ||
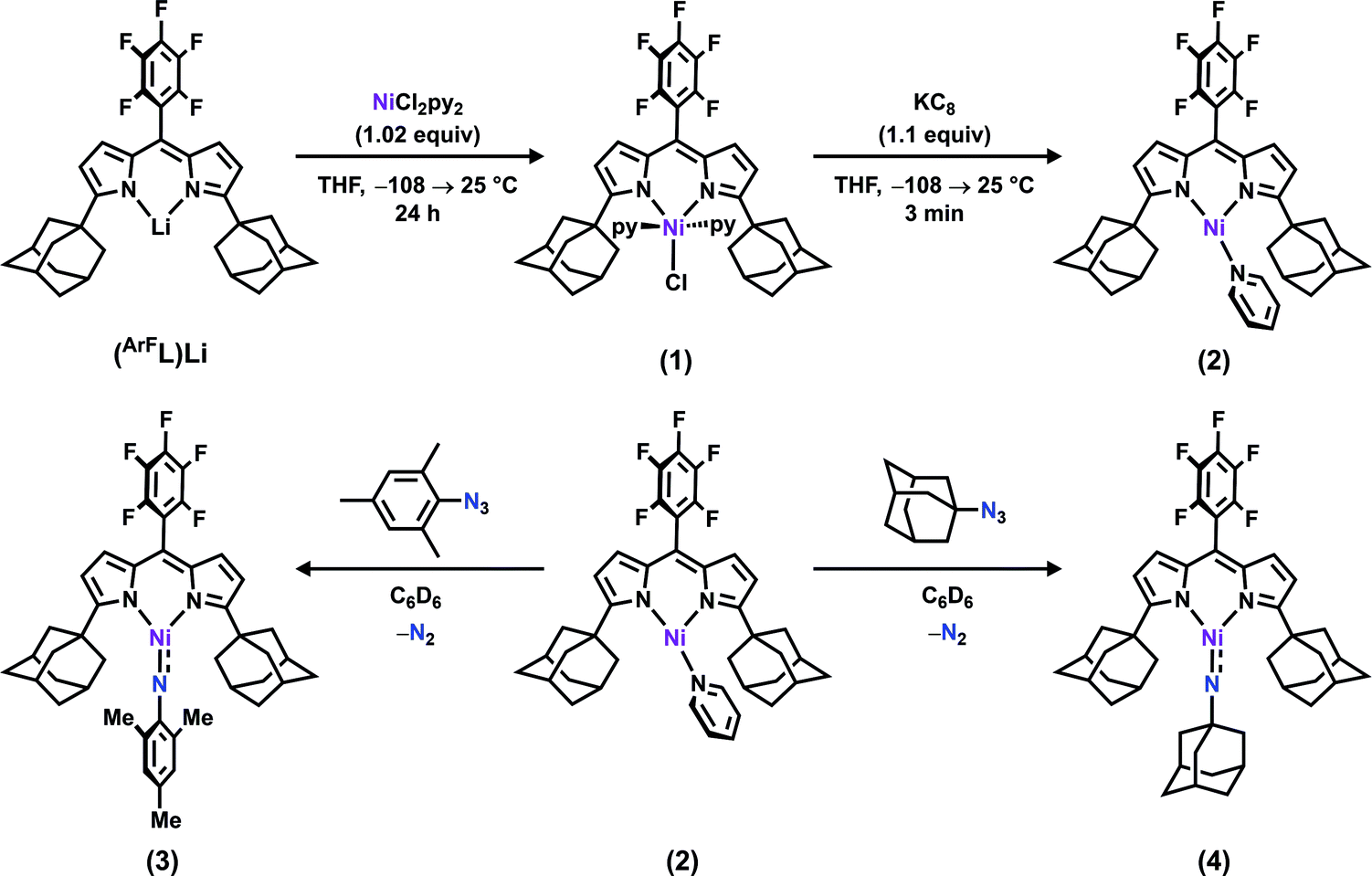 | ||
| Scheme 1 Synthesis of (AdFL)NiCl(py)2 (1), (AdFL)Ni(py) (2), (AdFL)Ni(NMes) (3), and (AdFL)Ni(NAd) (4). | ||
Chemical reduction of 1 with KC8 in thawing THF solution cleanly generated the pyridine adduct (AdFL)Ni(py) (2, Scheme 1) as a dark brown solid. Crystals of 2 suitable for single-crystal X-ray diffraction were obtained by storing a concentrated hexanes solution at −35 °C overnight (Fig. 2b). The solid-state structure of 2 unveils a pyramidally distorted T-shape geometry around the Ni center with an NL–Ni–Npy angle of 146.4(2)°, similar to the previously reported three-coordinate NiI β-diketiminate complex by Warren and coworkers (Fig. 1e).2 The EPR spectrum of 2 collected at 77 K in toluene indicates that the unpaired electron occupies a rhombic environment (g1 = 2.438, g2 = 2.121, g3 = 2.064; Fig. 2c).
2.1 Isolation and structural characterization of terminal nitrenoid complexes
The addition of 1 equiv. of either MesN3 or AdN3 to a benzene solution of 2 resulted in instantaneous color change from dark brown to dark pink accompanied by vigorous effervescence due to N2 release from the azide (Scheme 1). The 19F NMR spectra of the crude reaction mixtures showed total consumption of 2 with clean conversion to new paramagnetic products. Single crystals of the respective reaction products were obtained by storing concentrated hexanes solutions at −35 °C overnight. The solid-state structures of the reaction products (Fig. 3a and d) confirmed N2 extrusion from the organoazides to install terminal NMes and NAd functionalities onto Ni in (AdFL)Ni(NMes) (3) and (AdFL)Ni(NAd) (4), respectively.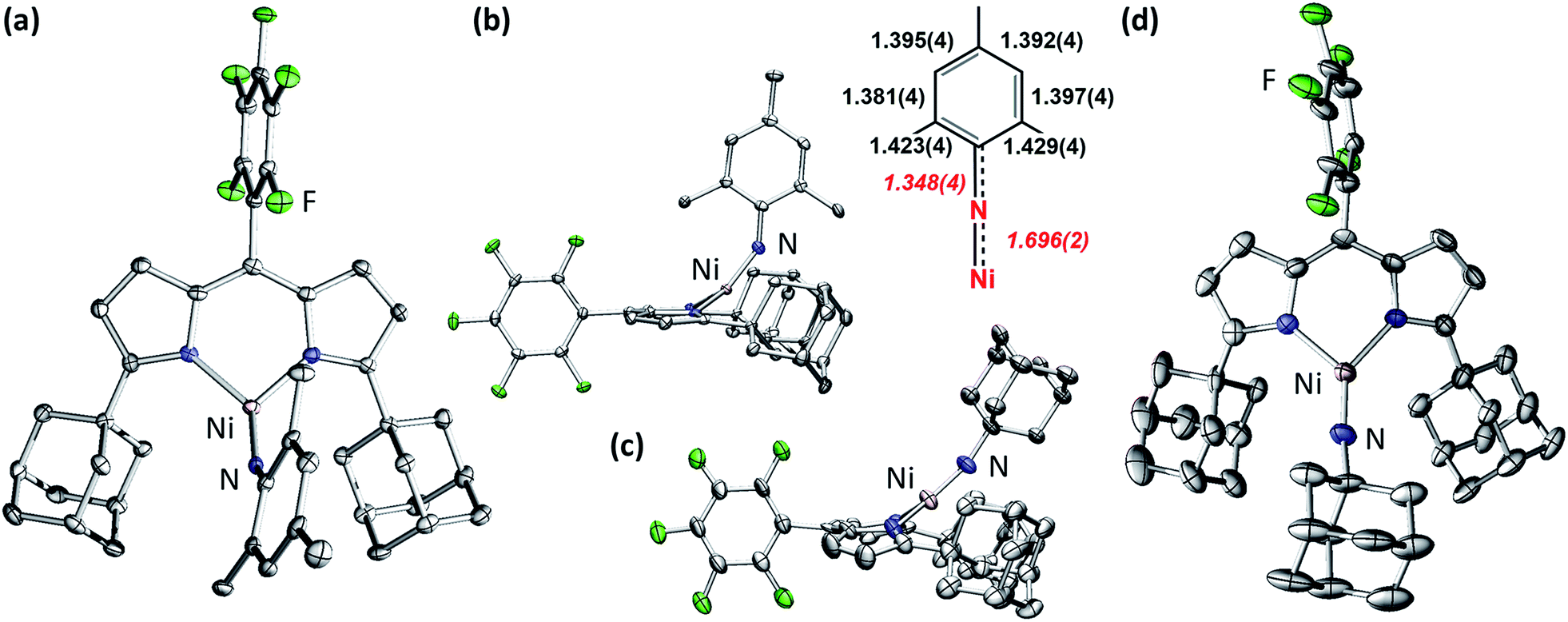 | ||
| Fig. 3 Solid-state molecular structures for the top-view (a) and side-view and aryl moiety bond metrics (b) of (AdFL)Ni(NMes) (3); the side-view (c) and top-view (d) of (AdFL)Ni(NAd) (4) with thermal ellipsoids at 50% probability level. Color scheme: Ni, pink; N, blue; C, gray; F, green. H atoms, solvent molecules, and positional disorder in 4 are omitted for clarity. | ||
The Ni–Nim distance of 1.696(2) Å in 3 is longer than the bis-phosphine supported cationic NiIII aryl imide [(dtbpe)Ni(NAr)]+ (1.674(3) Å)4 but similar to those three coordinated NiII aryl imides supported by the same ligand (1.697(2) → 1.703(4) Å)1,3,4 and bis-N-heterocylic carbene (NHC) ligand ((NHC2)Ni(NAr): 1.718(2) → 1.732(4) Å) (Fig. 1 and S7†).6 The imido linkage in 3 strongly deviates from linearity [∠(Ni–Nim–CMes): 146.5(2)°] in contrast to its linear bisphosphine supported analogue [(dtbpe)Ni(NAr)]+ [∠(Ni–Nim–CAr): 178.4(3)°]4 but similar to the bis-NHC system [(NHC2)Ni(NAr), ∠(Ni–Nim–CAr), Ar: 127.3(3)°, Mes; 170.0(2)°, DIPP] (Fig. 1 and S7†).6 The deviation from linearity for the Ni–N–CAr linkages occur most often with late transition metals featuring population of high-energy, antibonding (M–N)π* orbitals. Other factors that can contribute to distortions away from non-linearity include imido-group constraints incorporated into a chelate ring and crystal-packing effects within the crystalline lattice.6
Close inspection of the C–C bond lengths within the mesityl group of 3 (Fig. 3a and b) reveals the elongation of the Cipso–Cortho bonds (1.423(4), 1.429(4) Å), analogous to our previously reported FeIII iminyl species37–40 as well as all the previously reported Ni aryl imido complexes (Fig. S7†).1,3–6 The Nimido–CMes distance in 3 (1.348(4) Å) is in the range of a partial C–N double bond, indicating delocalization of electron density from Ni–Nimido π system to the mesityl aryl moiety.17
The Ni–Nim distance of 1.642(7) Å in 4 is consistent with reported three-coordinate NiIII alkyl imide supported by β-diketiminate ligand (1.662(2) Å)2 and Ni alkyl imides [(dtbpe)Ni(NAd)]n (n: +1, 1.673(2); n: 0, 1.657 (5) Å);3,4 and comparatively shorter than the Ni–aryl imido complexes (Fig. 1 and S7†). The shortened Ni–Nim bond is consistent across all ancillary ligand platforms, potentially due to the lack of π-delocalization into the imido aryl moiety. The alkyl imido linkage in 4 is more linear [∠(Ni–Nim–CAd): 164.8(17)°] compared to 3, and similar to all three reported Ni alkyl imides [∠(Ni–Nim–CAd): 163.0(2) → 165.2(4)°], regardless of ancillary ligand or oxidation state.2–4
2.2 Spectroscopic characterization of terminal nitrenoid complexes 3 and 4
The EPR spectrum of 3 (toluene, 77 K, Fig. 4a) displays two sets of rhombic, S = 1/2 signals (g11 = 2.238, g12 = 2.106, g13 = 1.940; g21 = 2.302, g22 = 2.128, g23 = 1.962), possibly due to two conformations of the mesityl group related by rotation, forming π-bonds with different metal based d-orbitals.1,3,12 The solid-state magnetometry measurement (Fig. 4c) confirmed that 3 remains low-spin (S = 1/2) up to 300 K. The EPR spectrum of 4 (toluene, 77 K, Fig. 4b) reveals a single rhombic absorption, supporting an S = 1/2 formulation (g1 = 2.185, g2 = 2.063, g3 = 1.924). The signal at g2 = 2.063 is split into a triplet with a hyperfine splitting constant of A(14N, I = 1) = 21.3 G. Similar spectroscopic features were observed in the NiIII β-diketiminate imides with a hyperfine splitting constant of A(14N, I = 1) = 22 G.2 The S = 1/2 low-spin ground states for 4 were further verified by SQUID magnetometry (Fig. S5†) up to 300 K, in contrast to the NiIII imide [(dtbpe)Ni(NAr)]+, which exhibits spin transition to high-spin S = 3/2 excited state.4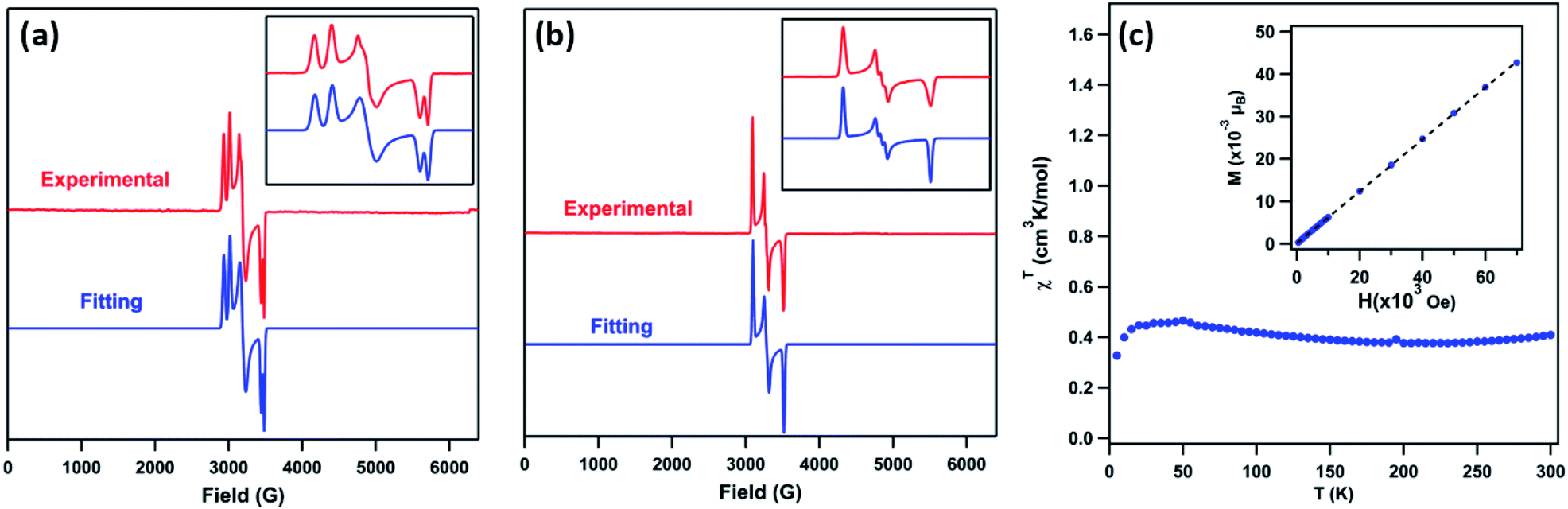 | ||
Fig. 4 (a) Frozen solution EPR spectrum of (AdFL)Ni(NMes) (3) collected at 77 K: S1 = 1/2, g11 = 2.238, g12 = 2.106, g13 = 1.940; S2 = 1/2, g21 = 2.302, g22 = 2.128, g23 = 1.962, relative weight = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (red); blue line represents a fit of the data using the program EasySpin;7 (inset) enlarged spectrum showing hyperfine splitting attributable to two mesityl ring orientations. (b) Frozen solution EPR spectrum of (AdFL)Ni(NAd) (4) collected at 77 K: S = 1/2, g1 = 2.185, g2 = 2.063, g3 = 1.924 (red); blue line represents a fit of the data using the program EasySpin;7,8 (inset) enlarged spectrum showing hyperfine splitting, A = 21.3 G (14N, I = 1).7,9 (c) Variable-temperature susceptibility data for of 3 collected at 1.0 T, with χMT = 0.41 cm3 K mol−1 at 295 K; (inset) solid-state magnetometry data for 3 of M vs. H at 100 K, showing the absence of ferromagnetic impurities. :1 (red); blue line represents a fit of the data using the program EasySpin;7 (inset) enlarged spectrum showing hyperfine splitting attributable to two mesityl ring orientations. (b) Frozen solution EPR spectrum of (AdFL)Ni(NAd) (4) collected at 77 K: S = 1/2, g1 = 2.185, g2 = 2.063, g3 = 1.924 (red); blue line represents a fit of the data using the program EasySpin;7,8 (inset) enlarged spectrum showing hyperfine splitting, A = 21.3 G (14N, I = 1).7,9 (c) Variable-temperature susceptibility data for of 3 collected at 1.0 T, with χMT = 0.41 cm3 K mol−1 at 295 K; (inset) solid-state magnetometry data for 3 of M vs. H at 100 K, showing the absence of ferromagnetic impurities. | ||
2.3 Redox stability of complex 3
The previously reported Ni imido complexes of the type [(dtbpe)Ni(NAr)]+/0 were accessible in both nominally NiII and NiIII oxidation levels (Fig. 1a and b).4 Cyclic voltammetry (CV) was performed to assess the redox stability of the dipyrrin aryl imido analogue 3 (Fig. 5). The CV examination of 3 in THF shows a quasi-reversible reduction event at −1.58 V versus [Cp2Fe]+/0, cathodically shifted from the same formally NiII/NiIII couple of the related bis-phosphine supported Ni imide (E1/2 = −0.76 V, vs. [Cp2Fe]+/0, THF).4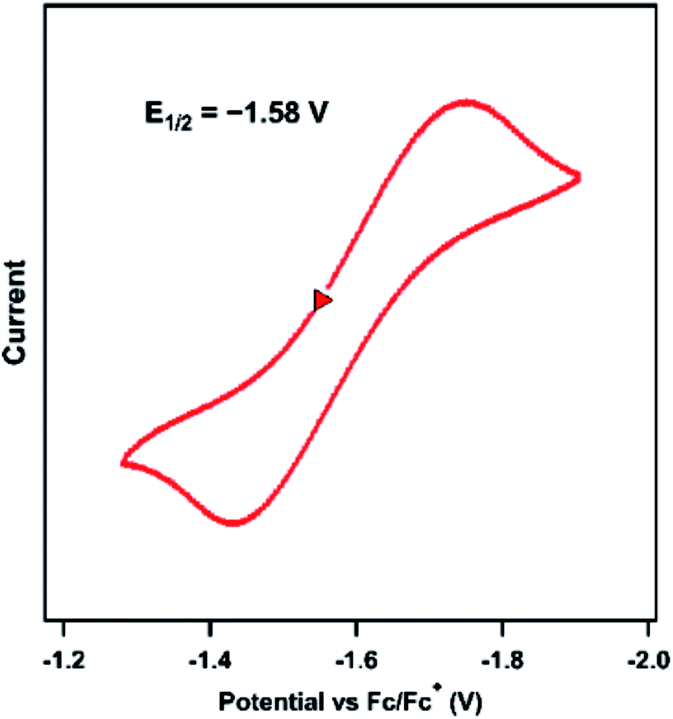 | ||
| Fig. 5 Cyclic voltammogram of 3 obtained in THF at 25 °C, with 0.1 M (nBu4N)(PF6); 100 mV s−1; referenced to [Cp2Fe]+/0 couple; OCP = −1.25 V; ΔE = 310 mV. | ||
The 0.8 V cathodic shift comparing [(dtbpe)Ni(NAr)]+versus neutral 3 likely largely reflects the change in molecular charge between the two species as opposed to effects originating with ligand field strength. Chemical reduction of 3 was attempted using decamethylcobaltacene 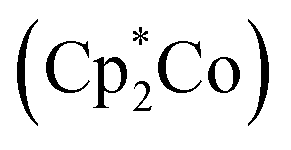 or KC8 as a reductant in frozen-thawing THF.46 In both cases, unfortunately, only inseparable mixtures of decomposition of 3 as ascertained by analysis of the crude reaction mixture by 19F NMR spectroscopy were obtained. Similar results were obtained when similar chemical reduction was attempted with alkyl imide 4.
or KC8 as a reductant in frozen-thawing THF.46 In both cases, unfortunately, only inseparable mixtures of decomposition of 3 as ascertained by analysis of the crude reaction mixture by 19F NMR spectroscopy were obtained. Similar results were obtained when similar chemical reduction was attempted with alkyl imide 4.
2.4 Ni and N K-edge X-ray absorption near edge spectroscopies of complexes 3 and 4
Given the challenges associated with metal oxidation assignment based solely on structural metrics, we investigated the suite of Ni complexes reported herein by multi-edge X-ray absorption spectroscopy (XAS). Formally NiI2 and the formally NiII (AdFL)Ni(NHAd) (5) (vide infra) were chosen as oxidation state references due to their similar coordination environments to those of complexes 3 and 4. The Ni K-edge (Fig. 6a) spectra reveal pre-edge absorption features for each of the complexes 2–5, conventionally assigned to Ni 1s → 3d excitations. The maxima for the pre-edge features increase in energy across the series from NiI2 (8332.0 eV), to formally NiII5 (8332.8 eV), to the terminal MLMB complexes 3 (8333.0 eV) and 4 (8333.2 eV). The pre-edge maximum shifts 0.8 eV on going from NiI2 to NiII5; while the terminal imidos reveal smaller shifts in pre-edge maxima from 5 (3, 0.2 eV; 4, 0.4 eV). Comparing the aryl and alkyl imides (3, 4), the pre-edge absorption feature of the alkyl species is shifted to higher energy relative to that of the aryl congener, likely resulting from conjugation of the aryl moiety in 3 to the singly occupied acceptor molecular orbital. A similar observation was made comparing aryl- and alkyl-substituted iminyl complexes on FeIII.40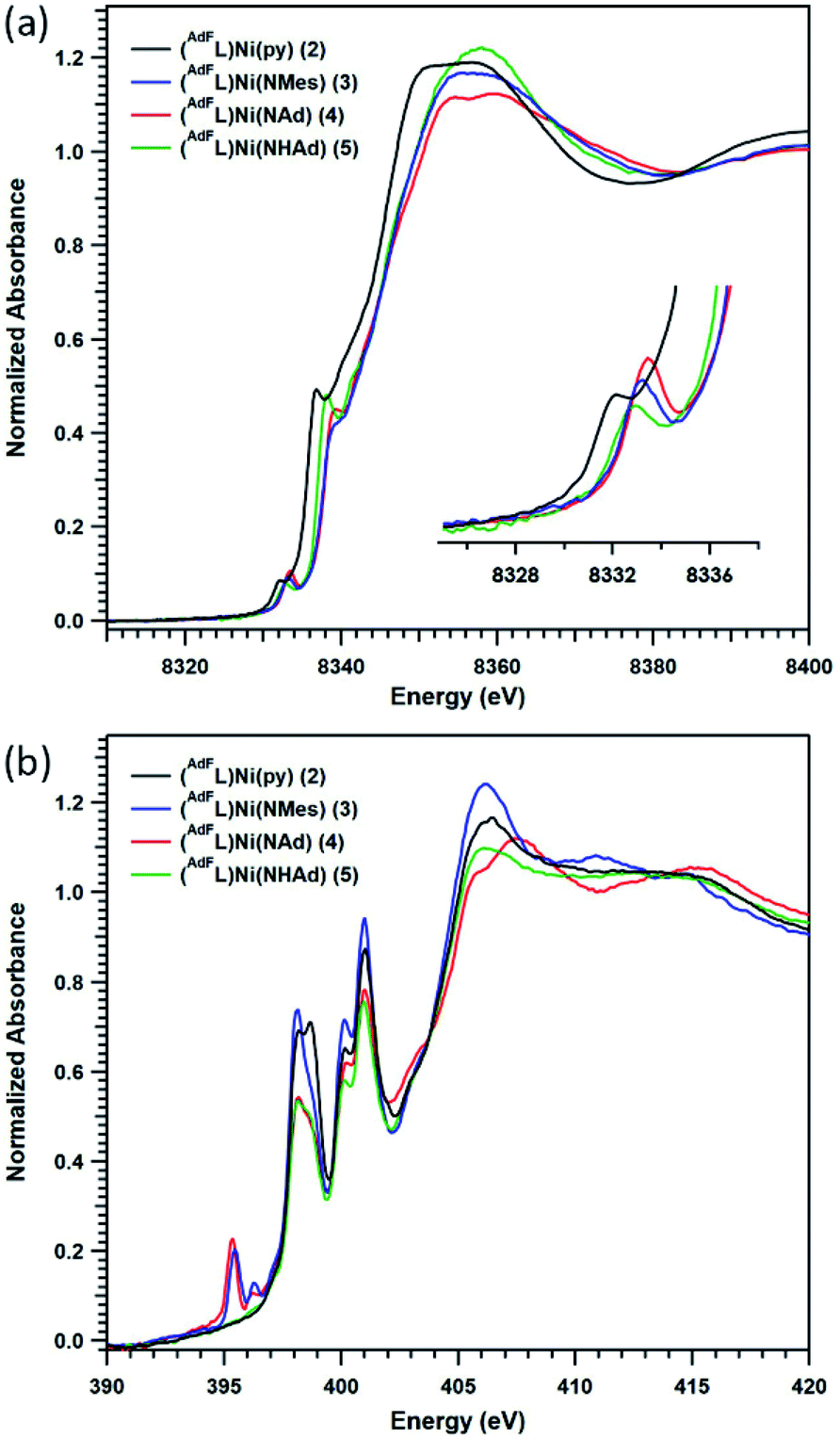 | ||
| Fig. 6 X-ray absorption spectroscopy: (a) Ni K-edge and (b) N K-edge absorption spectra of (AdFL)Ni(py) (2) (black), (AdFL)Ni(NMes) (3) (blue), (AdFL)Ni(NAd) (4) (red), and (AdFL)Ni(NHAd) (5) (green). | ||
To gain further insight into the redox states in the Ni/N pairs in 3 and 4, the nitrogen valence was probed directly using nitrogen K-edge XAS. As seen in Fig. 6b, pre-edge features at 398.0 and 400.9 eV are observed for all the complexes surveyed (2–5) that can be assigned as transitions into high-lying antibonding molecular orbitals of N 2p parentage.40,47 Two low-lying pre-edge absorptions are present for both 3 and 4 at 395.4 eV, accompanied by a smaller side-band at 396.3 eV. These peak energies are consistent with those encountered for the redox-active N-donor ligands in Fe-iminyl (394.5 and 394.8 eV),40 Ni–aminyl (397.0 eV),47 and Cu–nitrene48 (doublet with peaks at 395.3 and 395.9 eV) complexes. This tentative assignment of these transitions as N 1s into partially occupied, N-localized orbitals present in 3 and 4, respectively is further supported by time-dependent density functional theory (TDDFT) calculations (vide infra). However, quantitative covalency analysis was precluded by sensitivity of these samples to rapid photodamage that principally affected the low-energy pre-edge peaks of interest (Fig. S13†).
2.5 Theoretical analysis for the electronic structures of complexes 3 and 4
DFT calculations were carried out to probe the electronic structures of 3 and 4. Calculations were carried out on the experimental (crystallographic) structures using the BP8649,50 generalized gradient approximation (GGA) functional as well as the B3LYP51,52 hybrid density functional. The broken symmetry (BS)53–55 method was applied in both cases to ascertain whether antiferromagnetic coupling between a high-spin d8 NiII and a ligand radical (i.e., imido oxidized to an iminyl) could plausibly be invoked to yield the experimentally observed S = 1/2 ground states. No BS(2,1) solution was obtained for either species using the BP86 functional. Analogous calculations using B3LYP gave broken symmetry solutions with corresponding orbital overlap values near unity (3 = 0.68, 4 = 0.89) and whose total energies were effectively identical to those obtained using a standard unrestricted DFT approach.
To afford some justification for selecting between BP86 and B3LYP results, TDDFT calculations47,56,57 to model the N K-edge XAS obtained for 3 and 4 were carried out starting from either unrestricted single point solution. Spectra calculated using BP86 gave insufficient resolved peak structure to reliably correlate to the experimental data (Fig. S14†), whereas B3LYP-calculated spectra adequately reproduced pre-edge features, with a linear fit of experimental to calculated data giving R2 = 0.99 (Fig. 7). Agreement between calculated and experimental spectra is quite good in the case of 3, but less so in the case of 4.
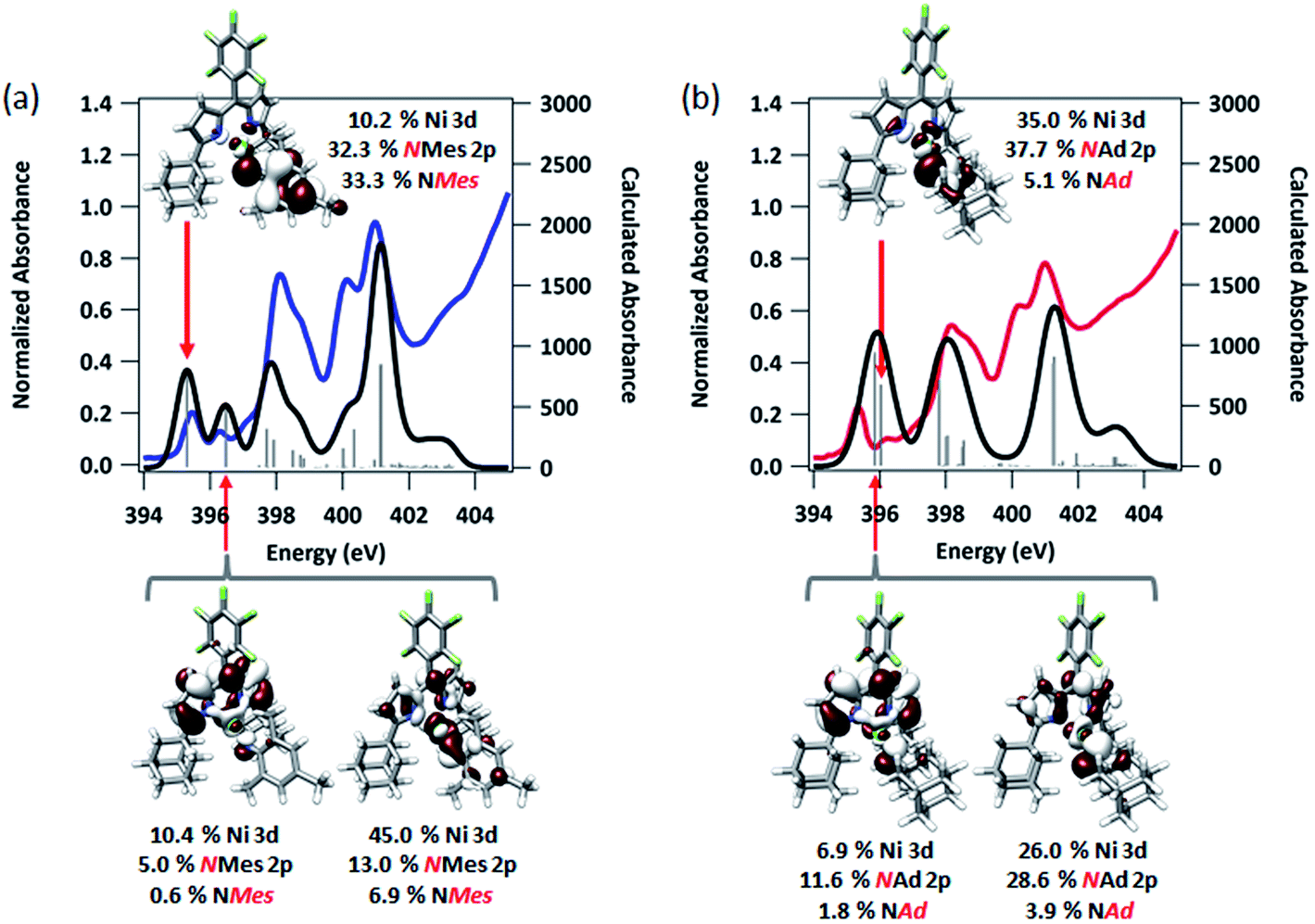 | ||
| Fig. 7 Experimental and TDDFT-calculated N K-edge XAS pre-edge regions for (a) (AdFL)Ni(NMes) (3), and (b) (AdFL)Ni(NAd) (4). Calculations were initiated from spin-unrestricted B3LYP single points using the CP(PPP) basis set on Ni and the ZORA-def2-TZVP(-f) basis set on all other atoms. Energies for the calculated spectra were adjusted using a linear correction presented as Fig. S15.† Unrestricted Kohn–Sham orbitals corresponding to the acceptor MOs involved the excitation giving the ca. 395 eV pre-edge peak are plotted at an isovalue of 0.03 au. | ||
The first peak in the N K-edge XAS of 3 is predicted as an excitation from N 1s to an MO dominated by iminyl character, because the MO is less than 50% Ni in character.58 This MO is delocalized over the NMes fragment and is comprised of 32.3% N 2p, 33.3% Mes arene π*, and 10.2% Ni 3d. The weaker, second pre-edge peak is assigned as an excitation involving a near equal mix of two acceptor MOs featuring large (10–20%) contributions from the orthogonal Nim 2p orbitals as well as dipyrrin π* contributions. In the case of 4, qualitatively similar excitations are involved, although the ordering is reversed from 3: the first excitation features contributions from 2 MOs, while the second excitation is analogous to the first excitation of 3. The estimated error in peak energy prediction from our prior N K-edge TDDFT study is on the order of 0.5 eV, so this discrepancy is not unexpected. Regardless, the pre-edge of 4 also features excitations to an MO with a large degree of Nim N 2p vacancy. Much less delocalization is possible in the imido fragment of 4, consequently this acceptor MO features 42.8% total contribution from NAd (37.7% N 2p), and 35.0% Ni 3d.
Quasi-restricted orbitals (QROs)59 were generated from the unrestricted B3LYP solutions to facilitate interpretation of the ground-state electronic structures of 3 and 4 (Fig. 8). The picture that emerges for both is that the unpaired electron resides in a MO of principally Ni 3d and Nim character reflecting a π* interaction. For 3, this MO comprises 58.5% Ni 3d and 24.3% NMes N 2p character. For 4, corresponding parentages are 33.0% Ni 3d and 46% NAd N 2p. The ca. 2-fold larger N 2p contribution to the SOMO of 4 is consistent with the resolved 14N HFC in its EPR spectrum—such fine structure is not resolved in the spectrum of 3. Each complex has one remaining unoccupied orbital featuring Ni 3d character, reflecting a π* interaction with Nim 2p. This is the LUMO for 3, which comprises 37.7% Ni 3d and 20.9% NMes N 2p. For 4, this is LUMO+1, which features 47.3% Ni 3d and 27.7% NAd N 2p. The ground state electronic configuration for 4 appears more consistent with a formal iminyl assignment, while the larger Ni parentage of the SOMO (58.5% Ni 3d) for 3 suggests 3 presents more imido-like character. Overall, the highly covalent Ni–Nim interactions can be viewed as total donation of a single electron from the Nim 2p orbitals involved in Ni–N bonding, giving rise to the iminyl character observed.
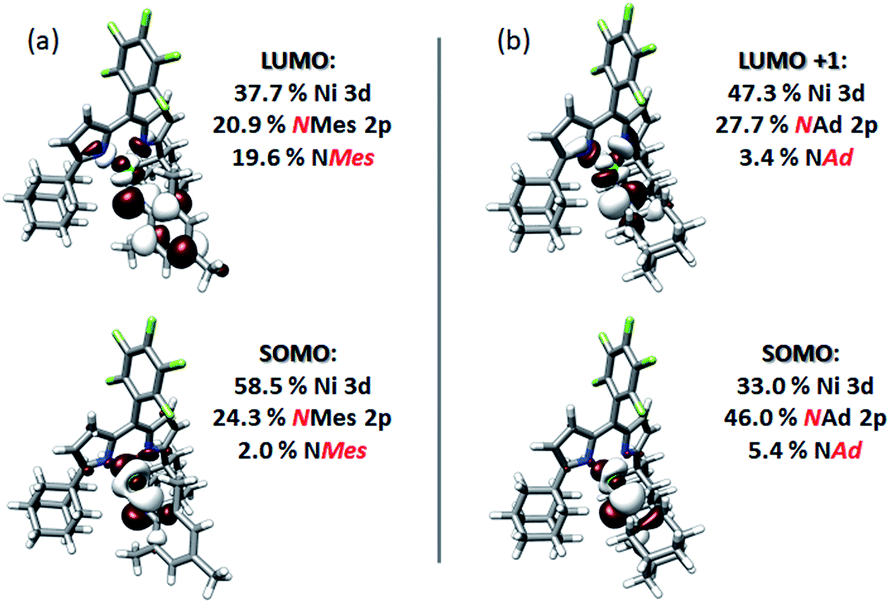 | ||
| Fig. 8 Frontier quasi-restricted orbitals (QROs) calculated for (a) (AdFL)Ni(NMes) (3), and (b) (AdFL)Ni(NAd) (4). QROs were generated from spin-unrestricted B3LYP orbitals produced using the CP(PPP) basis set on Ni and the ZORA-def2-TZVP(-f) basis set on all other atoms. Orbitals are plotted at an isovalue of 0.03 au. | ||
2.6 Reactivity of complexes 3 and 4
With the electronic structure of 3 and 4 described in detail, we next sought to explore their potency toward C–H amination and inspect the effects of substituents on the Nim in group transfer reactivity. Exposure of 4 to 0.5 equiv. of 1,4-cyclohexadiene (CHD) at room temperature resulted in instantaneous conversion into a new paramagnetic species as evidenced by the paramagnetically-shifted 1H and 19F NMR spectra (Scheme 2). Single crystals obtained from storing a concentrated hexanes solution of this reaction product at −35 °C overnight permitted structural determination by X-ray diffraction analysis. The product from reacting 4 with CHD is the corresponding amido species (AdFL)Ni(NHAd) (5, Fig. 9). The amide hydrogen atom was located in the difference map. Similar reactivity was observed in NiIII imides supported by β-diketiminate reported by Warren and coworkers.2,13 Amide 5 was not stable at elevated temperature as complete decomposition of 5 into free ligand was observed at 40 °C in 6 hours. Interestingly, treatment of 3 with 100 equiv. of CHD resulted in no reaction over the course of 3 hours at room temperature, whereas decomposition was observed above 60 °C. A similar reactivity discrepancy was also observed in our previously reported FeIII adamantyl and mesityl imido species, and could be explained by the lower radical density on Nim in 3 as well as steric-shielding of the electro-active orbital by the mesityl ortho-methyl units.39,40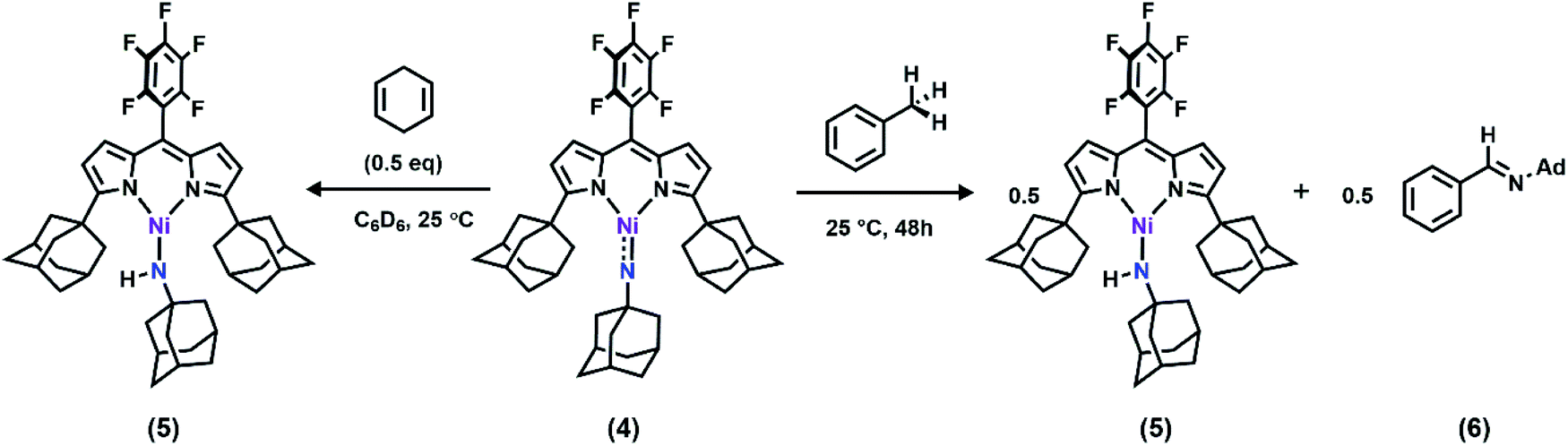 | ||
| Scheme 2 (Left) Reaction between 4 and 0.5 equiv. of CHD at 25 °C, quantitatively generating 5via facile H-atom abstraction; (right) reaction between 4 and toluene as solvent at 25 °C, generating 5 and 6via C–H functionalization followed by β-hydride elimination. | ||
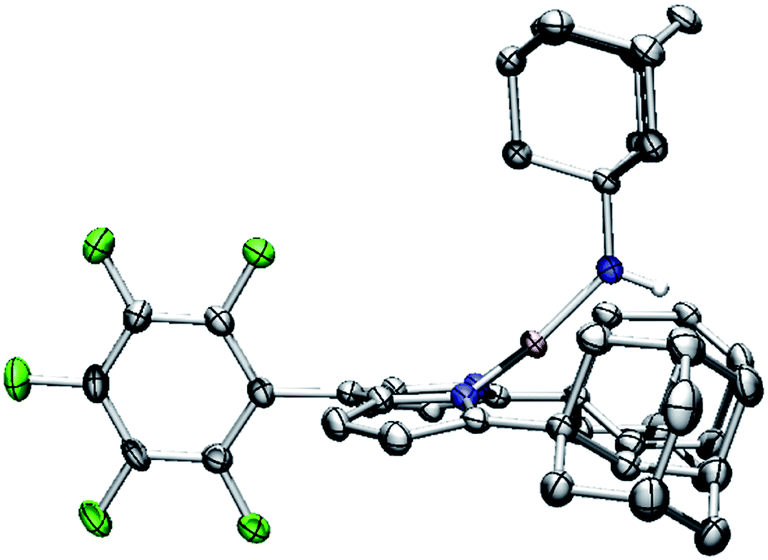 | ||
| Fig. 9 Molecular structure of (AdFL)Ni(NHAd) (5) with the ellipsoid probabilities set at 50%. C, gray; N, blue; H, white; Ni, pink; F green. | ||
Dissolving 4 in toluene resulted in slow conversion into 5 along with decomposition into ligand over the course of two days. The amount of 5 generated is quantified by 19F NMR spectrum to be 46% against an internal standard (trifluorotoluene), and the organic product is identified to be N-1-adamantyl-benzilidene (6) as evidenced by 1H NMR spectrum of the quenched reaction mixture (Scheme 2). The concentration of 4 in toluene was monitored by 19F NMR spectroscopy and can be fitted as a first-order decay (Fig. 10). An intermolecular KIE of 40 was measured over 80 days when d8-toluene was used as the solvent. However, due to the slow rate of the reaction, decomposition of 4 can also contribute to the large KIE value. Based on the C–H amination reaction mediated by previously reported NiIII β-diketiminate imides, we hypothesized that 4 can outcompete 5 (in situ) at capturing tolyl radical, giving rise to a secondary amido intermediate. This intermediate would then undergo β-hydride elimination and release the imine product. A similar mechanism was proposed by Warren et al. as a rationale towards their crystallographically characterized rearranged NiII product.2,13 An alternative mechanism would involve H-atom abstraction of benzyladamantylamine with a second equivalent of 4, generating both the imine product observed and 5 as a byproduct.
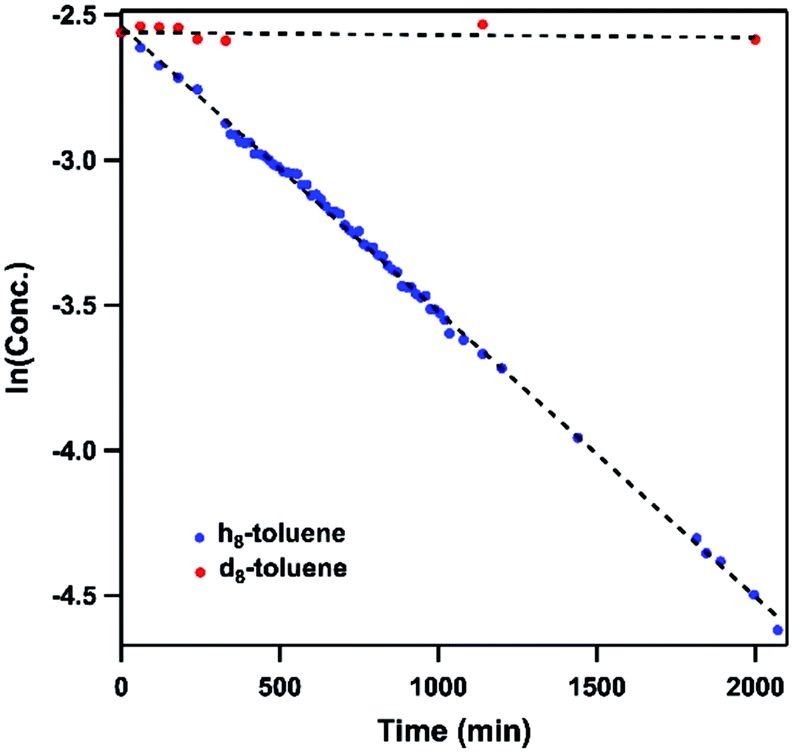 | ||
| Fig. 10 Concentration profile of 4 during reaction between (h8/d8-)toluene and 4 by 19F NMR spectroscopy. Trifluorotoluene is used as internal standard. | ||
In order to facilitate the radical recombination with the amido species formed in situ, the activated C–H bond was installed onto the azide. Addition of 1 equiv. of (4-azidobutyl)benzene (7)22 to a C6D6 solution of 2 resulted in instantaneous color change from dark brown to dark pink along with vigorous effervescence. The bubbling ceased after 10 min along with the solution reversion to a dark brown color, and 19F NMR spectrum revealed full regeneration of 2. However, rather than the anticipated cyclized pyrrolidine product,22,31,411H NMR analysis revealed the major organic product to be 4-phenylbutanenitrile (8, Scheme 3). To test the possibility of catalytically converting azide 7 to nitrile 8, 3–6 equivalents of azide 7 were added slowly to a C6D6 solution of 2. Full decomposition of catalyst 2 was observed when greater than five equivalents of the azide substrate 7 were used, potentially due to the high concentration of product nitrile 8. With 20 mol% catalyst loading of 2, azide 7 is catalytically converted to 4-phenylbutanenitrile 8, providing a moderate isolated yield of 78%. We propose the product is formed by iminyl generation following N2 extrusion from azide 7; β-H elimination (originating from the α-methylene unit) outcompetes intramolecular H-atom abstraction from the benzylic position to generate a Ni(hydride)(ketimide) intermediate. A rapid H2-elimination via deprotonation of the ketimide Ni(hydride) furnishes the final nitrile 8. Attempts to slow down this pathway by deutero-substitution at the gem position were ineffective.
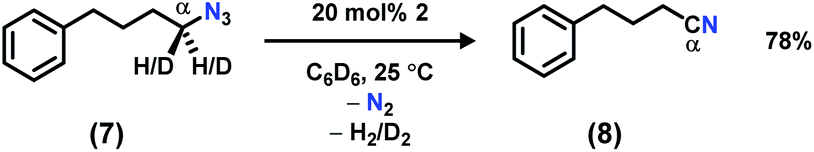 | ||
| Scheme 3 Catalytic conversion of 7 to 8 at 25 °C using 20 mol% of 2. | ||
3. Conclusions
Both NiII aryl and alkyl nitrenoid adducts were prepared and characterized. A significant amount of spin density is delocalized on to the nitrenoid fragment as determined by SC-XRD, EPR spectroscopy, XAS, and quantum calculations. On the continuum between imido (i.e., NiIII(NR)) and iminyl (i.e., NiII(2NR)) ligation, the alkyl nitrenoid 4 exhibits features consistent with an iminyl formulation whereas the singly occupied molecular orbital on 3 features more Ni character reminiscent of imido formulations. The nitrenoid bond distortions reported herein are similar to those observed in previously reported NiIII(NR), suggesting that the iminyl electronic structure proposed here may be applicable to those compounds as well.2,4,13 Indeed, the short Ni–NR bonds are commonly invoked for substantive metal–ligand multiple bond formation. However, the presence of low-energy absorption features in the N K-edge XAS data and corresponding assignments from TDDFT strongly evidence N 2p-localized vacancies. The C–H bond functionalization reactivities of these Ni(NR) species were closely examined. The Ni(NR) aryl congener is shown to be much less reactive than the alkyl variant, potentially owing to the greater spin concentration on Ni in the spin-bearing orbital. In the C–H bond amination reactions of Ni(NAd) with toluene, the corresponding imine was determined to be the major organic product, presumably due to recombination of the tolyl radical with the Ni(NAd) outcompeting that with the generated Ni(NHAd) amide. A subsequent β-hydride elimination would generate the secondary imine product. Future work will examine the efficacy of the (dipyrrin)Ni complexes to perform ring-closing C–H bond amination with substrates that cannot undergo α-hydride migration as observed here.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by a grant from the NIH (GM-115815), the Dreyfus Foundation (Teacher-Scholar Award to T. A. B.), and Harvard University. R. M. C. gratefully acknowledges an NSERC Postdoctoral Fellowship. K. M. L. acknowledges NSF (CHE-1454455) and the Alfred P. Sloan Foundation for support. XAS data were obtained at SSRL, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the Department of Energy's Office of Biological and Environmental Research, and by NIH/NIGMS (including P41GM103393). The authors wish to thank Dennis Nordlund for assistance with soft XAS data collection.Notes and references
- D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2001, 123, 4623–4624 CrossRef CAS PubMed
.
- E. Kogut, H. L. Wiencko, L. Zhang, D. E. Cordeau and T. H. Warren, J. Am. Chem. Soc., 2005, 127, 11248–11249 CrossRef CAS PubMed
- R. Waterman and G. L. Hillhouse, J. Am. Chem. Soc., 2008, 130, 12628–12629 CrossRef CAS
- V. M. Iluc, A. J. M. Miller, J. S. Anderson, M. J. Monreal, M. P. Mehn and G. L. Hillhouse, J. Am. Chem. Soc., 2011, 133, 13055–13063 CrossRef CAS PubMed
- C. A. Laskowski, A. J. M. Miller, G. L. Hillhouse and T. R. Cundari, J. Am. Chem. Soc., 2011, 133, 771–773 CrossRef CAS PubMed
- N. D. Harrold and G. L. Hillhouse, Chem. Sci., 2013, 4, 4011–4015 RSC
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS
- J. Du, L. Wang, M. Xie and L. Deng, Angew. Chem., Int. Ed., 2015, 54, 12640–12644 CrossRef CAS
- D. T. Shay, G. P. A. Yap, L. N. Zakharov, A. L. Rheingold and K. H. Theopold, Angew. Chem., Int. Ed., 2005, 44, 1508–1510 CrossRef CAS
- D. J. Mindiola and G. L. Hillhouse, Chem. Commun., 2002, 1840–1841 RSC
- R. Waterman and G. L. Hillhouse, J. Am. Chem. Soc., 2003, 125, 13350–13351 CrossRef CAS PubMed
- V. M. Iluc and G. L. Hillhouse, J. Am. Chem. Soc., 2010, 132, 15148–15150 CrossRef CAS PubMed
- S. Wiese, J. L. McAfee, D. R. Pahls, C. L. McMullin, T. R. Cundari and T. H. Warren, J. Am. Chem. Soc., 2012, 134, 10114–10121 CrossRef CAS PubMed
- C. T. Saouma and J. C. Peters, Coord. Chem. Rev., 2011, 255, 920–937 CrossRef CAS PubMed
- J. F. Berry, Comments Inorg. Chem., 2009, 30, 28–66 CrossRef CAS
- L. Zhang and L. Deng, Chin. Sci. Bull., 2012, 57, 2352–2360 CrossRef CAS
- A. I. O. Suarez, V. Lyaskovskyy, J. N. H. Reek, J. I. van der Vlugt and B. de Bruin, Angew. Chem., Int. Ed., 2013, 52, 12510–12529 CrossRef CAS
- L. Zhang, Y. Liu and L. Deng, J. Am. Chem. Soc., 2014, 136, 15525–15528 CrossRef CAS PubMed
- W. Liu and J. T. Groves, J. Am. Chem. Soc., 2010, 132, 12847–12849 CrossRef CAS PubMed
- H. Lu, H. Jiang, Y. Hu, L. Wojtas and X. P. Zhang, Chem. Sci., 2011, 2, 2361–2366 RSC
- H. Lu, Y. Hu, H. Jiang, L. Wojtas and X. P. Zhang, Org. Lett., 2012, 14, 5158–5161 CrossRef CAS PubMed
- E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591–595 CrossRef CAS
- E. N. Bess, R. J. DeLuca, D. J. Tindall, M. S. Oderinde, J. L. Roizen, J. Du Bois and M. S. Sigman, J. Am. Chem. Soc., 2014, 136, 5783–5789 CrossRef CAS
- E. T. Hennessy, R. Y. Liu, D. A. Iovan, R. A. Duncan and T. A. Betley, Chem. Sci., 2014, 5, 1526–1532 RSC
- H. Lu, C. Li, H. Jiang, C. L. Lizardi and X. P. Zhang, Angew. Chem., Int. Ed., 2014, 53, 7028–7032 CrossRef CAS PubMed
- W. Zhou, B. O. Patrick and K. M. Smith, Chem. Commun., 2014, 50, 9958–9960 RSC
- M. Goswami, V. Lyaskovskyy, S. R. Domingos, W. J. Buma, S. Woutersen, O. Troeppner, I. Ivanović-Burmazović, H. Lu, X. Cui, X. P. Zhang, E. J. Reijerse, S. DeBeer, M. M. van Schooneveld, F. F. Pfaff, K. Ray and B. de Bruin, J. Am. Chem. Soc., 2015, 137, 5468–5479 CrossRef CAS
- H. Lu, K. Lang, H. Jiang, L. Wojtas and X. P. Zhang, Chem. Sci., 2016, 7, 6934–6939 RSC
- B. Bagh, D. L. J. Broere, V. Sinha, P. F. Kuijpers, N. P. van Leest, B. de Bruin, S. Demeshko, M. A. Siegler and J. I. van der Vlugt, J. Am. Chem. Soc., 2017, 139, 5117–5124 CrossRef CAS PubMed
- D. Fujita, H. Sugimoto, Y. Shiota, Y. Morimoto, K. Yoshizawa and S. Itoh, Chem. Commun., 2017, 53, 4849–4852 RSC
- D. A. Iovan, M. J. T. Wilding, Y. Baek, E. T. Hennessy and T. A. Betley, Angew. Chem., Int. Ed., 2017, 56, 15599–15602 CrossRef CAS PubMed
- P. F. Kuijpers, M. J. Tiekink, W. B. Breukelaar, D. L. J. Broere, N. P. van Leest, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Chem.–Eur. J., 2017, 23, 7945–7952 CrossRef CAS PubMed
- C. K. Prier, R. K. Zhang, A. R. Buller, S. Brinkmann-Chen and F. H. Arnold, Nat. Chem., 2017, 9, 629–634 CrossRef CAS PubMed
- C. Li, K. Lang, H. Lu, Y. Hu, X. Cui, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2018, 57, 16837–16841 CrossRef CAS PubMed
- D. M. Spasyuk, S. H. Carpenter, C. E. Kefalidis, W. E. Piers, M. L. Neidig and L. Maron, Chem. Sci., 2016, 7, 5939–5944 RSC
- P. F. Kuijpers, J. I. van der Vlugt, S. Schneider and B. de Bruin, Chem.–Eur. J., 2017, 23, 13819–13829 CrossRef CAS PubMed
- E. R. King, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 4917–4923 CrossRef CAS PubMed
- D. A. Iovan and T. A. Betley, J. Am. Chem. Soc., 2016, 138, 1983–1993 CrossRef CAS PubMed
- M. J. T. Wilding, D. A. Iovan and T. A. Betley, J. Am. Chem. Soc., 2017, 139, 12043–12049 CrossRef CAS PubMed
- M. J. T. Wilding, D. A. Iovan, A. T. Wrobel, J. T. Lukens, S. N. MacMillan, K. M. Lancaster and T. A. Betley, J. Am. Chem. Soc., 2017, 139, 14757–14766 CrossRef CAS PubMed
- Y. Baek and T. A. Betley, J. Am. Chem. Soc., 2019, 141, 7797–7806 CrossRef CAS PubMed
- E. R. King, G. T. Sazama and T. A. Betley, J. Am. Chem. Soc., 2012, 134, 17858–17861 CrossRef CAS
- J. Cheng, J. Liu, X. Leng, T. Lohmiller, A. Schnegg, E. Bill, S. Ye and L. Deng, Inorg. Chem., 2019, 58, 7634–7644 CrossRef CAS
- E. R. King and T. A. Betley, Inorg. Chem., 2009, 48, 2361–2363 CrossRef CAS
- A. B. Scharf and T. A. Betley, Inorg. Chem., 2011, 50, 6837–6845 CrossRef CAS PubMed
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed
- J. T. Lukens, I. M. DiMucci, T. Kurogi, D. J. Mindiola and K. M. Lancaster, Chem. Sci., 2019, 10, 5044–5055 RSC
- K. M. Carsch, I. M. DiMucci, D. A. Iovan, A. Li, S.-L. Zheng, C. J. Titus, S. J. Lee, K. D. Irwin, D. Nordlund, K. M. Lancaster and T. A. Betley, Science, 2019, 365, 1138 CrossRef CAS
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS
- F. Neese, J. Phys. Chem. Solids, 2004, 65, 781–785 CrossRef CAS
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef CAS
- L. Noodleman and E. R. Davidson, Chem. Phys., 1986, 109, 131–143 CrossRef
- S. DeBeer George and F. Neese, Inorg. Chem., 2010, 49, 1849–1853 CrossRef CAS PubMed
- S. DeBeer George, T. Petrenko and F. Neese, Inorg. Chim. Acta, 2008, 361, 965–972 CrossRef CAS
- I. M. DiMucci, J. T. Lukens, S. Chatterjee, K. M. Carsch, C. J. Titus, S. J. Lee, D. Nordlund, T. A. Betley, S. N. MacMillan and K. M. Lancaster, J. Am. Chem. Soc., 2019, 141, 18508–18520 CrossRef CAS PubMed
- F. Neese, J. Am. Chem. Soc., 2006, 128, 10213–10222 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1953729–1953733. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04879k |
| This journal is © The Royal Society of Chemistry 2020 |