DOI:
10.1039/C9SC04849A
(Edge Article)
Chem. Sci., 2020,
11, 1636-1642
Readily accessible sp3-rich cyclic hydrazine frameworks exploiting nitrogen fluxionality†
Received
25th September 2019
, Accepted 23rd December 2019
First published on 2nd January 2020
Abstract
Increased molecular complexity correlates with improved chances of success in the drug development process. Here, a strategy for the creation of sp3-rich, non-planar heterocyclic scaffolds suitable for drug discovery is described that obviates the need to generate multiple stereogenic centers with independent control. Asymmetric transfer hydrogenation using a tethered Ru-catalyst is used to efficiently produce a range of enantiopure cyclic hydrazine building blocks (up to 99% ee). Iterative C–N functionalization at the two nitrogen atoms of these compounds produces novel hydrazine and hydrazide based chemical libraries. Wide chemical diversification is possible through variation in the hydrazine structure, use of different functionalization chemistries and coupling partners, and controlled engagement of each nitrogen of the hydrazine in turn. Principal Moment of Inertia (PMI) analysis of this small hydrazine library reveals excellent shape diversity and three-dimensionality. NMR and crystallographic studies confirm these frameworks prefer to orient their substituents in three-dimensional space under the control of a single stereogenic center through exploitation of the fluxional behavior of the two nitrogen atoms.
1. Introduction
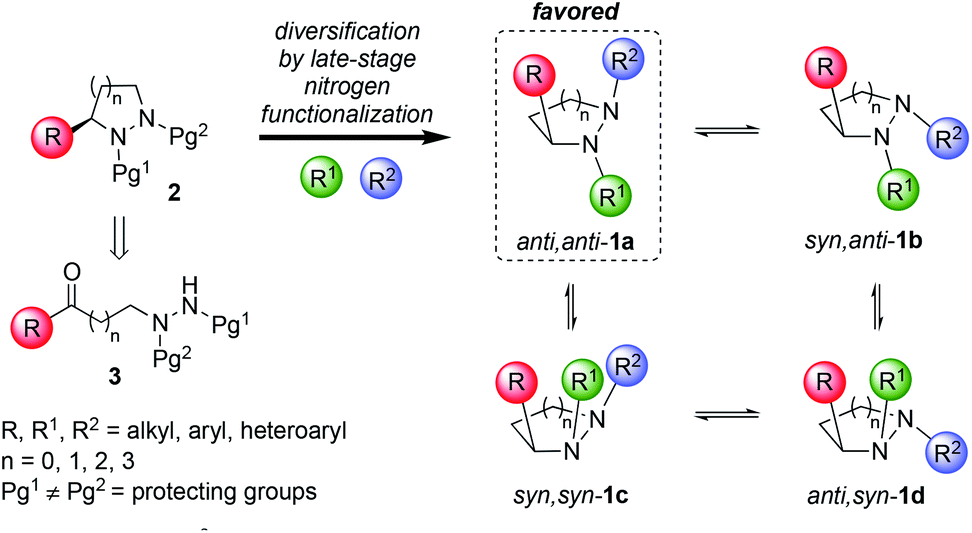
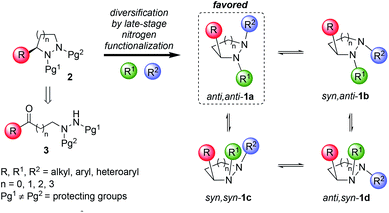
Libraries of small molecules capable of exploring new areas of three-dimensional chemical space are of considerable value in drug discovery. There is growing evidence that molecular complexity, as measured by parameters such as the fraction of saturated carbon atoms (Fsp3) and number of stereogenic centers, correlates with success rates in drug development.1,2 Despite these observations, analysis of current drug-like libraries such as the ChEMBL dataset reveals that they generally lack shape diversity and three-dimensionality.2 Hence, there is a need to create new stereochemically-rich compound libraries for use in drug-discovery programs that overcome these shortcomings. This is a challenging problem because it necessitates moving away from well-established synthetic methods (e.g. sp2–sp2 cross-couplings) toward chemistries that efficiently create non-planar scaffolds.3 In this Edge article, we report a way to create novel chemical libraries with considerable shape diversity without having to generate multiple stereogenic centers with independent control. The idea exploits the fluxional behavior of pyramidal nitrogen atoms within enantiopure cyclic hydrazines, to create new sp3-rich heterocyclic frameworks that display their chirality in an orchestrated way (Scheme 1).
 |
| | Scheme 1 New sp3-rich cyclic hydrazine scaffolds exploiting N-fluxionality. | |
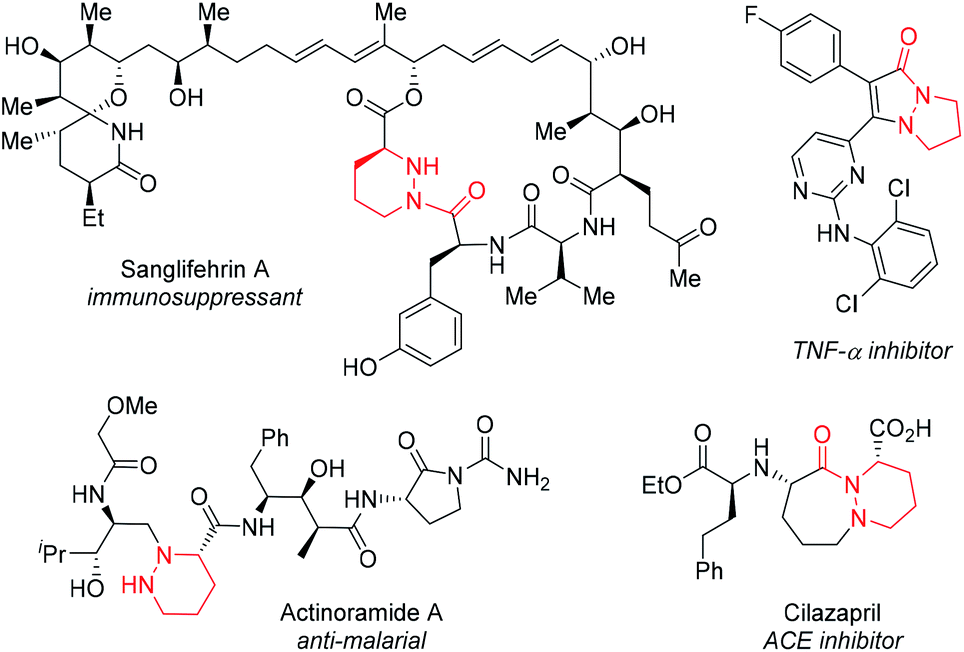
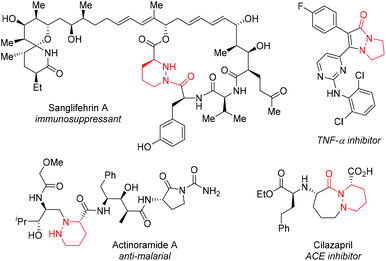
This approach has a number of attractive features. Cyclic hydrazines and hydrazides are well represented in medicinal chemistry.4 They are found in many bioactive compounds including natural products (e.g. actinoramide A,5 sanglifehrin A6), proprietary lead compounds7 and approved drugs (e.g. cilazapril8) (Fig. 1). Chemical libraries of 1 for discovery programs should be readily available by simple, late-stage diversification of 2 by iterative functionalization at each nitrogen atom. The availability of a plethora of C–N functionalization reactions combined with the high nucleophilicity of the hydrazine functional group make this disconnection highly attractive. By exploiting their dynamic behavior, it should be possible to display three substituents (R, R1 and R2) attached to carbon and nitrogen very precisely in three-dimensional space. Of the four conformational isomers 1a–d that arise from pyramidal inversion, 1a is expected to be dominant. Typically, cyclic hydrazines adopt an anti-configuration between the N-substituents to minimize non-bonded interactions between the lone pairs.9 Thus, the formation of 1b and 1c is expected to be disfavored. Furthermore, non-bonding interactions between adjacent syn-substituents should disfavor 1b–d relative to anti,anti-1a. Thus, control over only one stereogenic center is needed to produce predictable, non-planar frameworks. Finally, further fine-tuning of the substituent positioning should be possible by variation of the ring size.
 |
| | Fig. 1 Representative examples of bioactive molecules containing cyclic hydrazine and hydrazide frameworks (highlighted in red). | |
2. Results and discussion
2.1. Asymmetric synthesis of cyclic hydrazine frameworks
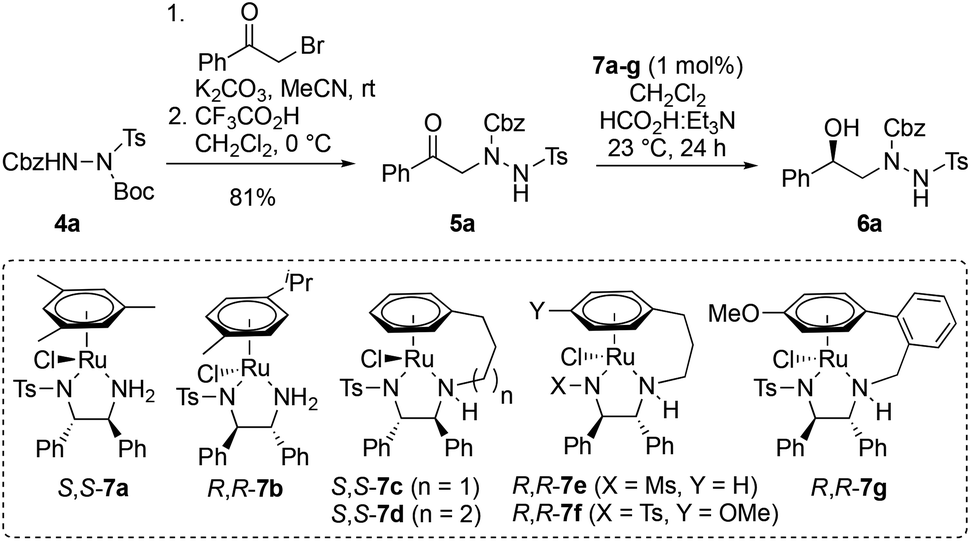
The first task was to identify an efficient, enantioselective route to the hydrazine building blocks, i.e.2 (Scheme 1). Key requirements for such a synthesis were that it: (i) allow the introduction of a variety of medicinally-relevant R substituents with high levels of enantiocontrol; (ii) provide access to different ring sizes; and (iii) yield differentially protected hydrazines 2 suitable for facile introduction of the R1 and R2 groups. While several methods for the asymmetric synthesis of cyclic hydrazines exist,10 none met our requirements. Consequently, a new two-step approach to 2 was devised based upon enantiocontrolled reduction of ketones 3 followed by ring closure. By varying the linker length between the ketone and hydrazine functional groups, the approach offers access to a range of different ring sizes. In choosing a method for the reduction, we were drawn to Ru-catalyzed asymmetric transfer hydrogenation (ATH) which typically delivers high levels of enantioselectivity, requires low catalyst loadings, has diverse functional group compatibility and is operationally simple to perform.11
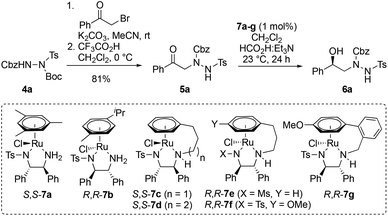
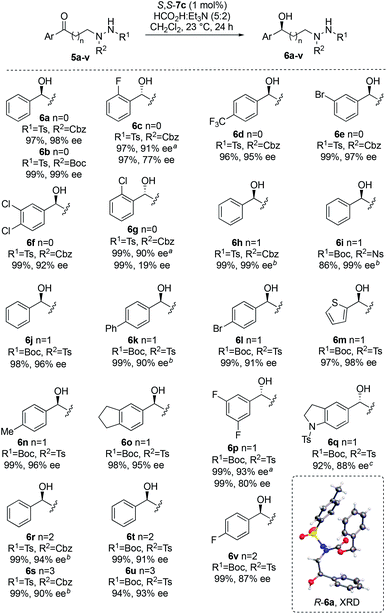
Initially, phenyl ketone 5a was prepared by alkylation of readily-available, triply-protected hydrazine 4a12 with 2-bromoacetophenone, followed by Boc deprotection (Table 1). Where doubly-protected CbzNHNHTs is used in these alkylations, the isomeric products in which the sulfonamide and carbamate groups are transposed can also be made (for full details, see ESI†). Next, ATH reduction of 5a was examined by screening a range of chiral diphenylethylenediamine (DPEN)/Ru(II) catalysts (7a–g) using formic acid and triethylamine (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 azeotrope) as hydrogen source. The majority of these reactions benefited from catalysts that were tethered between the arene and DPEN ligand.13 Variation in the catalyst structure has a greater impact on catalytic activity than enantioselectivity (Table 1), and chlorinated solvents were found to be most effective (for details, see ESI†). Under the optimized conditions, ketone 5a was efficiently converted into alcohol R-6a in 97% yield and 99% ee using 1 mol% of tethered ruthenium catalyst S,S-7c (Table 1, entry 3). The structure and absolute configuration of R-6a were confirmed by X-ray crystallography using the Flack parameter (Fig. 2, box insert).14 The sense of asymmetric induction fits established models,13a indicating that the hydrazine plays no active role in catalyst binding.
:2 azeotrope) as hydrogen source. The majority of these reactions benefited from catalysts that were tethered between the arene and DPEN ligand.13 Variation in the catalyst structure has a greater impact on catalytic activity than enantioselectivity (Table 1), and chlorinated solvents were found to be most effective (for details, see ESI†). Under the optimized conditions, ketone 5a was efficiently converted into alcohol R-6a in 97% yield and 99% ee using 1 mol% of tethered ruthenium catalyst S,S-7c (Table 1, entry 3). The structure and absolute configuration of R-6a were confirmed by X-ray crystallography using the Flack parameter (Fig. 2, box insert).14 The sense of asymmetric induction fits established models,13a indicating that the hydrazine plays no active role in catalyst binding.
Table 1 Optimization of Ru-catalyzed asymmetric transfer hydrogenation of ketone 5a
|
|
| Entry |
Catalyst |
Yielda [%] |
eeb [%] |
|
Isolated yield after column chromatography.
Determined by HPLC analysis using Chiralpak ADH.
Reaction run for 48 h.
Opposite S-enantiomer produced.
|
| 1 |
S,S-7a |
86 |
97 (R) |
| 2 |
R,R-7b |
16c |
91 (S)d |
| 3 |
S,S-7c |
97 |
99 (R) |
| 4 |
S,S-7d |
7 |
97 (R) |
| 5 |
R,R-7e |
97 |
97 (S)d |
| 6 |
R,R-7f |
80 |
99 (S)d |
| 7 |
R,R-7g |
16 |
98 (S)d |
 |
| | Fig. 2 Ru-catalyzed ATH of hydrazine-containing ketones. a Using R,R-7f. b ee determined after cyclization to 8. c Using R,R-7c. | |
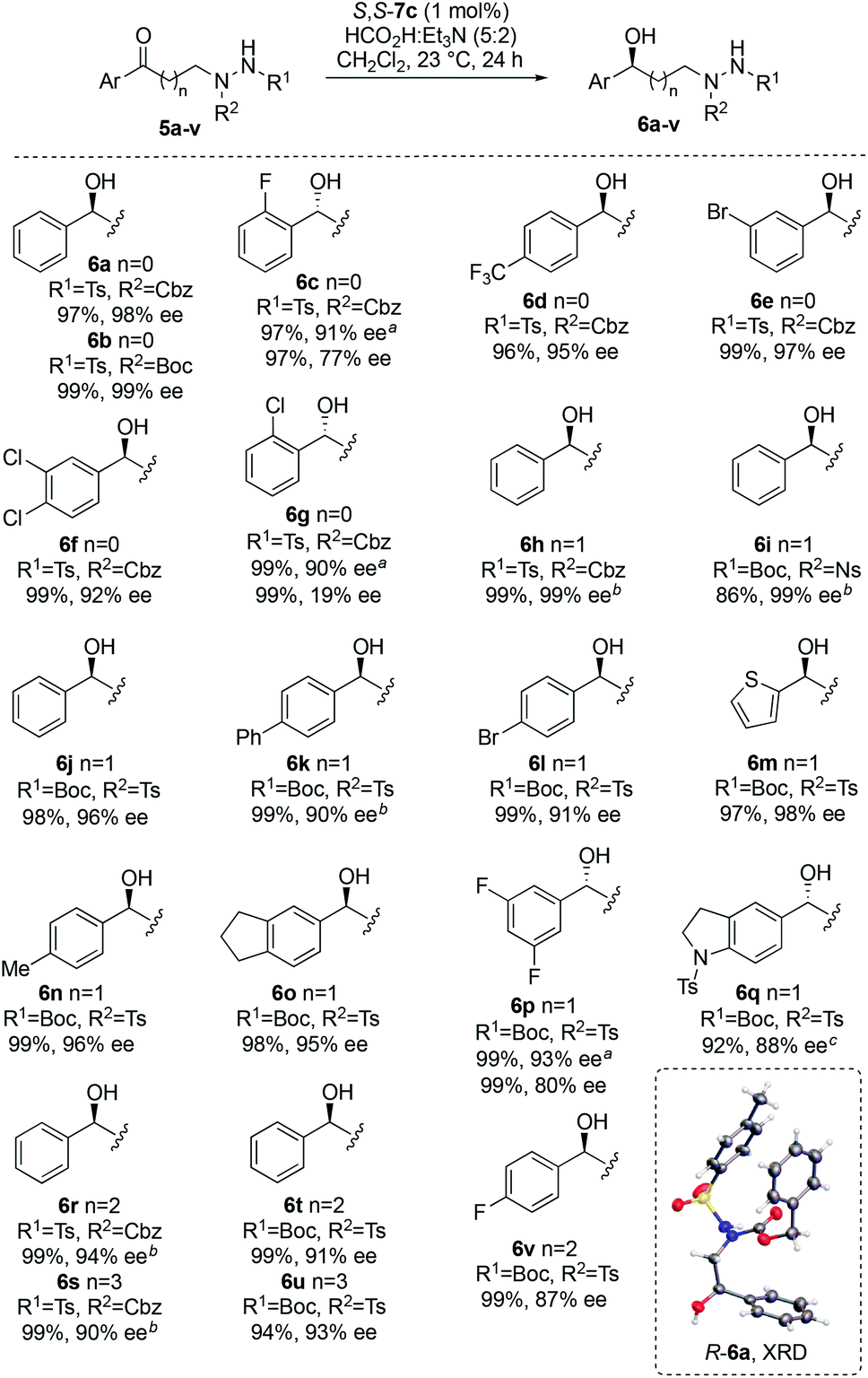
Next, the ATH of a broad range of ketones was examined using the optimized conditions (Fig. 2). Full details relating to the synthesis of substrates 5a–v are provided in the ESI.† High yields and enantioselectivities were achieved in all cases, with variations in the arene, nitrogen protecting groups and linker length well tolerated. The sense of asymmetric induction in these transformations was made by analogy to that seen for R-6a. For 2-substituted aryl derivatives 5c and 5g, and for 3,5-difluoro derivative 5p, poor levels of stereoinduction were obtained using 7c as catalyst. In these cases, use of 7f bearing an additional electron-rich methoxy group on the arene significantly improved the enantioselectivity.13b
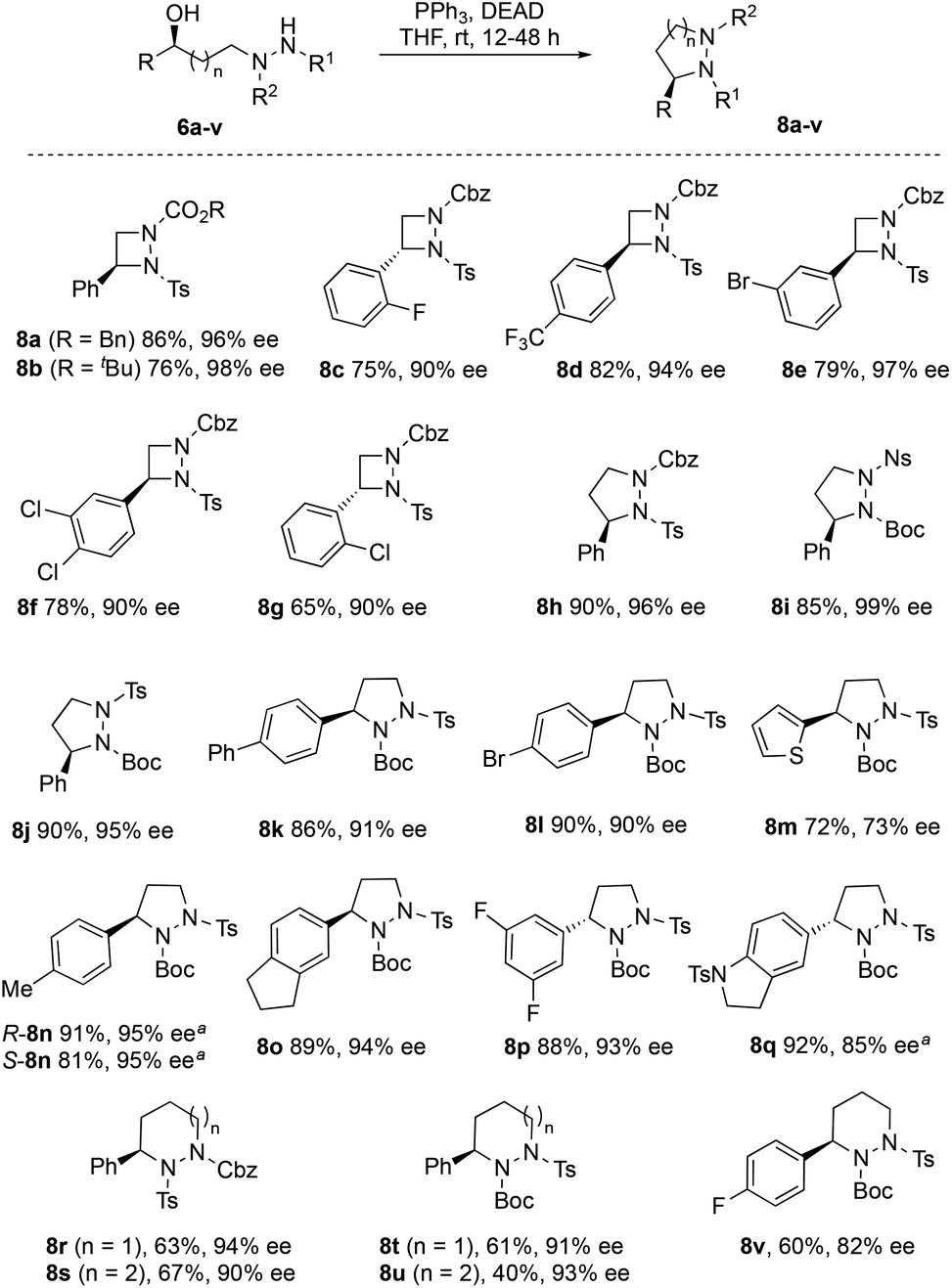
Ring closure of 6a–v using diethyl azodicarboxylate (DEAD) and triphenylphosphine under Mitsunobu conditions provided the corresponding cyclic hydrazines 8a–v in good to excellent yields (Fig. 3). It is possible to replace DEAD with less hazardous diisopropyl azodicarboxylate (DIAD), as illustrated by the cyclization of 6n to 8n and 6q to 8q with comparable efficiency. Furthermore, the methodology can be used to make either enantiomer of the cyclic hydrazine by use of catalyst R,R-7c or S,S-7c, as demonstrated by the synthesis of R-8n and S-8n (Fig. 3). Four- to seven-membered rings can conveniently be made using this method. In general, little or no loss in enantiopurity was seen during cyclization although for highly electron-rich thiophene 8m, partial racemization (73% ee) was detected. The chemistry can be performed preparatively as illustrated by the synthesis of pyrazolidine R-8j on >5 g scale (see ESI†). In this case, the catalyst loading was halved to 0.5 mol% in the ATH step. No loss in ee was observed at this lower catalyst loading although longer reaction times (72 h) were required. For 8d and 8h, the structures were verified by XRD to confirm that stereochemical inversion occurred during cyclization (see ESI†).14
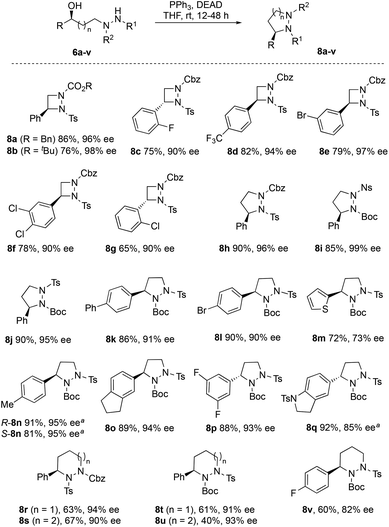 |
| | Fig. 3 Cyclic hydrazines 8a–v made by Mitsunobu ring closure. a Reaction with DIAD in place of DEAD (for full details, see ESI†). | |
2.2. Synthesis of sp3-rich hydrazine libraries by N-functionalization
To determine if these orthogonally-protected cyclic hydrazines could be used to generate sp3-rich heterocyclic libraries, we explored their functionalization. To make this study practicable, a subset of six cyclic hydrazines, namely 8b, 8j, 8k, and 8t–v, were used. The sequence involved: (i) deprotection of the Ts group from the less hindered nitrogen using Mg metal; (ii) functionalization by C–N bond formation to introduce a variety of R2 fragments; (iii) acidic deprotection of the Boc group from the second nitrogen; and (iv) addition of the R1 fragment using similar chemistries used in step (ii) (Fig. 4). The deprotection reactions were essentially quantitative and so product purification was only conducted after each C–N functionalization. Conventional acylation and reductive amination reactions could be conducted on either nitrogen in generally high yields across a range of ring sizes. However, high-throughput optimization was required to identify general conditions for the Pd-catalyzed Buchwald–Hartwig arylations (for full details, see ESI†). For the introduction of the R2 fragment at the least hindered nitrogen on 8j, XPhosPd(crotyl)Cl, NaOtBu and toluene proved the most effective combination of catalyst, solvent and base. Cross-coupling of the more hindered nitrogen in the pyrazolidine ring was achieved with a BippyPhosPd(allyl)OTf catalyst to provide 10d and 10g in good yields. For other substrates differing in ring size and aryl group, Pd(OAc)2 and Xantphos proved to be an effective catalyst system. Using this strategy, 14 derivatives (10a–k, 10m–o) were produced bearing a diverse set of N- and C-substituents across different hydrazine ring sizes. Additionally, by omitting the final cross-coupling, scaffolds bearing free amines can also be produced (10l, 10p and 10q).15 Four representative examples (10b, 10d, 10i, and 10o) confirmed that no erosion of ee occurred during these functionalization sequences.
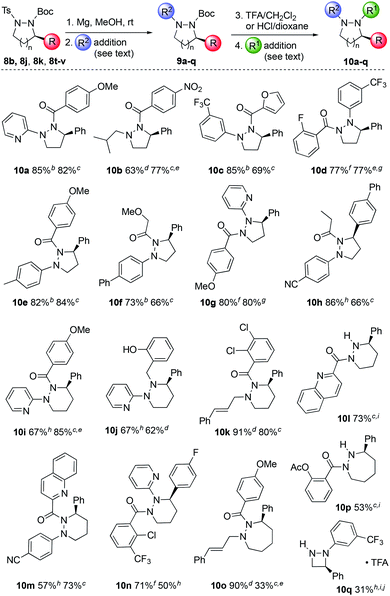 |
| | Fig. 4
N-Functionalization of enantioenriched cyclic hydrazines and hydrazides. a Isolated yields for two-step syntheses of 9 and 10 respectively. b ArBr, XPhosPd(crotyl)Cl (5 mol%), NaOtBu, toluene, 90 °C, 20 h. c RCOCl, DIPEA, CH2Cl2, 0 °C to rt, 18 h. d RCHO, NaBH(OAc)3, THF, rt, 20 h. e Enantiomeric excess determined by chiral HPLC. f RCOCl, CH2Cl2, rt, 18 h. g ArBr, BippyPhosPd(allyl)OTf (5 mol%), Cs2CO3, toluene, 90 °C, 20 h. h ArBr, Pd(OAc)2 (5–10 mol%), Xantphos (10–20 mol%), NaOtBu, toluene, 90 °C, 20 h. i Yield over 3 steps after Boc removal. j The Boc and Ts groups are transposed in the starting material 8b. | |
2.3. Molecular shape and dynamics of sp3-rich hydrazine scaffolds
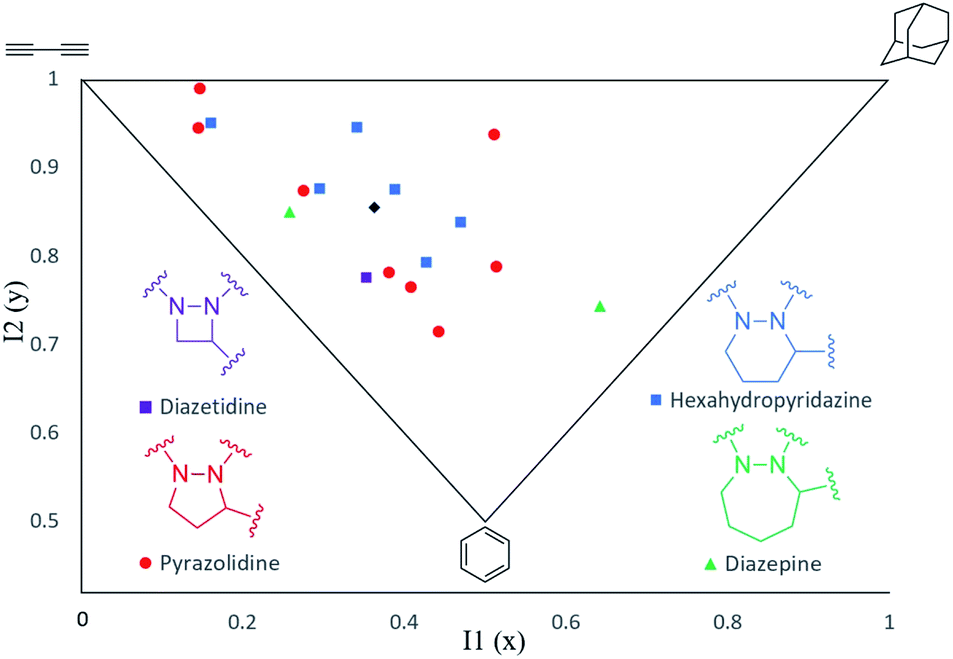
The shapes of the cyclic hydrazines have been studied using a combination of computational and spectroscopic tools. To assess the three-dimensionality of 10a–q in the 17-member hydrazine library, the LLAMA package16 was used to compute and display their principal moments of inertia (PMI) (Fig. 5).17 This tool randomly selects a number of 3D-conformers for each molecule, minimizes their energy and selects the lowest-energy one. The selected minimum energy conformers of 10a–q are provided in the ESI.† LLAMA then calculates the moments of inertia in the x, y and z axes. The PMI I1 coordinates are calculated by dividing inertia(x) by inertia(z). The I2 coordinates are calculated by dividing inertia(y) by inertia(z). This plot confirms that members of this small hydrazine library possess excellent shape diversity and three-dimensionality compared with conventional databases.2 The wide distribution of the heterocycle frameworks within this plot suggests that it is the nature of the appended substituents (R, R1 and R2), rather than the size of the hydrazine ring, that has the greatest influence on their overall shape (Fig. 5).
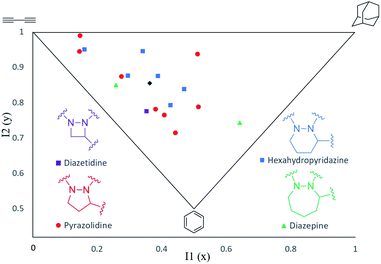 |
| | Fig. 5 Principal moments of inertia (PMI) plot of hydrazines 10a–q indicating their molecular shapes. Mean PMI indicated (◆). | |
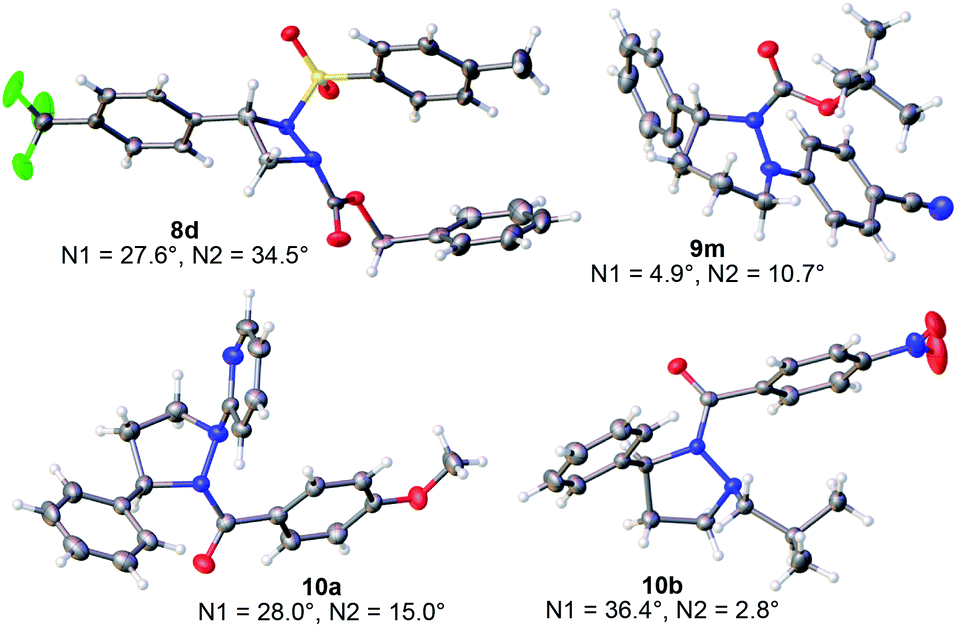
Next, the solution and solid-state structures of representative cyclic hydrazines and hydrazides were determined. A total of eight derivatives were studied by single crystal X-ray diffraction (XRD).14 The ORTEP depictions of 8d, 9m, 10a and 10b are given in Fig. 6. For four-membered 8d and five-membered 10a and 10b, the anti,anti-configuration of the R, R1 and R2 substituents is apparent. For hexahydropyridazine 9m where the conformational flexibility of the ring becomes more important, the R group still exerts a strong influence leading to a highly non-planar structure in which the R, R1 and R2 substituents are displayed in a predictable way on opposite faces of the hydrazine ring. The crystallographic data also provide insight into the extent of pyramidalization of the two nitrogen atoms within the ring,18 a factor influencing both their shapes and fluxional behavior. The values for the pyramidalization of each nitrogen are given in Fig. 6, with larger values indicating more sp3-character at the nitrogen center.
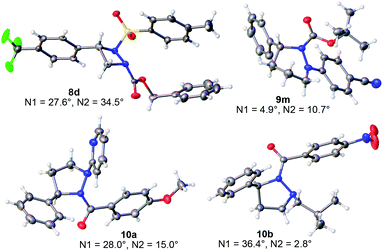 |
| | Fig. 6 Representative XRD structures of the cyclic hydrazines with measured values for the pyramidalization of the two ring nitrogens (N1 and N2).18 | |
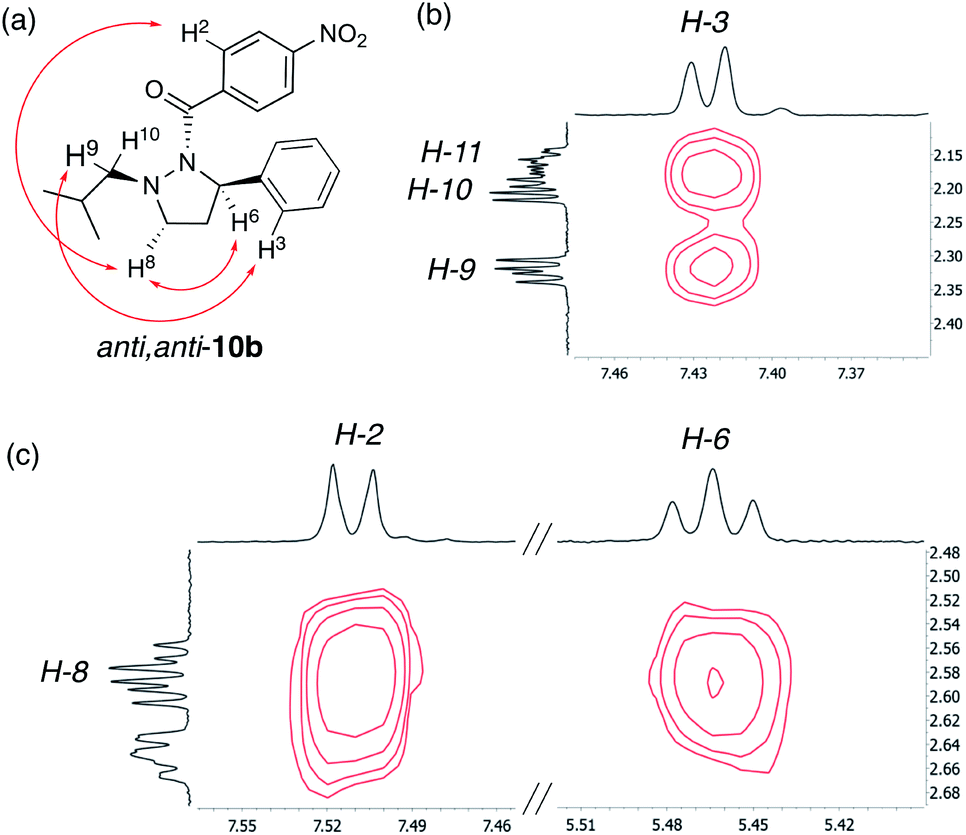
Next, the solution-state structure of 10b was determined in C6D6 at 343 K by 1H NOESY spectroscopy (600 MHz). These conditions were used to produce a single set of well resolved signals. The solution structure was assigned on the basis of NOESY cross-peaks between the ortho-hydrogens of the phenyl ring (H-3) and the methylene hydrogens of the isobutyl group (H-9 and possibly H-10) consistent with these substituents being on the same side of the hydrazine ring (Fig. 7b). Furthermore, the observation of a NOESY cross-peak between H-2 and H-8, and between H-6 and H-8 places all these atoms on the same side of the molecule and on the opposite face to the phenyl group (Fig. 7c). On this basis we assign an anti,anti-configuration to 10b in solution (Fig. 7a), similar to the solid-state structure (Fig. 6).14
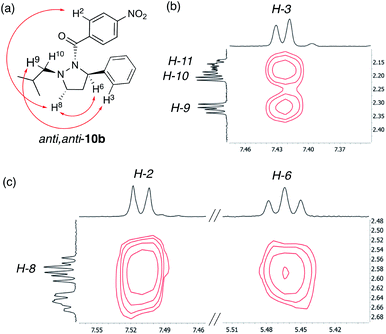 |
| | Fig. 7 Solution-state structure of 10b derived by 1H–1H NOESY spectroscopy (600 MHz, C6D6, 343 K): (a) anti,anti-representation of 10b with key NOEs indicated (red arrows). (b) Expansion of the NOESY spectrum highlighting cross-peaks between H-3 and H-9 and possibly H-10. (c) Expansion of the NOESY spectrum highlighting key cross-peaks between H-8 and H-2, and H-8 and H-6. | |
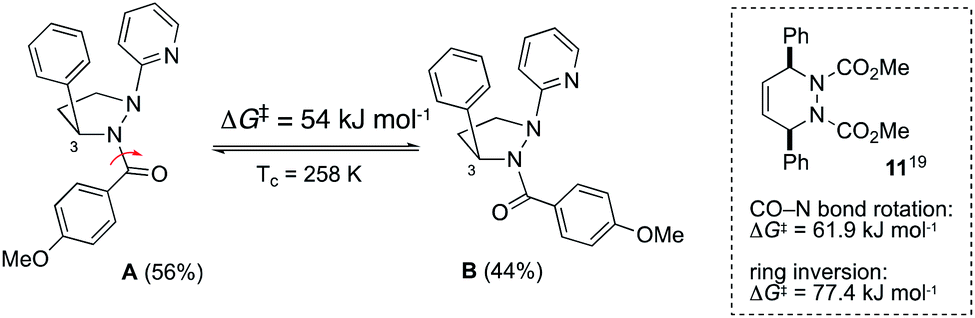
To explore the fluxional behavior of the cyclic hydrazines, variable temperature 1H NMR studies were conducted using 10a and 10b. For 10a, the 1H NMR spectrum resolved into two sharp sets of signals at 233 K in CDCl3, indicating the presence of two conformational isomers in a 56:44 ratio. In contrast, 10b exists predominately as a single conformational isomer at 233 K in CDCl3 although small amounts of two additional isomers are seen (84:11:5). Evidently, the fluxional behavior is highly dependent on the nature of the N-substituents. This is consistent with changes in the extent of N-pyramidalization seen in the solid-state (Fig. 6). Using the resolved methyl singlets of 10a, we were able to determine the barrier to interconversion of the two isomers at coalescence (Tc = 258 K, ΔG‡ = 54 kJ mol−1, see ESI†). The question arises as to whether the two isomers come from fluxionality at the hydrazine centers (Scheme 1), or from a process such as amide bond rotation within anti,anti-10a? On the available evidence, we suggest that only amide rotamers are being observed i.e.A and B (Scheme 2). Firstly, whilst low temperature NOESY experiments did not allow us to fully deduce the solution-state structure of 10a, we did observe a NOESY cross-peak between the methine hydrogen at H-3 and the ortho-hydrogens of the COAr group for A but not B consistent with this assignment. Secondly, the measured interconversion barrier is more consistent with that of an amide bond rotation in similar systems. For example, both types of fluxionality operate concurrently in 11 with a lower barrier for rotation about the N–CO bond than ring inversion (box insert, Scheme 2).19 Finally, the near equal population of A and B is inconsistent with N-inversion as 1b–d are expected to be higher energy species due to unfavorable non-bonded interactions (Scheme 1). Although N-fluxionality is not operating in 10a, the observation of a third set of signals for 10b at low temperature, suggests that conformational isomers arising from N-inversion are accessible for some systems, albeit in low concentrations.
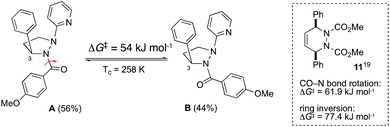 |
| | Scheme 2 Likely origin of fluxional behavior seen in 10a. | |
3. Conclusions
An efficient synthesis of differentially-protected cyclic hydrazines containing an adjacent stereogenic carbon atom has been devised using ATH as the key step. Commercially available tethered Ru-catalyst 7c, available in both enantiomeric forms, proved highly effective for the introduction of the asymmetric center by ketone reduction. In the few cases where low enantioselectivity was seen in the ATH, tethered catalyst 7f gave further improvements. The methodology was used to make 22 different cyclic hydrazines in up to 99% ee, through variation in ring size (4- to 7-membered rings), C-3 substituents and nitrogen protecting groups. The chemistry is operationally simple and can be performed on a preparative scale. Chemical library construction is possible from these hydrazines by iterative C–N bond formation. Wide chemical diversification is possible through variation in the hydrazine structure, use of different functionalization chemistries and coupling partners, and controlled engagement of each nitrogen of the hydrazine in turn. Principal moment of inertia (PMI) analysis revealed excellent shape diversity and three-dimensionality within the assembled library, indicating their potential value in drug discovery. The preference for the molecules to adopt anti,anti-orientations of the three stereocenters was established using a combination of XRD and NMR spectroscopy. The fluxional behavior of these compounds was further explored by VT-NMR and low temperature NOESY experiments, from which useful insights have emerged about how the molecules sample chemical space. Future work will examine the feasibility of making larger chemical libraries based on these non-planar scaffolds and the exploitation of this chemistry in drug discovery programs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Leverhulme Trust (RPG-2014-362), Eli Lilly and the University of Warwick for generous financial support, and Johnson Matthey for the gift of tethered catalysts 7c–e. Crystallographic data were collected using an instrument that received funding from the European Research Council under the European Union's Horizon 2020 research and innovation program (grant agreement No. 637313). We gratefully acknowledge the help of Dr Ivan Prokes (University of Warwick) with the NMR experiments.
Notes and references
- For reviews, see:
(a) A. Karawajczyk, F. Giordanetto, J. Benningshof, D. Hamza, T. Kalliokoski, K. Pouwer, R. Morgentin, A. Nelson, G. Müller, A. Piechot and D. Tzalis, Drug Discovery Today, 2015, 20, 1310–1316 CrossRef CAS PubMed;
(b) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed;
(c) T. J. Ritchie and S. J. F. Macdonald, Drug Discovery Today, 2009, 14, 1011–1020 CrossRef CAS PubMed.
- J. Meyers, M. Carter, N. Y. Mok and N. Brown, Future Med. Chem., 2016, 8, 1753–1767 CrossRef.
- For selected examples, see:
(a) B. L. Donnelly, L. D. Elliott, C. L. Willis and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2019, 58, 9095–9098 CrossRef CAS;
(b) A. Sveiczer, A. J. P. North, N. Mateu, S. L. Kidd, H. F. Sore and D. R. Spring, Org. Lett., 2019, 21, 4600–4604 CrossRef CAS PubMed;
(c) N. J. Flodén, A. Trowbridge, D. Willcox, S. M. Walton, Y. Kim and M. J. Gaunt, J. Am. Chem. Soc., 2019, 141, 8426–8430 CrossRef;
(d) T.-G. Chen, L. M. Barton, Y. Lin, J. Tsien, D. Kossler, I. Bastida, S. Asai, C. Bi, J. S. Chen, M. Shan, H. Fang, F. G. Fang, H.-W. Choi, L. Hawkins, T. Qin and P. S. Baran, Nature, 2018, 560, 350–354 CrossRef CAS PubMed;
(e) D. J. Foley, P. G. E. Craven, P. M. Collins, R. G. Doveston, A. Aimon, R. Talon, I. Churcher, F. von Delft, S. P. Marsden and A. Nelson, Chem.–Eur. J., 2017, 23, 15227–15232 CrossRef CAS PubMed;
(f) R. Gianatassio, J. M. Lopchuk, J. Wang, C.-M. Pan, L. R. Malins, L. Prieto, T. A. Brandt, M. R. Collins, G. M. Gallego, N. W. Sach, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, Science, 2016, 351, 241–246 CrossRef CAS PubMed;
(g) B. A. Chalmers, H. Xing, S. Houston, C. Clark, S. Ghassabian, A. Kuo, B. Cao, A. Reitsma, C.-E. P. Murray, J. E. Stok, G. M. Boyle, C. J. Pierce, S. W. Littler, D. A. Winkler, P. V. Bernhardt, C. Pasay, J. J. De Voss, J. McCarthy, P. G. Parsons, G. H. Walter, M. T. Smith, H. M. Cooper, S. K. Nilsson, J. Tsanaktsidis, G. P. Savage and C. M. Williams, Angew. Chem., Int. Ed., 2016, 55, 3580–3585 CrossRef CAS PubMed;
(h) M. U. Luescher and J. W. Bode, Org. Lett., 2016, 18, 2652–2655 CrossRef CAS PubMed.
- For a reviews, see:
(a) G. Le Goff and J. Ouazzani, Bioorg. Med. Chem., 2014, 22, 6529–6544 CrossRef CAS PubMed;
(b) E. Licandro and D. Perdicchia, Eur. J. Org. Chem., 2004, 665–675 CrossRef CAS.
- K. C.-C. Cheng, S. Cao, A. Raveh, R. MacArthur, P. Dranchak, G. Chlipala, M. T. Okoneski, R. Guha, R. T. Eastman, J. Yuan, P. J. Schultz, X.-Z. Su, G. Tamayo-Castillo, T. Matainaho, J. Clardy, D. H. Sherman and J. Inglese, J. Nat. Prod., 2015, 78, 2411–2422 CrossRef CAS PubMed.
- V. A. Steadman, S. B. Pettit, K. G. Poullennec, L. Lazarides, A. J. Keats, D. K. Dean, S. J. Stanway, C. A. Austin, J. A. Sanvoisin, G. M. Watt, H. G. Fliri, A. C. Liclican, D. Jin, M. H. Wong, S. A. Leavitt, Y.-J. Lee, Y. Tian, C. R. Frey, T. C. Appleby, U. Schmitz, P. Jansa, R. L. Mackman and B. E. Schultz, J. Med. Chem., 2017, 60, 1000–1017 CrossRef CAS and references cited therein.
- M. P. Clark, S. K. Laughlin, M. J. Laufersweiler, R. G. Bookland, T. A. Brugel, A. Golebiowski, M. P. Sabat, J. A. Townes, J. C. VanRens, J. F. Djung, M. G. Natchus, B. De, L. C. Hsieh, S. C. Xu, R. L. Walter, M. J. Mekel, S. A. Heitmeyer, K. K. Brown, K. Juergens, Y. O. Taiwo and M. J. Janusz, J. Med. Chem., 2004, 47, 2724–2727 CrossRef CAS PubMed.
- F. Deget and R. N. Brogden, Drugs, 1991, 41, 799–820 CrossRef CAS PubMed.
- For examples, see:
(a) M. Kamuf and O. Trapp, Chirality, 2013, 25, 224–229 CrossRef CAS PubMed;
(b) A. Perjéssy, P. Meyer, W.-D. Rudorf, D. Loos, E. Kolehmainen, K. Laihia, M. Nissinen, J. Koivisto and R. Kauppinen, J. Phys. Org. Chem., 2001, 14, 811–818 CrossRef;
(c) R. G. Kostyanovsky, G. K. Kadorkina, V. R. Kostyanovsky, V. Schurig and O. Trapp, Angew. Chem., Int. Ed., 2000, 39, 2938–2940 CrossRef CAS;
(d) J. E. Anderson, J. Am. Chem. Soc., 1969, 91, 6374–6380 CrossRef CAS.
- For selected examples, see:
(a) X. Kou, Q. Shao, C. Ye, G. Yang and W. Zhang, J. Am. Chem. Soc., 2018, 140, 7587–7597 CrossRef CAS PubMed;
(b) S. Rajkumar, G. J. Clarkson and M. Shipman, Org. Lett., 2017, 19, 2058–2061 CrossRef CAS PubMed;
(c) A.-S. Marques, M. Giardinetti, J. Marrot, V. Coeffard, X. Moreau and C. Greck, Org. Biomol. Chem., 2016, 14, 2828–2832 RSC;
(d) H.-J. Leng, F. Peng, S. Zingales, W. Huang, B. Wang, Q. Zhao, R. Zhou, G. He, C. Peng and B. Han, Chem.–Eur. J., 2015, 21, 18100–18108 CrossRef CAS PubMed;
(e) D.-Y. Zhang, L. Shao, J. Xu and X.-P. Hu, ACS Catal., 2015, 5, 5026–5030 CrossRef CAS;
(f) L. Lykke, B. D. Carlsen, R. S. Rambo and K. A. Jørgensen, J. Am. Chem. Soc., 2014, 136, 11296–11299 CrossRef CAS PubMed;
(g) M. Fernández, E. Reyes, J. L. Vicario, D. Badía and L. Carrillo, Adv. Synth. Catal., 2012, 354, 371–376 CrossRef;
(h) R. L. LaLonde, Z. J. Wang, M. Mba, A. D. Lackner and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 598–601 CrossRef CAS PubMed.
- For reviews, see:
(a) H. G. Nedden, A. Zanotti-Gerosa and M. Wills, Chem. Rec., 2016, 16, 2623–2643 CrossRef PubMed;
(b) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed;
(c) F. Foubelo, C. Nájera and M. Yus, Tetrahedron: Asymmetry, 2015, 26, 769–790 CrossRef CAS;
(d) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- L. Grehn, B. Nyasse and U. Ragnarsson, Synthesis, 1997, 1429–1432 CrossRef CAS.
-
(a) A. M. Hayes, D. J. Morris, G. J. Clarkson and M. Wills, J. Am. Chem. Soc., 2005, 127, 7318–7319 CrossRef CAS PubMed;
(b) R. Soni, K. E. Jolley, G. J. Clarkson and M. Wills, Org. Lett., 2013, 15, 5110–5113 CrossRef CAS;
(c) R. Hodgkinson, V. Jurčík, A. Zanotti-Gerosa, H. G. Nedden, A. Blackaby, G. J. Clarkson and M. Wills, Organometallics, 2014, 33, 5517–5524 CrossRef CAS;
(d) R. Soni, K. E. Jolley, S. Gosiewska, G. J. Clarkson, Z. Fang, T. H. Hall, B. N. Treloar, R. C. Knighton and M. Wills, Organometallics, 2018, 37, 48–64 CrossRef CAS.
- CCDC 1944266 (6a), 1944267 (8d), 1944268 (8h), 1944269 (10a), 1944270 (10b), 1944271 (9m), 1944272 (10l), 1944273 (9i), and 1955341 (10o) contain the supplementary crystallographic data for this paper.†.
- Double functionalization of four-membered diazetidines such as 8b has proven difficult in part because of low yields in the Pd-catalyzed cross-couplings. After N-deprotection, such substrates have more limited chemical stability.
- I. Colomer, C. J. Empson, P. Craven, Z. Owen, R. G. Doveston, I. Churcher, S. P. Marsden and A. Nelson, Chem. Commun., 2016, 52, 7209–7212 RSC.
- E. Lenci and A. Trabocchi, ChemBioChem, 2019, 20, 1115–1123 CrossRef CAS PubMed.
- The pyramidalization of the nitrogen atoms is defined as 360 − Σ where Σ is the sum of the valence angles of the nitrogen atom.
- J. E. Anderson and J. M. Lehn, Tetrahedron, 1968, 24, 137–149 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all new compounds, copies of HPLC traces, 1H and 13C NMR spectra, VT-NMR, PMI analysis and XRD structures. CCDC 1944266–1944273 and 1955341. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04849a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Sundaram
Rajkumar
a,
Stefan
Roesner
a,
Sundaram
Rajkumar
a,
Stefan
Roesner