
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA photo-regulated aptamer sensor for spatiotemporally controlled monitoring of ATP in the mitochondria of living cells†
Shanni
Hong
ab,
Xiaoting
Zhang
c,
Ryan J.
Lake
bd,
Gregory T.
Pawel
 bd,
Zijian
Guo
*c,
Renjun
Pei
*a and
Yi
Lu
*bd
bd,
Zijian
Guo
*c,
Renjun
Pei
*a and
Yi
Lu
*bd
aCAS Key Laboratory of Nano-Bio Interface, Suzhou Institute of Nano-Tech and Nano-Bionics, Chinese Academy of Sciences, Suzhou, 215123, China. E-mail: rjpei2011@sinano.ac.cn
bDepartment of Chemistry, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. E-mail: yi-lu@illinois.edu
cState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210093, China. E-mail: zguo@nju.edu.cn
dDOE Center for Advanced Bioenergy and Bioproducts Innovation, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
First published on 26th November 2019
Abstract
Fluorescent aptamer sensors have shown enormous potential for intracellular imaging of small molecule metabolites. Since metabolites distribute differently at different subcellular locations and their concentrations and locations fluctuate with time, methods are needed for spatiotemporally controlled monitoring of these metabolites. Built upon previous success in temporal control of aptamer-based sensors, we herein report an aptamer sensor containing a photocleavable linker and using DQAsomes to target mitochondria for spatiotemporally controlled monitoring of ATP in the mitochondria of living cells. The photocleavable modification on the DNA ATP aptamer sensor can prevent sensor activation before reaching mitochondria and the sensor can then be activated upon light irradiation. The sensor has a detection limit of 3.7 μM and high selectivity against other nucleotides, allowing detection of ATP concentration fluctuations in mitochondria induced by Ca2+ or oligomycin. This work represents the first successful delivery of a DNA aptamer sensor to mitochondria, providing a new platform for targeted delivery to subcellular organelles for monitoring energy producing processes, as well as mitochondrial dysfunction-related diseases in different cells.
Introduction
Aptamers are short, single stranded oligonucleotides that have emerged as a powerful candidate for detecting not only large biomolecules such as proteins,1,2 but also small organic metabolites,3–7 because target-specific aptamers can be obtained for a wide variety of different targets from a large nucleic acid library with 1015 different sequences using a process called Systematic Evolution of Ligands by EXponential enrichment (SELEX). As a result, many aptamer-based sensors have been reported for a wide variety of targets in environmental monitoring,3,8–12 medical diagnostics,13–16 and metabolite imaging.7,17–20 Among these aptamer sensors, fluorescent sensors have enjoyed the most success because of their fast response time and high signal intensity, allowing for real-time in situ detection and imaging.21,22Among the targets of aptamer sensors in cells, ATP is one of the most important, because it is the primary energy currency in living organisms and thus plays important roles in many biological processes, from energy conversion to production of bioproducts for many applications.23–27 ATP also serves as an indicator of cellular viability and injury.5,28 The mitochondria is the main location of ATP production, and the fluctuation of ATP levels may lead to mitochondrial dysfunction,29 which is related to neurodegenerative diseases,30 malignant cancers,31 and cardiac diseases.32 Therefore, it is important to develop a reliable method for imaging the ATP in mitochondria. Toward this goal, several methods have been developed to deliver fluorescent ATP aptamer sensors into cells, including using DNA nanostructures,33–35 DNA-functionalized nanomaterials,21,34,36–38 and DNA hydrogels39 for imaging ATP in cells.
Despite the current progress, most of these fluorescent aptamers are limited when applying them for intracellular imaging.40,41 Because these sensors employ an “always-on” design, they often exhibit fluorescence signals even during the cellular uptake process, which would inevitably result in inaccurate detection and high-background in images.41,42 To overcome this limitation, Heemstra and co-workers have demonstrated temporal control of an aptamer fluorescent sensor using a photocleavable linker for sensing ATP.43 While this novel concept has been demonstrated in test tubes, it was not applied to ATP imaging in living cells until Li and co-workers employed upconversion nanoparticles to convert irradiated near IR light into 365 nm light that allowed cleavage of a photocleavable linker designed into the ATP aptamer sensor.41 Similarly, the use of upconversion nanoparticles for photo-decaging of DNAzymes for imaging metal ions has also been demonstrated in living cells and zebrafish.44 While these methods have demonstrated temporal control of aptamer and DNAzyme sensors, they have not been applied to targeted imaging for spatial control at the same time. Since metabolites, such as ATP, have different subcellular distributions, it is important to detect ATP not only with temporal control, but also with spatial control.
Here, we report a method for the design of a photo-regulated aptamer sensor for spatiotemporally controlled monitoring of ATP in the mitochondria of living cells by employing a photo-cleavable (PC) aptamer for ATP with DQAsomes, which are liposome-like vesicles synthesized from dequalinium chloride that have been shown to target mitochondria, likely due to their highly positive charge.45 Such an ATP sensor with a PC group can be inactivated or caged before reaching the mitochondria and can be activated or decaged by light to detect ATP concentration fluctuations in mitochondria induced by Ca2+ or oligomycin.
Results and discussion
Design and characterization of photo-cleavable aptamer (PC-Apt) sensors
The design of photo-regulated ATP aptamer probes (PC-Apt) for the detection of mitochondrial ATP in living cells is illustrated in Fig. 1. The DNA aptamer for ATP (in red, Fig. 1A) is initially blocked for binding its target by hybridization to a partially complementary DNA strand containing a photocleavable (PC) linker consisting of a nitrobenzene group (Fig. 1B). To ensure efficient blocking of the DNA aptamer from binding ATP at the cellular temperature of 37 °C, the ATP aptamer is extended on the 5′ end by 5 bases so that the calculated melting temperature between the ATP aptamer strand and its complementary DNA strand, hereafter called the PC-Blocker, is 56.7 °C. At 37 °C, the two DNA strands will remain hybridized, positioning the Cy3 fluorophore at the 5′ end of the aptamer strand, directly opposite from the 3′ end of the PC-Blocker containing the Iowa Black FQ quencher, resulting in quenching of the fluorescence of Cy3 by the quencher. This hybridization between the PC-Blocker and the ATP aptamer is designed to be strong enough that even in the presence of ATP, the active aptamer conformation is not able to form, and so ATP will not be able to bind. Upon 365 nm light irradiation, the PC linker will undergo photolysis, splitting the PC-Blocker-Q into two DNA fragments. Since the calculated melting temperature between the shorter DNA fragment at the 5′ end of the PC-Blocker and its ATP aptamer is 31.6 °C, this fragment will dehybridize from the ATP aptamer at 37 °C, leaving the longer DNA fragment at the 3′ end of the PC-Blocker to remain hybridized to the ATP aptamer, because they have a calculated melting temperature of 45.8 °C. More importantly, the dehybridization of the shorter DNA fragment from the ATP aptamer exposes the ATP binding region, allowing the ATP aptamer to adopt its active conformation upon binding ATP, leading to the release of the longer PC-Blocker-Q fragment from the aptamer strand and thus resulting in a large increase in the fluorescence signal from Cy3.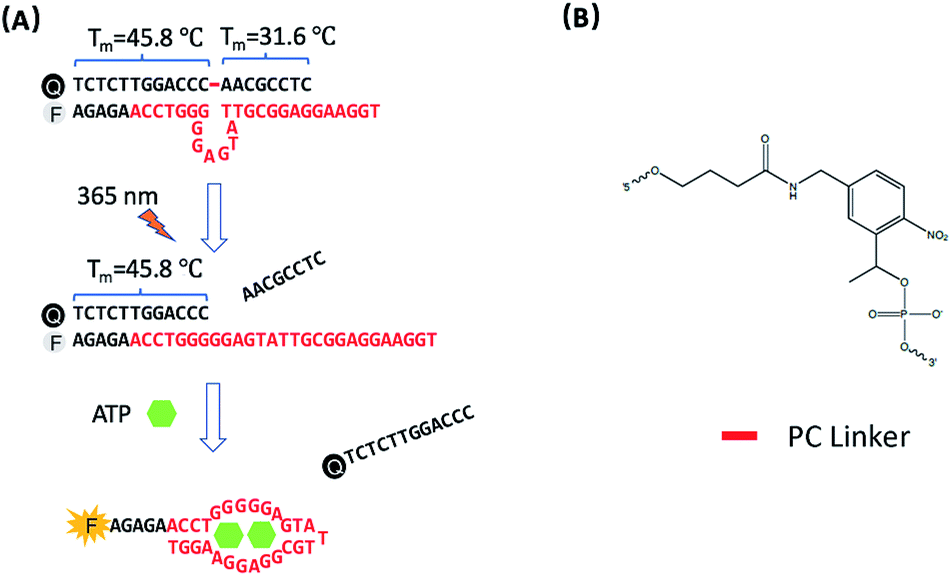 | ||
| Fig. 1 (A) Design of the photo-regulated ATP aptamer probe (PC-Apt) for the detection of mitochondrial ATP in living cells. (B) The chemical structure of the PC Linker. | ||
Before demonstrating the PC-Apt sensor design in living cells, we first tested it in test tubes. In order to simulate the pH of mitochondria, where a large proportion of cellular ATP resides, we chose Tris buffer at pH 8.0. We initially investigated the quenching efficiency of the PC-Blocker-Q when mixing different molar ratios of PC-Blocker-Q with Apt-Cy3. As shown in Fig. S1,† the fluorescence of 200 nM Apt-Cy3 decreased with increasing molar ratios of PC-Blocker-Q/Apt-Cy3 from 1.1 to 1.7, indicating more efficient quenching of the Apt-Cy3 by the PC-Blocker-Q. When the ratio reached 1.5, the fluorescence of Cy3 reached a minimum, with little further reduction in fluorescence with higher ratios. Therefore, we chose the 1.5 ratio for the following experiments.
Next, we studied the effect of photocleavage and its efficiency with the PC-Apt sensors. The photocleavage of PC-Apt sensors was first confirmed using native polyacrylamide gel electrophoresis (PAGE, Fig. S2†), and, as can be seen, irradiation with 365 nm light for 10 min resulted in a shorter DNA fragment. We further optimized the irradiation time and found that the fluorescence increased with longer 365 nm irradiation time of a solution containing the 200 nM PC-Apt sensor and 5 mM ATP (see the inset of Fig. 2A), reaching its maximum by 20 min. Furthermore, following the 20 min optimal irradiation time, the fluorescence intensity of PC-Apt samples increased proportionally with increasing concentrations of ATP from 0.05 mM to 5 mM (Fig. 2A). In the absence of light irradiation, minimal fluorescence was observed even when the ATP concentration was increased to 5 mM (Fig. 2B).
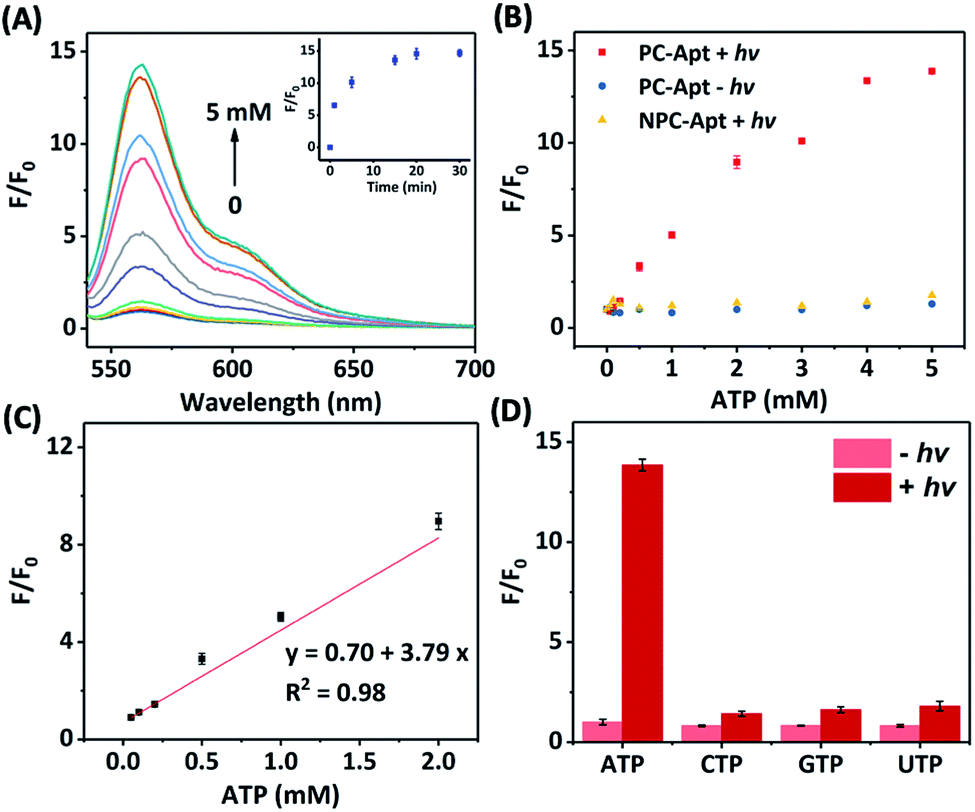 | ||
| Fig. 2 (A) Fluorescence spectra of the PC-Apt after 20 min 365 nm light irradiation in response to different ATP concentrations ([ATP] = 0, 0.05, 0.1, 0.2, 0.5, 1, 2, 3, 4, and 5 mM). Inset shows the corresponding response of 365 nm light irradiation time. Ex = 530 nm, Em = 540–700 nm. (B) Comparison of the fluorescence change between PC-Apt and NPC-Apt probes with/without 365 nm light (hv) exposure. [ATP] = 0, 0.05, 0.1, 0.2, 0.5, 1, 2, 3, 4, and 5 mM. Ex = 530 nm, Em = 565 nm. (C) Linear relationship between fluorescence and various concentrations of ATP after 365 nm light exposure. (D) Response of the PC-Apt probe to different nucleoside triphosphates with/without 365 nm light exposure, [Analytes] = 5 mM. F0 denotes the fluorescence intensity in the absence of ATP and light irradiation, F denotes the fluorescence intensity in the presence of ATP. Ex = 530 nm, Em = 565 nm. Error bars represent standard deviations from three repeated experiments. The concentration of probes was 200 nM and the 365 nm light irradiation time was 20 min. | ||
To verify that the fluorescence changes were indeed due to the cleavage of the PC linker activating the PC-Apt sensor, we designed a negative control called NPC-Apt, which has the same sequence as PC-Apt but without the PC linker and is not subject to photo-induced-cleavage. Indeed, in contrast to increasing fluorescence with increasing ATP concentrations, from 0.05 mM to 5 mM, for PC-Apt after 20 min light irradiation, the NPC-Apt did not respond to ATP under the same conditions (Fig. 2B). From these results, the detection limit for PC-Apt was calculated to be 3.7 μM (R2 = 0.98). Meanwhile, in the presence of CTP, GTP, or UTP, a minimal fluorescence signal was observed with and without light irradiation, indicating the high selectivity of the PC-Apt sensor for ATP against other nucleotide triphosphates (Fig. 2D).
Before exploring the PC-Apt sensor for cellular studies, we checked its stability and activity in cell lysate (Fig. S3†). After the sensors mixed with ATP were incubated in cell lysate buffer for 12 h, we did not observe any fluorescence intensity changes, suggesting that the PC-Apt sensor is stable in cell lysate and is not able to bind ATP without photo-decaging. Upon 365 nm light irradiation for 20 min, the fluorescence intensity was enhanced by over 10-fold, demonstrating that the PC-Apt sensors can be used as biosensors under cellular conditions and remain stable in the cell environment over the 12 h time period relevant for our studies.
Preparation and characterization of DQAsomes and PC-Apt/DQAsome complexes for targeted imaging in mitochondria
Since ATP is highly abundant in the mitochondria of all eukaryotic cells, measured in the range of 1–5 mM in HeLa cell mitochondria,46 we aim to deliver our PC-Apt sensors into mitochondria to image the ATP levels in this important energy-generating organelle of HeLa cells. Since DQAsomes, liposome-like cationic vesicles synthesized from dequalinium chloride, have been shown to be effective in targeting mitochondria, we synthesized DQAsomes according to a report by Weissig et al.45,47 Due to the positive charge of DQAsomes, the highly negatively charged backbone of DNA and DNA plasmids can be easily condensed with them through electrostatic interactions,48,49 thus helping to protect the DNA from DNase degradation47 and further allowing the delivery of PC-Apt into mitochondria (Fig. 3). As shown in Table 1, the size of the DQAsomes we synthesized, as measured by Dynamic Light Scattering (DLS), is 180 ± 34 nm, which is similar to those reported in the literature.50,51 Upon mixing the DQAsomes with PC-Apt, the size increased to 208 ± 32 nm, suggesting the successful condensation of PC-Apt sequences with the DQAsomes. The zeta potentials of the DQAsomes decreased from 52 ± 8 mV to 42 ± 8 mV after mixing with PC-Apt, further indicating the successful adsorption of the PC-Apt sensors to the DQAsome surface, forming the PC-Apt/DQAsome complex. Furthermore, in order to examine the effect that the DQAsomes would have on the sensing ability of PC-Apt, we tested the in vitro fluorescence of PC-Apt/DQAsomes against different concentrations of ATP after light irradiation (Fig. S4†). The results showed that the sensitivity of the PC-Apt/DQAsomes was slightly lower than that of PC-Apt only, which is likely caused by the interaction between PC-Apt and DQAsomes limiting the conformational change of the aptamer within the whole sensor construct. Despite this effect, the detection limit of the PC-Apt/DQAsome complex was calculated to be 9.0 μM (R2 = 0.99), with a linear range of 1–5 mM, which is suitable for detecting the HeLa cell mitochondrial ATP.46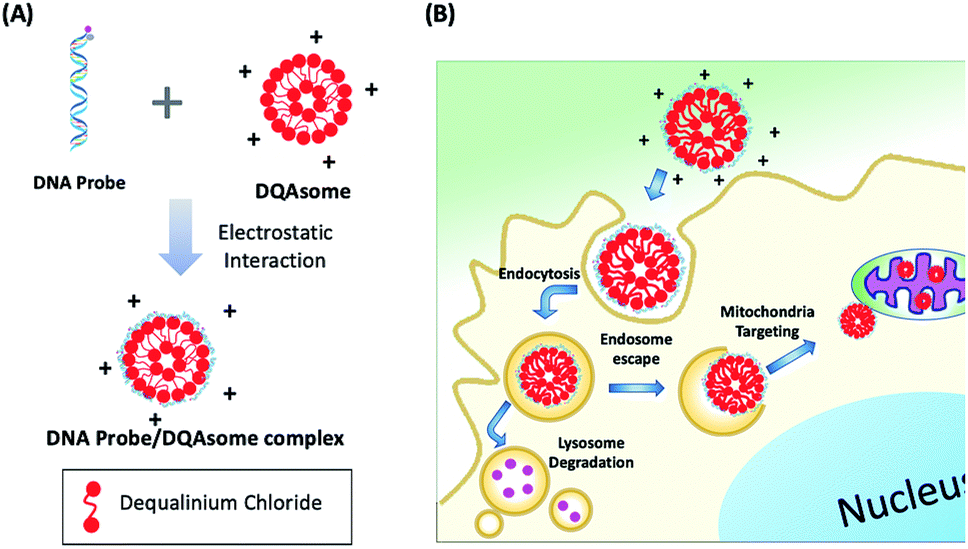 | ||
| Fig. 3 (A) Schematic of DNA probe/DQAsome complex formulation. (B) Schematic depiction of targeted delivery of DNA probe/DQAsomes to mitochondria. | ||
| DQAsomes | PC-Apt/DQAsomes | |
|---|---|---|
| Size (nm) | 180 ± 34 | 208 ± 32 |
| Zeta potential (mV) | 52 ± 8 | 42 ± 8 |
Mitochondrial colocalization studies with confocal microscopy imaging in living cells
To determine whether the DQAsomes can help to deliver the PC-Apt sensors to mitochondria selectively, we incubated the PC-Apt/DQAsome complexes with HeLa cells and observed their fluorescence using a confocal laser scanning microscope (CLSM, Fig. 4). At the same time, commercially available MitoTracker Green, which exhibits fluorescence in the green channel, was used to verify colocalization to mitochondria. As shown in Fig. 4A, the fluorescence in the red channel of the HeLa cells treated with PC-Apt/DQAsome complexes increased dramatically after 365 nm light irradiation owing to the photolysis of the PC linker. Furthermore, the PC-Apt red channel seems to overlap well with the MitoTracker Green channel, suggesting good co-localization to mitochondria. To further confirm this conclusion, the line profiling of the pixel intensity of interest suggests that the signals from the PC-Apt/DQAsome complexes closely match the MitoTracker Green staining (Fig. 4B). More detailed image analysis was performed to quantify the colocalization between the PC-Apt/DQAsome complexes and MitoTracker in mitochondria. Multiple images from independent experiments (n > 6) were analysed to obtain Pearson's correlation coefficient and Mander's coefficient,52 which are two common parameters to quantify the colocalization. As shown in Fig. 4C, Pearson's coefficient increased from 0.50 to 0.89, and Mander's coefficient increased from 0.66 to 0.92, confirming that the PC-Apt/DQAsome and MitoTracker Green have significant overlap.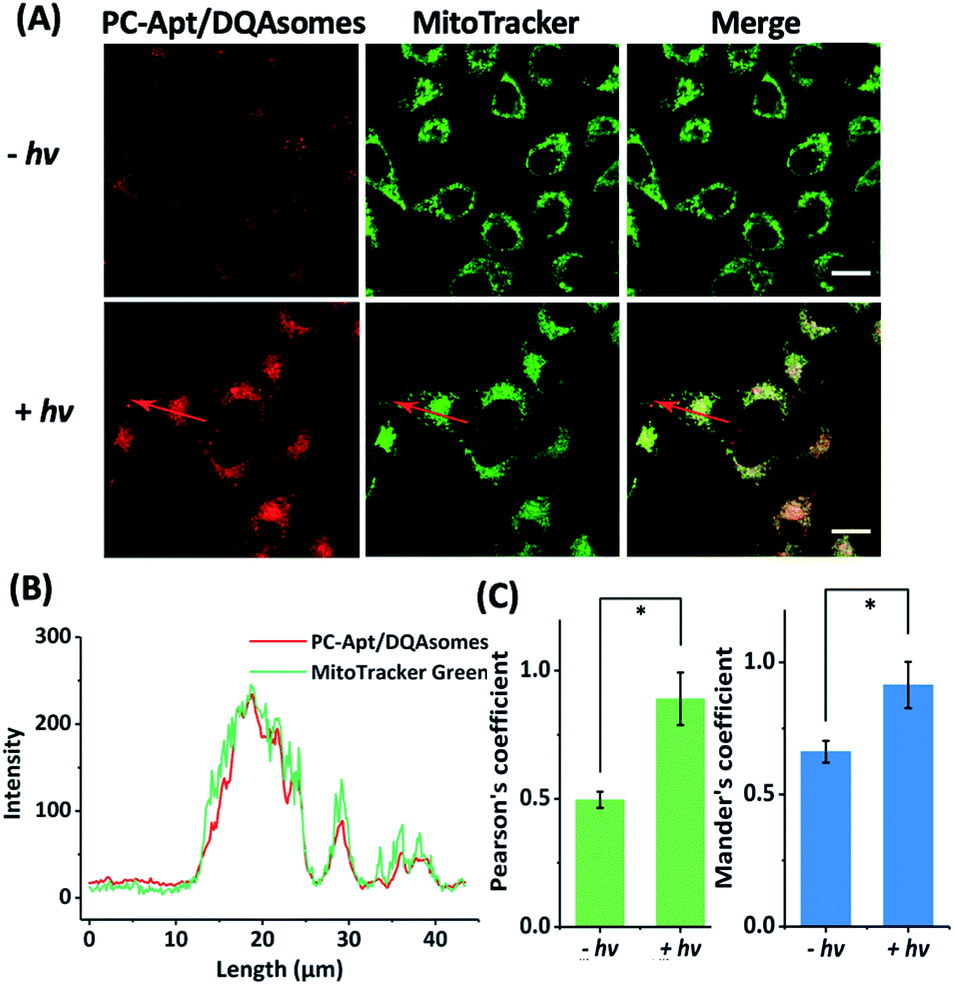 | ||
| Fig. 4 Confocal microscopy of human cells transfected with PC-Apt/DQAsome complexes. (A) Confocal microscopy of HeLa cells transfected with PC-Apt/DQAsomes (red) with (+hv) or without (−hv) 365 nm light irradiation. MitoTracker Green is used for visualization of the mitochondrial network (green). (B) Fluorescence intensity profile of regions of interest (red arrow in A). (C) Co-localization coefficient performance with (+hv) or without (−hv) light irradiation, which is analyzed by Pearson's correlation coefficient (indicating the correlation between PC-Apt/DQAsomes and mitochondrial signals) and by Mander's coefficient (representing the proportion of the PC-Apt/DQAsome signal overlapping with mitochondria). Scale bar 20 μm. *p < 0.05. | ||
To further confirm that the DQAsomes are accumulated in mitochondria, we studied the potential changes of the mitochondrial membrane after transfection with DQAsomes using the commercially available JC-1 mitochondrial membrane potential dye (Fig. S5†). For untreated cells (blank) with normal membrane potential, the JC-1 dye will accumulate in the negatively charged mitochondria and spontaneously change from a green fluorescence produced by the monomers to a red fluorescence upon formation of JC-1 aggregates.53 When the cells were treated with our DQAsomes, we found that the red fluorescence decreased, indicating that the membrane potential of the mitochondria became less negative because of the cationic DQAsome accumulation. Under this condition, the JC-1 dye is not able to reach a high enough concentration to form the JC-1 aggregates, thus retaining its original green fluorescence. In order to determine if the targeting efficiency of the DQAsomes was impacted by complexation with the negatively charged PC-Apt, we also compared the JC-1 mitochondrial membrane potential of the PC-Apt/DQAsome-transfected cells with that of DQAsome-transfected cells without PC-Apt (Fig. S5A and B†). Because we did not observe any significant difference in the membrane potentials between cells treated with DQAsomes and the PC-Apt/DQAsome complexes, these results suggest that the mitochondrial targeting ability of DQAsomes is similar with and without complexation with PC-Apt. On the basis of the above results, we conclude that DQAsomes can serve as a specific delivery reagent to deliver PC-Apt DNA probes to mitochondria.
Next, we optimized the delivery time of PC-Apt/DQAsome complexes to HeLa cells as monitored with CLSM (Fig. 5). The HeLa cells were transfected with PC-Apt/DQAsomes for different time periods up to four hours and the optimal transfection time was determined based on colocalization with MitoTracker Green. As shown in Fig. 5A, at the earlier stage of cellular uptake (2 h), the red fluorescence was relatively weak, and poorly colocalized with the staining signal of MitoTracker Green. This result may indicate that the PC-Apt/DQAsome has not yet had enough time to accumulate into the mitochondria. When the transfection time was increased to 4 h, the co-localization with MitoTracker Green significantly increased, as shown by the high values of Pearson's coefficient (0.89) and Mander's coefficient (0.92, Fig. 5B), suggesting that the PC-Apt-/DQAsomes were mostly accumulated in mitochondria.
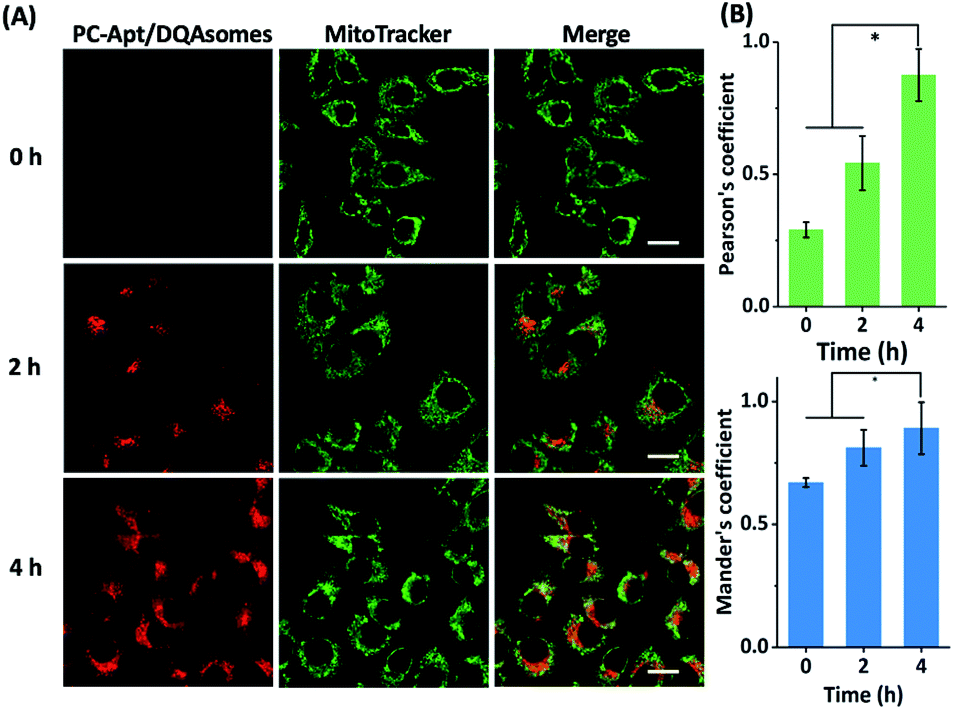 | ||
| Fig. 5 (A) Confocal microscopy of HeLa cells incubated with PC-Apt/DQAsomes for 0 h, 2 h, and 4 h followed by 20 min light irradiation. (B) Correlative co-localization coefficient at 0 h, 2 h, and 4 h, analysed by Pearson's correlation coefficient and Mander's coefficient. Scale bar 20 μm. *p < 0.05. | ||
Monitoring ATP concentration fluctuations in mitochondria
Since the mitochondrion is the primary organelle that produces ATP and the abnormal fluctuation of ATP concentrations in mitochondria would inevitably lead to changes in cellular activity,54 an assay to detect changes in ATP levels is essential to demonstrate the applicability of PC-Apt/DQAsomes for mitochondrial ATP sensing. According to previous reports, Ca2+ has been used to increase cellular ATP concentrations, while oligomycin is commonly used as an inhibitor of ATP synthase to decrease cellular ATP concentrations.42,55,56 Therefore, HeLa cells were treated with 10 μM oligomycin or 5 mM CaCl2 to regulate the cellular ATP levels before being incubated with the PC-Apt/DQAsome and compared with untreated cells. As shown in Fig. 6A, a significant fluorescence increase in the red channel was observed after light irradiation, with a higher red fluorescence signal for the Ca2+ treated samples and a lower red fluorescence signal for the oligomycin-treated samples in comparison with the untreated cells with endogenous ATP levels. In addition, the fluorescence intensity of the negative control NPC-Apt/DQAsome complexes in Fig. 6B was relatively weak in comparison with untreated cells (see Fig. 6A), regardless of whether or not 365 nm light was irradiated on the samples.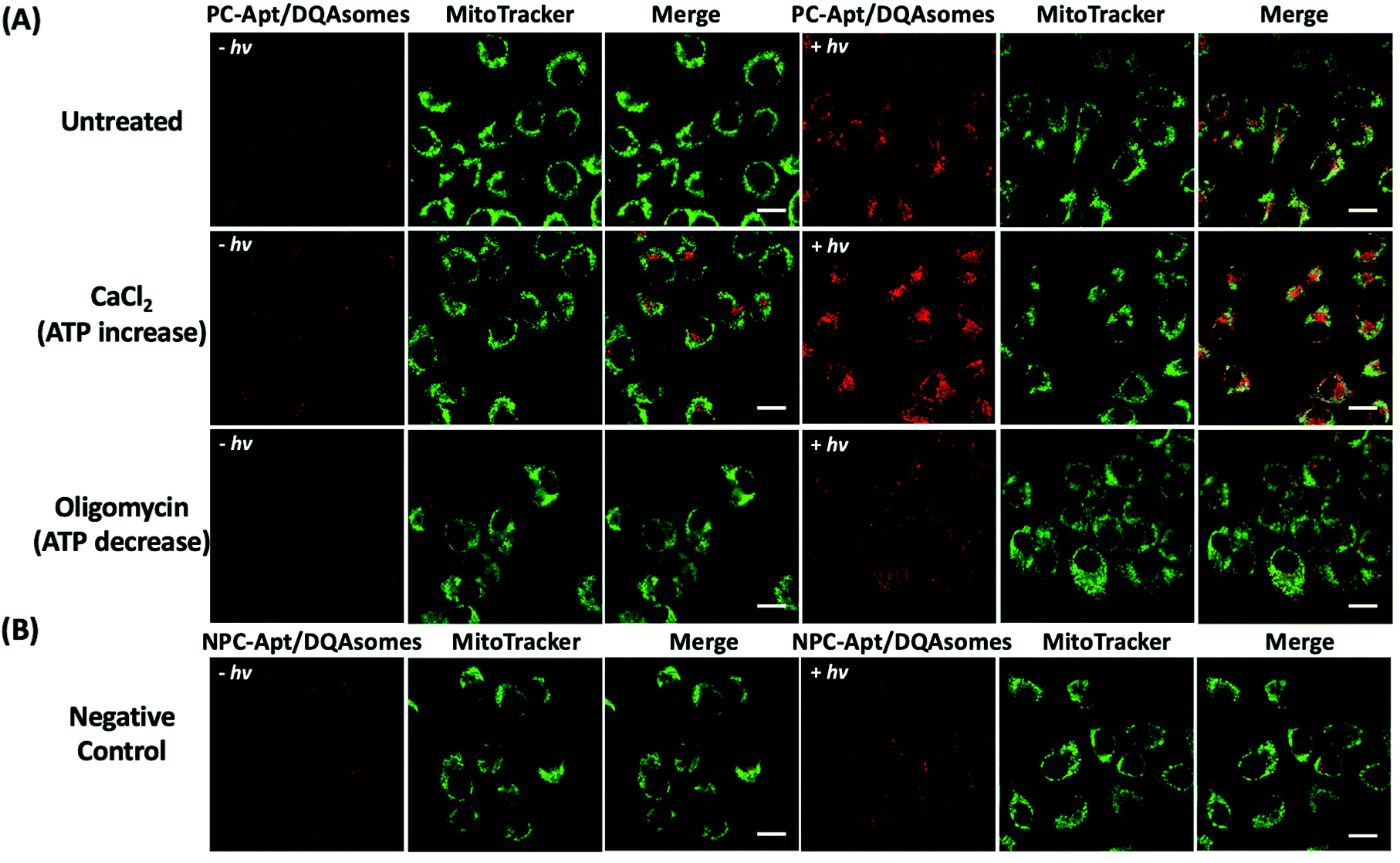 | ||
| Fig. 6 Fluorescence images of the mitochondrial ATP fluctuation with (+hv) or without (−hv) 365 nm light irradiation. (A) Fluorescence images of HeLa cells pre-treated with media (untreated), 5 mM CaCl2, or 10 μM oligomycin, followed by incubation with PC-Apt/DQAsome complexes for 4 h and finally with/without 20 min light irradiation. (B) Fluorescence images of negative control. HeLa cells were transfected with NPC-Apt/DQAsomes for 4 h with/without 20 min light irradiation. Scale bar 20 μm. | ||
To further confirm the CLSM results, flow cytometry was used to analyse the same four conditions. As shown in Fig. S6,† the intracellular fluorescence intensity of the untreated sample (endogenous ATP) with light irradiation was 2.7-fold higher than that of untreated samples without irradiation, confirming optical control of the PC-Apt sensors. Furthermore, the Ca2+ treated cells (increased ATP) with light irradiation showed a significant fluorescence increase, while the oligomycin-treated cells (decreased ATP) showed a decreased fluorescence signal. Meanwhile, the negative control using NPC-Apt sensors showed no obvious fluorescence change upon irradiation. Taken together, these results demonstrate the ability of Apt-PC/DQAsome sensors to monitor changes in mitochondrial ATP concentration caused by Ca2+ or oligomycin treatment.
Conclusions
In summary, we have demonstrated a photo-regulated ATP sensor coupled with cationic DQAsomes for spatiotemporally controlled imaging of ATP in the mitochondria of living cells. The in vitro results demonstrate that the sensor has high sensitivity and selectivity toward ATP over other nucleotides, as well as high stability under cellular conditions. The cellular results using confocal microscopy and flow cytometry demonstrate that the PC-Apt/DQAsome complex can target mitochondria to monitor differences in the ATP concentration of mitochondria in living cells. The mitochondrial targeting ability of this sensor is especially important for other DNA-based sensors, since the highly negatively charged phosphate backbone of DNA makes their delivery to mitochondria using other mitochondrial targeting moieties difficult. To the best of our knowledge, this work represents the first successful delivery of a DNA aptamer sensor to mitochondria. Furthermore, the PC-Apt DQAsome system demonstrated in this work provides a new platform for targeted delivery to subcellular organelles for monitoring energy producing processes, as well as mitochondrial dysfunction-related diseases in different cells.Experimental
Materials
All chemicals were purchased without further purification. Dequalinium chloride, oligomycin, adenosine 5′-triphosphate (ATP) disodium salt hydrate, cytidine 5′-triphosphate disodium salt (CTP), guanosine 5′-triphosphate sodium salt hydrate (GTP), uridine 5′-triphosphate tris salt (UTP), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT), sodium dodecyl sulfate (SDS), deoxycholic acid, phenylmethylsulfonyl fluoride (PMSF), and ethidium bromide (EB) were purchased from Sigma-Aldrich. PMSF Protease Inhibitor, MitoTracker Green, methanol, dimethyl sulfoxide (ACS certified) and JC-1 dye were purchased from Fisher Scientific. Phosphate-buffered saline (PBS) was purchased from Corning and Opti-MEM was purchased from Gibco. Dulbecco's modification of Eagle's medium (DMEM) was prepared by Dr Sandra McMasters of the University of Illinois School of Chemical Sciences Cell Media Facility. All experiments were performed at room temperature unless mentioned otherwise.DNA sequences
All DNA oligonucleotides were obtained from Integrated DNA Technologies (IDT). The DNA concentrations were accurately quantified through UV-vis spectroscopy. The melting temperature values labelled in Fig. 1A were calculated by the DINAMelt Web server (http://unafold.rna.albany.edu//?q=DINAMelt/Two-state-melting).Cy3 labelled aptamer strand (Apt-Cy3): 5′-Cy3-AGAGAACCTGGGGGAGTATTGCGGAGGAAGGT-3′.
Quencher labelled complementary strand with the PC linker (PC-Blocker-Q): 5′-CTCCGCAAPCCCCAGGTTCTCT-Iowa Black FQ-3′.
Quencher labelled complementary strand without the PC linker (NPC-Q): 5′-CTCCGCAACCCAGGTTCTCT-Iowa Black FQ-3′.
Preparation of the DQAsome and sensor/DQAsome complex
DQAsomes were prepared according to a previous report.45 26 mg dequalinium chloride was dissolved in 5 mL methanol in a 25 mL round-bottom flask, and then a rotary evaporator was used to remove methanol to obtain a dequalinium film at 37 °C for 1 h. The resulting film was hydrated with 5 mL HEPES buffer (5 mM HEPES, pH 7.4) and sonicated using a CPX2800 ultrasonic bath (Fisher Scientific) for 30 min until a clear opalescent solution of DQAsomes was obtained. The resulting preparation was then measured using a Malvern Nano ZS90 instrument to determine the size distribution and zeta-potential.The sensor/DQAsome complex was prepared by simply mixing 200 nM PC-Apt or FQ-Apt sensors with 20 μg mL−1 DQAsomes for 30 min in Opti-MEM.
Preparation of PC-Apt and NPC-Apt sensors
PC-Apt was obtained by hybridization of Apt-Cy3 with PC-Blocker-Q in Tris buffer (50 mM Tris, 100 mM NaCl, 5 mM MgCl2 pH 8) with a final ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5. NPC-Apt was obtained by hybridization of Apt-Cy3 with NPC-Blocker-Q in the same buffer and with the same ratio. The solutions were annealed at 95 °C for 5 min and stored at room temperature to allow full hybridization.
:1.5. NPC-Apt was obtained by hybridization of Apt-Cy3 with NPC-Blocker-Q in the same buffer and with the same ratio. The solutions were annealed at 95 °C for 5 min and stored at room temperature to allow full hybridization.
In vitro ATP sensing assay
The PC-Apt sensors were diluted to 200 nM for ATP sensing assays. Different concentrations of ATP (final concentrations of 0, 0.05, 0.1, 0.2, 0.5, 1, 2, 3, 4, and 5 mM) were mixed with 200 nM PC-Apt or NPC-Apt probes in Tris buffer (50 mM Tris, 100 mM NaCl, 5 mM MgCl2 pH 8) and incubated at room temperature for 20 min. For the photo-regulated sensing assay, the PC-Apt or NPC-Apt with different concentrations of ATP mixtures was irradiated with a 365 nm handheld lamp (UVGL-25, Analytikjena, US) for 20 min. For the selectivity assays, CTP, GTP, and UTP were used in comparison to ATP at a final concentration of 5 mM. The fluorescence spectra were collected with emission wavelengths from 540 to 700 nm at an excitation wavelength of 530 nm.Cell culture and cytotoxicity
HeLa cells were used in all mammalian cell experiments and were bought from American Type Culture Collection (ATCC). Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum, 100 U mL−1 penicillin, and 100 U mL−1 streptomycin was used to culture the cells in a 5% CO2, 37 °C incubator. A hemocytometer was used to determine cell density.The cell viability was determined using a standard MTT assay. HeLa cells were seeded at a density of 7000 cells per well in 96-well plates. The cells were grown to 60–70% confluency and incubated with 20 μg mL−1 DQAsomes and either 200 nM PC-Apt (PC-Apt/DQAsome complex) or 200 nM NPC-Apt (NPC-Apt/DQAsome complex) for 4 h. After irradiation with 365 nm light for 20 min, the cells were maintained for another 20 h. The MTT assays were performed to determine the cell viability and the absorbance at 570 nm was measured with a Spectramax M2 microplate reader. The untreated cells were used as a blank control for normalization.
Cell lysate preparation
Cell lysate was prepared according to previous methods.57,58 HeLa cells were collected at 80% confluency and 1 × 106 cells were dispensed in a 1.5 mL tube. After washing twice with cold PBS, the cells were resuspended in 500 μL cold cell lysis buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% deoxycholic acid and 0.1 mM PMSF), and then incubated on ice for 20 min. Finally, the resulting solution was centrifuged at 12000 rpm at 4 °C for 30 min, and the supernatant was collected in a new tube and stored at −20 °C for further use.
Confocal microscopy imaging of mitochondrial ATP
HeLa cells were seeded in 35 mm glass-bottomed culture dishes (MatTek Corporation, P35GC-1.5-14-C) at a density of 2 × 105 cells per dish and grown up to 60% confluency. The growth medium was then replaced with PC-Apt/DQAsomes in Opti-MEM. Cells without treatment were used as the blank control. A time series of cell culture was performed, including 0 h, 2 h, and 4 h incubation with PC-Apt/DQAsomes. To demonstrate the possibility of PC-Apt/DQAsomes for monitoring differences of mitochondrial ATP, the cells were pre-treated with Opti-MEM containing 10 μM oligomycin or 5 mM CaCl2 for 2 h, as described in previous studies.42 The cell samples were then incubated with PC-Apt/DQAsomes for another 4 h, followed by thorough washing with PBS. At the end of incubation, the samples were irradiated with 365 nm light for 20 min and incubated for another 1 h. Before imaging, the cell samples were stained with 100 nM MitoTracker Green (Ex: 490 nm, Em: 516 nm) for 15 min at 37 °C. After a washing step, the cell samples were imaged with a LSM 880 (Zeiss) confocal microscope with a 63× oil objective. The excitation wavelengths were 488 nm (Green Channel) and 561 nm (Red Channel). Acquired images were analysed by the software ImageJ (https://imagej.net/) using the JACop plugin (available at https://imagej.nih.gov/ij/plugins/track/jacop2.html). Pearson's and Mander's coefficients were calculated based on multiple cells and optical sections.Flow cytometry
HeLa cells were seeded in 12 well plates at a density of 1.5 × 105 cells per well and grown to 80% confluence. After washing with PBS, the cells were treated with Opti-MEM containing 10 μM oligomycin or 5 mM CaCl2 for 2 h, and then the cells were incubated with PC-Apt/DQAsomes or NPC-Apt/DQAsomes for 4 h. Finally, the cells were detached with 0.25% (w/v) trypsin and washed twice with PBS before analysis with flow cytometry (BD LSR II, BD Biosciences).Detection of mitochondrial membrane potential
The JC-1 dye was used for detecting the mitochondrial membrane potential after incubation with DQAsomes. The JC-1 dye could accumulate in mitochondria and indicate the mitochondrial membrane potential by the fluorescence ratio of green/red colours. After incubation with 20 μg mL−1 DQAsomes for 4 h, the HeLa cells were washed with PBS and stained with 10 μg mL−1 JC-1 for 20 min at 37 °C. The cell samples were washed with PBS three times before imaging. The images were collected with the green emission channel (500–540 nm, for JC-1 monomer) or the red emission channel (560–610 nm, for JC-1 aggregates), and the excitation wavelength was set at 488 nm. Fluorescence was measured with a Spectramax M2 microplate reader.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the DOE Center for Advanced Bioenergy and Bioproducts Innovation (U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under Award Number DE-SC0018420) and the CAS/SAFEA International Innovation Teams program. Any opinions, findings, and conclusions or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the views of the U.S. Department of Energy. We thank Dr Sandra McMasters of the University of Illinois School of Chemical Sciences Cell Media Facility for assistance with cell culturing, the Institute for Genomic Biology Core Facility for assistance with confocal microscopy, and the Roy J. Carver Biotechnology for assistance with flow cytometry and use of the fluorescence microplate reader. We also thank Zhenglin Yang, Yao Lin, Lu Chen, Mengyi Xiong, Quanbing Mou, and Yuan Ma for their helpful suggestions and assistance throughout this work.Notes and references
- J. S. Paige, T. Nguyen-Duc, W. Song and S. R. Jaffrey, Science, 2012, 335, 1194 CrossRef CAS PubMed.
- F. Li, J. Li, Y. Tang, C. Wang, X. F. Li and X. C. Le, Proteomes, 2017, 5, 1 CrossRef PubMed.
- Y. Du and S. Dong, Anal. Chem., 2017, 89, 189–215 CrossRef CAS PubMed.
- S. Song, L. Wang, J. Li, C. Fan and J. Zhao, Trends Anal. Chem., 2008, 27, 108–117 CrossRef CAS.
- C. Feng, S. Dai and L. Wang, Biosens. Bioelectron., 2014, 59, 64–74 CrossRef CAS PubMed.
- L. Xu, H. Zhang, X. Yan, H. Peng, Z. Wang, Q. Zhang, P. Li, Z. Zhang and X. C. Le, ACS Sens., 2018, 3, 2590–2596 CrossRef CAS PubMed.
- W. Zhong and J. T. Sczepanski, ACS Sens., 2019, 4, 566–570 CrossRef CAS.
- W. Zhao, W. Chiuman, M. A. Brook and Y. Li, ChemBioChem, 2007, 8, 727–731 CrossRef CAS.
- T. Li, S. Dong and E. Wang, J. Am. Chem. Soc., 2010, 132, 13156–13157 CrossRef CAS.
- C. L. Hamula, H. Zhang, L. L. Guan, X. F. Li and X. C. Le, Anal. Chem., 2008, 80, 7812–7819 CrossRef CAS.
- C. L. A. Hamula, H. Zhang, F. Li, Z. Wang, X. Chris Le and X. F. Li, Trends Anal. Chem., 2011, 30, 1587–1597 CrossRef CAS.
- H. Zhang, X. F. Li and X. C. Le, Anal. Chem., 2009, 81, 7795–7800 CrossRef CAS.
- M. Liu, Q. Yin, Y. Chang, Q. Zhang, J. D. Brennan and Y. Li, Angew. Chem., Int. Ed., 2019, 58, 8013–8017 CrossRef CAS.
- J. Liu, D. Mazumdar and Y. Lu, Angew. Chem., Int. Ed., 2006, 45, 7955–7959 CrossRef CAS.
- W. Zhou, P. J. Huang, J. Ding and J. Liu, Analyst, 2014, 139, 2627–2640 RSC.
- Y. Du, C. Chen, J. Yin, B. Li, M. Zhou, S. Dong and E. Wang, Anal. Chem., 2010, 82, 1556–1563 CrossRef CAS PubMed.
- T. T. Chen, X. Tian, C. L. Liu, J. Ge, X. Chu and Y. Li, J. Am. Chem. Soc., 2015, 137, 982–989 CrossRef CAS PubMed.
- Y. Helwa, N. Dave, R. Froidevaux, A. Samadi and J. Liu, ACS Appl. Mater. Interfaces, 2012, 4, 2228–2233 CrossRef CAS PubMed.
- H. Xu, Q. Li, L. Wang, Y. He, J. Shi, B. Tang and C. Fan, Chem. Soc. Rev., 2014, 43, 2650–2661 RSC.
- J. Liu and Y. Lu, Angew. Chem., Int. Ed., 2005, 45, 90–94 CrossRef.
- H. M. Meng, H. Liu, H. Kuai, R. Peng, L. Mo and X. B. Zhang, Chem. Soc. Rev., 2016, 45, 2583–2602 RSC.
- J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS PubMed.
- A. V. Gourine, E. Llaudet, N. Dale and K. M. Spyer, Nature, 2005, 436, 108–111 CrossRef CAS.
- J. Liu and Y. Lu, Anal. Chem., 2004, 76, 1627–1632 CrossRef CAS.
- Y. Xiang, A. Tong and Y. Lu, J. Am. Chem. Soc., 2009, 131, 15352–15357 CrossRef CAS.
- L. L. Li, P. Ge, P. R. Selvin and Y. Lu, Anal. Chem., 2012, 84, 7852–7856 CrossRef CAS PubMed.
- J. Liu and Y. Lu, Nat. Protoc., 2006, 1, 246–252 CrossRef CAS.
- J. N. Weiss and S. T. Lamp, J. Gen. Physiol., 1989, 94, 911 CrossRef CAS.
- D. R. Green and G. Kroemer, Science, 2004, 305, 626–629 CrossRef CAS PubMed.
- A. Johri and M. F. Beal, J. Pharmacol. Exp. Ther., 2012, 342, 619–630 CrossRef CAS.
- D. C. Wallace, Science, 1999, 283, 1482 CrossRef CAS.
- H. Yokoshiki, M. Sunagawa, T. Seki and N. Sperelakis, Am. J. Physiol.: Cell Physiol., 1998, 274, C25–C37 CrossRef CAS.
- H. M. Meng, X. Zhang, Y. Lv, Z. Zhao, N. N. Wang, T. Fu, H. Fan, H. Liang, L. Qiu, G. Zhu and W. Tan, ACS Nano, 2014, 8, 6171–6181 CrossRef CAS PubMed.
- C. Wu, T. Chen, D. Han, M. You, L. Peng, S. Cansiz, G. Zhu, C. Li, X. Xiong, E. Jimenez, C. J. Yang and W. Tan, ACS Nano, 2013, 7, 5724–5731 CrossRef CAS.
- J. Zhang, L. P. Smaga, N. S. R. Satyavolu, J. Chan and Y. Lu, J. Am. Chem. Soc., 2017, 139, 17225–17228 CrossRef CAS.
- H. Pei, L. Liang, G. Yao, J. Li, Q. Huang and C. Fan, Angew. Chem., Int. Ed., 2012, 51, 9020–9024 CrossRef CAS.
- H. Xing, N. Y. Wong, Y. Xiang and Y. Lu, Curr. Opin. Chem. Biol., 2012, 16, 429–435 CrossRef CAS PubMed.
- Z. Cao, R. Tong, A. Mishra, W. Xu, G. C. L. Wong, J. Cheng and Y. Lu, Angew. Chem., Int. Ed., 2009, 48, 6494–6498 CrossRef CAS.
- P. Song, D. Ye, X. Zuo, J. Li, J. Wang, H. Liu, M. T. Hwang, J. Chao, S. Su, L. Wang, J. Shi, L. Wang, W. Huang, R. Lal and C. Fan, Nano Lett., 2017, 17, 5193–5198 CrossRef CAS.
- S. Saha, V. Prakash, S. Halder, K. Chakraborty and Y. Krishnan, Nat. Nanotechnol., 2015, 10, 645 CrossRef CAS.
- J. Zhao, J. Gao, W. Xue, Z. Di, H. Xing, Y. Lu and L. Li, J. Am. Chem. Soc., 2018, 140, 578–581 CrossRef CAS PubMed.
- X. Zheng, R. Peng, X. Jiang, Y. Wang, S. Xu, G. Ke, T. Fu, Q. Liu, S. Huan and X. Zhang, Anal. Chem., 2017, 89, 10941–10947 CrossRef CAS.
- Z. Tan, T. A. Feagin and J. M. Heemstra, J. Am. Chem. Soc., 2016, 138, 6328–6331 CrossRef CAS.
- Z. Yang, K. Y. Loh, Y. T. Chu, R. Feng, N. S. R. Satyavolu, M. Xiong, S. M. Nakamata Huynh, K. Hwang, L. Li, H. Xing, X. Zhang, Y. R. Chemla, M. Gruebele and Y. Lu, J. Am. Chem. Soc., 2018, 140, 17656–17665 CrossRef CAS.
- V. Weissig, J. Lasch, G. Erdos, H. W. Meyer, T. C. Rowe and J. Hughes, Pharm. Res., 1998, 15, 334–337 CrossRef CAS.
- J. H. Liu, R. S. Li, B. Yuan, J. Wang, Y. F. Li and C. Z. Huang, Nanoscale, 2018, 10, 17402–17408 RSC.
- V. Weissig, C. Lizano and V. P. Torchilin, Drug Delivery, 2000, 7, 1–5 CrossRef CAS.
- G. G. D'Souza, R. Rammohan, S. M. Cheng, V. P. Torchilin and V. Weissig, J. Controlled Release, 2003, 92, 189–197 CrossRef.
- G. G. D'Souza, S. V. Boddapati and V. Weissig, Mitochondrion, 2005, 5, 352–358 CrossRef.
- Y. Bae, M. K. Jung, S. J. Song, E. S. Green, S. Lee, H. S. Park, S. H. Jeong, J. Han, J. Y. Mun, K. S. Ko and J. S. Choi, Mitochondrion, 2017, 37, 27–40 CrossRef CAS PubMed.
- S. Zupancic, P. Kocbek, M. G. Zariwala, D. Renshaw, M. O. Gul, Z. Elsaid, K. M. Taylor and S. Somavarapu, Mol. Pharm., 2014, 11, 2334–2345 CrossRef CAS.
- P. Venco, M. Bonora, C. Giorgi, E. Papaleo, A. Iuso, H. Prokisch, P. Pinton and V. Tiranti, Front. Genet., 2015, 6, 185 Search PubMed.
- F. Sivandzade, A. Bhalerao and L. Cucullo, Bio-Protoc., 2019, 9, e3128 Search PubMed.
- L. Wang, L. Yuan, X. Zeng, J. Peng, Y. Ni, J. C. Er, W. Xu, B. K. Agrawalla, D. Su, B. Kim and Y. T. Chang, Angew. Chem., Int. Ed., 2016, 55, 1773–1776 CrossRef CAS PubMed.
- E. K. Ainscow and G. A. Rutter, Diabetes, 2002, 51, S162 CrossRef CAS PubMed.
- Y. Gong, H. Sohn, L. Xue, G. L. Firestone and L. F. Bjeldanes, Cancer Res., 2006, 66, 4880 CrossRef CAS PubMed.
- Y. Chen, P. Wang, Y. Liu, T. Liu, Y. Xu, S. Zhu, J. Zhu, K. Ye, G. Huang and H. Dannong, Nanotechnology, 2018, 29, 035102 CrossRef.
- W. Wang, N. S. R. Satyavolu, Z. Wu, J. R. Zhang, J. J. Zhu and Y. Lu, Angew. Chem., Int. Ed., 2017, 56, 6798–6802 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04773e |
| This journal is © The Royal Society of Chemistry 2020 |