DOI:
10.1039/C9SC04294F
(Edge Article)
Chem. Sci., 2020,
11, 5339-5346
Observation of binding of carbon dioxide to nitro-decorated metal–organic frameworks†‡
Received
25th August 2019
, Accepted 26th February 2020
First published on 14th May 2020
Abstract
Metal–organic frameworks (MOFs) functionalised with amine, amide and hydroxyl groups show great promise for CO2 binding due to their ability to form hydrogen bonds to CO2. Herein we report the adsorption and selectivity of CO2 in four iso-reticular MOFs adopting the NbO topology. Functionalisation of the parent MOF, MFM-102, with –NO2, –NH2 and alkyl groups leads to an enhancement of CO2 adsorption of up to 36% for the NO2-decorated MOF and with raised selectivity. MFM-102-NO2 shows the highest adsorption capacity for CO2 (184 cm3 g−1 at 273 K and 1.0 bar) within this series, comparable to the best-behaving iso-reticular MOFs. At 298 K and 1.0 bar, MFM-102-NO2 shows a CO2/CH4 selectivity of 5.0. In situ inelastic neutron scattering and synchrotron FT-IR micro-spectroscopy were employed to elucidate the host–guest interaction dynamics within CO2-loaded MFM-102-NO2. Neutron powder diffraction enabled the direct observation of the preferred binding domains in MFM-102-NO2, and, to the best of our knowledge, we report the first example of CO2 binding to a –NO2 group in a porous MOF. Synergistic effects between the –NO2 group and the open metal sites lead to optimal binding of CO2 molecules within MFM-102-NO2via hydrogen bonding to C–H groups.
Introduction
Emissions of carbon dioxide (CO2) are causing significant environmental impacts.1 However, CO2 is also an inexpensive, non-toxic and a potentially abundant renewable C1 source, and, therefore, the development of cost-effective and efficient methods for carbon capture and storage (CCS) is a major target. State-of-the-art CCS is often based upon scrubbing systems that rely heavily upon the use of toxic and corrosive amines, and require high operational cost. In contrast, reversible physisorption of CO2 within porous materials is a promising approach with the potential of low running-cost and high efficiency. A wide spectrum of materials, such as zeolites, activated carbons, ionic liquids and silica-based materials have been investigated for CO2 adsorption.2–5
Metal–organic framework (MOF) materials show potential for gas adsorption and storage owing to their high porosity and designed pore size and structure reflecting their versatility via incorporation of active binding sites by decoration of the pores with functional groups and/or by incorporation of open metal sites.6,7 MOFs decorated with –NH2,8 –OH,9 –CONH–,10 piperazine,11 and pyridyl groups12 have been explored extensively for CO2 adsorption. The nitro group (−NO2) is one of the most powerful electron-withdrawing groups, which can polarise –CH groups on phenyl rings. We argued that potentially this might lead to enhanced interactions between –CH groups and adsorbed CO2 molecules, and moreover, the –NO2 group might itself serve as a binding site for CO2 molecules via dipole/quadrupole interactions.13,14 However, to date, experimental evidence of CO2 binding to NO2-decorated MOFs has not been reported. Herein, we report the adsorption and selectivity of CO2 in four iso-reticular NbO-type MOFs with different organic functional groups, namely –NO2, –NH2 and alkyl groups. The introduction of these functional groups inevitably reduces the pore volume of the parent MOF, but leads to notable enhancement in CO2 adsorption. In particular, the NO2-functionalised MOF MFM-102-NO2 shows a 36% increase in CO2 uptake compared to the parent MFM-102, confirming a positive effect on CO2 binding. A combination of static and dynamic experiments using in situ neutron powder diffraction (NPD), inelastic neutron scattering (INS) and synchrotron-based IR micro-spectroscopy have been applied to study the binding domains of adsorbed CO2 and the host–guest binding dynamics in MFM-102-NO2 as a function of CO2 loading to reveal fundamental insights into the mechanisms for enhanced CO2 adsorption.15
Experimental
Material and instrumentation
Chemicals and reagents were purchased from Fisher Scientific, Sigma Aldrich or Fluorochem and used as received without further purification. The detailed synthesis of MFM-102 has been reported previously.161H NMR and 13C NMR spectra were measured on a Bruker AV400 or AV500 spectrometer. High-resolution electrospray mass spectra were measured on a Bruker MicroTOF spectrometer with samples dissolved in MeOH, and scanning was conducted in both positive and negative modes. Infrared (IR) spectra were recorded in the 400–4000 cm−1 range in ATR sampling mode with a Thermo Scientific iD5 diamond ATR on a Nicolet iS5 FT-IR spectrometer, and elemental analysis was carried out on a CE-440 elemental analyser (EAI Company). TGA measurements were performed using a PerkinElmer TGA 7 Gravimetric Analyser under a flow of N2 (20 ml min−1) at a heating rate of 5 °C min−1. Powder X-ray diffraction (PXRD) patterns were obtained on a PANalytical X'Pert Pro MPD diffractometer in Bragg–Brentano geometry with Cu-Kα1 radiation (λ = 1.5406 Å) at 40 kV and 30 mA over 2θ range 3–40°. Samples were evenly dispersed on zero-background silicon plates with a cavity depth of 0.3 mm.
Neutron powder diffraction (NPD) of gas-loaded MFM-102-NO2
NPD data were collected at WISH beamline at ISIS Muon and Neutron facility. Acetone exchanged MFM-102-NO2 was loaded into vanadium cans of 11 mm diameter, and the sample was activated by heating at 393 K and at 1 × 10−7 mbar for 2 days. Data for the bare framework were collected after placing the sample can into a liquid He cryostat and cooling to 7 K. CO2 gas was dosed into the system after warming to 290 K. The gases were dosed volumetrically from a calibrated volume, and to ensure gases were fully adsorbed into the sample without condensation elsewhere the system was cooled to 7 K slowly over 3 h. Structure solutions of gas-loaded MFM-102-NO2 were solved by sequential difference Fourier analyses followed by Rietveld refinements (see ESI† Section 4).
Inelastic neutron scattering (INS) of CO2-loaded MFM-102-NO2 and MFM-102-NH2
INS data were collected on the TOSCA beamline at ISIS Muon and Neutron facility and on the VISION spectrometer at Spallation Neutron Source, Oak Ridge National Laboratory (USA). MFM-102-NO2 and MFM-102-NH2 were loaded into a vanadium can of 11 mm diameter and outgassed at 10−7 mbar at 393 K for 2 days. After placing the sample into a He cooled cryostat, INS data of the bare framework were collected at 7 K. CO2 was dosed volumetrically from a calibrated volume at room temperature and gradually cooled to 7 K to allow the gas to fully adsorb into MFM-102-NO2 and MFM-102-NH2. INS data of CO2-loaded MFM-102-NO2 and CO2-loaded MFM-102-NH2 were collected at 7 K. Experimentally obtained INS data were compared with modelled data obtained via density functional theory (DFT) calculations (see ESI† for further details).
Synchrotron FTIR micro-spectroscopy of CO2-loaded MFM-102-NO2
Single crystals of MFM-102-NO2 were loaded onto a ZnSe slide and placed into a Linkam FTIR600 variable temperature gas-tight cell fitted with ZnSe windows. The MOF sample was activated in situ under a flow of N2 whilst heating the Linkam stage to 413 K for 6 h. Partial pressures of zeolite dried gases N2 and CO2 were controlled by varying the volumetric flow of the two gases via separate mass flow controllers. FTIR spectra were collected at B22 MIRIAM beamline at Diamond Light Source using a highly bright synchrotron IR source connected to a Bruker Hyperion 3000 IR microscope with a 15× objective and MCT detector (liq. N2 cooled). Spectra (256 scans) were measured at room temperature with a 20 × 20 μm beam, in the spectral range of 4000–650 cm−1 (4 cm−1 resolution).
Results and discussion
Synthesis, structure and porosity
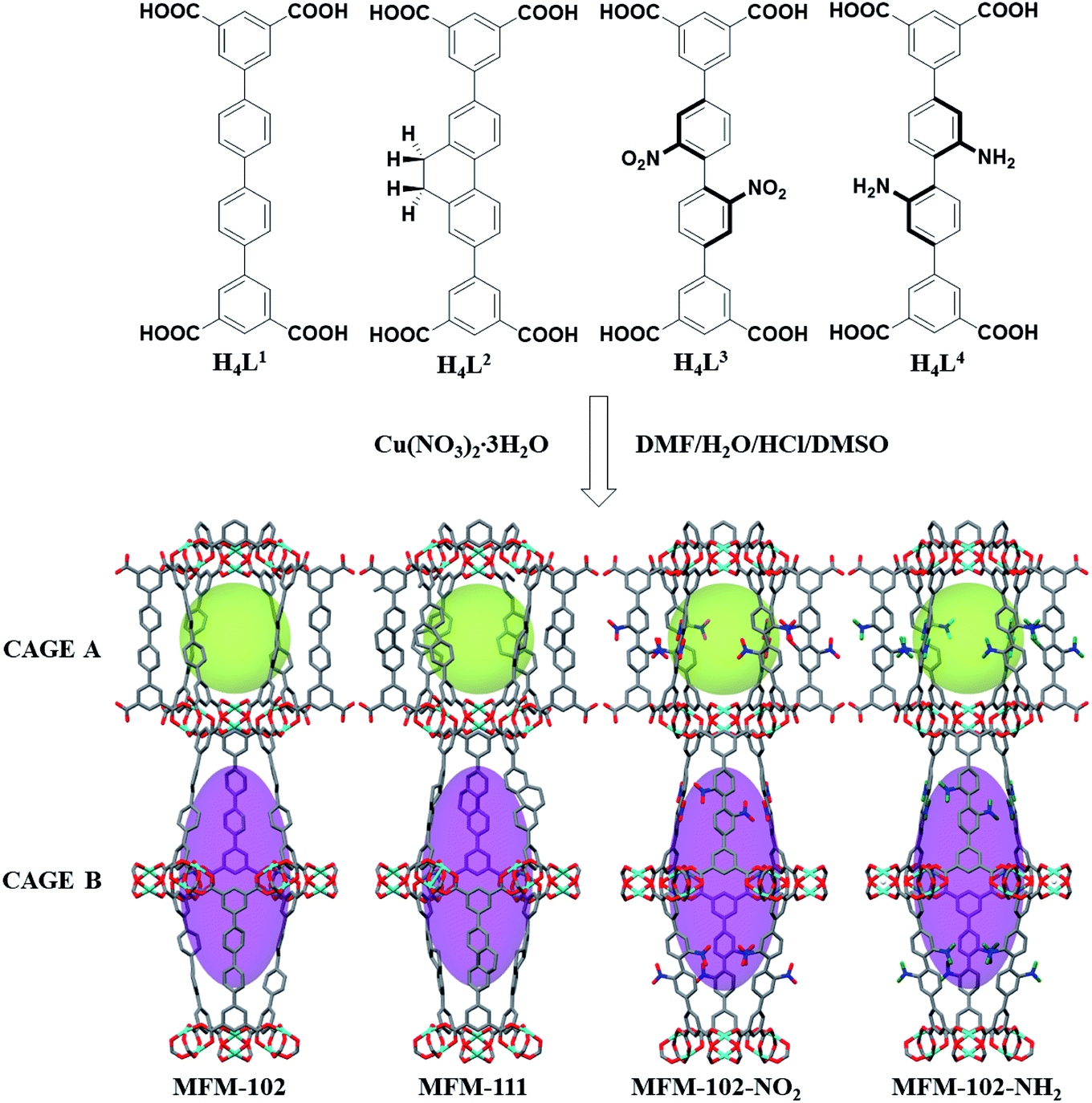
MFM-102, MFM-102-NH2, MFM-102-NO2 and MFM-111 were synthesised according to our previously reported methods,16–18 and the phase purity of all bulk materials was confirmed by powder X-ray diffraction (see ESI† Section 1). All four MOFs are iso-structural and crystallise in the trigonal space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. The framework is built from Cu2(O2CR)4 paddlewheel units in which two CuII cations are bridged by four carboxylate groups surrounded the [Cu⋯Cu] axis and two terminal H2O molecules at axial positions to form a [Cu2(O2CR)4(OH2)2] node. These Cu2(O2CR)4 units are bridged by the diisophthalate ligands to afford a 3D extended NbO-type network. Within each framework, there are two different types of metal–ligand cages (denoted as A and B) in a 1
m. The framework is built from Cu2(O2CR)4 paddlewheel units in which two CuII cations are bridged by four carboxylate groups surrounded the [Cu⋯Cu] axis and two terminal H2O molecules at axial positions to form a [Cu2(O2CR)4(OH2)2] node. These Cu2(O2CR)4 units are bridged by the diisophthalate ligands to afford a 3D extended NbO-type network. Within each framework, there are two different types of metal–ligand cages (denoted as A and B) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. 1). Cage A consists of six Cu2(O2CR)4 paddlewheels and six linkers to give a spherical shape of 15 Å diameter. Cage B is constructed of twelve Cu2(O2CR)4 paddlewheels residing at the vertices and six ligands on the faces to form an ellipsoid shaped cavity of 32 Å along the c axis and 18 Å across ab plane. Due to the alternative packing of cages A and B, the alkyl, nitro, and amino groups are directly grafted into the pore walls, providing an excellent platform to study their role in guest binding. The Brunauer–Emmett–Teller (BET) surface area and pore volume of MFM-102, MFM-111, MFM-102-NH2, MFM-102-NO2 were determined to be 3412, 2930, 2928, 2893 m2 g−1 and 1.29, 1.19, 1.12, 1.14 cm3 g−1, respectively, from N2 isotherms at 77 K. As expected, the porosity of decorated complexes reduces compared to the parent MOF, and so introduction of alkyl, –NH2 and –NO2 groups into MFM-102 leads to reduction in BET surface areas of 14%, 14% and 15%, respectively.
:1 ratio (Fig. 1). Cage A consists of six Cu2(O2CR)4 paddlewheels and six linkers to give a spherical shape of 15 Å diameter. Cage B is constructed of twelve Cu2(O2CR)4 paddlewheels residing at the vertices and six ligands on the faces to form an ellipsoid shaped cavity of 32 Å along the c axis and 18 Å across ab plane. Due to the alternative packing of cages A and B, the alkyl, nitro, and amino groups are directly grafted into the pore walls, providing an excellent platform to study their role in guest binding. The Brunauer–Emmett–Teller (BET) surface area and pore volume of MFM-102, MFM-111, MFM-102-NH2, MFM-102-NO2 were determined to be 3412, 2930, 2928, 2893 m2 g−1 and 1.29, 1.19, 1.12, 1.14 cm3 g−1, respectively, from N2 isotherms at 77 K. As expected, the porosity of decorated complexes reduces compared to the parent MOF, and so introduction of alkyl, –NH2 and –NO2 groups into MFM-102 leads to reduction in BET surface areas of 14%, 14% and 15%, respectively.
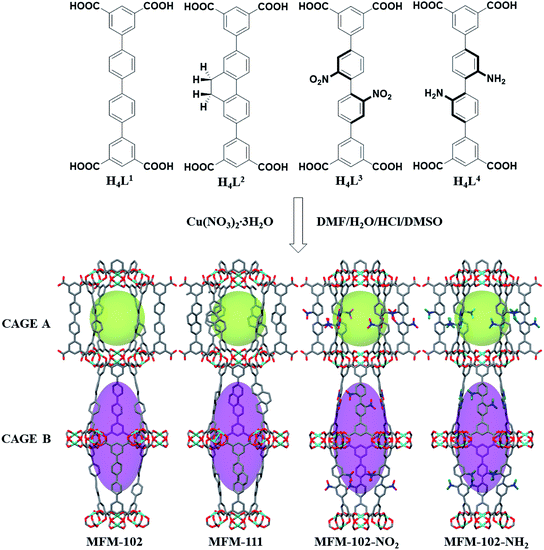 |
| | Fig. 1 Views of organic ligands used to construct the MFM-102 series of MOFs and the X-ray crystal structures of MFM-102, MFM-111, MFM-102-NO2 and MFM-102-NH2. The functional groups are highlighted in ball-stick mode. (C, dark grey; O, red; N, blue; Cu, turquoise; all coordinated H2O and hydrogen atoms are omitted for clarity, except for the hydrogen atoms on the –NH2 group, green). | |
Gas adsorption properties
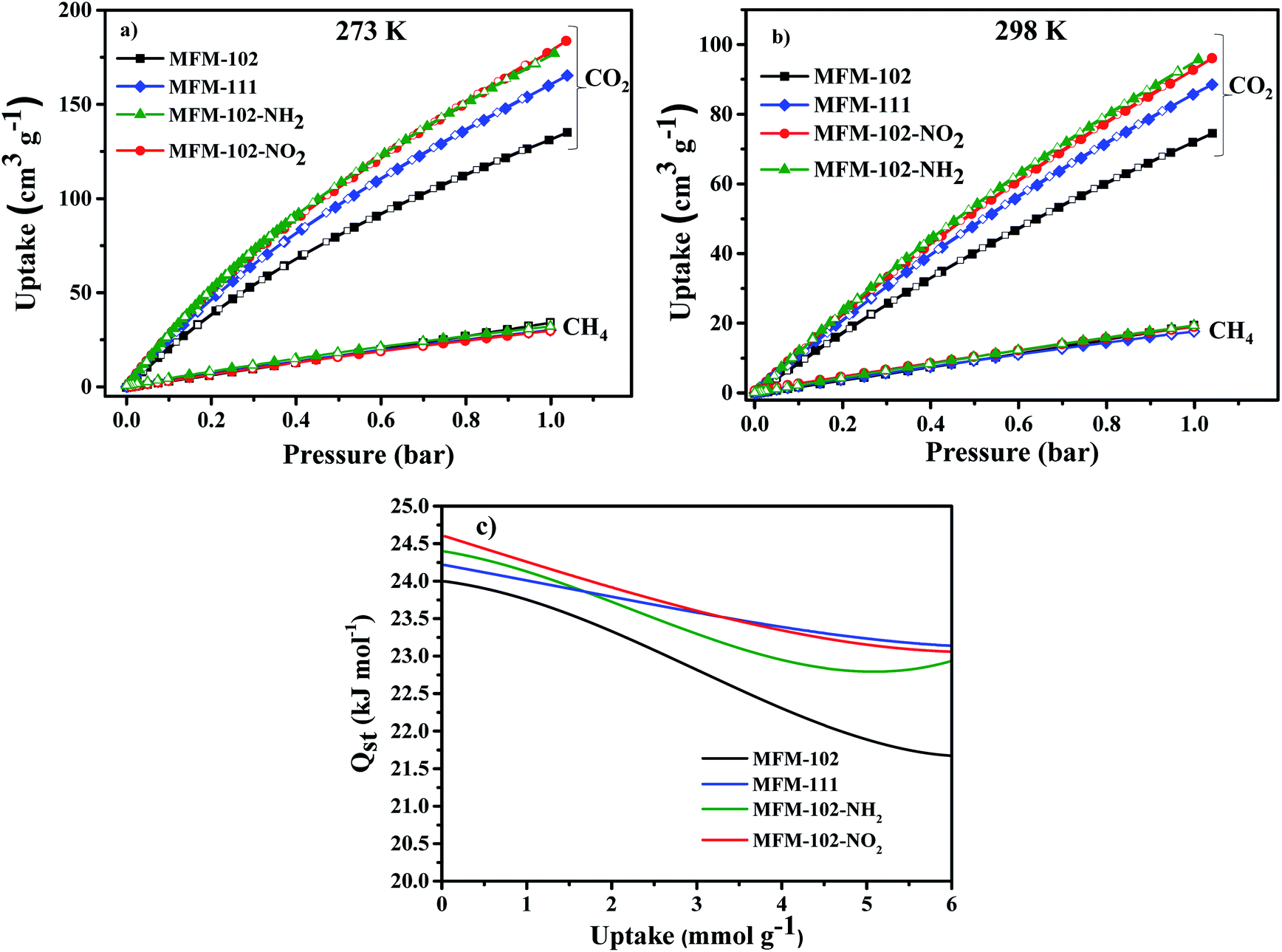
Adsorption isotherms of CO2 and CH4 were recorded at 273 and 298 K up to 1 bar. All isotherms are completely reversible, and show fast adsorption–desorption kinetics with equilibrium achieved typically within 3–5 min. Uptake of CO2 at 273 K and 1.0 bar were recorded as 184, 178, 165, 135 cm3 g−1 for MFM-102-NO2, MFM-102-NH2, MFM-111 and MFM-102, respectively (Fig. 2a). Interestingly, despite the reduction in porosity, the introduction of alkyl, –NH2 and –NO2 groups into MFM-102 leads to enhancement on the CO2 uptake of resultant MOFs by 22%, 32% and 36%, respectively. Notably, the CO2 uptake of MFM-102-NO2 is comparable to the best-performing iso-reticular MOF (NJU-Bai-14, 187 cm3 g−1) under the same conditions.19 Similar enhancement of CO2 adsorption was also observed at 298 K for the decorated MOFs (Fig. 2b), and these uptakes are comparable to other leading NbO-type MOFs (Table 1). In sharp contrast, these MOFs show similarly low uptakes of CH4 at 273 and 298 K and 1.0 bar (ranging from 30 cm3 g−1 to 20 cm3 g−1). This result indicates the potential of MFM-102-NO2 as a promising material for selective adsorption of CO2. The adsorption selectivities for equimolar mixtures of CO2/CH4 within these MOFs have been calculated using ideal adsorbed solution theory (IAST) using the experimental single–component isotherm data (Fig. S4 and S5†). At 298 K and 1 bar, the CO2/CH4 (50:50) selectivity is 5.0, which is comparable to those reported for other Cu-based MOFs with similar porosity, such as NJU-Bai12 (5.0),20 ZJNU-81(5.5),21 ZJNU-82 (5.4),21 ZJNU-83 (4.9),21 ZJU-15 (4.4) (ref. 22) and ZJNU-57 (5.5),23 but which do not show NbO topologies.
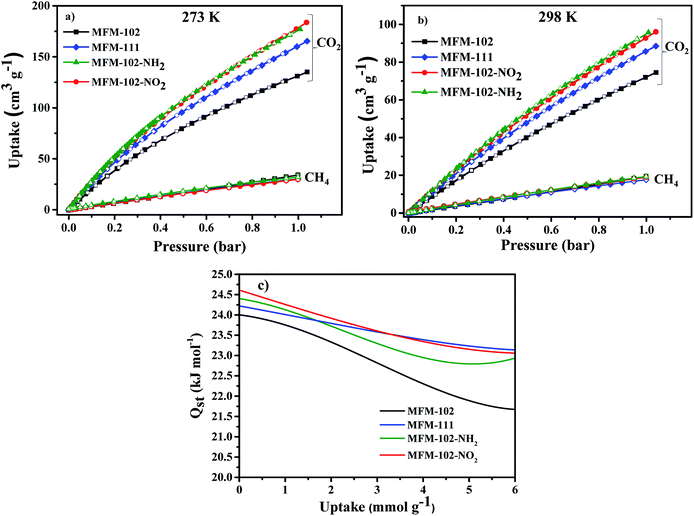 |
| | Fig. 2 Adsorption isotherms for desolvated MFM-102, MFM-111, MFM-102-NO2 and MFM-102-NH2 (a) 273 K, (b) 298 K. Solid and open symbols represent adsorption and desorption, respectively. (c) Heat of adsorption as a function of CO2 uptake derived by the virial method. | |
Table 1 Comparison of CO2 uptakes in selected Cu-MOFs of NbO topology
| MOFs |
CO2 uptake, 273 K (cm3 g−1) |
CO2 uptake, 298 K (cm3 g−1) |
V
p, pore volume (cm3 g−1) |
BET area (m2 g−1) |
Q
st (kJ mol−1) |
Ref. |
| ZJNU-44 |
175 |
116 |
0.894 |
2314 |
— |
24
|
| ZJNU-43 |
180 |
103 |
0.894 |
2243 |
— |
24
|
| ZJNU-45 |
163 |
107 |
0.877 |
2232 |
— |
24
|
| ZJNU-40 |
— |
86 |
0.997 |
2072 |
24.0 |
25
|
| ZJNU-41 |
— |
97 |
0.837 |
2530 |
23.5 |
26
|
| ZJU-8 |
175 |
98 |
1.022 |
2501 |
21.9 |
27
|
| NJU-Bai-14 |
187 |
100 |
0.942 |
2384 |
24.5 |
19
|
| MFM-101 |
164 |
86 |
1.080 |
2805 |
— |
25
|
| MFM-102 |
135 |
74 |
1.297 |
3412 |
24.0 |
This work |
| MFM-102-NO2 |
184 |
96 |
1.118 |
2893 |
24.6 |
This work |
| MFM-102-NH2 |
180 |
97 |
1.140 |
2928 |
24.4 |
This work |
| MFM-111 |
165 |
88 |
1.190 |
2930 |
24.2 |
This work |
| MFM-130 |
109 |
59 |
1.000 |
2173 |
26.0 |
28
|
The isosteric heats of adsorption (Qst) of CO2 were calculated using virial method based on isotherm data at 273 and 298 K (Fig. 2c). The values for Qst at low surface coverage range from 24.0 to 24.6 kJ mol−1, comparable to other MOFs of NbO topology with a high density of open Cu(II) sites (Table 1). With increasing CO2 loading, the values of Qst steadily reduce in all four cases. Interestingly, the Qst values of MFM-102 drop more rapidly than functionalised materials with increasing loading, indicating the presence of additional binding sites in the decorated MOFs.
In situ neutron powder diffraction (NPD) of CO2-loaded MFM-102-NO2
In situ NPD was employed to determine the binding domains for adsorbed CO2 molecules within desolvated MFM-102-NO2 at a loading of 2.0 CO2/Cu. NPD data for the desolvated complex confirm the complete removal of guest solvents, including coordinated H2O molecules on the Cu sites, and no significant structural distortion is observed compared to the parent solvated material. A loading of 2.0 CO2/Cu was used to assess the strongest binding sites within the material without involving notable adsorbate–adsorbate interactions. Fourier difference map analysis of the NPD patterns afforded the location of guest CO2 molecules, which after further development by Rietveld refinement, allowed unambiguous determination of gas positions, orientations and crystallographic occupancies within each sample.
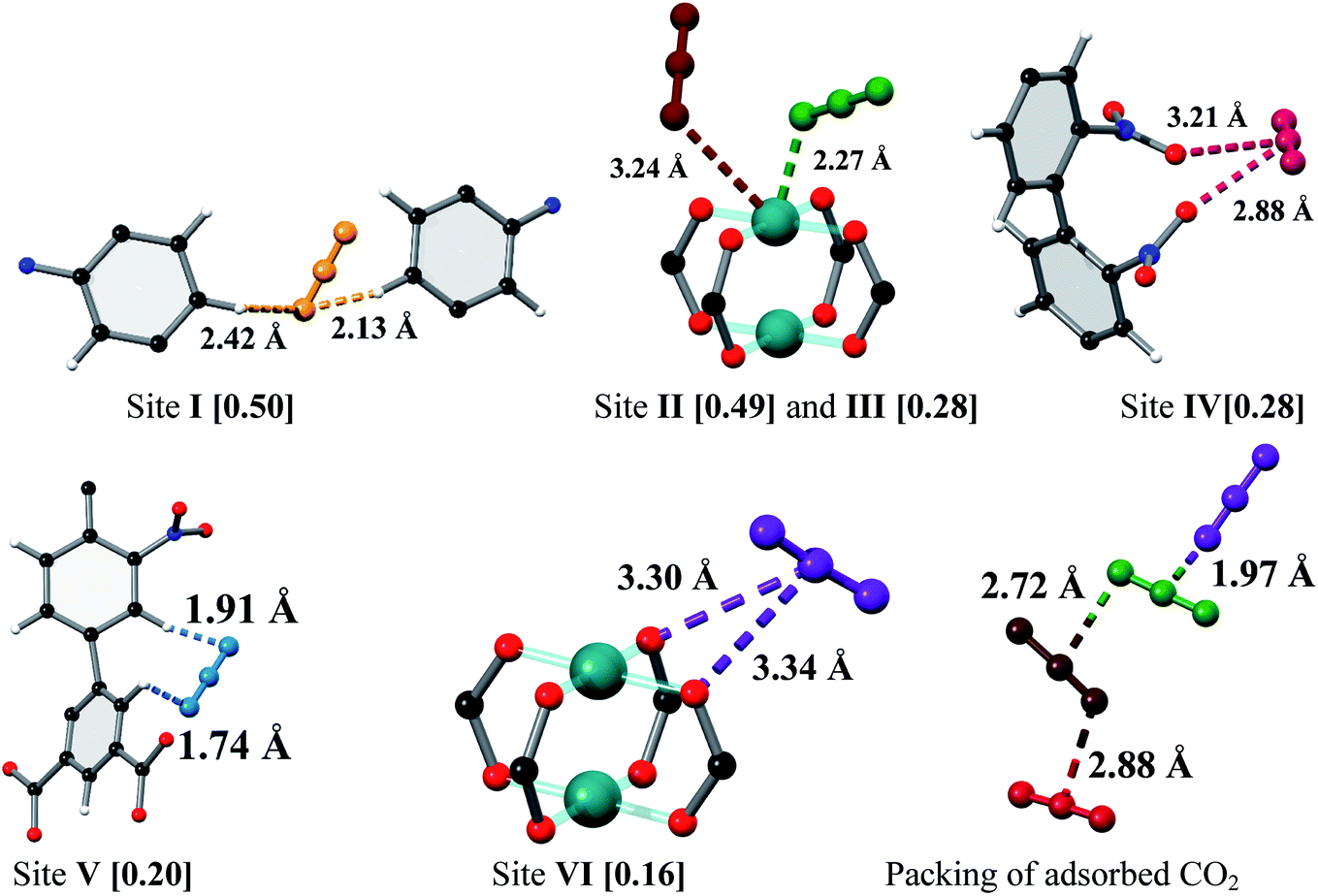
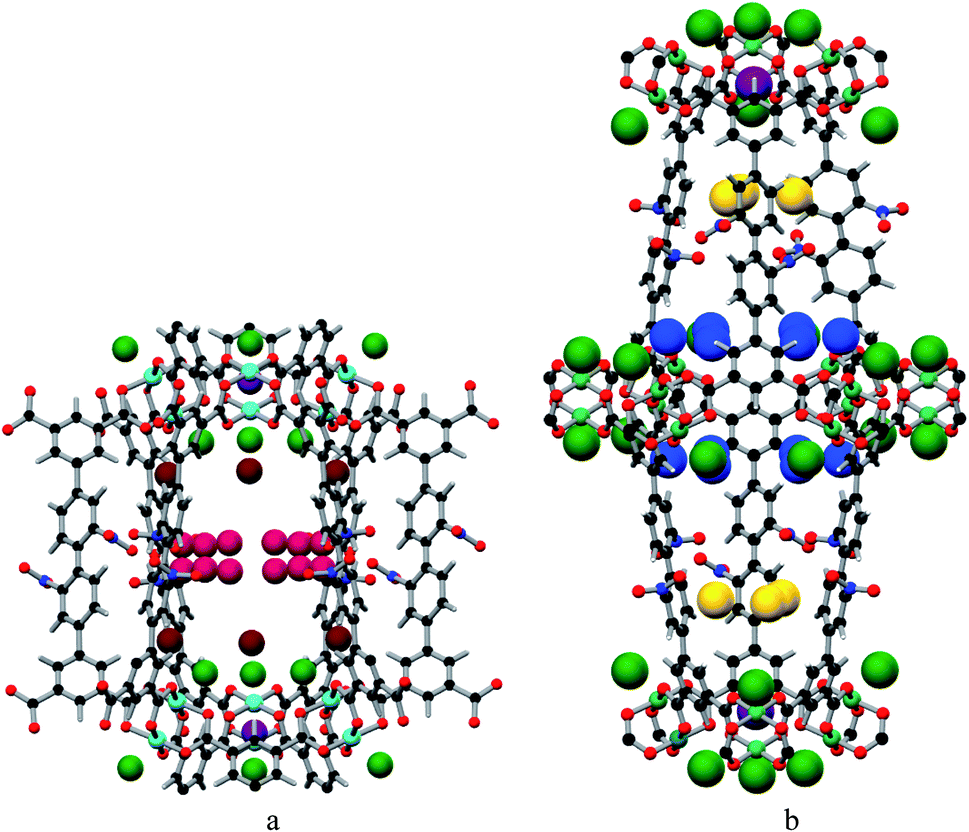
MFM-102-NO2 displays six binding sites for CO2 (denoted as I to VI with decreasing occupancies, Fig. 3 and 4). Significantly, site I (occupancy = 0.50) was found not to be at the open Cu(II) sites but is confined to the window between Cages A and B. At site I, CO2I forms supramolecular interactions to aromatic –CH group from two adjacent NO2-substituted phenyl rings [OCO2⋯![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) C– = 2.13(5), 2.42(8) Å]. It is noteworthy that –NO2 is a strong electron-withdrawing groups which leads to polarisation of adjacent –CH moieties. These –CH groups therefore show enhanced acidity and thus act as effective hydrogen bond donors to CO2 molecules. Site II and III (occupancy = 0.49 and 0.28, respectively) reside at the open Cu(II) centre [OCO2⋯Cu = 2.27(7) Å and 3.24(7) Å for site II and III, respectively]. Site IV (occupancy = 0.28) is stabilised by the dipole/quadrupole interaction between O(δ−) of the –NO2 group from 2 neighbouring phenyl rings and the C(δ+) centre of CO2 with distances of 3.20(6) and 2.87(9) Å. Site V (occupancy = 0.20) was found close to the isophthalate phenyl ring [OCO2⋯HC– = 1.72(9) Å] and the NO2-decorated phenyl ring [OCO2⋯HC– = 1.91(4) Å] stabilised by interactions with aromatic hydrogen atoms. At site VI (occupancy = 0.167), the C atom of CO2 is close to the two oxygen atoms from carboxylate groups of the [Cu2(O2CR)4] paddlewheel with distances of 3.30(1)–3.34(5) Å. Overall, the effects of –NO2 groups on CO2 binding are two-fold: (i) polarising neighbouring phenyl C–H bonds via the conjugate effect of the aromatic ring bound to electron-withdrawing –NO2 groups (site I), and (ii) creating additional adsorption sites via direct dipole/quadrupole interaction of –NO2 with CO2 (site IV).
C– = 2.13(5), 2.42(8) Å]. It is noteworthy that –NO2 is a strong electron-withdrawing groups which leads to polarisation of adjacent –CH moieties. These –CH groups therefore show enhanced acidity and thus act as effective hydrogen bond donors to CO2 molecules. Site II and III (occupancy = 0.49 and 0.28, respectively) reside at the open Cu(II) centre [OCO2⋯Cu = 2.27(7) Å and 3.24(7) Å for site II and III, respectively]. Site IV (occupancy = 0.28) is stabilised by the dipole/quadrupole interaction between O(δ−) of the –NO2 group from 2 neighbouring phenyl rings and the C(δ+) centre of CO2 with distances of 3.20(6) and 2.87(9) Å. Site V (occupancy = 0.20) was found close to the isophthalate phenyl ring [OCO2⋯HC– = 1.72(9) Å] and the NO2-decorated phenyl ring [OCO2⋯HC– = 1.91(4) Å] stabilised by interactions with aromatic hydrogen atoms. At site VI (occupancy = 0.167), the C atom of CO2 is close to the two oxygen atoms from carboxylate groups of the [Cu2(O2CR)4] paddlewheel with distances of 3.30(1)–3.34(5) Å. Overall, the effects of –NO2 groups on CO2 binding are two-fold: (i) polarising neighbouring phenyl C–H bonds via the conjugate effect of the aromatic ring bound to electron-withdrawing –NO2 groups (site I), and (ii) creating additional adsorption sites via direct dipole/quadrupole interaction of –NO2 with CO2 (site IV).
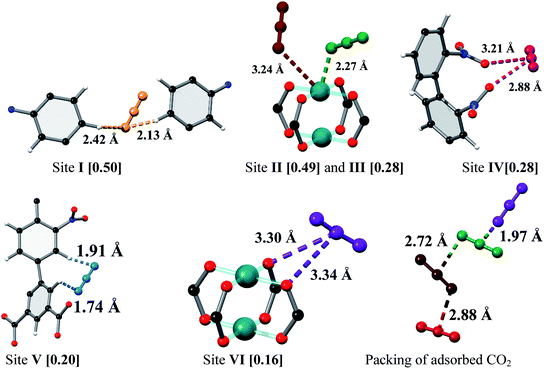 |
| | Fig. 3 View of the binding sites for adsorbed CO2 molecules in MFM-102-NO2 and the relative positions of these positions in the framework cages [C, dark grey; O, red; N, blue; Cu, turquoise and CO2 molecules at site I (yellow), site II (green), site III (brown), site IV (ruby), site V (blue) and site VI (purple)]. | |
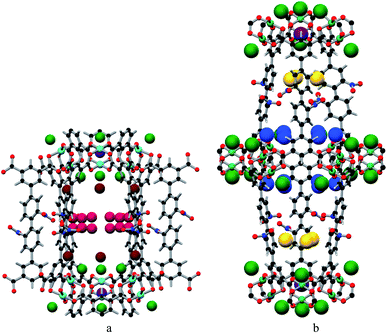 |
| | Fig. 4 The positions of the CO2 in the framework cage A (a) and B (b) of MFM-102-NO2. CO2 molecules are at site I (yellow), site II (green), site III (brown), site IV (ruby), site V (blue) and site VI (purple). | |
In situ synchrotron FT-IR micro-spectroscopy of MFM-102-NO2
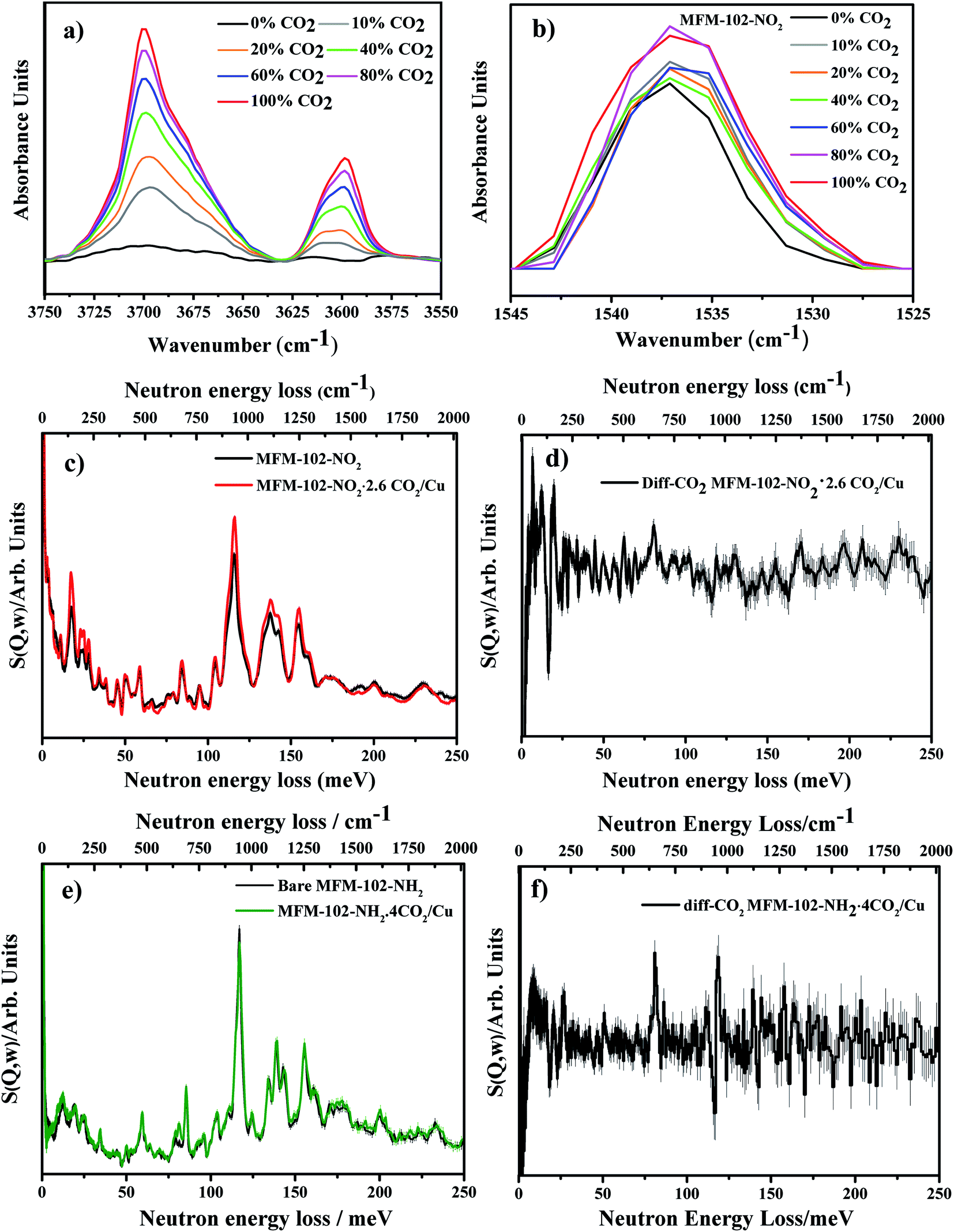
To investigate further the nature of host–guest interactions, in situ synchrotron FT-IR micro-spectroscopy was conducted on a single crystal of activated MFM-102-NO2. FTIR spectra were collected as a function of CO2-loading by increasing the partial pressure of CO2 in N2 from 0 to 1.0 bar (ppCO2) (Fig. 5a). Gaseous CO2 has three fundamental vibrational modes: the symmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode ν1 (1388 cm−1), a doubly-generate bending mode ν2 (667 cm−1) and an asymmetric stretching vibration ν3 (2349 cm−1) (Table S1†).29 The ν1 + ν3 (3714 cm−1) and 2ν2 + ν3 (3612 cm−1) combination bands of CO2 (Fig. 5a) were used to monitor CO2 sorption as the fundamental anti-symmetric stretch at ∼2348 cm−1 saturates at ppCO2 above 0.2 bar. On adsorption, a combination of electrostatic and dispersion forces between CO2 and the pore surface weakens the CO bonds in CO2, leading to the observed redshift of IR bands of adsorbed CO2 to lower frequencies (3700 and 3600 cm−1, respectively) compared to those observed in gas phase (Table S1†). If CO2 strongly coordinates to an open metal site, the molecular symmetry is lowered and the degeneracy of the ν2 bending mode is removed, splitting the band into two peaks.30 Three IR spectra of the activated, 10% CO2- and 100% CO2-loaded MFM-102-NO2 reveal the changes in the peak at 660 cm−1. This peak shows a significant increase in intensity at 10% CO2 loading, and broadens and splits into two peaks at 657 cm−1 and 663 cm−1 at 100% CO2 loading (Fig. S7a†). The IR band of the host framework reveals some interesting features. There is a continuous red shift of the band at 1670 cm−1 (the CO stretching mode of –COO groups31) with increasing CO2 loading, consistent with the interactions between CO2 molecules and [Cu2(O2CR)4] paddlewheel units (site VI, Fig. S7b†). An apparent change in the peak shape and intensity at 1537 cm−1 (N–O asymmetric stretching mode in the –NO2 group32) suggests dipole/quadrupole interaction between the –NO2 groups and adsorbed CO2 molecules (Fig. 5b). In the low energy regions, the peak at 1025 cm−1 (corresponding to C–H out-of-plane wagging33) gradually decreases in intensity with increasing CO2 loading, while the peak at 1005 cm−1 grows and shifts to lower energy (Fig. S7c†). These changes in FTIR spectra are in excellent agreement with the presence of multiple interactions between the framework and CO2 molecules as observed by the NPD experiment.
O stretching mode ν1 (1388 cm−1), a doubly-generate bending mode ν2 (667 cm−1) and an asymmetric stretching vibration ν3 (2349 cm−1) (Table S1†).29 The ν1 + ν3 (3714 cm−1) and 2ν2 + ν3 (3612 cm−1) combination bands of CO2 (Fig. 5a) were used to monitor CO2 sorption as the fundamental anti-symmetric stretch at ∼2348 cm−1 saturates at ppCO2 above 0.2 bar. On adsorption, a combination of electrostatic and dispersion forces between CO2 and the pore surface weakens the CO bonds in CO2, leading to the observed redshift of IR bands of adsorbed CO2 to lower frequencies (3700 and 3600 cm−1, respectively) compared to those observed in gas phase (Table S1†). If CO2 strongly coordinates to an open metal site, the molecular symmetry is lowered and the degeneracy of the ν2 bending mode is removed, splitting the band into two peaks.30 Three IR spectra of the activated, 10% CO2- and 100% CO2-loaded MFM-102-NO2 reveal the changes in the peak at 660 cm−1. This peak shows a significant increase in intensity at 10% CO2 loading, and broadens and splits into two peaks at 657 cm−1 and 663 cm−1 at 100% CO2 loading (Fig. S7a†). The IR band of the host framework reveals some interesting features. There is a continuous red shift of the band at 1670 cm−1 (the CO stretching mode of –COO groups31) with increasing CO2 loading, consistent with the interactions between CO2 molecules and [Cu2(O2CR)4] paddlewheel units (site VI, Fig. S7b†). An apparent change in the peak shape and intensity at 1537 cm−1 (N–O asymmetric stretching mode in the –NO2 group32) suggests dipole/quadrupole interaction between the –NO2 groups and adsorbed CO2 molecules (Fig. 5b). In the low energy regions, the peak at 1025 cm−1 (corresponding to C–H out-of-plane wagging33) gradually decreases in intensity with increasing CO2 loading, while the peak at 1005 cm−1 grows and shifts to lower energy (Fig. S7c†). These changes in FTIR spectra are in excellent agreement with the presence of multiple interactions between the framework and CO2 molecules as observed by the NPD experiment.
 |
| | Fig. 5 (a) IR spectra showing the adsorption of CO2 into MFM-102-NO2 and demonstrating the growth in ν1 + ν3 and 2ν2 + ν3 combination bands for CO2 near 3700 cm−1 and 3600 cm−1. (b) Changes in intensity and broadening of the NO asymmetric stretching vibration with CO2 loading. (c) INS spectra for activated MFM-102-NO2 and MFM-102-NO2 with CO2 loading. (d) Difference INS plot, derived by subtracting INS spectra for CO2-loaded MFM-102-NO2 and bare MFM-102-NO2 spectra. (e) and (f) are the same as (c) and (d), respectively, but replacing MFM-102-NO2 with MFM-102-NH2. | |
Inelastic neutron scattering (INS) studies of CO2-loaded MFM-102-NO2 and MFM-102-NH2
To gain further insight into the host–guest dynamics of CO2-loaded MFM-102-NO2, in situ INS has been investigated. The calculated INS spectrum of bare MOF was found to be in good agreement with the experimental INS data (Fig. S8†) leading to assignment of all the vibrational modes. In the INS spectrum of CO2-loaded MFM-102-NO2, all the INS peaks shift slightly to higher energy, attributed to a stiffening effect of the host–guest lattice on CO2 adsorption (Fig. 4c). There are changes in the peak intensity at 164 and 193 meV, corresponding to the symmetrical and asymmetrical stretching of –NO2 groups. The various distortion motions of the phenyl rings (83 meV), out-of-plane C–H bending on the isophthalate rings (116 meV) and in-plane C–H bending on phenyl rings (136 meV) increase in intensity. These results confirm the important role of –NO2 and –CH groups on the adsorption of CO2. Similarly, the difference INS spectra of CO2-adsorbed MFM-102-NH2 also show changes in intensity of the peaks at 80, 117, 119 meV, originating from various in-plane deformational vibrations of the phenyl rings and the wagging motion of aromatic C–H groups. These changes are attributed to the reduction of the motion of aromatic C–H groups with the introduction of CO2 into the framework.
Conclusions
We report a comprehensive investigation of the adsorption and binding of CO2 in four iso-reticular MOFs with varying functional groups. Introduction of –NO2 group to the parent MOF, MFM-102, leads a 15% reduction on the BET surface area but a 36% enhancement on the CO2 adsorption capacity at 273 K and 1.0 bar. NPD analysis of CO2-loaded MFM-102-NO2 reveals cooperative binding of CO2 to –CH hydrogen bond donors that is enhanced by the electron withdrawing effects of the –NO2 groups that decorate pore walls. In addition, direct interaction of CO2 with the open metal sites and the –NO2 groups is observed, thus enhancing further interaction of the MOF with CO2. The in situ spectroscopic studies using INS and FTIR spectroscopy establish that adsorbed CO2 molecules interact strongly with the –NO2 and –CH groups of MFM-102-NO2, with apparent shifts and changes of intensity of NO and C–H vibrational bands on CO2 loading. This work provides a fundamental understanding of the effect of the –NO2 group on CO2 adsorption, and the information gained here provides further insights into the development of materials showing improved gas binding via specific interaction with ligand sites within the MOF structure.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgements
We thank EPSRC (EP/I011870), the Royal Society and University of Manchester for funding. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no 742401, NANOCHEM). We thank ISIS/STFC, Diamond Light Source and Oak Ridge National Laboratory for access to Beamlines WISH, B22 and VISION, respectively. Neutron scattering experiments were performed at ORNL's Spallation Neutron Source, supported by the Scientific User Facilities Division, Office of Basic Energy Sciences, US DOE, under Contract No. DE-AC0500OR22725 with UT Battelle, LLC. The computing resources were made available through the VirtuES and the ICE-MAN projects, funded by Laboratory Directed Research and Development program and Compute and Data Environment for Science (CADES) at ORNL.
Notes and references
- D. W. Keith, Science, 2009, 325, 1654–1655 CrossRef CAS PubMed.
- E. David and J. Kopac, J. Anal. Appl. Pyrolysis, 2014, 110, 322–332 CrossRef CAS.
- T. D. Pham, R. Xiong, S. I. Sandler and R. F. Lobo, Microporous Mesoporous Mater., 2014, 185, 157–166 CrossRef CAS.
- M. Yang, Y. Song, L. Jiang, X. Wang, W. Liu, Y. Zhao, Y. Liu and S. Wang, J. Ind. Eng. Chem., 2014, 20, 322–330 CrossRef CAS.
- M. Ramdin, T. W. de Loos and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2012, 51, 8149–8177 CrossRef CAS.
- S. Kazemi and V. Safarifard, Polyhedron, 2018, 154, 236–251 CrossRef CAS.
- Y. Liao, L. Zhang, M. H. Weston, W. Morris, J. T. Hupp and O. K. Farha, Chem. Commun., 2017, 53, 9376–9379 RSC.
- A. Greenaway, B. Gonzalez-Santiago, P. M. Donaldson, M. D. Frogley, G. Cinque, J. Sotelo, S. Moggach, E. Shiko, S. Brandani, R. F. Howe and P. A. Wright, Angew. Chem., Int. Ed., 2014, 53, 13483–13487 CrossRef CAS PubMed.
- X. Duan, H. Wang, Z. Ji, Y. Cui, Y. Yang and G. Qian, J. Solid State Chem., 2016, 241, 152–156 CrossRef CAS.
- O. Benson, I. da Silva, S. P. Argent, R. Cabot, M. Savage, H. G. W. Godfrey, Y. Yan, S. F. Parker, P. Manuel, M. J. Lennox, T. Mitra, T. L. Easun, W. Lewis, A. J. Blake, E. Besley, S. Yang and M. Schröder, J. Am. Chem. Soc., 2016, 138, 14828–14831 CrossRef CAS PubMed.
- Q. Mu, H. Wang, L. Li, C. Wang, Y. Wang and X. Zhao, Chem.–Asian J., 2015, 10, 1864–1869 CrossRef CAS PubMed.
- Q.-Q. Dang, Y.-F. Zhan, L.-N. Duan and X.-M. Zhang, Dalton Trans., 2015, 44, 20027–20031 RSC.
- R. Banerjee, H. Furukawa, D. Britt, C. Knobler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 3875–3877 CrossRef CAS PubMed.
- S. Biswas, T. Ahnfeldt and N. Stock, Inorg. Chem., 2011, 50, 9518–9526 CrossRef CAS PubMed.
- T. L. Easun, F. Moreau, Y. Yan, S. Yang and M. Schröder, Chem. Soc. Rev., 2017, 46, 239–274 RSC.
- T. D. Duong, S. A. Sapchenko, I. da Silva, H. G. W. Godfrey, Y. Cheng, L. L. Daemen, P. Manuel, A. J. Ramirez-Cuesta, S. Yang and M. Schröder, J. Am. Chem. Soc., 2018, 140, 16006–16009 CrossRef CAS PubMed.
- X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2009, 131, 2159–2171 CrossRef CAS PubMed.
- S. Yang, X. Lin, A. Dailly, A.
J. Blake, P. Hubberstey, N. R. Champness and M. Schröder, Chem. - Eur. J., 2009, 15, 4829–4835 CrossRef CAS PubMed.
- M. Zhang, Q. Wang, Z. Lu, H. Liu, W. Liu and J. Bai, CrystEngComm, 2014, 16, 6287–6290 RSC.
- B. Zheng, R. Yun, J. Bai, Z. Lu, L. Du and Y. Li, Inorg. Chem., 2013, 52, 2823–2829 CrossRef CAS PubMed.
- M. He, Y. Wang, X. Gao, S. Li and Y. He, Dalton Trans., 2018, 47, 8983–8991 RSC.
- X. Duan, Y. Zhou, R. Lv, B. Yu, H. Chen, Z. Ji, Y. Cui, Y. Yang and G. Qian, J. Solid State Chem., 2018, 260, 31–33 CrossRef CAS.
- Y. Wang, M. He, Z. Tian, H. Zhong, L. Zhu, Y. Zhang, X. Zhang, D.-L. Chen and Y. He, Dalton Trans., 2018, 47, 2444–2452 RSC.
- C. Song, J. Hu, Y. Ling, Y. Feng, R. Krishna, D.-l. Chen and Y. He, J. Mater. Chem. A, 2015, 3, 19417–19426 RSC.
- C. Song, Y. He, B. Li, Y. Ling, H. Wang, Y. Feng, R. Krishna and B. Chen, Chem. Commun., 2014, 50, 12105–12108 RSC.
- C. Song, Y. Ling, L. Jin, M. Zhang, D.-L. Chen and Y. He, Dalton Trans., 2016, 45, 190–197 RSC.
- J. Cai, H. Wang, H. Wang, X. Duan, Z. Wang, Y. Cui, Y. Yang, B. Chen and G. Qian, RSC Adv., 2015, 5, 77417–77422 RSC.
- Y. Yan, M. Juríček, F.-X. Coudert, N. A. Vermeulen, S. Grunder, A. Dailly, W. Lewis, A. J. Blake, J. F. Stoddart and M. Schröder, J. Am. Chem. Soc., 2016, 138, 3371–3381 CrossRef CAS PubMed.
- S. Andonova, E. Ivanova, J. Yang and K. Hadjiivanov, J. Phys. Chem. C, 2017, 121, 18665–18673 CrossRef CAS.
- J. G. Vitillo, M. Savonnet, G. Ricchiardi and S. Bordiga, ChemSusChem, 2011, 4, 1281–1290 CrossRef CAS PubMed.
- N. R. Dhumal, M. P. Singh, J. A. Anderson, J. Kiefer and H. J. Kim, J. Phys. Chem. C, 2016, 120, 3295–3304 CrossRef CAS.
-
F. X. W. Robert, M. Silverstein, D. J. Kiemle and D. L. Bryce, Spectrometric Identification of Organic Compounds, 8th edn, 2014 Search PubMed.
- F. P. Ureña, M. F. Gómez, J. J. L. González and E. M. n. Torres, Spectrochim. Acta, Part A, 2003, 59, 2815–2839 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization and additional analysis of crystal structures. Single crystal data of CO2-loaded MFM-102-NO2 is deposited at Cambridge Crystallographic Data Centre (CCDC) 1860478. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04294f |
| ‡ The crystal data for MFM-102-NO2, MFM-102-NH2, and bare MFM-102-NO2, CCDC 1857304, 1857305 and 1857873, respectively, were previously reported in ref. 16. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  d,
Harry G. W.
Godfrey
a,
Yongqiang
Cheng
e,
Luke L.
Daemen
e,
Pascal
Manuel
d,
Mark D.
Frogley
d,
Harry G. W.
Godfrey
a,
Yongqiang
Cheng
e,
Luke L.
Daemen
e,
Pascal
Manuel
d,
Mark D.
Frogley