
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZn-mediated decarboxylative carbagermatranation of aliphatic N-hydroxyphthalimide esters: evidence for an alkylzinc intermediate†
Wei-Tao
Jiang
,
Shuo
Yang
,
Meng-Yu
Xu
,
Xiu-Ying
Xie
and
Bin
Xiao
 *
*
Department of Chemistry, University of Science and Technology of China, Hefei 230026, China. E-mail: binxiao@ustc.edu.cn
First published on 14th November 2019
Abstract
Alkyl nucleophiles synthesized by decarboxylation of the corresponding N-hydroxyphthalimide esters (NHP esters) would inherit the complex structure of natural carboxylic acids and result in useful cross-coupling fragments. Herein, we report the synthesis of alkyl carbagermatranes via Zn-mediated decarboxylation of NHP esters without Ni catalysis or photocatalysis. Mechanistic studies indicate that an alkyl zinc intermediate was involved; however, the generation of alkyl zinc will be inhibited in the presence of Ni. Hence, this study provides valuable resolution to the perplexing problem about whether organozinc was involved in recently emerging catalytic systems of NHP ester–Zn. Meanwhile, alkyl zinc reagents from NHP esters are compatible with aryl/alkyl bromides and iodides; therefore the scope of carbagermatranation in this work precedes that of in situ-generated organozinc from alkyl halides.
Introduction
In the realm of cross-coupling reactions, while the innovation of catalytic systems has been occurring constantly, another major concern should be the synthesis of nucleophiles. In recent years, numerous methods have been extensively developed for the synthesis of alkyl nucleophiles.1 In particular, synthesis of alkyl nucleophiles by means of decarboxylation would help incorporate complex structures that are derived from naturally abundant carboxylic acid resources. Baran's group has already reported the synthesis of diversified alkyl boronates via decarboxylation of aliphatic NHP esters under nickel or copper catalysis (Scheme 1a).2 In addition, Aggarwal’s group and Li's group also managed to synthesize alkyl boronates through decarboxylation induced by light (Scheme 1a).3 By this strategy, diversified alkyl silanes were also readily synthesized by Oestreich's group (Scheme 1b).4–6 It's noted that decarboxylative functionalization has received increasing attention these years;7 decarboxylation for the synthesis of other kinds of nucleophiles remains to be further explored.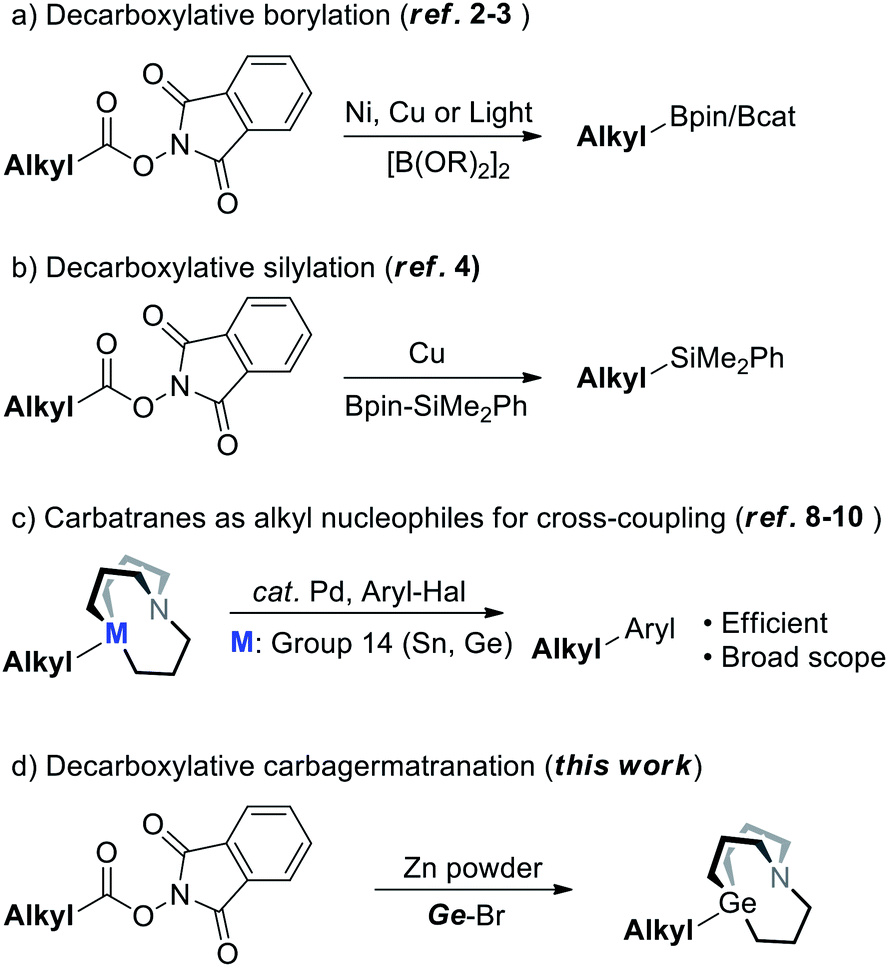 | ||
| Scheme 1 Synthesis of alkyl nucleophiles by decarboxylation of the corresponding NHP esters. | ||
Meanwhile, carbatranes of group 14 elements (Scheme 1c) were proved to be efficient alkyl cross-coupling reagents back in 1992 and further demonstrated in recent years6c,8,9 (i.e.M = Sn) by Vedejs' and Biscoe's groups, respectively. Our group has subsequently reported the synthesis of alkyl carbagermatranes (i.e.M = Ge)10 from in situ-generated alkyl Grignard reagents and alkyl zinc reagents. While carbatranes exhibit not only superior cross-coupling reactivity but also promising orthogonality with organoboron reagents, methods of the synthesis of alkyl carbatranes are fewer than those of alkylboron.11 Hence, expanding the library of alkyl carbatranes with a complex structure is attractive to synthetic chemistry.
Herein, we reported a Zn-mediated decarboxylative carbagermatranation reaction using aliphatic NHP esters as alkylation reagents (Scheme 1d). In this manner, we were able to acquire a wide range of alkyl carbagermatrane reagents and also avoid the pre-preparation of alkyl halides. Furthermore, aryl/alkyl halides were all well tolerated in this protocol. Above all, it's proved that alkyl zinc was generated in the system, but only in the absence of nickel.
Results and discussion
We envisaged that reductive cross-coupling of 1-bromo-carbagermatranes (GeBr, 1) and aliphatic NHP esters would be a feasible idea. However, when we conducted a blank control experiment, in which only two starting reagents and Zn powder were added, surprising results were obtained. Simply stirring GeBr and n-butanoic acid NHP ester (2a) with Zn powder in DMF at room temperature already gave n-propyl carbagermatrane (3a) in nearly quantitative yield (entry 1, Table 1). By replacing Zn powder with other commonly used reductants in the reductive cross-coupling reaction, such as Mg, Mn and TDAE (tetrakis(dimethylamino)ethylene),12 no desired product was detected (entry 2–4). Changing solvent to THF and DMAc afforded 57% and 56% yield, respectively (entry 5 and 6). Lowering the load of 2a resulted in lower yield (entry 7 and 8). Then we applied the optimized condition for 2a to 2-methylbutyric acid NHP ester (2b), and only a trace amount of s-butyl carbagermatrane (3b) was detected (entry 9). Increasing the reaction temperature to 60 °C would improve the yield to 32% (entry 10). Swapping solvent to DMAc only gave 8% yield (entry 11). Nevertheless, using THF as solvent gave considerably improved 68% yield (entry 12). In addition, extending the reaction time to 18 hours made little difference in yield (entry 13). Ultimately, using 3.0 equivalent of Zn powder gave a satisfactory yield of 80% (entry 14).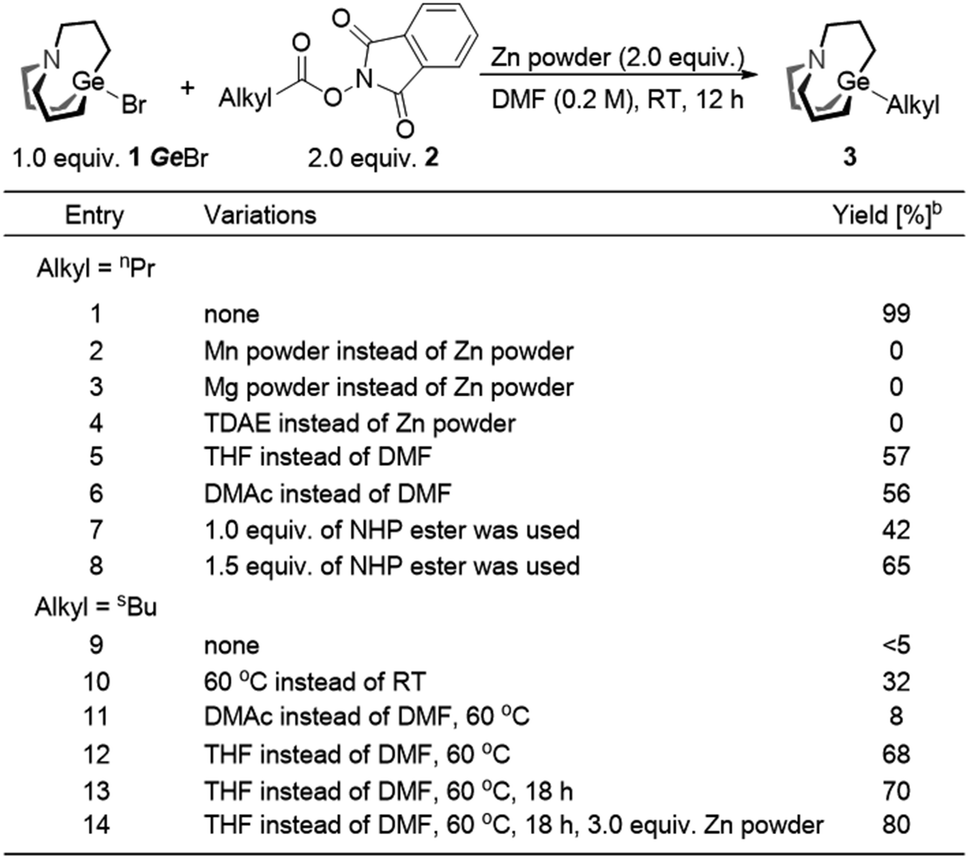
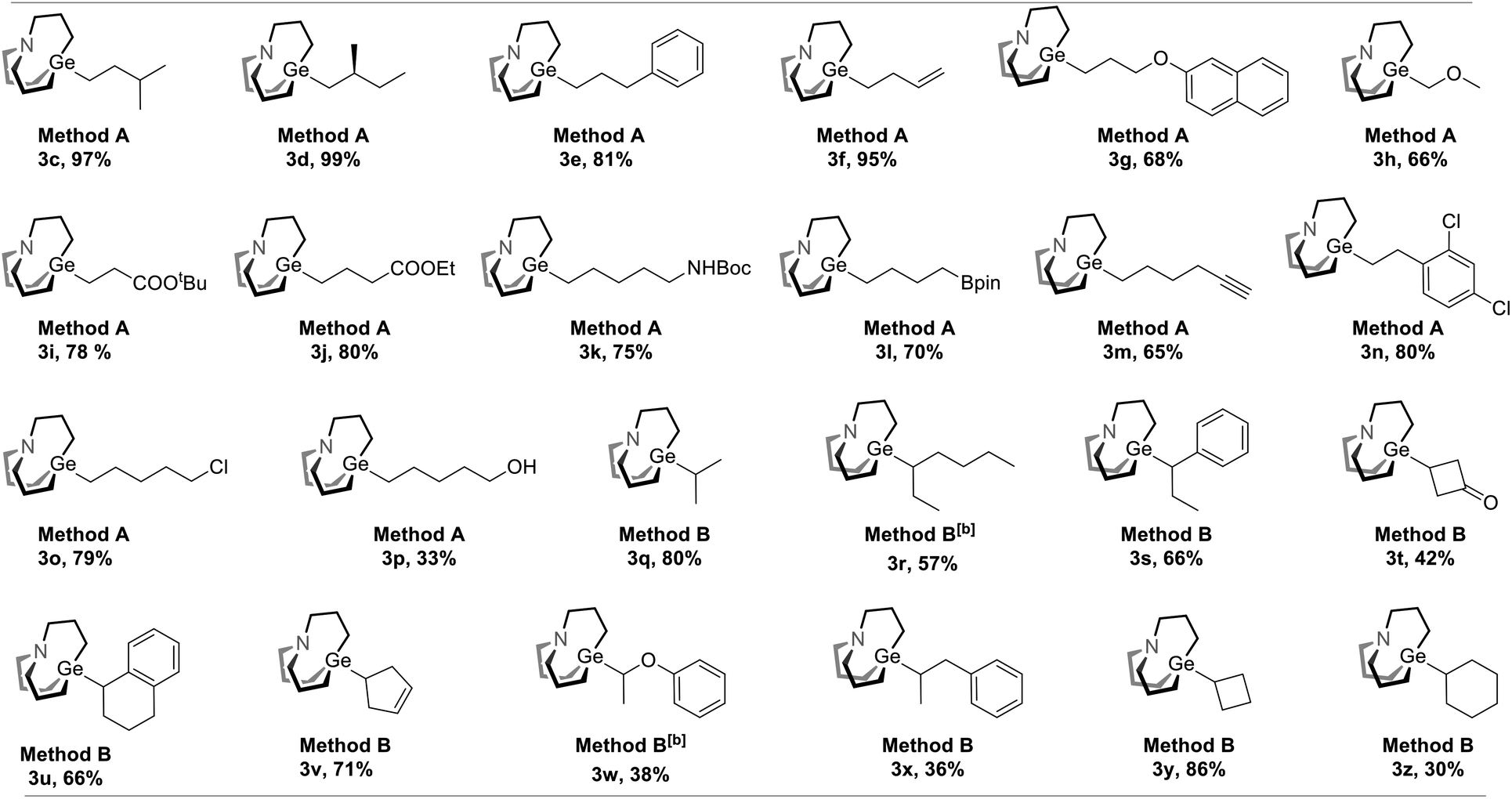
With the optimal condition in hand, the scope of decarboxylative carbagermatranation was examined (Table 2). Conventional aliphatic NHP esters were successfully converted to alkyl carbagermatranes with good to excellent yield (3c–h). Esters (3i–j), protected amine (3k) and boronate (3l) were also well tolerated in the system. Notably, unprotected terminal alkynes did not interfere the reaction, and alkyne-containing alkyl carbagermatrane (3m) was obtained in 65% yield. Chloride containing substrates were obtained in good yield (3n–o). We next examined the scope of secondary and tertiary substrates.13 Isopropyl (3q) and hept-3-yl (3r) carbagermatranes were readily synthesized in good to excellent yield. Secondary benzyl carbagermatranes (3s and 3u) were also obtained in good yield. In addition, cyclic strained products (3t and 3y) were successfully accessed.
| a All reactions were performed on a 0.1 mmol scale, and yields were determined by isolation unless otherwise noted. Method A: 1 (0.1 mmol), 2 (0.2 mmol), Zn powder (0.2 mmol), DMF (0.5 mL), RT, 12 hours. Method B: 1 (0.1 mmol), 2 (0.2 mmol), Zn powder (0.3 mmol), THF (0.5 mL), 60 °C, 18 hours. b Conducted at 90 °C instead. |
|---|
|
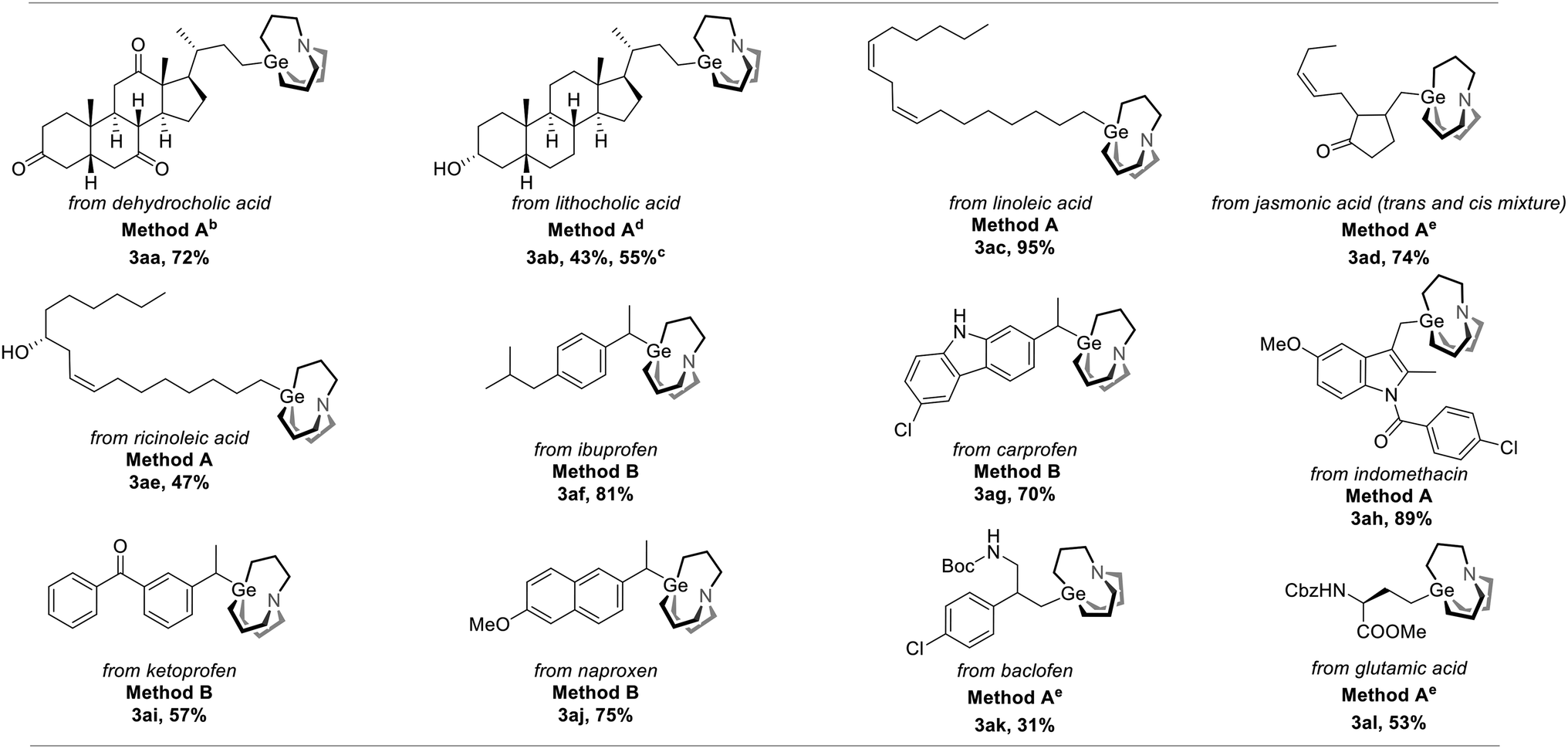
To further demonstrate the utility of this strategy, we applied it to drug and nature product-derived substrates and various complex structures were successfully incorporated. Dehydrocholic acid NHP ester was converted to the corresponding carbagermatrane (3aa) in 72% yield. Lithocholic acid and linolenic acid-derived carbagermatranes (3ab and 3ae) were also successfully accessed with the unprotected secondary alcohol still remaining. However, for the case of 3ab, it was isolated in an inseparable mixture of the corresponding carbagermatrane and protonation byproduct with a NMR ratio of 2.1/1. All primary and secondary benzyl carbagermatranes derived from indomethacin (3ah), racemic ibuprofen (3af), ketoprofen (3ai) and N-unprotected carprofen (3ag), were obtained in good to excellent yield. Notably, racemization occurred when enantiomerically pure naproxen was applied (3ai). At last, glutamic acid-derived carbagermatrane (3al) was also obtained (Table 3).
| a All reactions were performed on a 0.1 mmol scale, and yields were determined by isolation unless otherwise noted. Method A: 1 (0.1 mmol), 2 (0.2 mmol), Zn powder (0.2 mmol), DMF (0.5 mL), RT, 12 hours. Method B: 1 (0.1 mmol), 2 (0.2 mmol), Zn powder (0.3 mmol), THF (0.5 mL), 60 °C, 18 hours. b 1.0 mL DMF was used as solvent instead. c 1H-NMR yield using mesitylene as an internal standard. d 0.5 mL THF was used as solvent instead. e Conducted at 60 °C instead. |
|---|
|
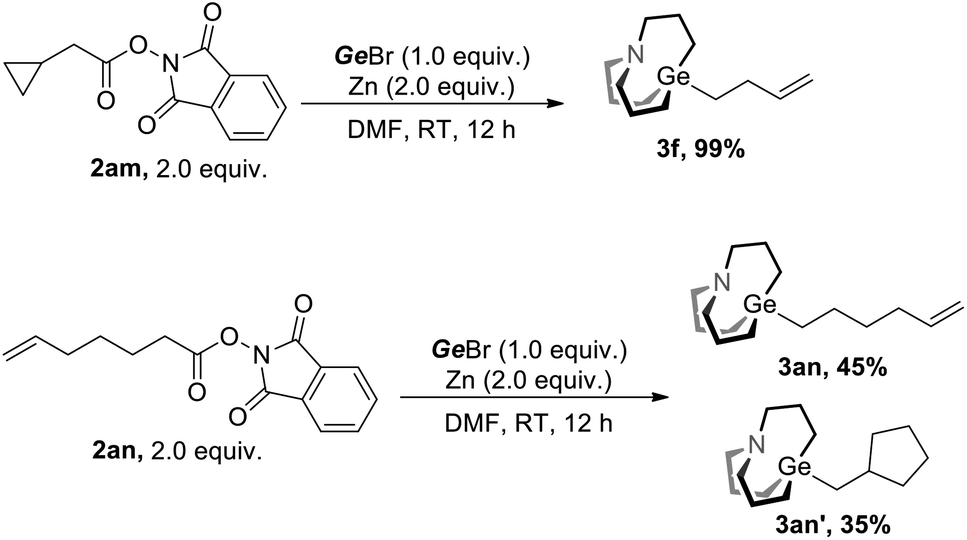
We next investigate the mechanism of this strategy. Radical opening along with the radical cyclization experiment was conducted (Scheme 2). Under the conditions of Method A, 2am was converted to fully opening product 3f in nearly quantitative yield, and 2an was converted to non-cyclization product 3an and cyclization product 3an′ with a ratio of 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35. These experiments suggest that radicals were involved in the reaction.
:35. These experiments suggest that radicals were involved in the reaction.
 | ||
| Scheme 2 Radical probe experiments. | ||
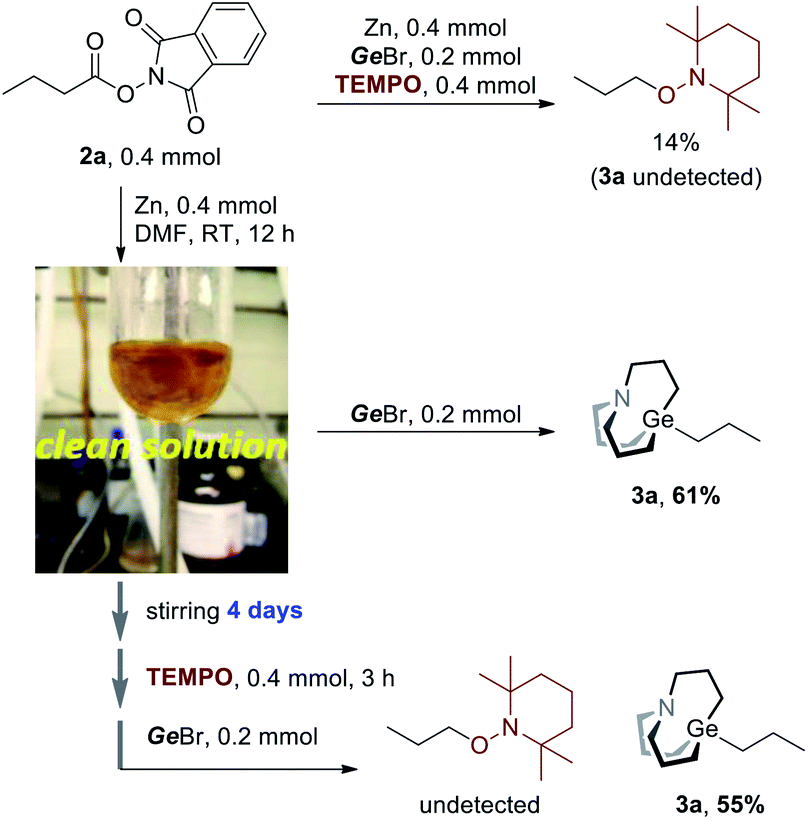
However, we still don't know whether radicals were directly involved in the Ge–C bond formation step. Hence, more evidence should be collected to gain clear insight into the mechanism. When TEMPO was added to the reaction (Scheme 3, upper), the TEMPO-captured product was detected in 14% GC yield, and no carbagermatranation product was detected. Following this, 2a was stirred with equimolar Zn powder in DMF at room temperature and Zn powder was almost dissolved in 12 hours to give a clear yellow solution, after which GeBr was added and further stirred for 12 hours to give product 3a in 61% isolated yield (Scheme 3a, middle). It's unlikely that radicals would survive for a very long time since they tend to quench with each other or solvent after they were generated. Therefore, a more reliable experiment was conducted. The clear solution was stirred for a very long time (i.e. 4 days) before being quenched by TEMPO, and then GeBr was added (Scheme 3, under). Surprisingly, 3a was obtained in a comparable yield of 55% (vs. 61%, Scheme 3, middle) and no TEMPO-captured product was detected. The results clearly rule out that radicals were involved in the Ge–C bond formation step.14
 | ||
| Scheme 3 Ruling out radical involvement in the Ge–C bond formation step. | ||
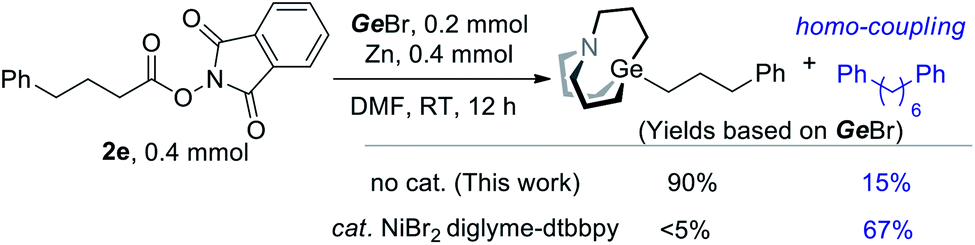
The above results indicated that interaction of NHP esters and Zn might lead to the generation of alkyl zinc. It has been a controversial issue in the regard of a recently popular NHP–Zn–Ni system, namely whether alkyl zinc works as an intermediate.15 To understand the behavior of radicals under a catalytic or non-catalytic system, we also conducted a comparative experiment with or without a nickel catalyst. When a nickel catalyst was added, a significant amount of alkyl homo-coupling product and a trace amount of alkyl carbagermatrane were detected (Scheme 4). In the presence of Ni, radicals are rather reduced by Ni than Zn. In contrast, radicals are reduced by low valent Zn+ to carbanion in the absence of Ni.15e,16 The distinct selectivity shown in Scheme 4 indicates that the organozinc intermediate may not be involved in the NHP–Zn–Ni system.
 | ||
| Scheme 4 Interference experiment with the Ni catalyst. | ||
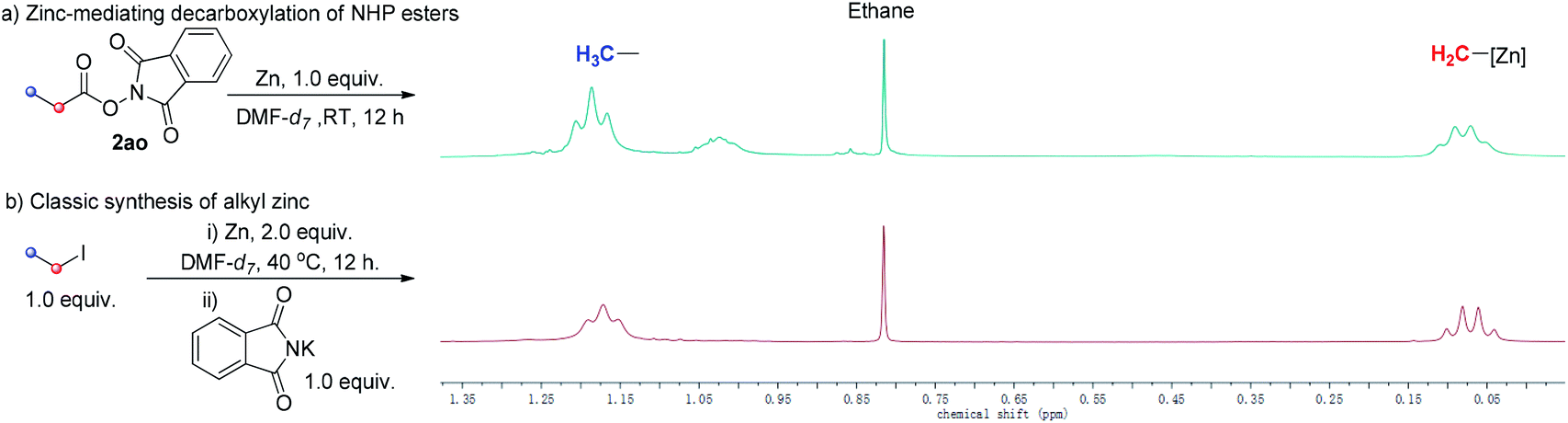
To observe directly, 1H-NMR spectra of the alkyl zinc reagent of our strategy and classic alkyl zinc regent were compared. 1H-NMR spectra of both gave a set of clear signals of the ethyl group (Scheme 5). The chemical shift of Zn-attaching CH2 was approaching 0 ppm, which is in accordance with previous reports.17 These highly matched spectra further support our proposal that the alkyl zinc reagent was generated in situ via Zn-mediated decarboxylation of aliphatic NHP esters.
 | ||
| Scheme 5 Comparison of 1H-NMR spectra of the alkyl zinc reagent of our strategy and classic alkyl zinc reagent. | ||
To be distinguished from the synthesis of organozinc using halides, Br/I-containing substrates were also tested (Scheme 6). Primary and secondary bromide-containing carbagermatranes (3ap–aq) were obtained in excellent yield. An iodide-containing substrate was obtained in reasonable low yield (3ar). Meanwhile, aromatic bromide and iodide were all well compatible (3as–au). These Br/I-containing carbagermatranes bear both nucleophilic sites and electrophilic sites, which allow for further diversified derivatizations. Indeed, selective Suzuki cross-coupling took place using 3av (Scheme 6, under) as a partner. Cross-coupling product 3aw was acquired in excellent 90% isolated yield, which means that the Ge–C bond was perfectly preserved under Suzuki-reaction conditions and reveals the orthogonality with other reactions of alkyl carbagermatranes. Furthermore, intramolecular cyclization of 3av was also conducted. By using iPr-Xuphos as the ligand,10 cyclization product 4a was obtained in 70% yield. By simply swapping the ligand with less hindered Me-Xuphos, a satisfactory yield of 82% was obtained. Notably, unsubstituted benzo[b]oxocanes or those lacking gem-dimethyl groups have not been accessed through C–C bond cross-coupling before.
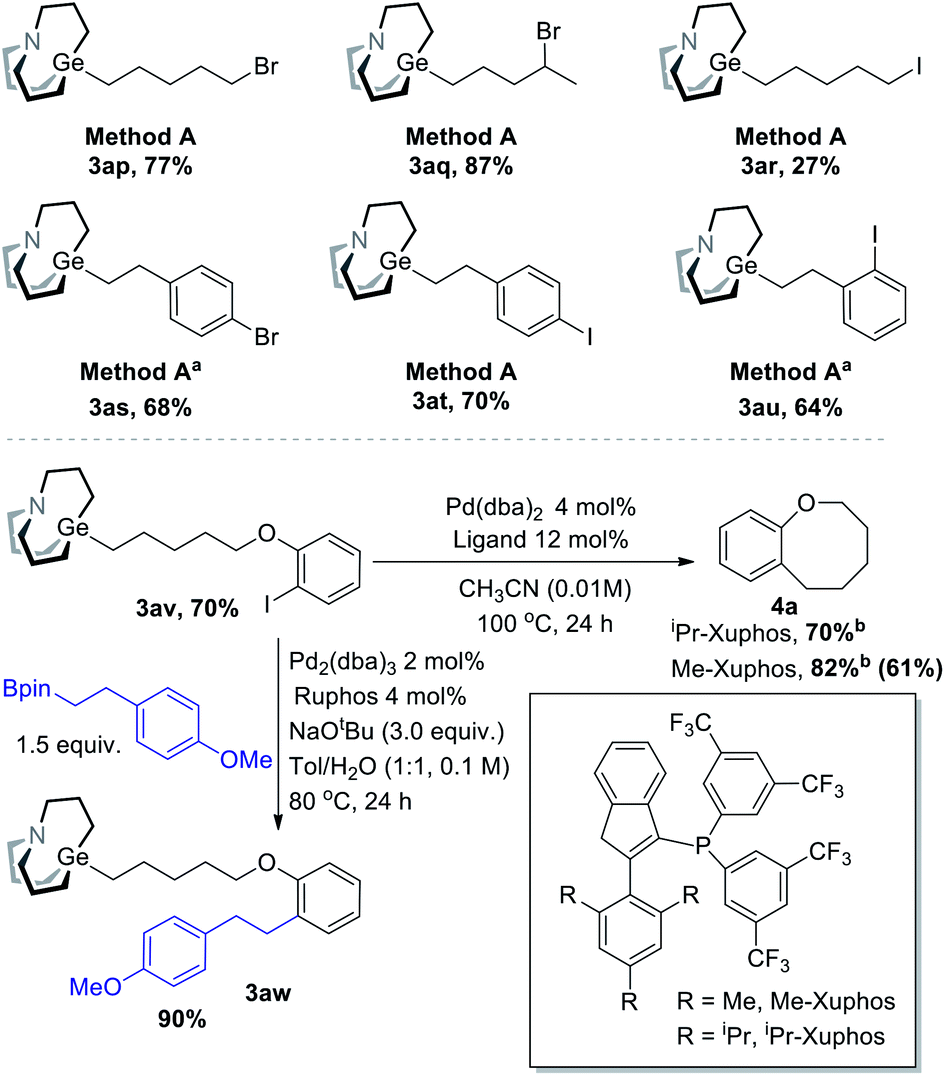 | ||
| Scheme 6 Br/I-containing substrates and orthogonal experiment with Suzuki cross-coupling reaction. All reactions were performed on a 0.1 mmol scale, and yields were determined by isolation unless otherwise noted. a 0.5 mL THF was used as solvent instead. b 1H-NMR yield using mesitylene as an internal standard. Slightly lower isolated yield (shown in parentheses) is due to its low boiling point. | ||
Conclusions
Various alkyl carbagermatranes were synthesized via Zn-mediated decarboxylation of aliphatic NHP esters, which enrich the categories and methodologies of the synthesis of alkyl nucleophiles from natural carboxylic acid. It's proved that alkyl zinc reagents were generated when no other radical acceptor (e.g. Ni) exists in a simple system of aliphatic NHP esters and Zn powder. Meanwhile, generation of alkyl zinc reagents using NHP esters can be distinguished from alkyl halides, which was responsible for the tolerance of aryl and alkyl halides. Our study provides more insight into the decarboxylative chemistry of aliphatic NHP esters, and the new generation of organozinc may have further applications in other fields.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank the NSFC (21871239) and Youth Innovation Pro-motion Association of the Chinese Academy of Sciences (2015371) for financial support.Notes and references
- For selected reviews, see: (a) B. S. Collins, C. M. Wilson, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 11700–11733 CrossRef CAS; (b) D. Hemming, R. Fritzemeier, S. A. Westcott, W. L. Santos and P. G. Steel, Chem. Soc. Rev., 2018, 47, 7477–7494 RSC; (c) E. C. Neeve, S. J. Geier, I. A. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS; (d) J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS; (e) S. Bähr, W. Xue and M. Oestreich, ACS Catal., 2018, 9, 16–24 CrossRef.
- (a) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, W. Y. Antony, K. A. Johnson and A. K. Chatterjee, Science, 2017, 356, eaam7355 CrossRef . The catalyst was further expanded to Cu, see; (b) J. Wang, M. Shang, H. Lundberg, K. S. Feu, S. J. Hecker, T. Qin, D. G. Blackmond and P. S. Baran, ACS Catal., 2018, 8, 9537–9542 CrossRef CAS.
- (a) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS; (b) D. Hu, L. Wang and P. Li, Org. Lett., 2017, 19, 2770–2773 CrossRef CAS PubMed.
- W. Xue and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 11649–11652 CrossRef CAS.
- For other selected examples of photoinduced, copper-catalysed decarboxylation of NHP esters, see: (a) W. Zhao, R. P. Wurz, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 12153–12156 CrossRef CAS; (b) A. Tlahuext-Aca, L. Candish, R. A. Garza-Sanchez and F. Glorius, ACS Catal., 2018, 8, 1715–1719 CrossRef CAS; (c) R. Mao, A. Frey, J. Balon and X. Hu, Nat. Catal., 2018, 1, 120–126 CrossRef CAS; (d) D. Wang, N. Zhu, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 15632–15635 CrossRef CAS; (e) C. Wang, M. Guo, R. Qi, Q. Shang, Q. Liu, S. Wang, L. Zhao, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2018, 130, 16067–16072 CrossRef; (f) X.-L. Lyu, S.-S. Huang, H.-J. Song, Y.-X. Liu and Q.-M. Wang, Org. Lett., 2019, 21, 5728–5732 CrossRef CAS.
- For selected examples of synthesis of 14 group element nucleophiles from halides using metal reagents, see: (a) W. Xue, W. Mao, L. Zhang and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 6440–6443 CrossRef CAS; (b) W. Xue, R. Shishido and M. Oestreich, Angew. Chem., Int. Ed., 2018, 57, 12141–12145 CrossRef CAS; (c) C. Y. Wang, G. Ralph, J. Derosa and M. R. Biscoe, Angew. Chem., Int. Ed., 2017, 56, 856–860 CrossRef CAS.
- For selected reviews of decarboxylative functionalization, see: (a) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS; (b) J. Xuan, Z. G. Zhang and W. J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS; (c) N. Rodriguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC.
- (a) E. Vedejs, A. R. Haight and W. O. Moss, J. Am. Chem. Soc., 1992, 114, 6556–6558 CrossRef CAS; (b) L. Li, C.-Y. Wang, R. Huang and M. R. Biscoe, Nat. Chem., 2013, 5, 607–612 CrossRef CAS.
- Non-palladium catalysed reactions of stannatranes, see: X. Ma, M. Diane, G. Ralph, C. Chen and M. R. Biscoe, Angew. Chem., Int. Ed., 2017, 129, 12837–12841 CrossRef.
- M.-Y. Xu, W.-T. Jiang, Y. Li, Q.-H. Xu, Q.-L. Zhou, S. Yang and B. Xiao, J. Am. Chem. Soc., 2019, 141, 7582–7588 CrossRef CAS PubMed.
- For review of group 14 nucleophiles and carbatranes, see: (a) A. C. Spivey, C. J. G. Gripton and J. P. Hannah, Curr. Org. Synth., 2004, 1, 211–226 CrossRef CAS; (b) G. Ralph and M. R. Biscoe, Organometallics, 2019, 38, 3912–3915 CrossRef CAS.
- For selected examples of using Mn and TDAE as reductants, see: (a) L. Huang, A. M. Olivares and D. J. Weix, Angew. Chem., Int. Ed., 2017, 56, 11901–11905 CrossRef CAS; (b) N. Suzuki, J. L. Hofstra, K. E. Poremba and S. E. Reisman, Org. Lett., 2017, 19, 2150–2153 CrossRef CAS.
- Tertiary and aryl substrates failed to give the corresponding carbagermatranes using the same strategy.
- The lack of reactivity of GeBr with radicals may be attributed to the high BDE value of the Ge-Br bond and extraordinary stability of the carbatrane cations, see: (a) Y. Luo and J. Cheng, Bond Dissociation Energies, in CRC Handbook of Chemistry and Physics, 99th ed. (Internet Version 2018), ed. J. R. Rumble, CRC Press/Taylor & Francis, Boca Raton, FL, 2018, pp. 9–72 Search PubMed; (b) A. Kavoosi and E. Fillion, Angew. Chem., Int. Ed., 2015, 54, 5488–5492 CrossRef CAS . On the other hand, a related electrochemical oxidation study attests that even if a SET process took place and results in radical cations, the unpaired electron tends to reside at the N atom; (c) V. Romanovs, V. Sidorkin, E. Belogolova and V. Jouikov, Dalton Trans., 2017, 46, 8849–8854 RSC; (d) P. Simidzija, M. J. Lecours, R. A. Marta, V. Steinmetz, T. B. McMahon, E. Fillion and W. S. Hopkins, Inorg. Chem., 2016, 55, 9579–9585 CrossRef CAS.
- For selected examples of involving NHP esters/Zn/Ni, see: (a) J. Wang, H. Lundberg, S. Asai, P. Martin-Acosta, J. S. Chen, S. Brown, W. Farrell, R. G. Dushin, C. J. O'Donnell, A. S. Ratnayake, P. Richardson, Z. Liu, T. Qin, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6404–E6410 CrossRef CAS; (b) T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. E. Novak, J. Z. Zhong, R. B. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 260–265 CrossRef CAS; (c) X. Lu, X.-X. Wang, T.-J. Gong, J.-J. Pi, S.-J. He and Y. Fu, Chem. Sci., 2019, 10, 809–814 RSC; (d) K. M. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, J. Am. Chem. Soc., 2016, 138, 5016–5019 CrossRef CAS; (e) L. Yu, M.-L. Tang, C.-M. Si, Z. Meng, Y. Liang, J. Han and X. Sun, Org. Lett., 2018, 20, 4579–4583 CrossRef CAS PubMed; (f) J. Wang, B. P. Cary, P. D. Beyer, S. H. Gellman and D. J. Weix, Angew. Chem., Int. Ed., 2019, 58, 12081–12085 CrossRef CAS.
- For an insightful discussion of molecules that facilitate electron transfer could promote magnesium insertion, see: Z.-L. Shen and P. Knochel, ACS Catal., 2015, 5, 2324–2328 CrossRef CAS.
- For selected examples of the organozinc NMR spectrum, see: (a) A. Guerrero, D. L. Hughes and M. Bochmann, Organometallics, 2006, 25, 1525–1527 CrossRef CAS; (b) J. E. Fleckenstein and K. Koszinowski, Organometallics, 2011, 30, 5018–5026 CrossRef CAS; (c) R. Cariou and J. W. Shabaker, ACS Catal., 2015, 5, 4363–4367 CrossRef CAS; (d) K. Jess, K. Kitagawa, T. K. S. Tagawa and S. A. Blum, J. Am. Chem. Soc., 2019, 141, 9879–9884 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04288a |
| This journal is © The Royal Society of Chemistry 2020 |