
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA general method for site-selective Csp3–S bond formation via cooperative catalysis†
Yuman
Qin‡
,
Yujie
Han‡
,
Yongzhen
Tang
,
Junfa
Wei
and
Mingyu
Yang
 *
*
Key Laboratory of Applied Surface and Colloid Chemistry of MOE, School of Chemistry and Chemical Engineering, Shaanxi Normal University, 620 West Chang'an Ave, Xi'an, 710119, China. E-mail: yangmy05@snnu.edu.cn; yangmy05@gmail.com
First published on 6th December 2019
Abstract
Herein, we report a copper-catalysed site-selective thiolation of Csp3–H bonds of aliphatic amines. The method features a broad substrate scope and good functional group compatibility. Primary, secondary, and tertiary C–H bonds can be converted into C–S bonds with a high efficiency. The late-stage modification of biologically active compounds by this method was also demonstrated. Furthermore, the one-pot preparation of pyrrolidine or piperidine compounds via a domino process was achieved.
Introduction
The sp3 carbon–sulfur bond plays a pivotal role in modern organic synthesis, natural products and pharmaceuticals. For example, Butoconazole is an imidazole antifungal used in gynaecology, and Nelfinavir is an antiretroviral drug which was used in the treatment of the human immunodeficiency virus (HIV) (Scheme 1). Clinical trials demonstrated that the introduction of Csp3–S moieties could improve their biological activities, such as anti-tumour, anti-inflammatory and immunomodulatory properties.1 Besides that, the Csp3–S bond can also be found in the natural amino acid, such as cysteine, and facilitate the metabolism process of the related protein in the organism.2 Given the importance of Csp3–S containing compounds, methods that can directly assemble the Csp3–S bond through direct C–H functionalization with a high efficiency and selectivity are highly demanded. Moreover, these methods could facilitate the late-stage functionalization of complex molecules that enable the rapid diversification and construction of a library of molecule analogues.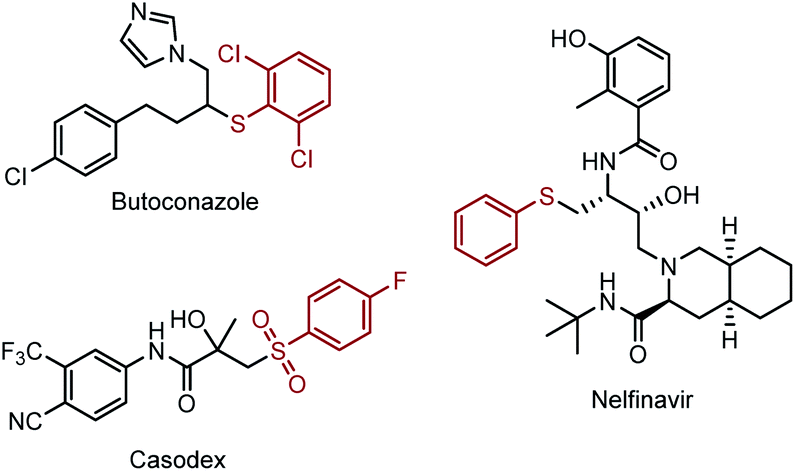 | ||
| Scheme 1 Pharmaceutical molecules containing sp3 C–S thiol moieties. | ||
Csp3–S bonds are usually constructed by nucleophilic substitution of the alkyl halides with metal thiolate (Scheme 2a). Also, similar to other kinds of nucleophilic substitution reactions, these methods are sensitive to the sterics.3 With the development of transition-metal catalysis, more efficient protocols to introduce the Csp3–S bond were realized via the transition-metal catalysed cross-coupling of organo halides/alkene and thiols/thioethers.4 For example, Li reported the palladium catalysed cross-coupling of aryl halides and thiols; however, only primary and secondary thiols were shown to be suitable substrates.5 Furthermore, the preparation of alkyl halide substrates was tedious and was not step- and atom-economic (Scheme 2b). Alternatively, hydrothiolation of alkenes has been developed to construct the Csp3–S bond. However, pre-functionalization of the substrates is necessary, and the regioselectivity greatly depends on the structure of the substrate (Scheme 2c).6 Recently, the direct thiolation via the alpha-sp3 C–H bonds of ether and amine was achieved by Li,7 Yuan and Xiang,8 independently (Scheme 2d). The thiolation of Csp3–H was also developed, facilitated by a directing group strategy (Scheme 2e).9 However, to date, a general method to realize the C–H bond thiolation remains a significant challenge.
 | ||
| Scheme 2 The strategies to construct the Csp3–S bond: (a) Csp3–S bond formation via nucleophilic substitution. (b) Transition-metal catalysed cross-coupling. (c) Catalytic addition to unsaturated double bond. (d) Direct Csp3–H bond thiolation. (e) Directing group assisted Csp3–H bond thiolation. (f) Site selective Csp3–S bond formation. | ||
Inspired by the Hofmann–Löffler–Freytag (HLF) reaction,10 C–H bond functionalization via a radical pathway provides a new route to construct different types of targeted molecules.11 Elegant related works have been documented by Suárez,12 Muñiz13 and Nagib14 independently by using the stoichiometric amounts of iodine, catalytic iodine, and triiodide for the HLF type reaction. More recently, a breakthrough of metallaphotoredox-catalysed remote functionalization of amides was demonstrated by the groups of Knowels,15 Rovis,16 and Meggers.17 In addition, Cu-modified HLF-type reactions with a simple NH substrate are particularly valuable in primary C–H bond amination.18 Although the generated amidyl radical induced 1,5-HAT (HLF reaction) process is well established, further transformation of C-centred radicals is limited to the halogenation, cyclization or interceptions by alkenes or heteroaryls.19 The metal mediated C-centred radical resulted in the functionalization of unactivated Csp3–H bond selectively and efficiently, a great challenge. We proposed that by using a N–F precursor, the low tendency toward N–F homolysis and instability of the fluorine radical, can enable the 1,5-HAT process to facilitate Csp3–H bond functionalization over C–F bond formation. Previously, and independently, Zhu20 and Nagib21 have reported a remote arylation of an aliphatic amine via activation of the N–F precursors. Very recently, Muñiz demonstrated a cyclization reaction of an aliphatic amine via N–F bond activation by a copper catalyst, albeit under a high temperature.22 Li has successfully achieved a copper catalysed trifluoromethylation by using N–F precursors.23 Given the importance of thiol moieties in functional molecules, here we describe a protocol for the site-selective thiolation of aliphatic amines through the combination of light-driven and copper-facilitated 1,5-hydrogen atom transfer (Scheme 2f). This strategy enables the transformation of primary, secondary and tertiary C–H bonds into C–S bonds with a high efficiency. Meanwhile, a late-stage modification of biologically active compounds was demonstrated to show the synthetic utility of such a methodology.
Results and discussion
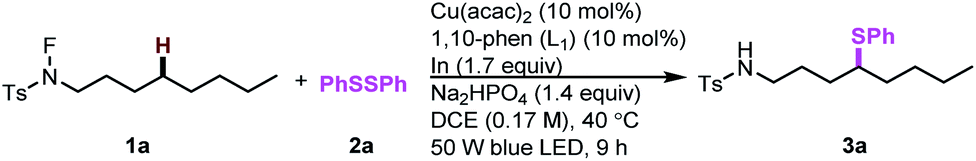
To initiate our proposal, N-fluoro-tosylamide 1a and phenyl disulfide 2a were chosen as the model substrates. After extensive optimization, we found that a cocktail containing 10 mol% of Cu(acac)2, 10 mol% of 1,10-phenanthroline (L1), 1.4 equivalent Na2HPO4 and 1.7 equivalent of indium powder as additives could promote the desired δ thiolated product 3a in 75% isolated yield in the presence of blue LED. Control experiments were conducted to determine the role of each ingredient (Table 1, entries 2–5). Without a copper catalyst or ligand, the yield decreased significantly (Table 1, entries 2 and 3). The desired product was only observed in 36% yield, indicating the pivotal role of light in this transformation (Table 1, entry 4). Moreover, a lower yield was obtained in the absence of indium powder and base (Table 1, entry 5). Other copper complexes were also tested in this reaction, resulting in low yields (Table 1, entries 6–8). The aliphatic amines are generally regarded as good reducing reagents in photoredox reactions. However, the addition of triethylamine inhibited this reaction (Table 1, entry 9). To determine the role of copper complexes in this transformation,24 we replaced the copper with an Ir complex or Ru complex, and no desired results were obtained, indicating the role of copper is more than as a photoredox catalyst in this reaction (Table 1, entries 11 and 12).|
|
||
|---|---|---|
| Entry | Variations from “standard” conditions | Yieldb (%) |
| a Reaction was conducted on a 0.1 mmol scale. b Yields were determined by 1H NMR analysis versus 1,1,2,2-tetrachloroethane as the internal standard. Isolated yield in parentheses. c cat.[Cu]1 is Cu(CH3COCHCOCF3)2. d cat.[Cu]2 is Cu(CF3COCHCOCF3)2. | ||
| 1 | None | 78 (75) |
| 2 | Without Cu(acac)2 | 27 |
| 3 | Without L1 | 13 |
| 4 | Under dark | 36 |
| 5 | Without In powder and Na2HPO4 | 65 |
| 6 | Cu(OTf)2 instead of Cu(acac)2 | 47 |
| 7 | cat.[Cu]1c instead of Cu(acac)2 | 31 |
| 8 | cat.[Cu]2d instead of Cu(acac)2 | 20 |
| 9 | Et3N instead of Na2HPO4 | 0 |
| 10 | 2,6-Lutidine instead of Na2HPO4 | 19 |
| 11 | Ir(ppy)3 instead of Cu(acac)2 | 10 |
| 12 | Ru(1,10-phen)3Cl2 instead of Cu(acac)2 | 7 |
|
||
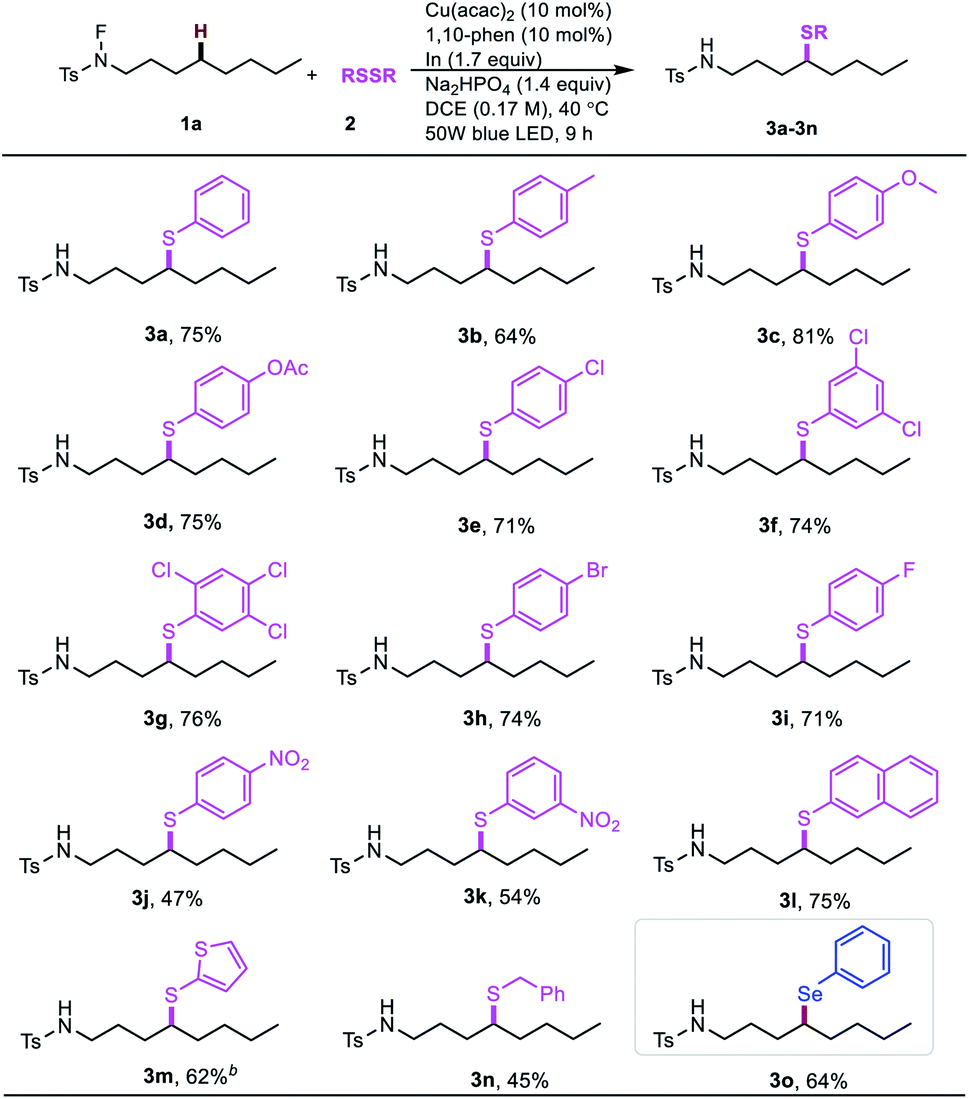
With the optimal conditions in hand, we examined the scope of different disulfides with N-fluoro-tosylamide 1a (Table 2). The substituted disulfides with electron-donating groups, such as methyl (3b), methoxyl (3c) and acetoxyl (3d), were all well tolerated. Moreover, the halide-substituted aryl disulfides also underwent this thiolation reaction (3e–3i), even with multi-halides or fluoro substituents. Remarkably, the electron-withdrawing nitro-group on the disulfide led to a moderate drop in reactivity (3j, 3k). Interestingly, the thiophenyl group generally has special properties in optoelectronic materials, and can afford the desired product in an acceptable yield (3m). Furthermore, to our satisfaction, the benzyl disulfide can give the δ benzyl thioether in a slightly compromised yield (3n, 45%). Interestingly, the δ-selenation could also be achieved when using diselenide instead of disulfide (3o).
This C–H thiolation protocol was also successfully applied to functionalize a wider range of electronically and sterically diverse primary, secondary and tertiary aliphatic C–H bonds (Table 3). The thiolation yield for primary C–H bond is slightly increased with a N-fluorotosylamide substrate containing a substituent at the α-position (4a, 4b). N-Fluorocarboxamides are also good substrates for this transformation (4c, 4d). Notably, functionalization of the secondary C–H bond afforded a series of thiolether derivatives selectively. Even with longer carbon chains in the substrates (4e–4g), the reaction occurred only at the δ position. Surprisingly, a gem-dialkyl group on the linear N-fluoroamide substrates at the β-position does not improve the product yield (4f). The biologically relevant functionalities, such as Nphth (4h), carboxylic ester (4i) and OTBDMS (4j, 4k), as well as OBz (4l) are well tolerated. Specifically, exclusive δ-selectivity was observed in the case of 4h and 4j bearing a heteroatom near the reaction centres. Cycloalkanes with various ring sizes ranging from four to seven carbons were thiolated in good yields (4m–4q, 4s–4t). The adamantyl group can be functionalized selectively on secondary C–H bonds rather than tertiary ones (4r). Obviously, in heteroatom-containing cycles, functionalized products were obtained at the α-position with a better diasteroselectivity for the six-member ring than the five-member ring (4u, 4v). Disappointingly, the steric hindered substituents at the α- or β-position of N-fluoroamides led to a poor diasteroselectivity (4w–4y). Both the 1,5-HAT and 1,6-HAT processes occurred in a substrate which involved a tertiary C–H bond at the ε-position (4z). Finally, this transformation can be successfully applied to construct the quaternary carbon centre with a thiol group, highlighting the synthetic utility of this methodology (4aa, 4ab).
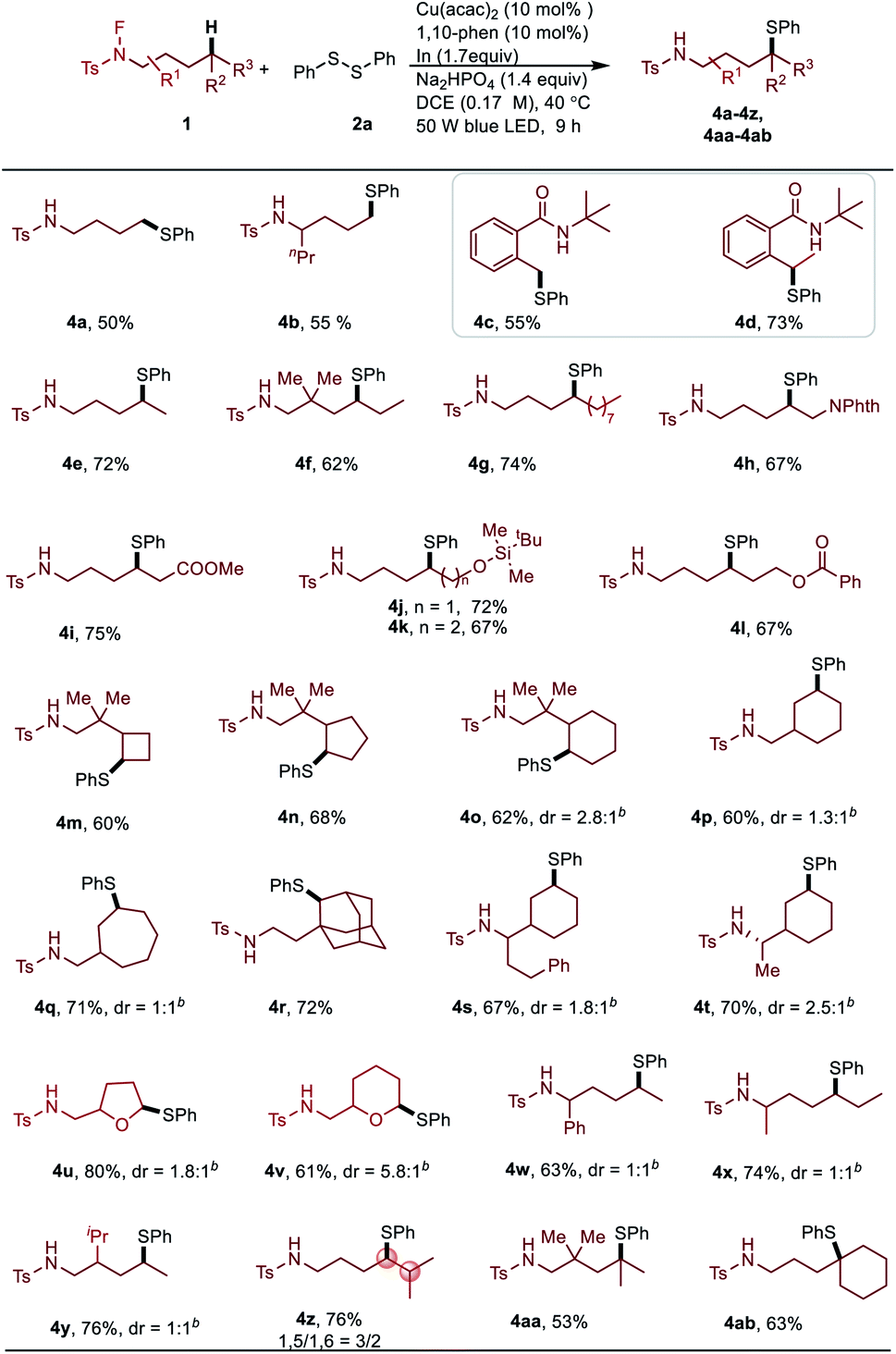
Having demonstrated the applicability of this remote Csp3–H thiolation protocol, late-stage functionalization of biologically active molecules was carried out (Table 4). The amino alcohol derivatives can be coupled with varieties of disulfides, which greatly demonstrated the powerful utility of this method (5a–5g). These functionalized amino alcohols have a great potential in the synthesis of biological compounds. A non-symmetric thiolation of the methyl group occurred on the menthol derived N-fluoroamide substrate (5h). It's important to note that pharmaceutical molecular (5i) and complex natural products (5j, 5k) can be readily modified with excellent regioselectivity.
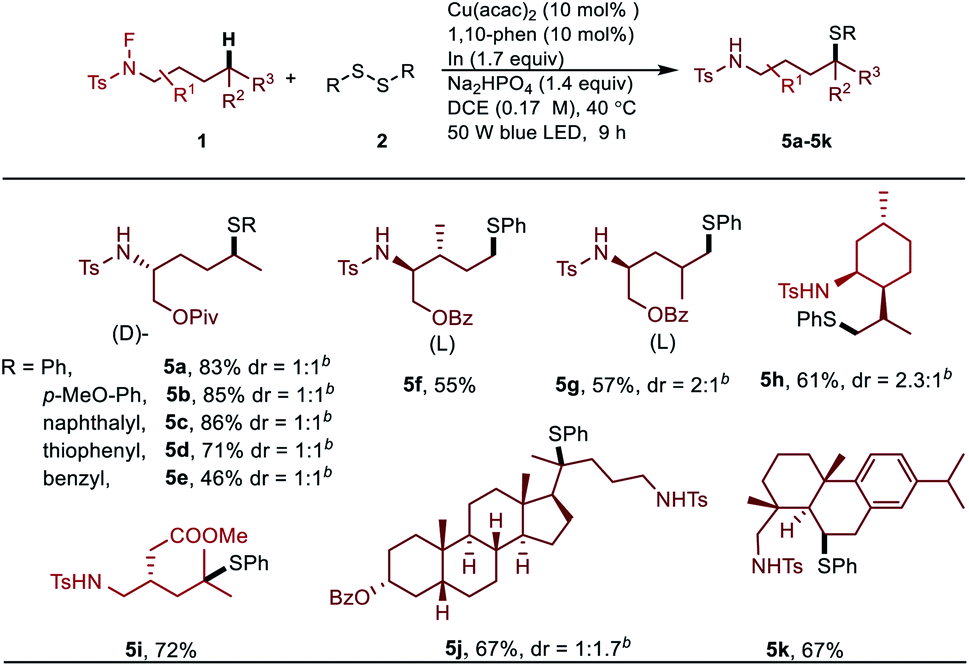
Furthermore, to illustrate the utility of this new transformation with respect to constructing drug scaffolds, we tested a type of substrates which can proceed by a domino process involving the 1,5-HAT followed by cyclization and sequence thiolation, affording thiol substituted pyrrolidine or piperidine derivatives (Scheme 3).
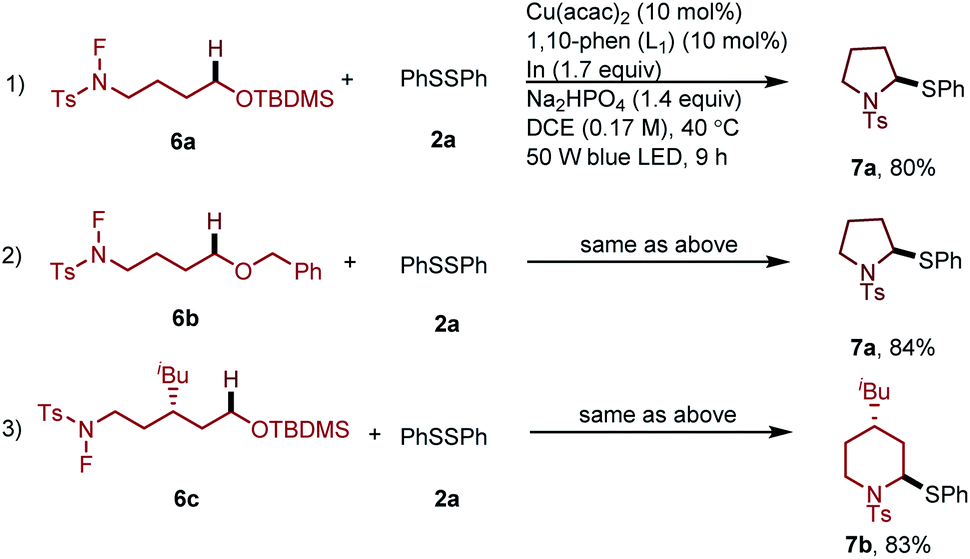 | ||
| Scheme 3 Preparation of pyrrolidine or piperidine derivatives via remote thiolation. | ||
To gain insight into the reaction mechanism, competition experiments between 1°/2° C–H bonds, 2°/3° C–H bonds and 1°/3° C–H bonds were carried out independently (Scheme 4a). The total reactivity of the hydrogen atoms decreased in the order tertiary > secondary > primary. Furthermore, to test the selectivity of thiolation reagents, the mixed disulfides were subjected to this transformation (Scheme 4b). For an alkyl phenyl disulfide, only the phenyl thioether product was observed. For a mixed aryl disulfide with an electron-donating group OMe and an electron-withdrawing group CF3, the thiolation occurred only with the electron-donating OMe substituted thiol group.25 These selectivities may result from the electron density on different sulfur moieties. Based on this selectivity, we developed a direct cross-coupling between two biologically interesting molecules to give the corresponding conjugated compounds (Scheme 5a). Subsequently, the thioether group can be directly oxidized to a sulphone group, which broadly exist in pharmaceuticals (Scheme 5b).
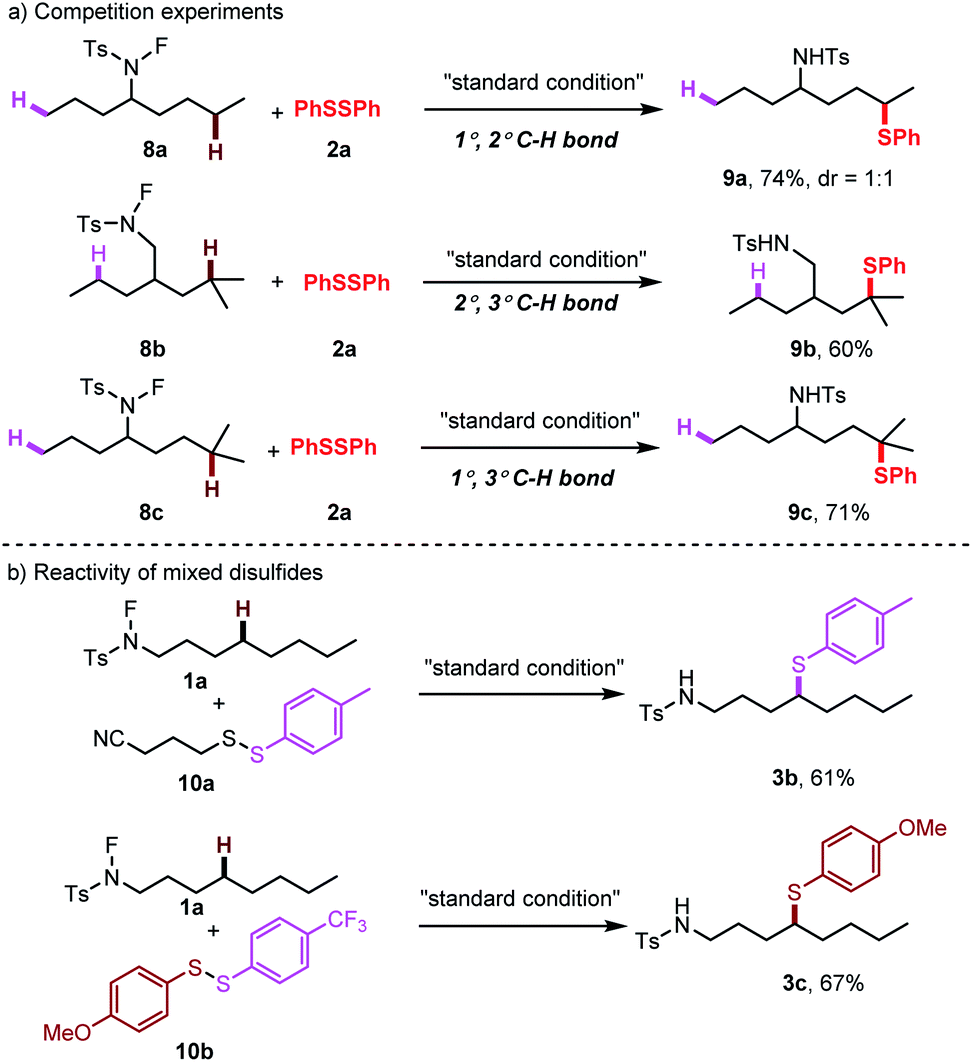 | ||
| Scheme 4 Control experiments: (a) competition experiments; (b) reactivity of mixed disulfides. | ||
 | ||
| Scheme 5 Late-stage functionalization of biological active compounds: (a) cross-coupling with biological active compounds; (b) oxidized thioether to sulphone. | ||
Moreover, radical control experiments were carried out to uncover evidence for the presence of radical intermediates. Radical-clock experiments were conducted (Scheme 6a), the observation of products 15a, 17a and 19a indicating the formation of a carbon-centred radical in the transformation. Especially for 19a, the observed linear selectivity deduced that a η1- or η3 metal allylic intermediate maybe involved in this process. Subsequently, the radical process was further demonstrated by the results in the presence of TEMPO and BHT. Furthermore, the experiments of PhSH and In(SPh)3 were conducted to test the possible active species of this thiolation reaction. The results indicated that by using PhSH, 20% of the desired product was obtained, while no desired product was observed by using In(SPh)3 as the thiolation reagent (Scheme 6b). Finally, blank experiments were carried out to confirm the effective role of each additive in this transformation (details in the ESI†).
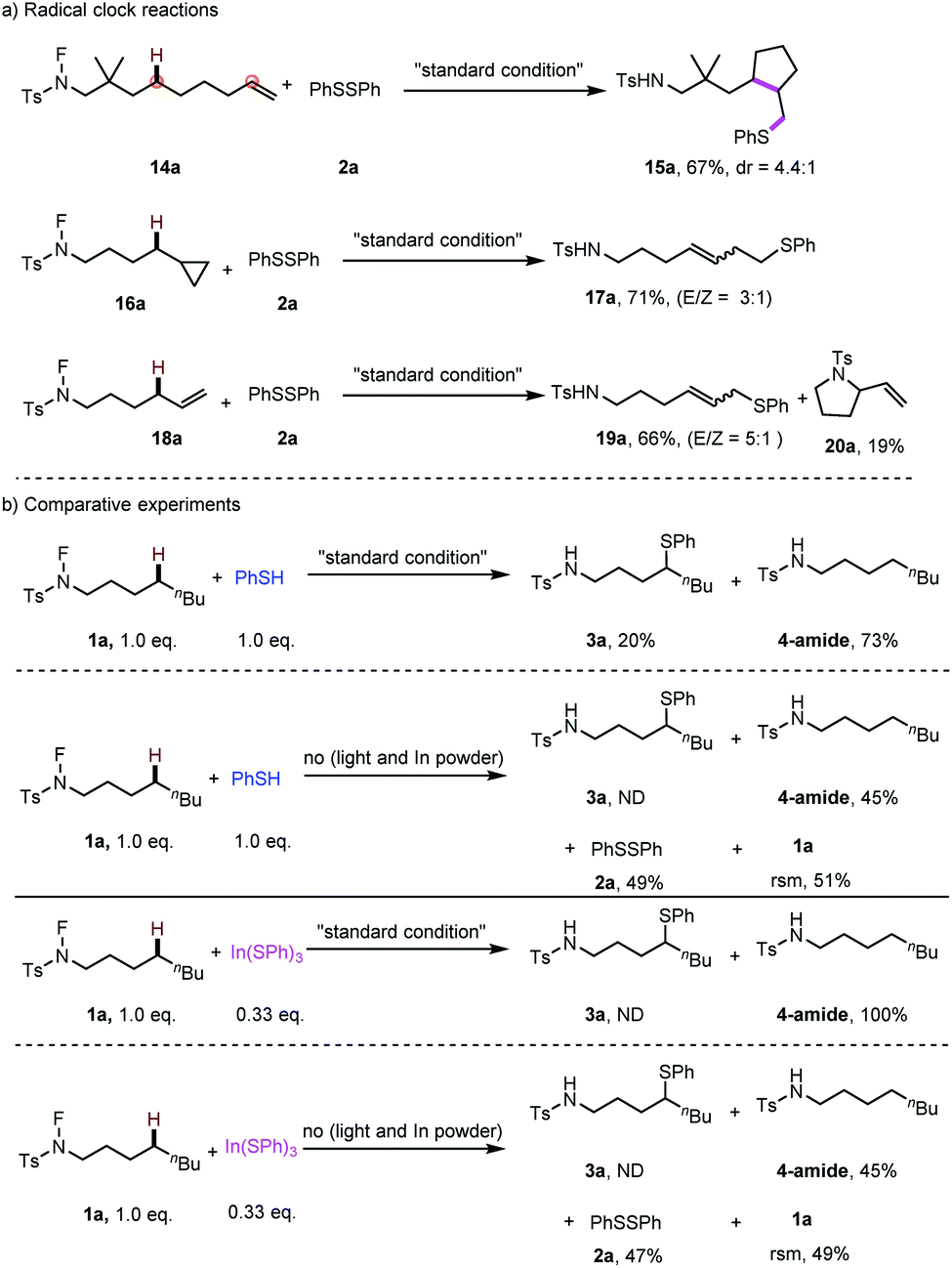 | ||
| Scheme 6 Mechanistic investigation: (a) radical clock reactions; (b) comparative experiments. | ||
Based on these results, we propose that this transformation is preferred by a light-driven, copper initiated 1,5-HAT process (Scheme 7). Ligand exchange between an in situ generated Cu(I) catalyst and disulfide occurred to generate Cu(I)–I species, which could be excited by visible light followed by reducing the N–F amide A to afford the N-centred radical B and Cu(II)–II intermediate. The Cu(II)–II released one molecular of F-SAr26 through a metathesis process to afford the Cu(II)–III. The selective formation of F-SAr is likely to result from the coordination ability of the sulfur group. The electron-rich sulfur group coordinated to form the Cu(I)–I species. This selectivity is in conjunction with the result of the mixed disulfide. Translocation of the N-centred radical B to the C-centred radical Cvia 1,5-HAT occurred, and then the δ C-radical was intercepted by the Cu(II)–III to form the Cu(III)–IV intermediate. Finally, the reductive elimination of Cu(III)–IV gave the desired product D and regenerated active Cu(I) catalyst. Furthermore, efforts to expand the scope of this transformation are underway.
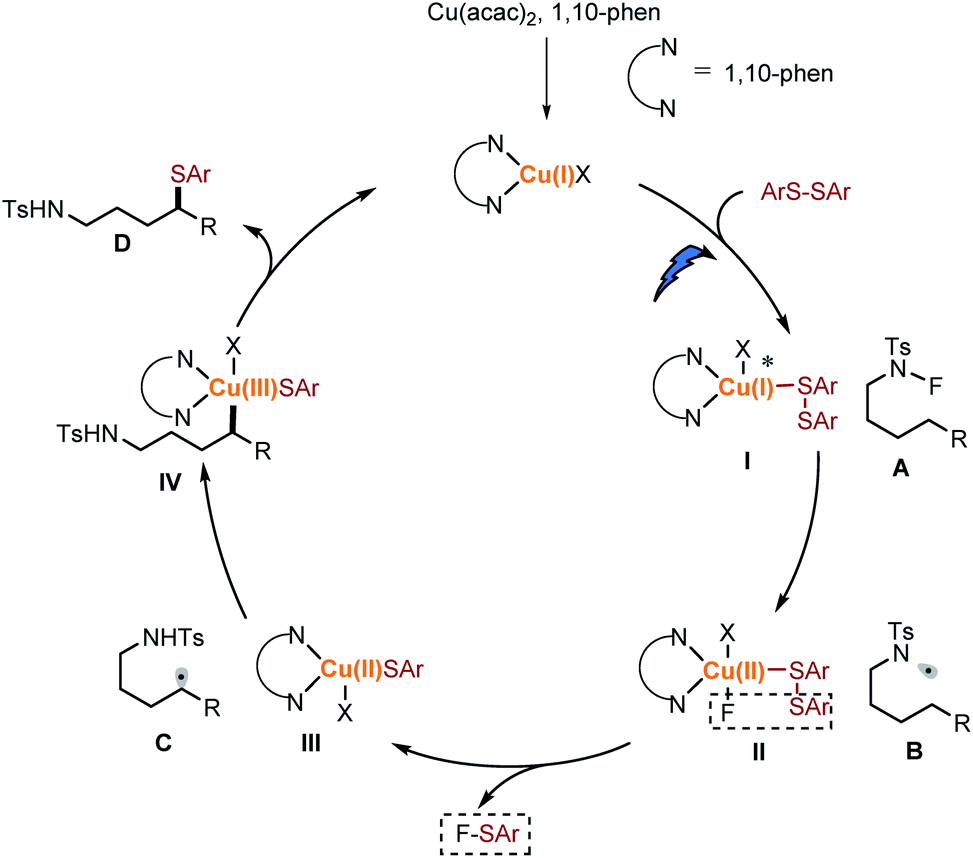 | ||
| Scheme 7 Proposed mechanism. | ||
Conclusions
In summary, we have developed a light-driven, copper-catalysed site-selective thiolation of Csp3–H bonds for aliphatic amines. Primary, secondary and tertiary C–H bonds can all be successfully converted into C–S bonds. The broad amine scope, good functional compatibility, and late-stage modification of biologically active intermediates underscore the unique synthetic potential of this method.27Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We gratefully acknowledge the financial support by the National Natural Science Foundation of China (21572124, 21602128), Fundamental Research Funds for the Central Universities (GK201802008), and start-up funds from Shaanxi Normal University, Shaanxi Provincial Natural Science Foundation (2018JM2010).Notes and references
- (a) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef CAS; (b) D. Xiao, Y.-J. Wang, X.-B. Hu, W.-J. Kan, Q. Zhang, X. Jiang, Y.-B. Zhou, J. Li and W. Lu, Eur. J. Med. Chem., 2019, 176, 419 CrossRef CAS PubMed; (c) C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355 RSC; (d) H. Guo, B. Sun, H. Gao, X. Chen, S. Liu, X. Yao, X. Liu and Y. Che, J. Nat. Prod., 2009, 72, 2115 CrossRef CAS; (e) J.-M. Wang, G.-Z. Ding, L. Fang, J.-G. Dai, S.-S. Yu, Y.-H. Wang, X.-G. Chen, S.-G. Ma, J. Qu, S. Xu and D. Du, J. Nat. Prod., 2010, 73, 1240 CrossRef CAS PubMed.
- (a) J. Zhao and X. Jiang, Chin. Chem. Lett., 2018, 29, 1079 CrossRef CAS; (b) M. C. Bagley, J. W. Dale, E. A. Merritt and X. Xiong, Chem. Rev., 2005, 105, 685 CrossRef CAS PubMed; (c) X. Just-Baringo, F. Albericio and M. Álvarez, Angew. Chem., Int. Ed., 2014, 53, 6602 CrossRef CAS.
- (a) Q. W. Decker and H. W. Post, J. Org. Chem., 1957, 22, 1957 CrossRef; (b) F. M. Menger and L. Shi, J. Am. Chem. Soc., 2006, 128, 9338 CrossRef CAS PubMed.
- (a) T. Kondo and T.-A. Mitsudo, Chem. Rev., 2000, 100, 3205 CrossRef CAS PubMed; (b) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed; (c) H. Liu and X. Jiang, Chem.–Asian J., 2013, 8, 2546 CrossRef CAS PubMed.
- (a) G. Y. Li, Angew. Chem., Int. Ed., 2001, 40, 1513 CrossRef CAS; (b) G. Y. Li, G. Zhang and A. F. Noonan, J. Org. Chem., 2001, 66, 8677 CrossRef CAS.
- (a) T. Tamai, K. Fujiwara, S. Higashimae, A. Nomoto and A. Ogawa, Org. Lett., 2016, 18, 2114 CrossRef CAS PubMed; (b) X.-H. Yang, R. T. Davison, S.-Z. Nie, F. A. Cruz, T. M. McGinnis and V. M. Dong, J. Am. Chem. Soc., 2019, 141, 3006 CrossRef CAS PubMed.
- R.-Y. Tang, Y.-X. Xie, Y.-L. Xie, J.-N. Xiang and J.-H. Li, Chem. Commun., 2011, 47, 12867 RSC.
- S.-R. Guo, Y.-Q. Yuan and J.-N. Xiang, Org. Lett., 2014, 15, 4654 CrossRef PubMed.
- (a) X. Wang, R. Qiu, C. Yan, V. P. Reddy, L. Zhu, X. Xu and S.-F. Yin, Org. Lett., 2015, 17, 1970 CrossRef CAS PubMed; (b) S.-Y. Yan, Y.-J. Liu, B. Liu, Y.-H. Liu, Z.-Z. Zhang and B.-F. Shi, Chem. Commun., 2015, 51, 7341 RSC.
- (a) A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1883, 16, 558 CrossRef; (b) A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1885, 18, 5 CrossRef; (c) K. Löffler and C. Freytag, Ber. Dtsch. Chem. Ges., 1910, 42, 3427 CrossRef; for selected review, see: (d) M. E. Wolff, Chem. Rev., 1963, 63, 55 CrossRef CAS; (e) R. S. Neal, Synthesis, 1971, 1 CrossRef.
- For selected examples on remote C–H bond activation via N-radical chemistry, see: (a) Z. Li, L. Song and C. Li, J. Am. Chem. Soc., 2013, 135, 4640 CrossRef CAS PubMed; (b) B. J. Groendyke, D. I. AbuSalim and S. P. Cook, J. Am. Chem. Soc., 2016, 138, 12771 CrossRef CAS; (c) S. P. Morcillo, E. M. Dauncey, J. H. Kim, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 12945 CrossRef CAS; (d) E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 744 CrossRef CAS PubMed; (e) E. A. Wappes, K. M. Nakafuku and D. A. Nagib, J. Am. Chem. Soc., 2017, 139, 10204 CrossRef CAS; (f) C. Zhu, Y. Liang, X. Hong, H. Sun, W.-Y. Sun, K. N. Houk and Z. Shi, J. Am. Chem. Soc., 2015, 137, 7564 CrossRef CAS; (g) Y. Tang, Y. Qin, D. Meng, C. Li, J. Wei and M. Yang, Chem. Sci., 2018, 9, 6374 RSC; (h) L. Wang, Y. Xia, K. Bergander and A. Studer, Org. Lett., 2018, 20, 5817 CrossRef CAS; (i) W. Shu and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1881 CrossRef CAS; (j) H. Zhang, Y. Zhou, P. Tian and C. Jiang, Org. Lett., 2019, 21, 1921 CrossRef CAS; (k) X.-Q. Mou, X.-Y. Chen, G. Chen and G. He, Chem. Commun., 2018, 54, 515 RSC; (l) L. M. Stateman, E. A. Wappes, K. M. Nakafuku, K. M. Edwards and D. A. Nagib, Chem. Sci., 2019, 10, 2693 RSC; (m) Y. Kumar, Y. Jaiswai and A. Kumar, Org. Lett., 2018, 20, 4964 CrossRef CAS; (n) H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 1692 CrossRef CAS; (o) F. Wang and S. S. Stahl, Angew. Chem., Int. Ed., 2019, 58, 6385 CrossRef CAS PubMed; (p) Y.-R. Gu, X.-H. Duan, L. Chen, Z.-Y. Ma, P. Gao and L.-N. Guo, Org. Lett., 2019, 21, 917 CrossRef CAS. For selected review, see: (q) S. Z. Zard, Chem. Soc. Rev., 2008, 37, 1603 RSC; (r) L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Synthesis, 2018, 50, 1569 CrossRef CAS PubMed; (s) J. Davies, S. P. Morcillo, J. J. Douglas and D. Leonori, Chem.–Eur. J., 2018, 24, 12154 CrossRef CAS PubMed.
- (a) R. Hernández, A. Rivera, J. A. Salazar and E. Suárez, J. Chem. Soc., Chem. Commun., 1980, 958 RSC; (b) C. Betancor, J. I. Concepcion, R. Hernandez, J. A. Salazar and E. Suárez, J. Org. Chem., 1983, 48, 4430 CrossRef CAS; (c) C. G. Francisco, A. J. Herrera and E. Suárez, J. Org. Chem., 2003, 68, 1012 CrossRef CAS PubMed.
- (a) C. Martínez and K. Muńiz, Angew. Chem., Int. Ed., 2015, 54, 8287 CrossRef PubMed; (b) C. Q. O'Broin, P. Fernández, C. Martínez and K. Muńiz, Org. Lett., 2016, 18, 436 CrossRef PubMed; (c) P. Becker, T. Duhamel, C. J. Stein, M. Reiher and K. Muñiz, Angew. Chem., Int. Ed., 2017, 56, 8004 CrossRef CAS PubMed; (d) H. Zhang and K. Muñiz, ACS Catal., 2017, 7, 4122 CrossRef CAS; (e) T. Duhamel, C. J. Stein, C. Martínez, M. Reiher and K. Muñiz, ACS Catal., 2018, 8, 3918 CrossRef CAS.
- E. A. Wappes, S. C. Fosu, T. C. Chopko and D. A. Nagib, Angew. Chem., Int. Ed., 2016, 55, 9974 CrossRef CAS.
- G. C. Choi, Q. Zhu, D. Miller, C. Gu and R. R. Knowels, Nature, 2016, 539, 268 CrossRef CAS.
- J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272 CrossRef.
- W. Yuan, Z. Zhou, L. Gong and E. Meggers, Chem. Commun., 2017, 53, 8964 RSC.
- (a) C. Yamamoto, K. Takamatsu, K. Hirano and M. Miura, J. Org. Chem., 2016, 81, 7675 CrossRef CAS PubMed; (b) F. Pan, B. Wu and Z.-J. Shi, Chem.–Eur. J., 2016, 22, 6487 CrossRef CAS PubMed.
- For selected examples on amidyl radical induced HAT, see: for halogenation, see: (a) Q. Qin and S. Yu, Org. Lett., 2015, 17, 1894 CrossRef CAS; (b) T. Liu, M. C. Myers and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 129, 313 Search PubMed; (c) M. A. Short, J. M. Blackburn and J. L. Roizen, Angew. Chem., Int. Ed., 2017, 57, 296 CrossRef PubMed. For cyclization, see: (d) R. Fan, D. Pu, F. Wen and J. Wu, J. Org. Chem., 2007, 72, 8994 CrossRef CAS PubMed; (e) P. Becker, T. Duhamel, C. Martínez and K. Muńiz, Angew. Chem., Int. Ed., 2018, 57, 5166 CrossRef CAS; (f) N. R. Paz, D. Rodíguez-Sosa, H. Valdés, R. Marticorena, D. Melián, M. B. Copano, C. C. González and A. J. Herrera, Org. Lett., 2015, 17, 2370 CrossRef CAS; (g) M. Yang, B. Su, Y. Wang, K. Chen, X. Jiang, Y.-F. Zhang, X.-S. Zhang, G. Chen, Y. Cheng, Z. Cao, Q.-Y. Guo, L. Wang and Z.-J. Shi, Nat. Commun., 2014, 5, 4707 CrossRef CAS; (h) D. Meng, Y. Tang, J. Wei, X. Shi and M. Yang, Chem. Commun., 2017, 53, 5744 RSC. Reacted with alkenes, see: (i) M. A. Ashley, C. Yamauchi, J. C. K. Chu, S. Otsuka, H. Yorimitsu and T. Rovis, Angew. Chem., Int. Ed., 2019, 58, 4002 CrossRef CAS; (j) S. T. Nguyen, Q. Zhu and R. R. Knowles, ACS Catal., 2019, 9, 4502 CrossRef CAS; (k) Z.-Y. Ma, L.-N. Guo, Y. You, F. Yang, M. Hu and X.-H. Duan, Org. Lett., 2019, 21, 5500 CrossRef CAS PubMed. Reacted with heteroaryl, see: (l) N. Tang, X. Wu and C. Zhu, Chem. Sci., 2019, 10, 6915 RSC.
- Z. Li, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2018, 57, 13288 CrossRef CAS PubMed.
- Z. Zhang, L. M. Stateman and D. A. Nagib, Chem. Sci., 2019, 10, 1207 RSC.
- D. Bafaluy, J. M. Muñoz-Molina, I. Funes-Ardoiz, S. Herold, A. J. de Aguirre, H. Zhang, F. Maseras, T. Belderrain, P. J. Pérez and K. Muñiz, Angew. Chem., Int. Ed., 2019, 58, 8912 CrossRef CAS PubMed.
- Z. Liu, H. Xiao, B. Zhang, H. Shen, L. Zu and C. Li, Angew. Chem., Int. Ed., 2019, 58, 2510 CrossRef CAS.
- For selected copper photoredox catalysis, see: (a) Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu., Science, 2016, 351, 681 Search PubMed; (b) D. B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B. M. Bhanage and O. Reiser, Angew. Chem., Int. Ed., 2015, 54, 6999 CrossRef CAS.
- The results was detected by MS-ESI.
- For the preparation and characterization of F-SPh, see: (a) F. Seel, R. Budenz, R. D. Flaccus and R. Staab, J. Fluorine Chem., 1978, 12, 437 CrossRef CAS; (b) H. Poleschner and K. Seppelt, Chem.–Eur. J., 2004, 10, 6565 CrossRef CAS; (c) S. Liu, J. Chem. Phys., 2014, 141, 194109 CrossRef.
- When our manuscript is in revision, a similary type reaction was reported: A. Modak, E. N. Pinter and S. P. Cook, J. Am. Chem. Soc., 2019, 141, 18405 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, compound characterization. See DOI: 10.1039/c9sc04169a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |