
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed enantioselective arylalkynylation of alkenes†
Guangyue
Lei
,
Hanwen
Zhang
,
Bin
Chen
,
Meichen
Xu
and
Guozhu
Zhang
 *
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Center for Excellence in Molecular Synthesis, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P. R. China. E-mail: guozhuzhang@sioc.ac.cn
First published on 8th January 2020
Abstract
A copper-catalyzed enantioselective arylalkynylation of alkenes with diaryliodonium salt and a monosubstituted alkyne is reported. The three-component coupling reactions proceed under mild reaction conditions with a broad substrate scope, leading to synthetically valuable 1,2-diaryl-3-butynes. The key to the success of this chemistry is the employment of the chiral bisoxazoline-phenylaniline (BOPA) ligand. A novel reaction pathway involving the phenyl radical generation under thermal copper catalysis is proposed according to mechanistic studies.
Introduction
Alkenes, alkynes, and arenes are three ubiquitous unsaturated substructures in numerous compounds. Chemical reactions that link alkenes, alkynes, and arenes are highly useful in both industry and academic research, because the products of their union could provide a versatile platform for the synthesis of bioactive and medicinally relevant molecules, fine chemicals, and monomers for polymerization, as exemplified by Heck, ring-closing metathesis, Sonogashira, and related reactions.1 The dual functionalization of alkenes is currently considered as one of the most straightforward approaches for the construction of multi-functional compounds.2 However, the catalytic, enantioselective, and three-component arylalkynylation of alkenes is rare.3Due to the high oxidation state of the iodine center, hypervalent diaryliodonium salts readily interact with transition metals in redox processes and are widely used in the formation of aryl–carbon and aryl–heteroatom bonds,4 an important field known as arylation chemistry.5 In those studies, a combination of a Cu catalyst and diaryliodonium salts has recently emerged as a very useful and efficient strategy for arylation via a high oxidation state Cu(III)–aryl intermediate that behaves as an activated aryl cation.6 Despite great advances, the arylation-initiated double functionalization of alkenes with diaryliodonium salt remains relatively unexplored.7 Taking advantage of the unique electrophilic property of copper(III)–aryl species, in 2012, Gaunt and co-workers reported the first example of Cu-catalyzed arylation of simple alkenes with diaryliodonium salts.7a The same group further realized the cascade enantioselective arylation and subsequent cyclization of allylic amide; a regiodivergent outcome was observed when using the electron-poor iodonium aryl group.7b Very recently, they reported a copper-catalyzed enantioselective arylative semipinacol rearrangement of allylic alcohols using diaryliodonium salts (Fig. 1, eqn (a)).7c Notably, in those cases, the privileged bisoxazoline (BOX) ligand imparts excellent enantio-induction in the arylation step.7d,7e Recently, Liu reported an elegant example of copper catalyzed enantioselective radical relay trifluoromethyl alkynlylation of styrene derivatives (Fig. 1, eqn (b)). In addition, [Cu(dpp)2] +PF6 (dpp = 2,9-diphenyl-1,10-phenanthroline) complexes could serve as photocatalyst and promote the aryl radical generation from diaryl iodonium salt under green LED.7f We envisaged that, a proper combination of copper, ligand and substrate, a reduction of diaryl iodonium salt with generation of aryl radical under thermal conditions might be viable.
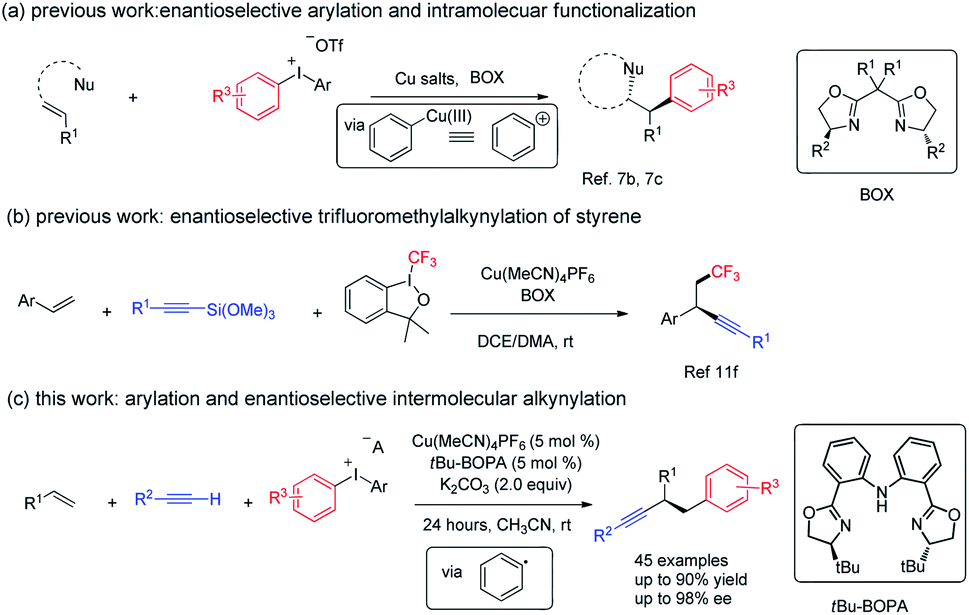 | ||
| Fig. 1 Copper-catalyzed enantioselective arylalkynylation of styrenes. | ||
We have been interested in the radical difunctionalization of alkenes including feedstock 1,3-butadiene.8C2-symmetric bisoxazoline-carbazole (Cbzbox), a tridentate monoanionic ligand known as one class of “pincer” ligands, which have been found to effectively stabilize other 3d transition metals, such as Cr, Fe, Ni, Cu, and Zn. The resulting metal complexes exhibit high activities and stereoselectivities for a broad range of asymmetric reactions, featuring Lewis acid catalysis in most cases;9 Cbzbox–chromium complex could reduce carbohalides to alkyl radicals for Nozaki–Hiyama–Kishi reactions.8a We hypothesized that, Cbzbox–copper(I)-complex could serve as a single-electron reductant to reduce diphenyliodonium salt to generate a phenyl radical to initiate the alkene difunctionalization. Herein, we describe a novel copper-catalyzed three-component coupling reaction involving a mono-substituted alkyne, styrene, and diaryliodonium salt, enabled by a bisoxazoline-phenylaniline (tBu-BOPA)–copper complex (Fig. 1, eqn (c)). A broad range of substrates efficiently participate in this coupling reaction, leading to valuable enantio-enriched diarylated propargylic compounds, a privileged segment observed in pharmaceutical agents,10 and an advanced intermediate leading to functional materials not easily accessible through other methods.11 Preliminary mechanistic studies support a radical pathway. This process represents a valuable alternative reaction mode to existing methods of Cu-catalyzed arylfunctionalization of alkene with diaryliodonium salt.7,12
Results and discussion
We selected p-methyl styrene, diaryliodonium salt, and a terminal alkyne to test the three-component coupling reactions by varying different reaction parameters. After extensive experiments of screening various multidentate ligands, we found that reaction of 1a with diphenyliodonium hexafluorophosphate 3a and phenyl acetylene 2a, with terpyridine as the ligand and 10 mol% of Cu(MeCN)4PF6 as the catalyst, gave a moderate yield of racemic 4aa (Table 1, entry 2). Gratifyingly, using iPr-Cbzbox L1,134aa was obtained in comparable yield with an enantiomeric excess (ee) of 67% (Table 1, entry 3). Inspired by this, we tested a variety of Cbzbox with different substituents (L2–L6). However, no better results in terms of ee were obtained (Table 1, entries 4–8). Surprisingly, bisoxazoline-phenylaniline (iPr-BOPA) L7 with a flexible backbone led to product in equal level of ee (60%) (Table 1, entry 9).14 Further adjustment of the substituents on the BOPA ligand resulted in a reaction catalyzed by tBu-BOPA L9 that gave a 78% yield of 4aa in 86% ee (Table 1, entry 1).|
|
|||
|---|---|---|---|
| Entrya | Deviation | Yieldb (%) | eec (%) |
| a The reaction conditions were styrene (0.2 mmol), diphenyl iodonium salt (0.25 mmol), alkyne (0.4 mmol), Cu(I) (5 mol%), base (0.4 mmol), ligand (5 mol%), at 20 °C unless noted otherwise. b Determined by 1HNMR analysis with internal standard (diethyl phthalate). c Determined by chiral HPLC analysis, the absolute configuration was assigned by single crystal X-ray analysis of ent-4f, see ESI for further details. d Isolated yield. | |||
| 1 | None | 78d | 86d |
| 2 | 2,2,2-Tripyridine | 68 | — |
| 3 | L1 | 53 | 67 |
| 4 | L2 | 60 | 52 |
| 5 | L3 | 52 | 57 |
| 6 | L4 | 32 | 8 |
| 7 | L5 | 35 | 35 |
| 8 | L6 | Trace | — |
| 9 | L7 | 69 | 60 |
| 10 | L8 | 49 | 55 |
| 11 | L10 | — | — |
| 12 | L11 | 59 | 0 |
| 13 | Cs2CO3 | 65 | 85 |
| 14 | Na2CO3 | 57 | 84 |
| 15 | CuI | 59 | 86 |
| 16 | CuBr | 63 | 86 |
| 17 | Toluene | 34 | 85 |
| 18 | THF | 49 | 83 |
| 19 | 0 °C | 45 | 86 |
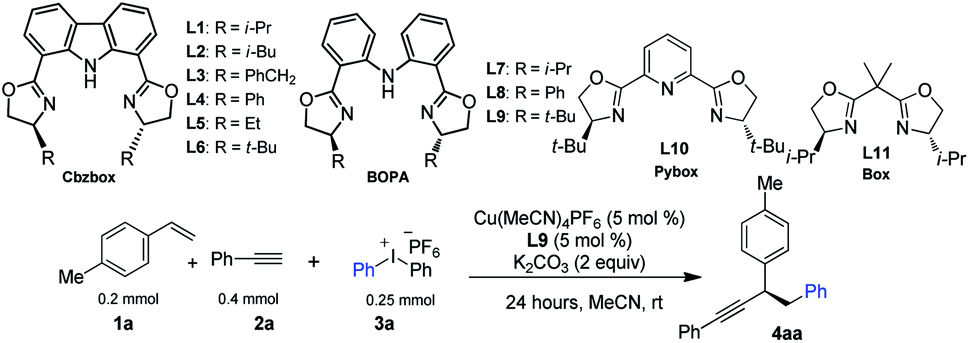
Other privileged chiral ligands, such as Pybox and Box, were also briefly examined, yet they are not effective for the current reaction, and neither gives promising enantiocontrol (entries 11 and 12). A systematic investigation of other parameters revealed that changing the solvent to something other than acetonitrile and using other bases in place of K2CO3 and other Cu-catalysts except Cu(MeCN)4PF6 all had a deleterious effect on both the yield and ee of the product (entries 13–18). Lowering the reaction temperature did not improve the enantioselectivity (entry 19). Notably, under the current reaction conditions, side-products derived from two-component reactions, e.g., the Heck reaction7a or Sonogashira reaction, are negligible.
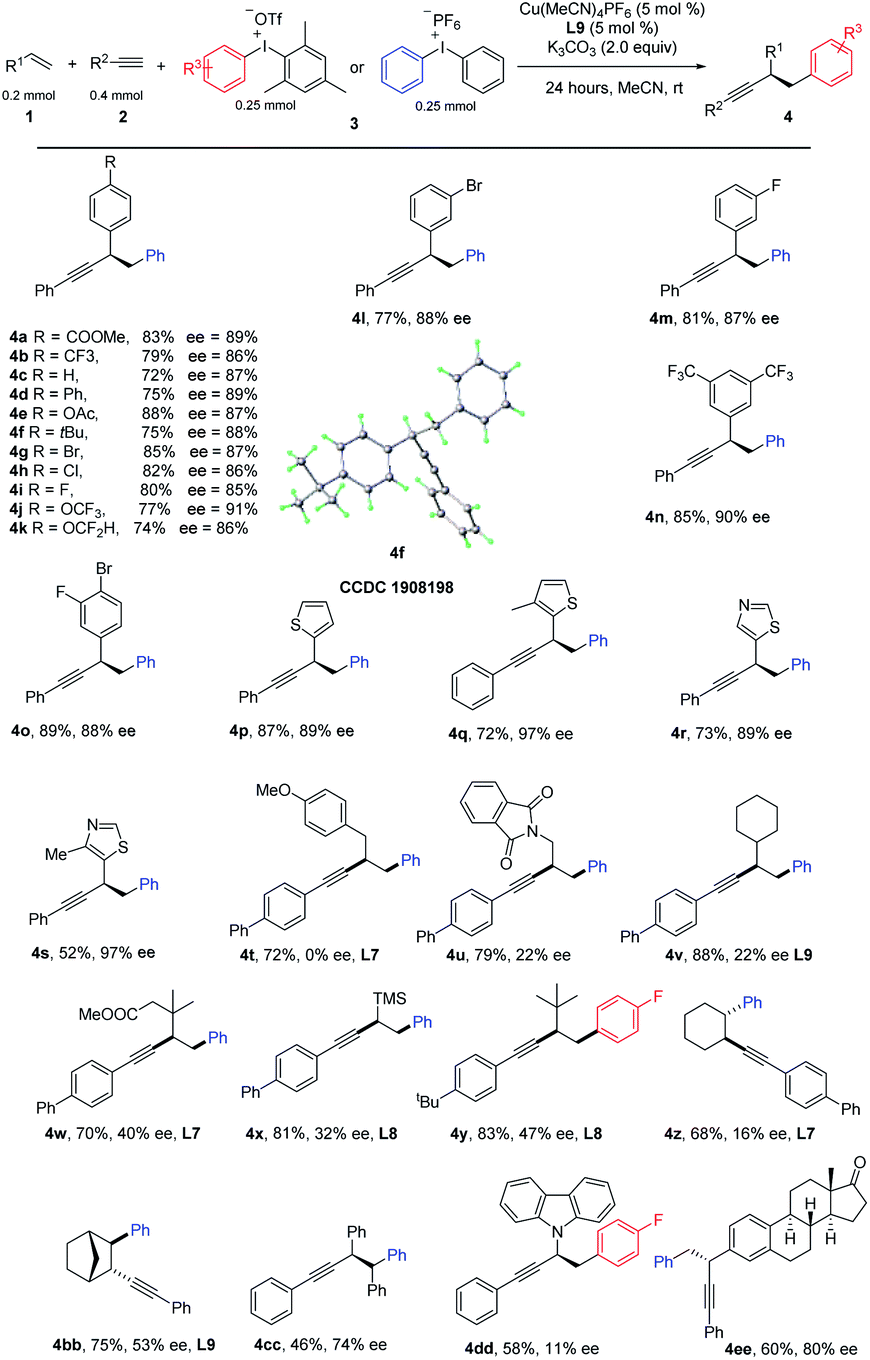
Having optimized the reaction conditions, we next investigated the scope of the copper-catalyzed three-component coupling with regard to the styrene partner (Table 2). We were delighted to find that styrene with mono para-substituted aryl groups that were electron deficient (4a, 4b), electron rich (4c–4f), and halogen-containing (4g–4i) reacted well. Interestingly, medicinally relevant CF3O and HCF2O could also be tolerated (4j, 4k). Br and F could be allowed at the meta-position (4l, 4m). A further substrate scope study revealed that double substitutions at different positions did not affect the reaction efficiency (4n, 4o). Heterocyclic functionality including thiophene and thiazole also worked well (4p, 4r). 4-Methyl thiazole and 3-methyl-thiophene with a slightly increased steric hindrance gives a much higher ee (4q and 4s). However, for ortho-substituted styrenes, such as 1-fluoro-2-vinylbenzene and 1-vinylnaphthalene, either a low yield or ee was observed. We further tested the performance of nonactivated alkenes in the difunctionalization. A variety of alkenes bearing different functional groups participated in this aryl alkynylation efficiently, leading to products in moderate to good yields. For sterically unbiased products, the product was racemic (4t); we reasoned that, it's difficult for chiral ligand to discriminate two vicinal methylene groups. To our delight, with sterically demanding substitutions, enantiomeric induction was observed (4u–4x). By careful tuning of the steric and electronic properties of substrates, and condition optimization, ee was improved to 47% with product 4x. Interestingly, trimethyl(vinyl)silane was good substrate as well, allowing a facile introduction of TMS at the propargylic position (4x), other heteroatoms such as nitrogen could be tolerated, 9-vinyl-9H-carbazole reacted well to give the desired product (4dd). Internal alkenes, including cyclohexene and norbornylene reacted well, giving products in moderate yields with excellent trans-selectivity (4z, 4bb). Surprisingly, an enantiomeric excess was obtained for 4bb. After thorough survey of chiral ligands, L9 gave the best result, 53% ee was obtained. Reaction of (E)-1,2-diphenylethene proceeded well to give 4cc in moderate yield and fare ee. Alkene derived from Estrone could be arylalkynylated in equal efficiency (4ee).
|
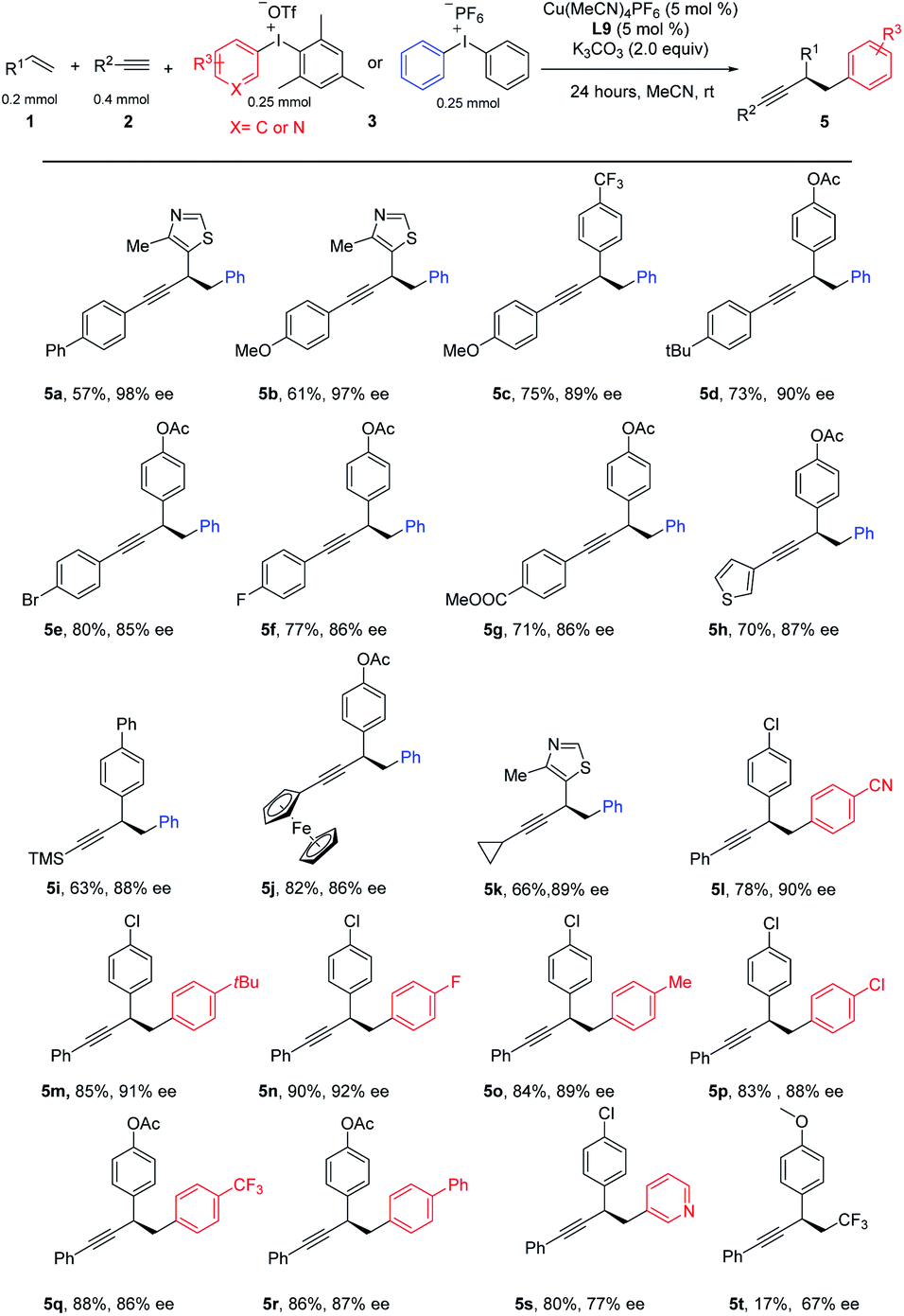
This three-component coupling proceeded efficiently with a range of readily available mono-arylated ethynes. Benzeneethynes with either electron-donating groups, such as Ph, MeO, and tBu (Table 3, 5a–5d), or electron-withdrawing, such as Br, F, and CO2Me (5e–5g), all gave desired products in good yields and good-to-excellent ees. Hetero aryl alkynes such as 2-ethynylthiophene were suitable substrates as well (5h). To our delight, ethynyltrimethylsilane was successfully engaged in these three-component coupling reactions, leading to 5i in moderate yield and a useful level of ee, and further functionalization is possible by removing the TMS. Interestingly, a ferrocene could be introduced into the arylated propargylic system in equal efficiency (5j). A systematic examination of aliphatic alkynes revealed that a moderate ee was obtained for most cases, except cyclopropyl acetylene, which yielded the desired product at a useful level with excellent ee (5k). Finally, we tested the catalytic system with a series of diaryliodonium salts. A number of aryl-substituted iodonium salts proved to be good substrates for this reaction, leading to diversified products (5l–5r). Iodonium salt bearing a heteroaryl group such as pyridine was good substrate, leading to product 5s in good yield and moderate ee. We tested other hypervalent iodonium salts with various transfer functionalities including cyano, alkyne, vinyl and CF3, however, only Togni reagent provides the desired product in low yield and moderate ee.
|
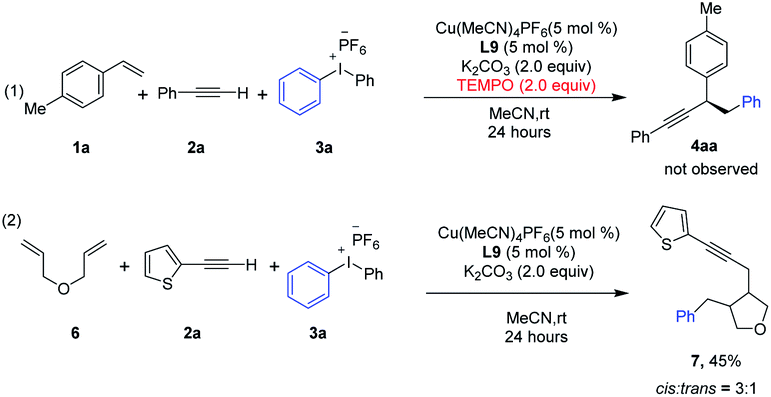
Control experiments were conducted to probe the possible mechanism of this copper catalyzed three-component coupling reaction (Scheme 1). Upon the addition of radical trapping agent TEMPO, the reaction was totally shut down, indicating a radical mechanism might be possible (eqn (1)). A cyclization experiment with diene 6 was further conducted (eqn (2)). 6 was subjected to the standard conditions in place of styrene, and ring-closing took place leading to the cyclized product 7 with 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cis/trans selectivity, a typical reactivity and stereoselectivity that have been reported for the cyclization of the derived secondary alkyl radical.15
:1 cis/trans selectivity, a typical reactivity and stereoselectivity that have been reported for the cyclization of the derived secondary alkyl radical.15
 | ||
| Scheme 1 Control experiments for radical probe. | ||
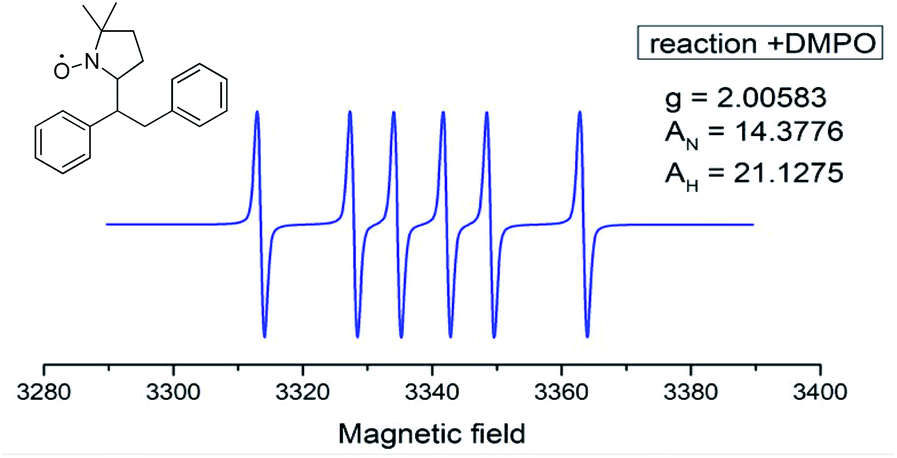
Next, the reaction was monitored by electron paramagnetic resonance spectroscopy (EPR) with and without 5,5-dimethyl-1-pyrroline N-oxide (DMPO), and the corresponding EPR spectrum indicated the existence of copper(II)16a and benzylic radical species, a benzylic DMPO adduct more likely formed based on spin coupling parameters (Fig. 2).16b These results suggest an aryl radical, instead of a Cu(III)–aryl species, is likely involved in the reaction pathway.
 | ||
| Fig. 2 EPR spectra of reaction in the presence of DMPO. | ||
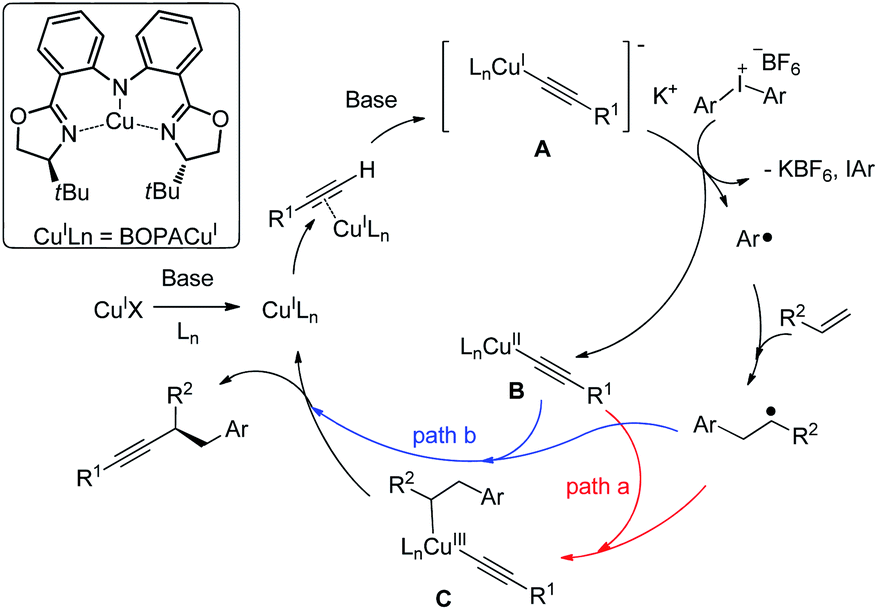
Based on the literature12 and these findings, we proposed a reaction mechanism (Fig. 3). Cu(I)X undergoes ligand exchange with Ln in the presence of base leading to complex Cu(I)Ln.17 A subsequent interaction with alkyne in presence of base through a π copper(I) alkyne complex yields [LnCu(I)(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR)]− K+ (A), which could be oxidized by diaryliodonium salt to provide a copper(II) species LnCu(II)(CCR) (B) with a simultaneous formation of an aryl radical.
CR)]− K+ (A), which could be oxidized by diaryliodonium salt to provide a copper(II) species LnCu(II)(CCR) (B) with a simultaneous formation of an aryl radical.
 | ||
| Fig. 3 Proposed reaction mechanism. | ||
Moreover, the cyclic voltammogram of copper acetylide show a reversible wave at −2.42 V versus the saturated calomel electrode (SCE) corresponding to the Cu(I)/Cu(II) redox couple.16c The Ph2I+/PhI + Ph. have reduction potential E = −0.7 versus SCE.18 Those results suggest that the SET process form copper acetylide to diaryl iodonium salt may be viable.16 Notably, [Cu(dpp)2][PF6] (dpp = 2,9-diphenyl-1,10-phenanthroline) could promote aryl radical generation from diaryl iodonium salt under light irridiation.12 The organic radical was trapped by the styrene to generate a benzylic radical. Two possible pathways might be as follows: in path a, the benzylic radical reacts with copper(II) delivering a copper(III) complex (C), which undergoes reductive elimination to generate the product, with the regeneration of the catalyst. Alternatively, in path b, the benzylic radical could undergo direct out-of-cage bond formation with the copper(II) species to provide the same result. Related examples of this so-called radical relay strategy have been extensively studied by Liu,19,11f Liu20 and Xiao.21 Alternatively, the reaction might proceed through a electrophilic Cu(III)–Ph intermediate, although less likely, it cannot be ruled out.7a-c
In this study, beside the unusual BOPA ligand employed for enantioselective C–C bond formation, the observation of moderate 53% ee for 4bb suggests a possible interaction of phenyl radical species with chiral copper complex, which could potentially explain the selectivity when unsymmetrical diarylidonium salts were used. Aryl group with less steric hindrance prefer to transfer, which is in contrast to free aryl radical species.12a Further improvement of the interesting asymmetric aryl alkynylation of nonactivated alkene relies on the detailed mechanistic study by elucidating the identity of enantiodetermining step and ligand development.
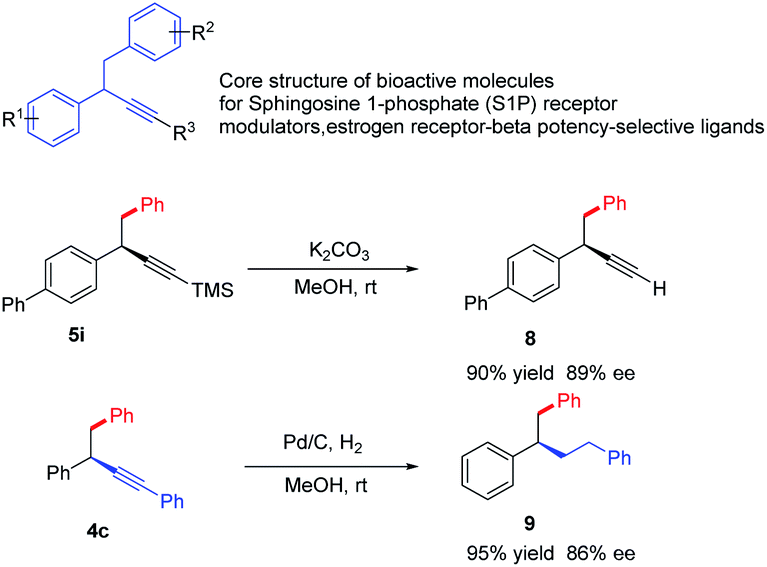
1,2-Diaryl-3-butynes are core structure of bioactive molecules for sphingosine 1-phosphate (S1P) receptormodulators, estrogen receptor-beta potency-selective ligands.10 Traditional approach towards this motif required multistep synthesis, the current strategy offer a straightforward and divergent entry to enantioenriched form (Scheme 2). Meanwhile, propargylic compounds are valuable intermediates in organic synthesis, and they can participate in a wide range of transformations at the carbon–carbon triple bond. We briefly demonstrate the potential utilization of these products. The simple cleavage of the trimethylsilyl group provides a free alkyne, which could serve as the coupling partners in both Sonogashira and click reactions.22 Hydrogenation of the triple bond leads to saturated products in excellent yields.
 | ||
| Scheme 2 Utilities of diarylated propargylic compounds. | ||
Conclusions
In summary, we report here a copper-catalyzed enantioselective arylalkynylation of styrene using diaryliodonium salt enabled by a BOPA–Cu(I) complex. Under mild reaction conditions, an array of alkynes, alkenes, and diaryliodonium salts are good substrates leading to valuable propargylic products with good to excellent enantioselectivity. In contrast to the existing copper-catalyzed aryl difunctionalization of an alkene with diaryliodonium salt through an electrophilic aryl–Cu(III) species, BOPA–Cu catalysis is more likely via a phenyl radical pathway based on preliminary mechanistic studies. This novel asymmetric coupling reaction holds potential for further expansion and application in functional molecule synthesis.Conflicts of interest
Shanghai Institute of Organic Chemistry, CAS has filed a patent application for this technology (Chinese patent application number 202010016009.7).Acknowledgements
We are grateful to NSFC-21772218, 21821002, XDB20000000, the “Thousand Plan” Youth program, State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, and the Chinese Academy of Sciences.Notes and references
- (a) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320 CrossRef CAS; (b) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed; (c) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef; (d) R. R. Schrock, J. S. Murdzek, G. C. Bazan, J. Robbins, M. DiMare and M. O'Regan, J. Am. Chem. Soc., 1990, 112, 3875 CrossRef CAS; (e) R. H. Grubbs, S. J. Miller and G. C. Fu, Acc. Chem. Res., 1995, 28, 446 CrossRef CAS.
- for selected reviews, (a) E. Hiromichi and S. Mikiko, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef PubMed; (b) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413 CrossRef CAS PubMed; (c) J.-S. Zhang, L. Liu, T. Chen and L.-B. Han, Chem. - Asian J., 2018, 13, 2277 CrossRef CAS PubMed; (d) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed; (e) H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483 CrossRef CAS.
- W. Li, J. K. Boon and Y. Zhao, Chem. Sci., 2018, 9, 600 RSC.
- (a) X. Wang and A. Studer, Acc. Chem. Res., 2017, 50, 1712 CrossRef CAS PubMed; (b) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328 CrossRef CAS PubMed; (c) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052 CrossRef CAS PubMed; (d) N. R. Deprez and M. S. Sanford, Inorg. Chem., 2007, 46, 1924 CrossRef CAS PubMed; (e) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523 CrossRef CAS PubMed.
- L. Ackermann, Modern Arylation Methods, Wiley-VCH, Weinheim, 2009 Search PubMed.
- (a) P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123 CrossRef CAS PubMed; (b) F.-M. Martín, Synthesis, 2017, 49, 1905 CrossRef.
- (a) R. J. Phipps, L. McMurray, S. Ritter, H. A. Duong and M. J. Gaunt, J. Am. Chem. Soc., 2012, 134, 10773 CrossRef CAS PubMed; (b) E. Cahard, H. P. J. Male, M. Tissot and M. J. Gaunt, J. Am. Chem. Soc., 2015, 137, 7986 CrossRef CAS PubMed; (c) D. H. Lukamto and M. J. Gaunt, J. Am. Chem. Soc., 2017, 139, 9160 CrossRef CAS PubMed. Other related examples: (d) A. Bigot, A. E. Williamson and M. J. Gaunt, J. Am. Chem. Soc., 2011, 133, 13778 CrossRef CAS PubMed; (e) J. S. Harvey, S. P. Simonovich, C. R. Jamison and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 13782 CrossRef CAS PubMed; (f) A. Baralle, L. Fensterbank, J.-P. Goddard and C. Ollivier, Chem.–Eur. J., 2013, 19, 10809 CrossRef CAS PubMed; (g) M. Prakash, S. Muthusamy and V. Kesavan, J. Org. Chem., 2014, 79, 7836 CrossRef CAS PubMed; (h) Y. Yang, J. Han, X. Wu, S. Mao, J. Yu and L. Wang, Synlett, 2014, 25, 1419 CrossRef; (i) X. Li, J. Xu, P. Zhang, Y. Gao, J. Wu, G. Tang and Y. Zhao, Synlett, 2014, 25, 2009 CrossRef CAS.
- (a) Y. Xiong and G. Zhang, J. Am. Chem. Soc., 2018, 140, 2735 CrossRef CAS PubMed; (b) Y. Xiong, Y. Sun and G. Zhang, Org. Lett., 2018, 20, 6250 CrossRef CAS PubMed.
- For a review, (a) Q. H. Deng, R. L. Melen and L. H. Gade, Acc. Chem. Res., 2014, 47, 3162 CrossRef CAS PubMed. For selected recent examples on Lewis acid catalysis: (b) J. Peng and D. M. Du, Eur. J. Org. Chem., 2012, 40, 4042 CrossRef; (c) F. Tan, L.-Q. Lu, Q.-Q. Yang, W. Guo, Q. Bian, J.-R. Chen and W.-J. Xiao, Chem.–Eur. J., 2014, 20, 3415 CrossRef CAS PubMed; (d) J. C. K. Chu, D. M. Dalton and T. Rovis, J. Am. Chem. Soc., 2015, 137, 4445 CrossRef CAS PubMed; (e) C. Despotopoulou, S. C. McKeon, R. Connon, V. Coeffard, H. Müller-Bunz and P. J. Guiry, Eur. J. Org. Chem., 2017, 45, 6734 CrossRef; (f) J.-Q. Zhao, X.-J. Zhou, Y. Zhou, X.-Y. Xu, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2018, 20, 909 CrossRef CAS PubMed. For recent examples on redox catalysis: (g) T. Niwa and M. Nakada, J. Am. Chem. Soc., 2012, 134, 13538 CrossRef CAS PubMed; (h) T. Inagaki, L. T. Phong, A. Furuta, J.-i. Ito and H. Nishiyama, Chem.–Eur. J., 2010, 16, 3090 CrossRef CAS PubMed.
- (a) M. J. Meyers, J. Sun, K. E. Carlson, G. A. Marriner, B. S. Katzenel lenbogen and J. A. Katzenellenbogen, J. Med. Chem., 2001, 44, 4230 CrossRef CAS PubMed; (b) S. C. Sinha, S. S. Bhat, E. G. Corpuz, K. Chow, W. K. Fang and W. B. Im, WO2012/74926.A1, 2012; (c) M. A. Islam, R. Pal, T. Hossain, A. Mukherjee and A. Saha, Med. Chem. Res., 2012, 21, 2652 CrossRef CAS.
- For selected reviews, (a) P. J. Stang and F. Diederich, Modern Acetylene Chemistry, WileyVCH, Weinheim, 1995 CrossRef; (b) F. Diederich, P. J. Stang and R. R. Tykwinski, Acetylene Chemistry: Chemistry, Biology, and Material Science, WileyVCH, Weinheim, 2005 Search PubMed; (c) B. M. Trost and C.-J. Li, Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations, Wiley-VCH, New York, 2014 Search PubMed; For recent examples of acetylene synthesis; (d) X.-Y. Bai, W.-W. Zhang, Q. Li and B.-J. Li, J. Am. Chem. Soc., 2018, 140, 506 CrossRef CAS PubMed; (e) X.-Y. Bai, Z.-X. Wang and B.-J. Li, Angew. Chem., Int. Ed., 2016, 55, 9007 CrossRef CAS PubMed; (f) L. Fu, S. Zhou, X. Wan, P. Chen and G. Liu, J. Am. Chem. Soc., 2018, 140, 10965 CrossRef CAS PubMed; (g) Y. Xu, Z. Wu, J. Jiang, Z. Ke and C. Zhu, Angew. Chem., Int. Ed., 2017, 56, 4545 CrossRef CAS PubMed.
- For example through aryl radical mechanism under thermal conditions (a) J. Xu, P. Zhang, Y. Gao, Y. Chen, G. Tang and Y. Zhao, J. Org. Chem., 2013, 78, 8176 CrossRef CAS PubMed; Examples under photo irradication conditions, (b) Y. Li, T. Koike and M. Akita, Synlett, 2016, 27, 736 CrossRef CAS; (c) Y. Chen, C. Shu, F. Luo, X. Xiao and G. Zhu, Chem. Commun., 2018, 54, 5373 RSC; (d) D. Sun, K. Yin and R. Zhang, Chem. Commun., 2018, 54, 1335 RSC.
- (a) T. Suzuki, A. Kinoshita, H. Kawada and M. Nakada, Synlett, 2003, 4, 570 Search PubMed; (b) M. Inoue, T. Suzuki and M. Nakada, J. Am. Chem. Soc., 2003, 125, 1140 CrossRef CAS PubMed; For our modified new synthesis of this ligand: (c) W. Chen, Q. Yang, T. Zhou, Q. Tian and G. Zhang, Org. Lett., 2015, 17, 5236 CrossRef CAS PubMed.
- (a) H. A. McManus and P. J. Guiry, J. Org. Chem., 2002, 67, 8566 CrossRef CAS PubMed; (b) H. A. McManus, P. G. Cozzi and P. J. Guiry, Adv. Synth. Catal., 2006, 348, 551 CrossRef CAS; (c) G. C. Hargaden, H. A. McManus, P. G. Cozzi and P. J. Guiry, Org. Biomol. Chem., 2007, 5, 763 RSC.
- J. Lusztyuk, B. Maillard, S. Deycard, D. A. Lindsay and K. U. Ingold, J. Org. Chem., 1987, 52, 3509 CrossRef CAS.
- (a) S. Tang, Y. Liu, X. Gao, P. Wang, P. Huang and A. Lei, J. Am. Chem. Soc., 2018, 140, 6006 CrossRef CAS PubMed; (b) G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259–303 CrossRef CAS; (c) A. Sagadevan, A. Pagupathi and K. C. Hwang, Angew. Chem., Int. Ed., 2015, 54, 13896 CrossRef CAS PubMed and see the ESI† for details.
- Q.-H. Deng, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2012, 134, 2946 CrossRef CAS PubMed.
- P. P. Romańczyk and S. S. Kurek, Electrochim. Acta, 2017, 255, 482 CrossRef.
- (a) D. Wang, L. Wu, F. Wang, X. Wan, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 6811 CrossRef CAS PubMed; (b) L. Wu, F. Wang, P. Chen and G. Liu, J. Am. Chem. Soc., 2019, 141, 1887 CrossRef CAS PubMed; (c) D. Wang, F. Wang, P. Chen, Z. Lin and G. Liu, Angew. Chem., Int. Ed., 2017, 56, 2054 CrossRef CAS PubMed.
- J.-S. Lin, T.-T. Li, J.-R. Liu, G.-Y. Jiao, Q.-S. Gu, J.-T. Cheng, Y.-L. Guo, X. Hong and X.-Y. Liu, J. Am. Chem. Soc., 2019, 141, 1074 CrossRef CAS PubMed.
- F.-D. Lu, D. Liu, L. Zhu, L.-Q. Lu, Q. Yang, Q.-Q. Zhou, Y. Wei, Y. Lan and W.-J. Xiao, J. Am. Chem. Soc., 2019, 141, 6167 CrossRef CAS PubMed.
- (a) H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS PubMed; (b) W. H. Binder and R. Sachsenhofer, omol. Rapid Commun., 2007, 28, 15 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1908198. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04029c |
| This journal is © The Royal Society of Chemistry 2020 |