
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgrammable adenine deamination in bacteria using a Cas9–adenine-deaminase fusion†
Ya
Zhang
ab,
Hongyuan
Zhang
ab,
Zhipeng
Wang
ab,
Zhaowei
Wu
a,
Yu
Wang
c,
Na
Tang
ab,
Xuexia
Xu
de,
Suwen
Zhao
 de,
Weizhong
Chen
*a and
Quanjiang
Ji
*a
de,
Weizhong
Chen
*a and
Quanjiang
Ji
*a
aSchool of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China. E-mail: quanjiangji@shanghaitech.edu.cn; chenwzh@shanghaitech.edu.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
cCollege of Life Science and Engineering, Jiangxi Agricultural University, Nanchang 330045, China
diHuman Institute, ShanghaiTech University, Shanghai 201210, China
eSchool of Life Science and Technology, ShanghaiTech University, Shanghai 201210, China
First published on 6th January 2020
Abstract
Precise genetic manipulation is vital to studying bacterial physiology, but is difficult to achieve in some bacterial species due to the weak intrinsic homologous recombination (HR) capacity and lack of a compatible exogenous HR system. Here we report the establishment of a rapid and efficient method for directly converting adenine to guanine in bacterial genomes using the fusion of an adenine deaminase and a Cas9 nickase. The method achieves the conversion of adenine to guanine via an enzymatic deamination reaction and a subsequent DNA replication process rather than HR, which is utilized in conventional bacterial genetic manipulation methods, thereby substantially simplifying the genome editing process. A systematic screening targeting the possibly editable adenine sites of cntBC, the importer of the staphylopine/metal complex in Staphylococcus aureus, pinpoints key residues for metal importation, demonstrating that application of the system would greatly facilitate the genomic engineering of bacteria.
Introduction
Precise genetic manipulation is a key approach for studying bacterial physiology and relies heavily on homologous recombination (HR) in most bacterial species. Despite the substantial importance, genetic manipulations in some bacterial species are difficult to induce due to the extremely weak intrinsic HR capacity and lack of a compatible exogenous HR system, such as phage-derived λ-Red and RecET systems that are widely adopted for genetic recombineering in some Gram-negative bacteria.1,2The recently discovered clustered regularly interspaced short palindromic repeats (CRISPRs) and CRISPR-associated (Cas) proteins have been engineered for robust genetic manipulation in a variety of organisms, including eukaryotes and diverse bacterial species, owing to their programmable and strong DNA-cleavage capacity.3–8 Given that most bacterial species lack the non-homologous end joining (NHEJ) double-strand DNA break repair mechanism, genome cleavage by CRISPR/Cas systems is lethal to most bacteria. Therefore, CRISPR/Cas systems have been explored as effective selection agents to eliminate unedited cells, dramatically simplifying the genetic manipulation processes in bacteria.9 Despite their simplicity and high efficiency, CRISPR/Cas-based genome editing methods still rely on HR in bacteria to achieve precise genetic manipulation and are therefore difficult to establish in some bacteria that lack a strong HR system, such as Mycobacterium tuberculosis.
More recently, the development of deaminase-mediated base editing systems has provided new strategies for precise genetic manipulation in biology.10–12 The base editing systems directly convert target bases using a deamination reaction and a subsequent DNA replication process rather than HR, which is utilized in the aforementioned CRISPR/Cas-based genome editing methods. Two major types of base editing systems have been established: cytosine base editors (CBEs)10,11 and adenine base editors (ABEs).12,13 CBEs have been widely adopted for programmable cytosine to thymine conversions in a variety of organisms including eukaryotes10,11,14–17 and some bacterial species,17–22 and ABEs are mostly established in eukaryotes, such as mammalian cells12,23 and plants,24,25 for precise adenine to guanine conversion. Recently, an ABE system named CRISPR-aBEST has been developed in streptomycetes.13 In addition, programmable adenosine to inosine and cytidine to uridine RNA editors have also been developed.26,27
In this study, we report the establishment of an adenine base editor in Escherichia coli and Staphylococcus aureus using the fusion of an adenine deaminase12 and a Cas9 nickase. We applied the adenine base editing system to screen the functional residues of CntBC, a staphylopine/metal complex transporter in S. aureus, pinpointing four key residues for metal importation. The establishment and application of the adenine base editing system would facilitate genetic engineering in bacteria and provide insights for adenine base editing system development in other bacteria.
Results and discussion
To examine whether ABEs are applicable for genetic manipulation in bacteria, we designed and constructed an adenine base editing system pABE (Fig. 1A) and sought to assess the capacity of pABE to convert adenine to guanine in two distinct bacterial species: Escherichia coli (a major Gram-negative model bacterium) and Staphylococcus aureus (a major Gram-positive human pathogen). pABE contained two different replication origins ColE1 and repF, allowing for its replication in both E. coli and S. aureus. In addition, it contained an expression cassette encoding a fusion protein comprising a Cas9 nickase from Streptococcus pyogenes (spCas9 D10A) and an evolved adenine deaminase (ABE7.10)12 as well as an expression cassette encoding the corresponding small-guide RNA (sgRNA). With the guidance of sgRNA/Cas9, the deaminase could catalyze an adenine deamination reaction at a target locus (Fig. 1B), resulting in the formation of hypoxanthine12 (Fig. 1C). After DNA replication, the deaminated product hypoxanthine was permanently converted to guanine, and a precise adenine to guanine substitution was thus achieved.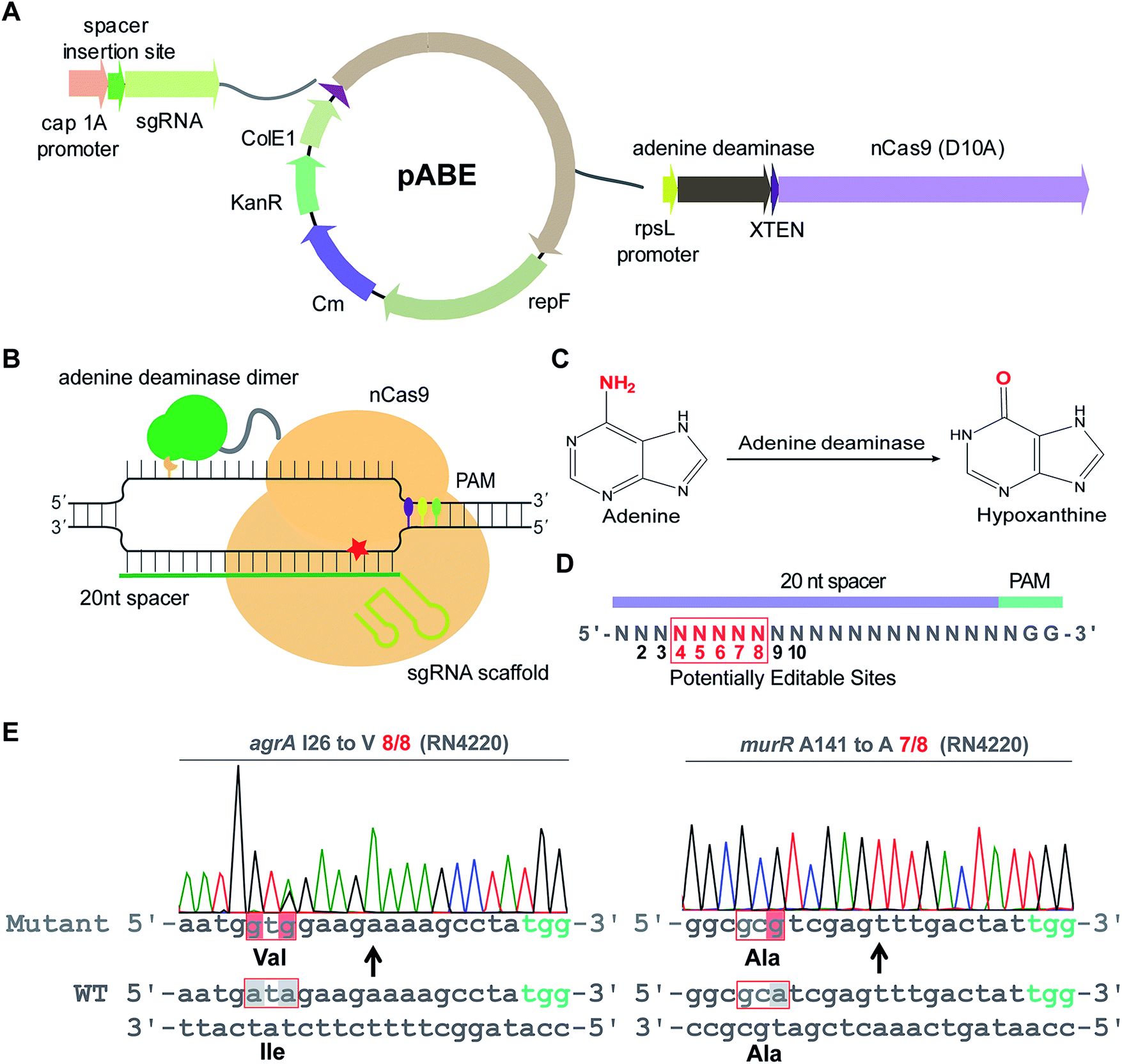 | ||
| Fig. 1 pABE enabled highly efficient adenine to guanine conversion in S. aureus. (A) Map of the adenine base editing system pABE. KanR: the kanamycin-resistant marker for selection in E. coli; Cm: the chloramphenicol-resistant marker for selection in S. aureus; ColE1: a replication origin in E. coli; repF: a replication origin in S. aureus; cap 1A promoter: the promoter drives the expression of sgRNA; rpsL promoter: the promoter drives the expression of the fusion protein. (B) A schematic model for Cas9–adenine deaminase base editor. (C) The adenine deamination reaction catalyzed by an adenine deaminase. (D) The editable window of a mammalian adenine base editor. (E) Highly efficient conversions of adenine to guanine by pABE in the S. aureus RN4220 strain. | ||
We first sought to test the editing efficiency of the pABE system in the common laboratory S. aureus strain RN4220. Because mammalian ABEs have an editable window from position 4 to 8 (Fig. 1D), four different spacers containing potentially editable As were selected and cloned into the pABE plasmid. As shown in Fig. 1E, S1A and B,† high editing efficiencies of adenine to guanine conversion were achieved at the tested sites using the pABE system, as determined by the ratios of successfully edited colonies to the total numbers of randomly picked colonies. We also noticed that when multiple As were present in a target region, they might have different editing efficiencies (Fig. S1A and B†). Because the RN4220 strain contains numerous mutations and the editing efficiency of the pABE system may vary in different strains with distinct genetic background, we examined the editing efficiency of the pABE system in two clinically isolated S. aureus strains Newman and N315. As shown in Fig. S2A–D,† high editing efficiencies of the pABE system were observed for all the tested spacers of both the Newman and N315 strains, similar to that in the RN4220 strain. Together, these results demonstrated the strong capacity of the pABE system to precisely convert adenine to guanine in S. aureus.
The editable windows of some bacterial CBEs are different from those of mammalian CBEs.21 To systematically investigate the editable window of pABE in bacteria, we selected 9 different spacers containing A(s) at different positions (from position 2 to 10) and inserted them individually into the pABE plasmid. The plasmids were individually transformed into the RN4220 strain to test the editing efficiency. As shown in Fig. S3,† the adenine sites from positions 4 to 8 were successfully edited with efficiencies from 4/8 to 8/8, whereas almost no editing events were observed at positions 2, 3, 9, or 10, concluding that the editable window of pABE in S. aureus is from position 4 to position 8.
Next, we sought to probe the editing efficiency of the pABE system in E. coli. Two different spacers from the MG1655 strain and one spacer from the DH5α strain were inserted into the pABE plasmid individually. As shown in Fig. 2A and S4,† high editing efficiencies were observed in all the tested spacers. To comprehensively analyze the editing efficiency of pABE in E. coli, we attempted to use the lacZ gene as the reporter system. The lacZ gene in the MG1655 strain was first inactivated by introducing a premature stop codon at Q51 using the bacterial CBE system pBECKP,21 rendering the cells unable to produce a blue color in the presence of 5-bromo-4-chloro-3-indolyl β-D-galactopyranoside (X-gal) (Fig. 2B). Second, the premature stop codon was changed back to Q51 using the pABE system, thereby restoring the blue color producing capacity of the cells in the presence of X-gal (Fig. 2B). Consistent with our expectation, numerous blue colonies appeared after transformation with an approximate ratio of 85% (blue colonies/total colonies) (Fig. 2C). In addition, we collected all the colonies from the plate and sent them out for sequencing to examine the conversion efficiency. An approximate 71% conversion efficiency was determined based on the sequencing results (Fig. 2C), demonstrating that the pABE system possesses a strong capacity for editing adenine in E. coli.
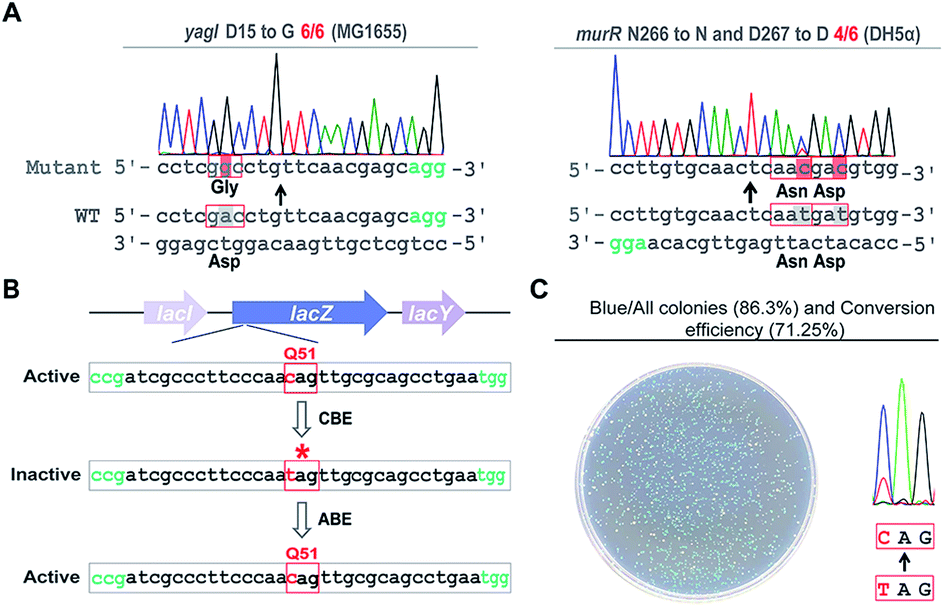 | ||
| Fig. 2 pABE enabled highly efficient adenine to guanine conversion in E. coli. (A) Highly efficient conversions of adenine to guanine by pABE in the E. coli MG1655 and DH5α strains. (B) The reporter gene lacZ was first inactivated by installing a premature stop codon using a cytosine base editor (CBE) and then mutated back to the active form by pABE to evaluate its adenine editing efficiency. (C) Highly efficient editing of the lacZ gene was achieved using the pABE system, confirmed by both the phenotypical assay and the sequencing result. | ||
In addition to single-spacer editing, we tested the capacity of the pABE system for multiplex editing. We assembled two individual spacers into a single pABE plasmid and tested the editing in the S. aureus RN4220 strain. As shown in Fig. S5,† two genes were simultaneously edited with high efficiencies.
To evaluate the unbiased editing efficiency of the pABE system in bacteria, five editable sites from 4 spacers in the S. aureus RN4220 strain were selected for deep sequencing. Compared with the wild-type strain, unexpected nucleotide changes (A to T or A to C) in all the target sites of mutants could be negligible (Fig. S6 and Table S1†). In addition, we further subjected two edited RN4220 strains for whole-genome sequencing to evaluate the genome-wide off-target effect of the pABE system. We examined all of the potential off-target sites which contain the identical sequences to the PAM proximal 1–8 nt of the targets,28 and did not find any off-target event (Table S2†). These data demonstrated the high editing accuracy and DNA fidelity of the pABE system, which is consistent with the previous studies.13,29
By taking advantage of the high-throughput oligo synthesis technique and the high efficiency of NHEJ-mediated DNA repair, CRISPR/Cas-based genome-wide screening strategies have been widely applied in eukaryotes for key gene and pathway identification.30–32 In most bacterial species, because of the lack of NHEJ, the relatively low efficiency of HR-based DNA recombineering, and the requirement of long repair templates for HR, precise CRISPR/Cas-based genetic screening is time-consuming and laborious in bacteria. The developed base editing systems in bacteria can efficiently catalyze the conversion of target bases without utilizing repair templates, rendering them usable for high-throughput genetic screening in bacteria.
As a proof of concept, we sought to use the pABE system to screen key residues of CntBC for staphylopine (a recently identified metallophore)/metal complex transportation33 by comprehensively mutating the editable adenine sites of the cntBC genes. CntBC is an essential transporter that possibly forms a heterodimer for staphylopine/transition metal complex acquisition in S. aureus (Fig. 3A). Comprehensively screening the key CntBC residues would further our understanding of the transportation mechanism at the molecular level. To achieve this goal, we assembled 38 spacers targeting different cntBC sites into the pABE system individually (Fig. S7†). The assembled plasmids were transformed into the S. aureus RN4220 strain separately for editing (Fig. 3B). Successful editing of the target sites was confirmed individually by Sanger sequencing, resulting in 42 mutant strains that were subjected to a growth-curve analysis. Because overload of a high concentration of cobalt is toxic to S. aureus33 and mutations of the key CntBC residues may relieve the toxicity, growth-curve analysis was a rapid and convenient assay to probe the functionality of the residues. In agreement with our expectation, growth of the wild-type RN4220 strain was arrested in the presence of a high concentration of cobalt, whereas the repression was relieved when cntB or cntC was deleted. Intriguingly, the growth repression of four CntBC single-amino-acid mutants (C5 [F143L], C10 [C113R], B6 [E253G], and B10 [V19A]) created by the pABE system was also relieved (Fig. 3C and D), implicating the key role of these residues in CntBC function.
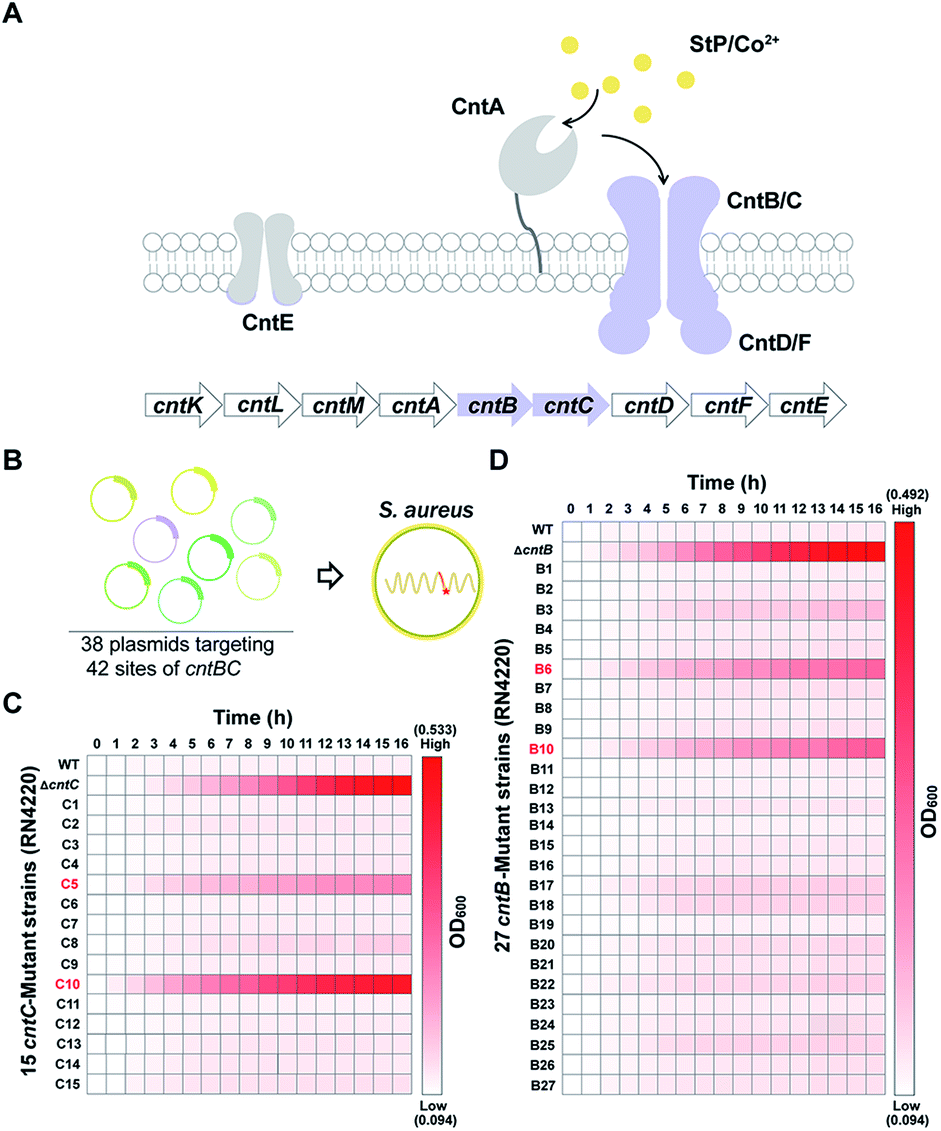 | ||
| Fig. 3 A pABE-based screening identified key residues for CntBC-mediated metal acquisition. (A) Scheme of the processes for StP/metal acquisition in S. aureus. StP is synthesized by the cntKLM gene cluster and is transported out of cells by CntE. After chelating with transition metals, such as Co2+, the StP/metals complexes are recognized by CntA and imported into cells by CntBC. (B) Thirty-eight plasmids targeting 42 sites of cntBC were constructed and transformed into S. aureus to screen key functional residues of CntBC. (C) Heat map of the growth of the 15 cntC-mutant strains in the presence of 3 mM Co2+. (D) Heat map of the growth of the 27 cntB-mutant strains in the presence of 3 mM Co2+. | ||
To further confirm the importance of these residues, we utilized the CRISPR/Cas9-based genome editing method pCasSA9 and pKOR1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34 to precisely mutate these residues to potentially nonfunctional residues. In total, we created four mutant strains: F143A and C113A of CntC and E253A and V19S of CntB. Next, we subjected the mutant strains to growth-curve analysis. The mutants constructed by the pCasSA and pKOR1 system exhibited growth similar to that of the mutants created by the pABE system (Fig. 4A), further confirming that the four residues, F143 and C113 of CntC and E253 and V19 of CntB, are critical for CntBC function. Furthermore, inductively coupled plasma-optical emission spectrometry (ICP-OES) was used to measure the intracellular contents of cobalt ions in various strains (WT, ΔcntB, V19A, and E253G). In agreement with the growth experiment, the complete deletion of cntB and the single amino acid mutation of the residues (V19A and E253G) reduced metal accumulation (Fig. S8†). In addition, we performed protein structure modeling of CntBC, revealing that V19 and C113 located at the dimerization interface between CntB and CntC and E253 and F143 stayed at the solute entrance channel (Fig. 4B). Mutation of V19 or C113 would disrupt CntB and CntC dimerization and mutation of E253 or F143 might terminate the interaction of the residues with staphylopine/metal complexes, thereby inactivating the solute-transportation function of CntBC. Together, these experiments demonstrated that the adenine base editing system is an effective genetic screening tool in bacteria.
34 to precisely mutate these residues to potentially nonfunctional residues. In total, we created four mutant strains: F143A and C113A of CntC and E253A and V19S of CntB. Next, we subjected the mutant strains to growth-curve analysis. The mutants constructed by the pCasSA and pKOR1 system exhibited growth similar to that of the mutants created by the pABE system (Fig. 4A), further confirming that the four residues, F143 and C113 of CntC and E253 and V19 of CntB, are critical for CntBC function. Furthermore, inductively coupled plasma-optical emission spectrometry (ICP-OES) was used to measure the intracellular contents of cobalt ions in various strains (WT, ΔcntB, V19A, and E253G). In agreement with the growth experiment, the complete deletion of cntB and the single amino acid mutation of the residues (V19A and E253G) reduced metal accumulation (Fig. S8†). In addition, we performed protein structure modeling of CntBC, revealing that V19 and C113 located at the dimerization interface between CntB and CntC and E253 and F143 stayed at the solute entrance channel (Fig. 4B). Mutation of V19 or C113 would disrupt CntB and CntC dimerization and mutation of E253 or F143 might terminate the interaction of the residues with staphylopine/metal complexes, thereby inactivating the solute-transportation function of CntBC. Together, these experiments demonstrated that the adenine base editing system is an effective genetic screening tool in bacteria.
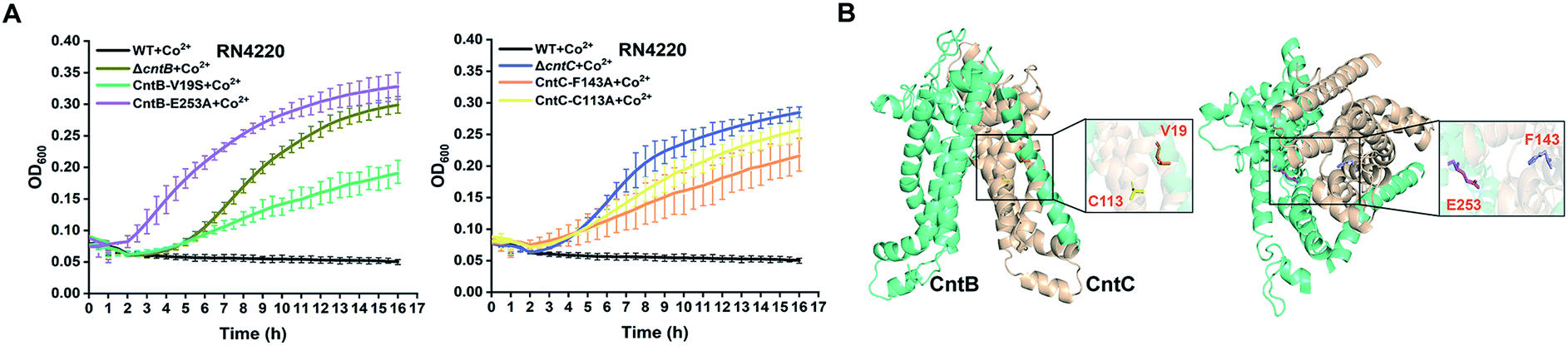 | ||
| Fig. 4 The critical roles of the identified four key residues for metal acquisition were confirmed by structural modeling and CRISPR/Cas9 and pKOR1-based genome editing experiments. (A) Growth curves of four mutant strains (V19S, E253A of CntB and C113A, F143A of CntC) in the presence of 2 mM Co2+. (B) A modeled structure of CntBC. The residues V19, E253 of CntB and C113, F143 of CntC were highlighted. | ||
Conclusions
We have established a highly efficient and robust adenine base editing system in bacteria using the fusion of a Cas9 nickase and an adenine deaminase. In addition to single nucleotide substitution, the system is applicable for large-scale or even genome-wide screening to probe functional genes and new mechanisms. Further engineering of this system with PAM-expanded Cas9 nucleases, such as xCas9,35,36 Cas9-NG,37 and other PAM-expanded Cas9 nucleases,38 would greatly expand the targeting scope of the pABE system, thereby allowing for extensive profiling of key residues and genes. The successful development of the pABE system in E. coli and S. aureus will provide critical insights for adenine base editing system development in other bacteria. Future applications of the adenine base editing system will dramatically facilitate genome engineering in bacteria.Experimental
Bacterial strains, plasmids, primers and growth conditions
All the bacterial strains used in this study are listed in Table S3.† The plasmids used in this study are listed in Table S4.† The primers used in this study are listed in Table S5.† The E. coli strains were cultured in Luria–Bertani broth (LB). The S. aureus strains were cultured in tryptic soy broth (TSB). Antibiotics were used at the following concentrations: kanamycin 50 μg mL−1 for E. coli; chloramphenicol 7.5 μg mL−1 for S. aureus RN4220 strain, 10 μg mL−1 for S. aureus Newman and N315 strains.Construction of the pABE plasmid
The pABE plasmid was constructed using the following steps: the Cas9 nickase gene and part of the S. aureus temperature-sensitive repF origin were amplified from the pnCasSA–BEC19 plasmid. The rest part of the repF origin, the ColE1 origin, the chloramphenicol-resistance marker, the kanamycin-resistance marker, the sgRNA expression cassette, and the rpsL promoter were amplified from the pnCasSA–BEC plasmid. E. coli codon optimized adenine deaminase dimer gene ABE7.10 was synthesized by GENEWIZ (Suzhou, China). The three fragments were assembled by Gibson assembly. The successful construction of the pABE plasmid was verified by PCR, enzyme digestion, and sequencing.For multiplex genome editing, the second sgRNA expression cassette was PCR-amplified and inserted into the XbaI/XhoI sites of a former constructed pABE plasmid, which harbored the first sgRNA cassette. Thus two sgRNA cassette were assembled into one pABE plasmid. The successful construction of the plasmid was verified by PCR, enzyme digestion, and sequencing.
Competent cell preparation and electroporation
The competent cells of the S. aureus strains RN4220, Newman, and N315 were prepared using the following steps. A single colony of a S. aureus strain was inoculated into 3 mL TSB and incubated at 30 °C for 12 h. The cells were 1:100 diluted into 100 mL fresh TSB medium and incubated at 30 °C. When the optical density at 600 nm (OD600) of the culture reached 0.3 to 0.4, the bacterial solution was chilled on ice for 10 min. Then, the cells were harvested by centrifugation (5000 rpm) at 4 °C for 5 min, and washed twice with 20 mL sterile ice-cold sucrose (0.5 M). Finally, the cells were resuspended into 0.5 mL of sucrose (0.5 M). 50 μL aliquots were frozen in liquid nitrogen and stored at −80 °C.
For electroporation, 50 μL of the competent cells were thawed on ice for 5 min. Next, 1–2 μg of the pABE plasmid were added and mixed with the cells. After incubation on ice for 5 min, the mixture was transferred into a 1 mm electroporation cuvette (Bio-Rad, USA). After pulsed at 21 kV cm−1, 100 Ω, 25 μF, the cells were mixed with 1 mL of TSB and incubated at 30 °C for 1.5 h. Finally, the cells were plated onto a TSB agar plate containing chloramphenicol and the plate was incubated at 30 °C for 24 h.
The competent cells of the E. coli MG1655 strain were prepared using the following steps. A fresh single colony of an E. coli strain was inoculated into 3 mL LB and incubated at 37 °C for 12 h. The cells were 1:100 diluted into 100 mL fresh LB medium. When the optical density at 600 nm (OD600) of the culture reached 0.4 to 0.5, the culture was chilled on ice for 10 min. Then, the cells were harvested by centrifugation (4000 rpm) at 4 °C for 5 min, and washed twice with 20 mL sterile and ice-cold 10% glycerol. Finally, the cells were resuspended into 1 mL of 10% glycerol. 50 μL aliquots were frozen in liquid nitrogen and stored at −80 °C.
For electroporation, 50 μL of competent cells were thawed on ice for 5 min. Next, 500 ng of the pABE plasmid were added and mixed with the cells. After incubation on ice for 5 min, the mixture was transferred into a 1 mm electroporation cuvette (Bio-Rad, USA). After pulsed at 18 kV cm−1, 200 Ω, 25 μF, the cells were mixed with 1 mL of LB and incubated at 30 °C for 1.5 h. Finally, the cells were plated onto a LB agar plate containing kanamycin and incubated at 30 °C for 24 h.
Adenine base editing
A pair of oligonucleotide DNAs for the construction of the targeting gRNA vector pABE was designed as follows: 5′-gaaa-(target sequence)-3′ and 5′-aaac-(reverse complement of the target sequence)-3′. The pair of the oligonucleotide DNAs was phosphorylated, annealed, and ligated into BsaI-digested pABE plasmid via Golden Gate assembly. The successful constructions of spacer-introduced pABE plasmids were verified by PCR, enzyme digestion, and sequencing. For base editing in S. aureus, a plasmid extracted from the E. coli DH5α strain was modified in the IM08B39 strain before being electroporated into S. aureus. The S. aureus cells harboring the pABE plasmid were plated onto a TSA plate containing chloramphenicol and incubated at 30 °C for 24 h. For base editing in E. coli, a plasmid extracted from the E. coli DH5α strain was directly transformed into the target E. coli strain. The cells were plated onto a LB agar plate containing kanamycin and incubated at 30 °C for 24 h. After editing, the mutants were further purified by dilution series, and the fully edited colonies were selected and confirmed by Sanger sequencing before being used for subsequent experiments.Editing efficiency evaluation
The genomic DNA of each colony was individually extracted and used as the template for PCR amplification. The PCR products covering the editable region were sent out for Sanger sequencing. The editing efficiency was calculated either by the ratio of successfully edited colonies to the total numbers of randomly picked colonies or by the conversion efficiency of A to G using EditR.40Gene editing using the pBECKP,21 pnCasSA–BEC,19 pKOR1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) 34 and pCasSA9 systems
34 and pCasSA9 systems
Spacers were designed and inserted into the pBECKP, pnCasSA–BEC plasmids to generate premature stop codons. Correctly constructed pBECKP-spacer, pnCasSA–BEC-spacer were verified by PCR, enzyme digestion, and sequencing. The constructed plasmids were transformed into the target cells to perform gene editing by following the detailed protocols of the pBECKP21 and pnCasSA–BEC19 methods. All the base editing experiments were performed in triplicate.
Spacer near the desired mutation sites was designed and inserted into the pCasSA plasmid. Correctly constructed pCasSA-spacer was verified by PCR, enzyme digestion, and sequencing. Then pCasSA-spacer plasmid was digested with XbaI and XhoI. Amplification of the upstream (∼1000 bp) and downstream (∼1000 bp) was carried out by using primers containing the desired mutation. The three fragments (upstream, downstream, and the digested plasmid) were assembled by Gibson assembly. The successful construction of the pCasSA-derived plasmid was verified by PCR, enzyme digestion, and sequencing. Gene editing by the pCasSA plasmid was carried out by following the detailed protocol of the pCasSA9 method.
Primers containing attB1 and attB2 sites were designed for PCR of the pKOR1 backbone. Amplification of the upstream (∼1000 bp) and downstream (∼1000 bp) was carried out by using primers containing the desired mutation. The three fragments were assembled by Gibson assembly. The successful construction of the pKOR1-derived plasmid was verified by PCR, enzyme digestion, and sequencing. Gene editing by the pKOR1 plasmid was carried out by following the detailed protocol of the pKOR1 method.34
Plasmid curing
The editing plasmid was cured in S. aureus RN4220 strain before the grow-curve measurement. In brief, 3 μL of an overnight culture of cells containing the desired mutation were 1:1000 diluted into 3 mL TSB and incubated at 42 °C for 12 h without antibiotics. After that, 10 μL cells were streaked onto a TSA plate without antibiotics and incubated at 37 °C for 12 h. A single colony was picked and diluted into 10 μL ddH2O. A fraction of the diluted cells was plated onto a TSA plate without chloramphenicol and another fraction was plated onto a TSA plate containing chloramphenicol. The cells whose plasmid was successfully cured could only grow on the plate without chloramphenicol.
lacZ assay
The lacZ gene of the MG1655 strain was first inactivated by introducing a premature stop codon at Q51 (CAG to TAG) using a bacterial CBE pBECKP by following the detailed procedures described in the literature.21 Then, the cells containing the inactivated lacZ gene was transformed with a pABE plasmid targeting the stop codon. The cells were plated onto a LB agar plate containing 0.1 mg mL−1 X-gal and 3 × 10−4 mol L−1 IPTG. Successfully edited cells were blue while the unedited cells were white. The experiments were performed in triplicate.Growth curves assay
Single colonies of various mutant strains were individually picked and inoculated into 3 mL of chemically defined medium (CDM).33 The cells were incubated at 30 °C for 16 h. Then the cells were 1:100 diluted into 200 μL CDM supplemented with 3 mM Co2+. The cells were transferred into a micro-well plate and the growth of the cells were measured using the automated microbe growth analysis system BioScreen C (OY Growth Curves Ltd, Finland). The optical density at 600 nm (OD600) was measured by BioScreen every 0.5 h lasting for 16 h. The experiments were performed in triplicate.
Deep sequencing
Genomes of four edited S. aureus RN4220 strains were extracted using the Rapid Bacterial Genomic DNA Isolation Kit (Sangon Biotech, Shanghai, China). The isolated genomes were used as templates for PCR-amplification of ∼200 bp DNA fragments containing the edited sites. The barcode sequences were included on the 5′ terminus of the forward primers. Purified DNA products were sent out for sequencing using the Illumina Hiseq X Ten (2 × 150) platform at GENEWIZ (Suzhou, China). Fifty thousand reads per sample were analyzed. The experiments were performed in triplicate. The wild-type S. aureus RN4220 strain was performed following the same process as control.Whole-genome sequencing
Genomes of the wild-type and two edited S. aureus RN4220 strains were extracted using the Rapid Bacterial Genomic DNA Isolation Kit (Sangon Biotech, Shanghai, China) and sent out for whole-genome sequencing by the BGISEQ PE150 platform at Beijing Genomics Institute. The paired-end fragment libraries were sequenced according to the Illumina HiSeq 4000 system's protocol. Raw reads of low quality from paired-end sequencing were discarded. The assembly scaffolds were aligned with reference sequence (GCA_000212435.2) for detection of single nucleotide polymorphism (SNP).Co2+-content measurement using ICP-OES
The samples for ICP-OES were prepared following the protocols described in literatures.41,42S. aureus RN4220 strains were cultured at 30 °C in 20 mL of CDM (supplemented with 0.5 μM CoCl2). When the cells grew to exponential phase (OD600 ∼ 0.6–0.8), the cultures were chilled on ice for 10 min and then harvested by centrifugation at 7000 g at 4 °C for 10 min. Pellets were washed 3 times using 1 mL of 2 mM EDTA solution, followed by a final wash with 1 mL MilliQ H2O. Next, cells were dried at 100 °C and then digested by 300 μL nitric acid (66–68% HNO3) for 12 h at 80 °C. After diluted to final volumns of 3 mL using MilliQ H2O, the samples were used for element measurements by using the inductively coupled plasma-optical emission spectrometry (iCAP 7000 Plus Series, Thermo Scientific™). Concentrations of cobalt ions of each sample were determined by standard solutions and was calculated assuming 4 × 108 cfu for an OD600 of 1 unit. Each experiment was repeated three times.CntB–CntC heterodimer modeling
To build the model for CntB and CntC heterodimer, we did a blast against PDB to find possible templates. We failed to find any close homologs of CntB and CntC that have solved structures. Then we used HHpred43 to find distant homologs of CntB and CntC that have structures. The top hit 2ONK (sequence identity 14%, in an inward-facing conformation) was used as the template to build the heterodimer model of CntB and CntC. We used Prime in Schrodinger Suite 2019-132 to build the dimer, using the sequence alignments from the results of HHpred.
Data availability
The sequence of the pABE plasmid (accession number MN735463), deep sequencing data (accession number PRJNA598507), and whole-genome sequencing data of various RN4220 strains (accession numbers: PRJNA598506, PRJNA598514, PRJNA598516) have been deposited at the NCBI.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21922705, 91753127, and 31700123), the Shanghai Committee of Science and Technology, China (19QA1406000, 17ZR1449200, and 18YF1416500).References
- K. A. Datsenko and B. L. Wanner, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6640–6645 CrossRef CAS PubMed.
- Y. Zhang, F. Buchholz, J. P. Muyrers and A. F. Stewart, Nat. Genet., 1998, 20, 123–128 CrossRef CAS PubMed.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini and F. Zhang, Science, 2013, 339, 819–823 CrossRef CAS PubMed.
- W. Jiang, D. Bikard, D. Cox, F. Zhang and L. A. Marraffini, Nat. Biotechnol., 2013, 31, 233–239 CrossRef CAS PubMed.
- P. Mali, L. Yang, K. M. Esvelt, J. Aach, M. Guell, J. E. DiCarlo, J. E. Norville and G. M. Church, Science, 2013, 339, 823–826 CrossRef CAS PubMed.
- H. Wang, M. La Russa and L. S. Qi, Annu. Rev. Biochem., 2016, 85, 227–264 CrossRef CAS PubMed.
- Z. Wu, Y. Wang, Y. Zhang, W. Chen, Y. Wang and Q. Ji, Small Methods, 2019 DOI:10.1002/smtd.201900560.
- W. Chen, Y. Zhang, W.-S. Yeo, T. Bae and Q. Ji, J. Am. Chem. Soc., 2017, 139, 3790–3795 CrossRef CAS PubMed.
- A. C. Komor, Y. B. Kim, M. S. Packer, J. A. Zuris and D. R. Liu, Nature, 2016, 533, 420–424 CrossRef CAS PubMed.
- K. Nishida, T. Arazoe, N. Yachie, S. Banno, M. Kakimoto, M. Tabata, M. Mochizuki, A. Miyabe, M. Araki, K. Y. Hara, Z. Shimatani and A. Kondo, Science, 2016, 353(6305), aaf8729 CrossRef PubMed.
- N. M. Gaudelli, A. C. Komor, H. A. Rees, M. S. Packer, A. H. Badran, D. I. Bryson and D. R. Liu, Nature, 2017, 551, 464–471 CrossRef CAS PubMed.
- Y. Tong, C. M. Whitford, H. L. Robertsen, K. Blin, T. S. Jørgensen, A. K. Klitgaard, T. Gren, X. Jiang, T. Weber and S. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 20366–20375 CrossRef CAS PubMed.
- P. Billon, E. E. Bryant, S. A. Joseph, T. S. Nambiar, S. B. Hayward, R. Rothstein and A. Ciccia, Mol. Cell, 2017, 67, 1068–1079 CrossRef CAS PubMed.
- Z. Shimatani, S. Kashojiya, M. Takayama, R. Terada, T. Arazoe, H. Ishii, H. Teramura, T. Yamamoto, H. Komatsu, K. Miura, H. Ezura, K. Nishida, T. Ariizumi and A. Kondo, Nat. Biotechnol., 2017, 35, 441–443 CrossRef CAS PubMed.
- Y. Zong, Y. Wang, C. Li, R. Zhang, K. Chen, Y. Ran, J. L. Qiu, D. Wang and C. Gao, Nat. Biotechnol., 2017, 35, 438–440 CrossRef CAS PubMed.
- L. Yang, A. W. Briggs, W. L. Chew, P. Mali, M. Guell, J. Aach, D. B. Goodman, D. Cox, Y. Kan and E. J. N. C. Lesha, Nat. Commun., 2016, 7, 13330 CrossRef CAS PubMed.
- S. Banno, K. Nishida, T. Arazoe, H. Mitsunobu and A. Kondo, Nat. Microbiol., 2018, 3, 423–429 CrossRef CAS PubMed.
- T. Gu, S. Zhao, Y. Pi, W. Chen, C. Chen, Q. Liu, M. Li, D. Han and Q. Ji, Chem. Sci., 2018, 9, 3248–3253 RSC.
- W. Chen, Y. Zhang, Y. Zhang, Y. Pi, T. Gu, L. Song, Y. Wang and Q. Ji, iScience, 2018, 6, 222–231 CrossRef CAS PubMed.
- Y. Wang, S. Wang, W. Chen, L. Song, Y. Zhang, Z. Shen, F. Yu, M. Li and Q. Ji, Appl. Environ. Microbiol., 2018, 84, e01834-18 CrossRef PubMed.
- Y. Wang, Z. Wang, Y. Chen, X. Hua, Y. Yu and Q. Ji, Cell Chem. Biol., 2019, 26, 1732–1742 CrossRef CAS PubMed.
- S. M. Ryu, T. Koo, K. Kim, K. Lim, G. Baek, S. T. Kim, H. S. Kim, D. E. Kim, H. Lee, E. Chung and J. S. Kim, Nat. Biotechnol., 2018, 36, 536–539 CrossRef CAS PubMed.
- K. Hua, X. Tao, F. Yuan, D. Wang and J. K. Zhu, Mol. Plant, 2018, 11, 627–630 CrossRef CAS PubMed.
- C. Li, Y. Zong, Y. Wang, S. Jin, D. Zhang, Q. Song, R. Zhang and C. Gao, Genome Biol., 2018, 19, 59 CrossRef PubMed.
- O. O. Abudayyeh, J. S. Gootenberg, B. Franklin, J. Koob, M. J. Kellner, A. Ladha, J. Joung, P. Kirchgatterer, D. B. T. Cox and F. Zhang, Science, 2019, 365, 382–386 CrossRef CAS PubMed.
- D. B. T. Cox, J. S. Gootenberg, O. O. Abudayyeh, B. Franklin, M. J. Kellner, J. Joung and F. Zhang, Science, 2017, 358, 1019–1027 CrossRef CAS PubMed.
- S. Banno, K. Nishida, T. Arazoe, H. Mitsunobu and A. J. N. M. Kondo, Nat. Microbiol., 2018, 3, 423–429 CrossRef CAS PubMed.
- S. Jin, Y. Zong, Q. Gao, Z. Zhu, Y. Wang, P. Qin, C. Liang, D. Wang, J.-L. Qiu, F. Zhang and C. Gao, Science, 2019, 364, 292–295 CAS.
- O. Shalem, N. E. Sanjana, E. Hartenian, X. Shi, D. A. Scott, T. Mikkelson, D. Heckl, B. L. Ebert, D. E. Root, J. G. Doench and F. Zhang, Science, 2014, 343, 84–87 CrossRef CAS PubMed.
- T. Wang, J. J. Wei, D. M. Sabatini and E. S. Lander, Science, 2014, 343, 80–84 CrossRef CAS PubMed.
- Y. Zhou, S. Zhu, C. Cai, P. Yuan, C. Li, Y. Huang and W. Wei, Nature, 2014, 509, 487–491 CrossRef CAS PubMed.
- G. Ghssein, C. Brutesco, L. Ouerdane, C. Fojcik, A. Izaute, S. Wang, C. Hajjar, R. Lobinski, D. Lemaire, P. Richaud, R. Voulhoux, A. Espaillat, F. Cava, D. Pignol, E. Borezee-Durant and P. Arnoux, Science, 2016, 352, 1105–1109 CrossRef CAS PubMed.
- T. Bae and O. Schneewind, Plasmid, 2006, 55, 58–63 CrossRef CAS PubMed.
- J. H. Hu, S. M. Miller, M. H. Geurts, W. Tang, L. Chen, N. Sun, C. M. Zeina, X. Gao, H. A. Rees, Z. Lin and D. R. Liu, Nature, 2018, 556, 57–63 CrossRef CAS PubMed.
- W. Chen, H. Zhang, Y. Zhang, Y. Wang, J. Gan and Q. Ji, PLoS Biol., 2019, 17, e3000496 CrossRef PubMed.
- H. Nishimasu, X. Shi, S. Ishiguro, L. Gao, S. Hirano, S. Okazaki, T. Noda, O. O. Abudayyeh, J. S. Gootenberg, H. Mori, S. Oura, B. Holmes, M. Tanaka, M. Seki, H. Hirano, H. Aburatani, R. Ishitani, M. Ikawa, N. Yachie, F. Zhang and O. Nureki, Science, 2018, 361, 1259–1262 CrossRef CAS PubMed.
- B. P. Kleinstiver, M. S. Prew, S. Q. Tsai, V. V. Topkar, N. T. Nguyen, Z. Zheng, A. P. Gonzales, Z. Li, R. T. Peterson, J. R. Yeh, M. J. Aryee and J. K. Joung, Nature, 2015, 523, 481–485 CrossRef PubMed.
- I. R. Monk, J. J. Tree, B. P. Howden, T. P. Stinear and T. J. J. M. Foster, mBio, 2016, 7, 00308–00315 CrossRef PubMed.
- M. G. Kluesner, D. A. Nedveck, W. S. Lahr, J. R. Garbe, J. E. Abrahante, B. R. Webber and B. S. Moriarity, CRISPR J., 2018, 1, 239–250 CrossRef CAS PubMed.
- L. Song, Y. Zhang, W. Chen, T. Gu, S.-Y. Zhang and Q. Ji, Proc. Natl. Acad. Sci. U. S. A., 2017, 115, 3942–3947 CrossRef PubMed.
- G. Ghssein, C. Brutesco, L. Ouerdane, C. Fojcik, A. Izaute, S. Wang, C. Hajjar, R. Lobinski, D. Lemaire and P. J. S. Richaud, Science, 2016, 352, 1105–1109 CrossRef CAS PubMed.
- L. Zimmermann, A. Stephens, S. Z. Nam, D. Rau, J. Kubler, M. Lozajic, F. Gabler, J. Soding, A. N. Lupas and V. Alva, J. Mol. Biol., 2018, 430, 2237–2243 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03784e |
| This journal is © The Royal Society of Chemistry 2020 |