
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biosynthesis of plant tetrahydroisoquinoline alkaloids through an imine reductase route†
Lu
Yang
ab,
Jinmei
Zhu
b,
Chenghai
Sun
b,
Zixin
Deng
a and
Xudong
Qu
 *ab
*ab
aState Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: quxd@whu.edu.cn
bKey Laboratory of Combinatorial Biosynthesis and Drug Discovery Ministry of Education, School of Pharmaceutical Sciences, Wuhan University, Wuhan 430071, China
First published on 18th November 2019
Abstract
Herein, we report a biocatalytic approach to synthesize plant tetrahydroisoquinoline alkaloids (THIQAs) from dihydroisoquinoline (DHIQ) precursors using imine reductases and N-methyltransferase (NMT). The imine reductase IR45 was engineered to significantly expand its substrate specificity, enabling efficient and stereoselective conversion of 1-phenyl and 1-benzyl 6,7-dimethoxy-DHIQs into the corresponding (S)-tetrahydroisoquinolines (S-THIQs). Coclaurine N-methyltransferase (CNMT) was able to further efficiently convert these (S)-THIQ intermediates into (S)-THIQAs. By assembling IRED, CNMT, and glucose dehydrogenase (GDH) in one reaction, we effectively constituted two artificial biosynthetic pathways in Escherichia coli and successfully applied them to the production of five (S)-THIQAs. This highly efficient (100% yield from DHIQs) and easily tailorable (adding other genes) biosynthetic approach will be useful for producing a variety of plant THIQAs.
Introduction
Tetrahydroisoquinoline alkaloids (THIQAs) are a class of important bioactive natural products.1 Molecules in this family typically contain benzyl (benzylisoquinoline alkaloids, BIAs) or phenyl (phenylisoquinoline alkaloids, PIAs) groups at the C-1 position and show a remarkable spectrum of biological activity (Scheme 1).1,2 Currently, more than a dozen of naturally occurring or semisynthetic THIQAs have been used in the clinic for treating a diverse range of diseases.1–3 As most THIQAs are produced by plants, significant efforts have been devoted in the past few decades to develop synthetic approaches for their preparation.1 However, the unsatisfactory enantioselectivity and high cost of asymmetric catalysis make chemical approaches still commercially unfeasible.1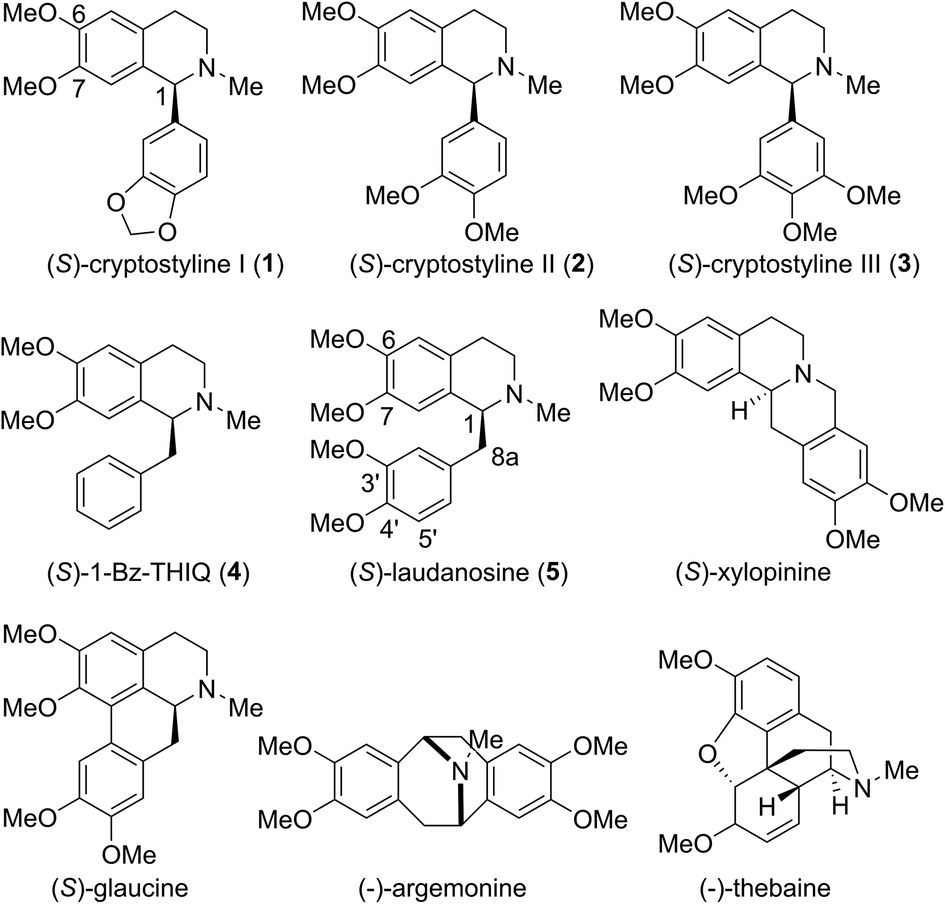 | ||
| Scheme 1 Representative THIQAs. | ||
In natural products, THIQ scaffolds are biosynthesized through a Pictet–Spengler reaction.4 The major enzymes which catalyze these reactions are norcoclaurine synthases (NCSs). NCSs condense dopamine with 4-hydroxyphenylacetaldehyde (4-HPAA) or 3,4-hydroxyphenylacetaldehyde (3,4-HPAA) to give rise to (S)-norcoclaurine or (S)-norlaudanosoline.5 Following decorations by methyltransferases (MTs) or P450s, 4-HPAA and 3,4-HPAA can be converted into (S)-reticuline (Scheme 2), a key intermediate of ∼2500 BIAs.5 With the growing understanding of BIA biosynthesis, reconstruction of plant biosynthetic pathways in microbial hosts has become a feasible strategy for producing BIAs. By incorporation of partial or complete biosynthetic pathways in microbial hosts, a number of BIAs (i.e. opiates, sanguinarine and noscapine) have been successfully synthesized from simple precursors or glucose.6 Such biosynthetic approaches can become more economical and easy-to-operate than chemical synthesis, making biosynthesis a highly attractive alternative.
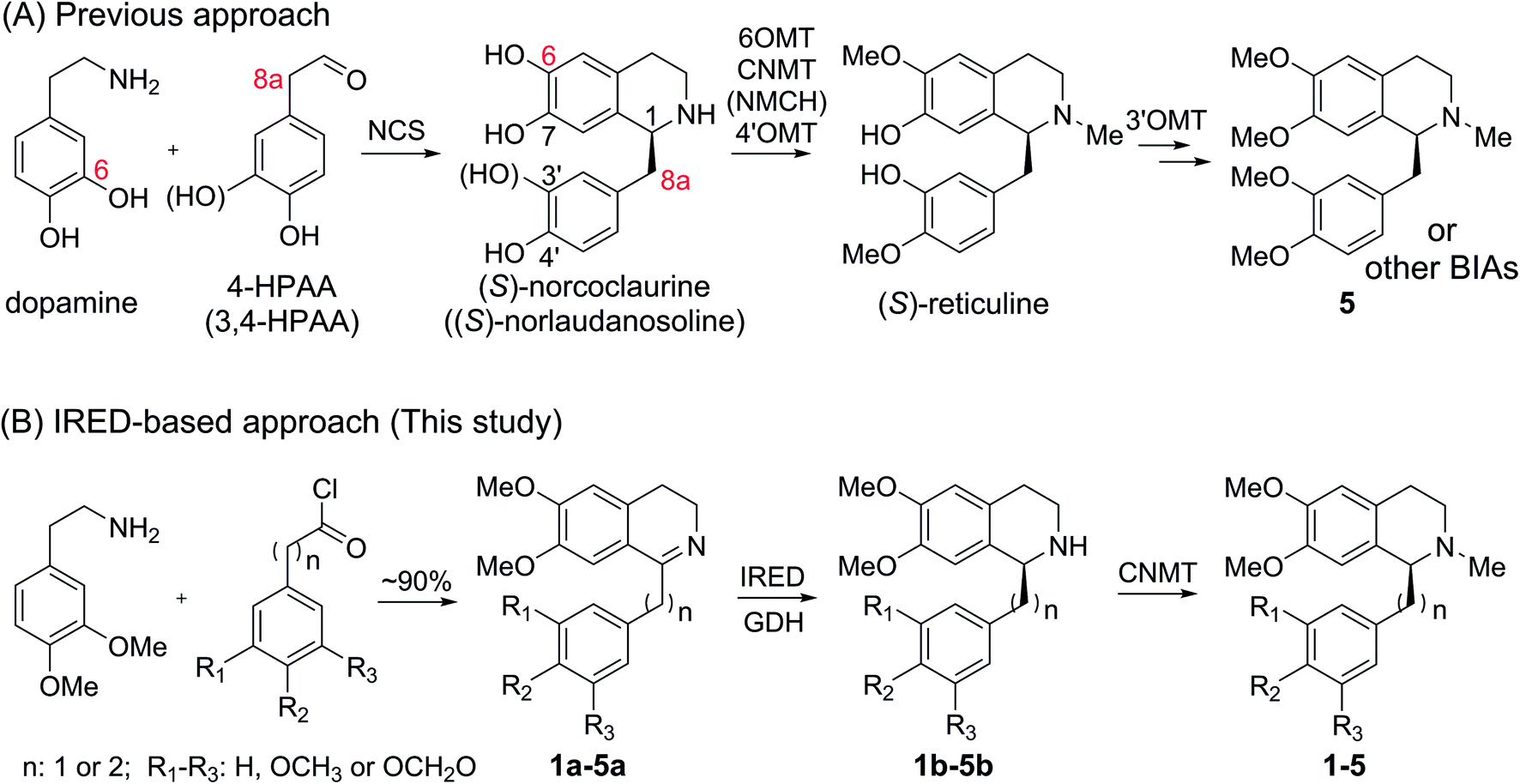 | ||
| Scheme 2 Approaches for plant THIQA biosynthesis. | ||
Although great success has been achieved, there still remain many challenges for microbial production of plant THIQAs:7 (i) the biosynthesis of PIAs and many BIAs remain elusive, and thus, knowledge of essential genes for reconstitution of their biosynthetic pathways is lacking. (ii) Most valuable THIQAs are highly modified, i.e. bearing methoxy groups at the C6, C7, C3′ and C4′ positions. (iii) Reconstitution of the biosynthetic pathways in microbes requires the introduction of a large numbers of genes, which brings a heavy metabolic burden on the microbe. Moreover, many plant enzymes such as OMTs and P450s are poorly expressed or show low reactivity (NCSs) in microbial systems.6e,k,8 Therefore, production of plant THIQAs in microbes often results in very low yields.3 To avoid these challenges, appropriately substituted precursors can be fed into simplified biosynthetic pathways. However, such an approach is restricted either by the narrow substrate scope of NCSs, whose activity is strictly dependent on the C6-hydroxyl and C8a-methylene groups (Scheme 2)9 or the stereochemistry at C1, which is challenging to prepare via practical chemical synthesis.1
Recently, our lab and another group identified imine reductases (IREDs) that are capable of reducing sterically hindered 1-phenyl dihydroisoquinolines (DHIQs) to yield the corresponding chiral 1-phenyl-THIQs.10 In contrast to NCSs, IREDs have a distinct catalytic mechanism, which does not rely on the C6-hydroxyl or C8a-methylene group, and therefore can accept highly modified DHIQ precursors. Moreover, IREDs from bacterial sources can be well overexpressed and show good activity in Escherichia coli. Thus, by combining IREDs with other modification enzymes, it is potentially feasible to produce THIQAs through feeding DHIQ precursors. Herein, we demonstrate this concept by using IREDs, N-methyltransferases (NMTs), and glucose dehydrogenase (GDH) to construct two minimal biosynthetic pathways. By incorporating these artificial pathways into E. coli, we successfully synthesize a group of pharmaceutically valuable PIAs and BIAs. Our results demonstrate that this strategy is an efficient approach to the biosynthesis of plant THIQAs.
Results
Engineering the substrate specificity of IR45 for the synthesis of 1-phenyl 6,7-dimethoxyl-THIQs
Cryptostyline I, II and III (1–3, Scheme 1) are three PIAs isolated from the plant Cryptostylis fulva.11 They are used as pharmacological probes for understanding the pathophysiological roles of peptides in the nervous system.12 Unlike BIAs, the biosynthesis of PIAs is still poorly understood, and currently the biosynthetic genes of 1–3 are not known. Given their valuable pharmaceutical utility and representative roles for the PIAs, these three products were chosen as the targets for this study.Three 1-phenyl-6,7-dimethoxy-DHIQ precursors (1a–3a, Scheme 3) corresponding to cryptostyline I–III (1–3) were obtained in good yields (90–91%) by chemical synthesis (see the ESI†). In a previous study, we identified an S-selective IRED, IR45, which showed good tolerance toward 1-phenyl-DHIQs.10a Therefore, this enzyme was selected for activity assays with the new substrates. Encouragingly, IR45 effectively reduced 1a to the (S)-1-methylenedioxyphenyl-6,7-dimethoxy-THIQ (1b, Scheme 2) with 100% conversion and >99% ee (Table 1), although it showed no activity toward the two bulkier substrates (2a and 3a). To gain a more detailed understanding of its substrate specificity, the structure of IR45 was modelled based on the IRED Q1EQE0 (64% identity, PDB: 3ZHB).13 Docking of 1a into the binding pocket of IR45 indicated that the substrate is closely surrounded by the conserved aspartic acid (D183) and NADPH (Fig. 1). The imine bond is positioned close to the si-face of NADPH, consistent with the observed S-selectivity in the product. The methylenedioxyphenyl group points to the right cleft, which consists of residues from two subunits of the enzyme (V81, S107, E253′ and H257′). On the other side of the active site, the 6,7-dimethoxy aryl group is surrounded by two residues, W191 and F190. These two bulky residues form the bottom of the binding pocket, with F190 being the major contributor to the pocket's overall shape.
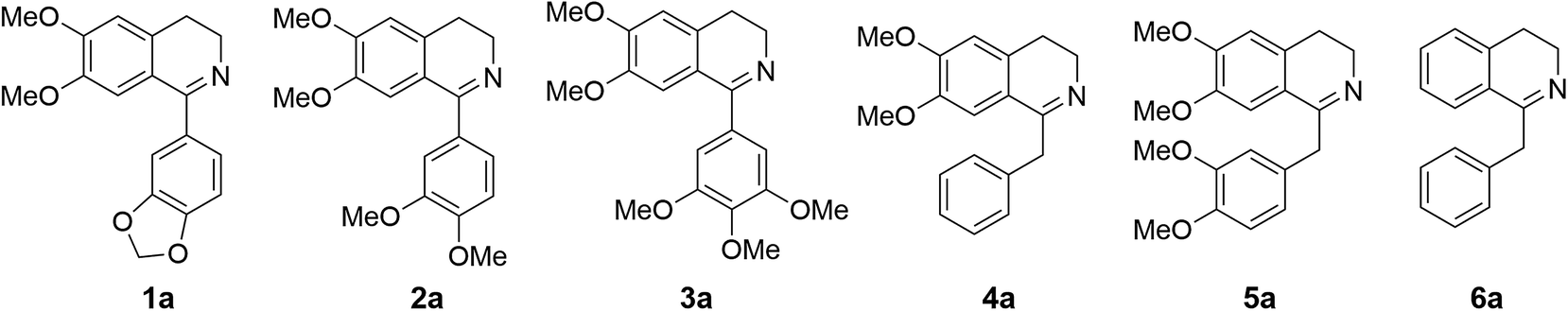 | ||
| Scheme 3 DHIQ substrates prepared and used in this study. | ||
| Subs. | Conversion (%) and enantiomeric excess (ee)a (%) | |||
|---|---|---|---|---|
| IR45 | W191F | F190L–W191F | F190M–W191F | |
| a First value shown represents conversion and the second value represents the ee value. Stereospecificity is indicated by the superscripts. Dash indicates data not tested. All reactions were performed at 30 °C for 24 h. | ||||
| 1a | 100; >99S | — | 100; >99S | 100; >99S |
| 2a | 0 | 3; — | 75; >99S | 0 |
| 3a | 0 | 2; — | 100; >99S | 0 |
| 4a | 100; >99S | — | 100; >99S | 100; >99S |
| 5a | 100; >99S | — | 100; >99S | 100; >99S |
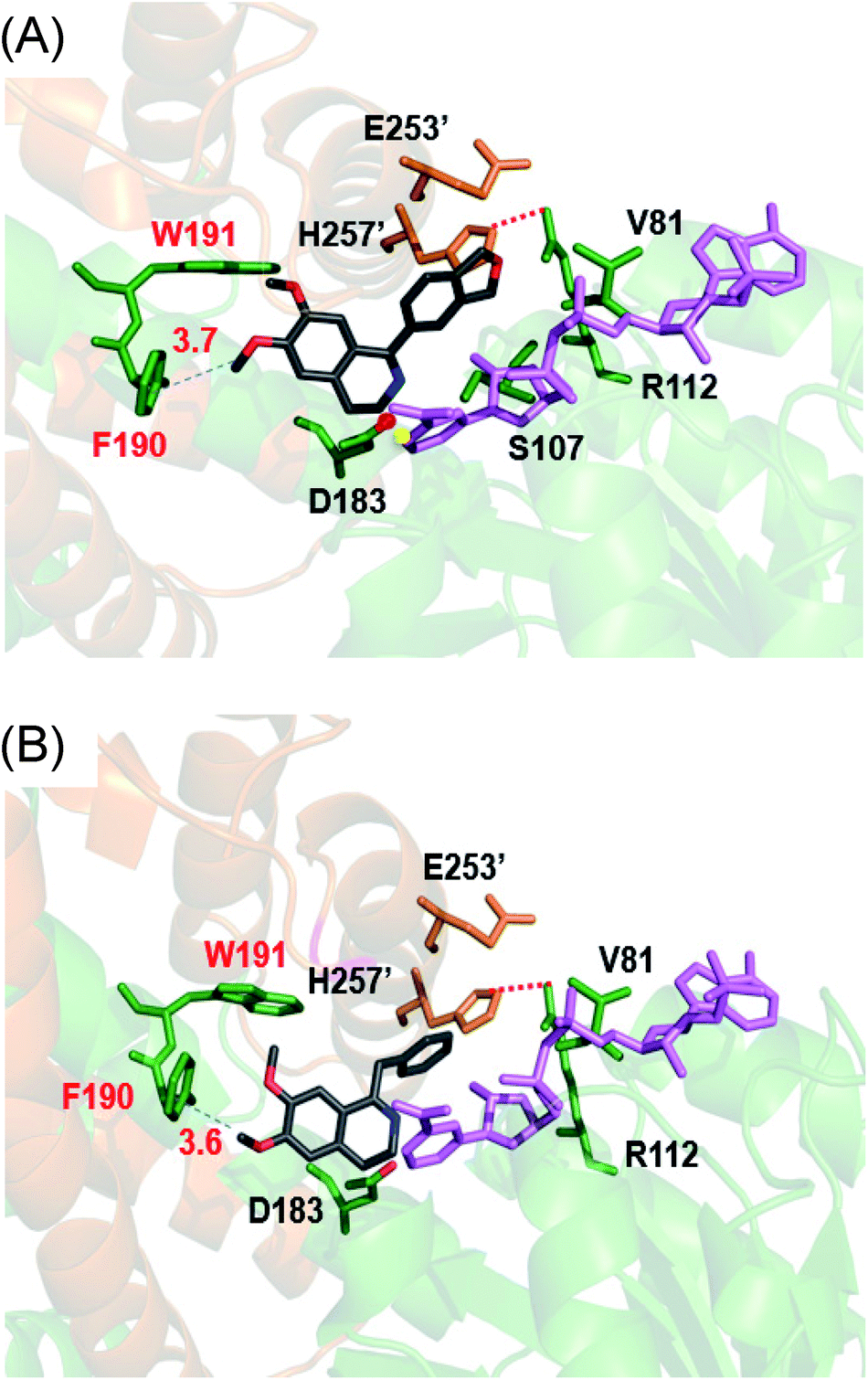 | ||
| Fig. 1 In silico model of IR45 with substrates 1a and 4a. The structure of IR45 is modelled based on Q1EQE0 (PDB: 3zhb) and docked with 1a or 4a (grey) and NADPH (pink); critical residues in the binding cavity are labelled. Protein backbones and residues from different subunits are indicated by yellowish brown and green colours. (A) IR45 with 1a; (B) IR45 with 4a. | ||
From the model, it can be seen that the right cleft forms the major constraint for the accommodation of the methylenedioxyphenyl group (Fig. 1). Engineering this area, however, may significantly impair the enzyme's activity and stability, as residues in this region are crucial for inter-subunit interactions, i.e. H257′ forms a stabilizing salt bridge with R112. Thus, we envisioned enlarging the binding pocket by engineering the side of the pocket comprising F190 and W191. By mutating these sterically demanding residues, we thought that the substrate could be moved slightly deeper into the left cavity, thus potentially relieving the steric hindrance between the right cleft and substrates 2a and 3a.
In our previous studies, we found that W191 plays important roles in substrate binding and maintaining the enzyme's catalytically active conformation. Mutation of W191 to alanine or leucine reduces the kcat and Km for the substrate 1-phenyl-DHIQ (6a, Scheme 3).10a To identify optimal mutations, W191 was further mutated to the remaining 17 amino acids. These variants together with the W191A and W191L were assayed for the reduction of 2a and 3a. Although most mutants were inactive, one mutant in particular, W191F, showed marginal but clear activity toward these substrates (3% and 2% for 2a and 3a respectively, Table 1 and Fig. S1A†). To unveil the detailed effects of these mutations on the IRED's activity, we used the more active substrate 6a for activity analysis. Similarly, W191F shows a clear improvement in the conversion of 1-phenyl-DHIQ (1.5-fold), while the others all drastically reduce or even abolish the catalytic activity (Fig. S2†).
Kinetic analysis of the most active mutants, including W191F, W191C, W191M, W191H, W191N, and W191S as well as previously obtained W191A and W191L revealed that their kcat and Km values both have been reduced (Table S1 and Fig. S3A†). However, unlike the other enzymes whose kcat is significantly reduced by one to two orders of magnitude, the kcat value of W191F is decreased by only 1.8-fold (Table S1†), suggesting that W191F has the least negative impact on the enzyme's catalytic conformation. As W191 is highly conserved, we assume that this mutation should be beneficial for many other IREDs.
The encouraging results with W191F inspired us to further engineer the enzyme by targeting the adjacent residue F190. We mutated all 19 other residues on top of W191F and then assayed these double mutants for activity on the bulkier DHIQ substrates. To our gratification, one mutant F190L–W191F shows drastically improved activity toward both 2a and 3a (75% and 100% conversion within 24 hours respectively) with very high stereoselectivity for the S-products 2b and 3b (Scheme 2 and Table 1). Kinetic analysis confirmed that both 2a and 3a are good substrates for F190L–W191F (kcat/Km values are 0.011 and 0.087 s−1 mM−1, respectively, Table 2). The identical Km values (0.307 mM for 2a and 0.372 mM for 3a) yet different kcat values (0.003 s−1 for 2a and 0.032 s−1 for 3a) suggest that binding conformation accounts for the difference in their activity. This indicates that other residues influencing the binding conformation in the cavity could be engineered to further improve the enzyme's catalytic activity.
| Subs. | IR45 | F190L–W191F | F190M–W191F | ||||||
|---|---|---|---|---|---|---|---|---|---|
| K m [mM] | k cat [s−1] | k cat/Km [s−1 mM−1] | K m [mM] | k cat [s−1] | k cat/Km [s−1 mM−1] | K m [mM] | k cat [s−1] | k cat/Km [s−1 mM−1] | |
| a NA indicates no activity. | |||||||||
| 1a | 0.154 | 0.020 | 0.129 | 0.123 | 0.009 | 0.076 | 0.108 | 0.031 | 0.288 |
| 2a | NA | NA | NA | 0.307 | 0.003 | 0.011 | NA | NA | NA |
| 3a | NA | NA | NA | 0.372 | 0.032 | 0.087 | NA | NA | NA |
| 4a | 0.148 | 0.021 | 0.146 | 0.252 | 0.049 | 0.196 | 0.251 | 0.248 | 0.989 |
| 5a | 0.518 | 0.090 | 0.174 | 0.353 | 0.041 | 0.117 | 0.178 | 0.054 | 0.306 |
Through protein engineering, we have successfully broadened the substrate specificity and enabled IR45 to effectively convert very sterically hindered DHIQ precursors into their corresponding THIQs. In addition, the successful expansion of the substrate specificity of IR45 demonstrates that alteration of the sites that do not directly repel the substrate is indeed an effective way to optimize an enzyme's substrate specificity. This strategy will be useful for engineering other proteins, in which certain substrate–protein interactions are critical for maintaining catalytic activity or active conformation.
Employing IR45 mutants to synthesize 1-benzyl 6,7-dimethoxyl-THIQs
Because BIAs make up the remainder of the THIQA family, we were interested in applying IR45 to their synthesis as well. (S)-Laudanosine (5, Scheme 1) is a pharmaceutically important BIA, naturally found in opium in minute amounts (0.1%).14 Its semisynthetic derivative atracurium is medically used for skeletal muscle relaxation during surgery and mechanical ventilation.3 By mild chemical oxidation, (S)-laudanosine can be readily converted into the antitussive drug (S)-glaucine.15 Production of racemic (R,S)-laudanosine in E. coli has been previously achieved through feeding (R,S)-norlaudanosoline to a strain expressing O-MTs and NMTs.16 However, the poor activities of O-MTs result in low yields of (R,S)-laudanosine, and the accumulation of many methylated intermediates (e.g. (R,S)-reticuline, (R,S)-codamine and (R,S)-laudanine) complicates its isolation (Fig. S4†). We chose to test our IRED approach in the synthesis of 5 given its pharmaceutical value and (S)-1-benzyl-6,7-dimethoxyl-N-methyl-THIQ (BzTHIQ, 4 in Scheme 1) given its prototypical structure.Two corresponding DHIQ substrates 4a and 5a were chemically synthesized (yields 95 and 93%) following a procedure similar to that used in the synthesis of 1a–3a. As W191 and F190 are critical for substrate binding, wild-type IR45 along with the library of F190X–W191F (X denotes other 19 amino acids) variants was tested for activity. To our gratification, wild-type IR45 effectively converted 4a and 5a into the corresponding (S)-THIQ products (100% conversion and >99% ee). The best mutant F190M–W191F showed a further 6.8- and 1.8-fold improvement in catalytic activity for 4a and 5a, respectively (kcat/Km values for 4a and 5a are 0.989 and 0.306 s−1 mM−1, Table 2).
It is intriguing that the wild-type IR45 can accept 4a and 5a whose planar frameworks are larger than those of the inactive substrates 2a and 3a. To explore how differences in binding in the enzyme cavity may account for this disparity in activity, we docked 4a into IR45 (Fig. 1B). In silico analysis revealed that the DHIQ plane of 4a takes an identical conformation to that of 1a. On the other hand, the benzyl group of 4a is bent toward the large cavity underneath the right cleft unlike the 1-phenyl group of 1a which points directly towards the right cleft. This difference in positioning may result in less steric hindrance for the otherwise ‘larger substrate’ 4a. This indicates that the rigid frameworks of 1-phenyl DHIQs are indeed the most challenging substrates for IREDs. Fortunately, our engineered mutant F190L–W191F is able to effectively convert the very sterically hindered substrates 2a and 3a. In addition to the elevated activity on 4a and 5a, F190M–W191F also shows a 2.23- and 3.79-fold improvement (kcat/Km) on 1a compared to wild-type IR45 and F190L–W191F, respectively (Table 2). This outstanding activity makes the imine reduction step highly efficient, ensuring the success of this strategy.
Conversion of THIQs into THIQAs by CNMT
After overcoming the bottlenecks associated with imine reduction, we turned our attention to the N-methylation step. Coclaurine N-methyltransferase (CNMT) is a key enzyme in the pathway of (S)-reticulene, introducing the N-methyl substituent into coclaurine.17 CNMT from Coptis japonica is known to have a relaxed substrate specificity and is able to methylate THIQ products.17,18 Therefore, it was assayed for N-methylation activity on THIQ substrates 1b–5b. To improve expression, the CNMT gene was codon optimized for E. coli. As expected, following optimization, CNMT was robustly expressed in E. coli.To our gratification, CNMT efficiently converted all five of the S-THIQ substrates into their corresponding N-methylated products (100% conversion, Fig. S5A†). Kinetic analysis revealed that CNMT showed similar activity to all 1-phenyl and 1-benzyl substrates except for the most active substrate 4b (Table S2†). As these substrates are much bulkier than previously known substrates of CNMT, we were interested in understanding how they bind to the active site. Two representative substrates 1b and 5b were individually docked into the crystal structure of CNMT (Fig. S6†).18 Similar to the conformation of 4a in IR45, 5b is bent, allowing it to be well accommodated in the binding cavity. The substrates are bound in an identical orientation with their methylenedioxy and methoxy groups in close proximity to residues F322 and W329.18 Previous work showed that two mutants, F322A and W329A, had significantly reduced catalytic activity, with W329A having the most negative impact.18 In order to maintain these hydrophobic interactions while enlarging the binding cavity, we mutated W329 into phenylalanine. However, like W329A, W329F showed drastically reduced methylation activity toward 1–5 (Fig. S7A†), confirming that W329 is indeed critical for maintaining the enzyme's activity.
Production of THIQAs from DHIQ precursors through the biosynthetic pathway of IRED–NMT–GDH
Armed with efficient imine reduction and N-methylation enzymes, we proceeded to combine these two steps into one biocatalytic reaction. The purified F190L–W191F and F190M–W191F were coupled with CNMT and glucose dehydrogenase (GDH, for recycling NADPH from glucose) for the synthesis of 2–3 and 1, 4–5 respectively. As expected, these enzyme cascades can efficiently convert 1a–5a into THIQAs with 100% conversion (within 24 h, Fig. S7B†). Moreover, we observed that N-methylation improved the conversion of substrates 2a and 3a into their corresponding THIQAs. We hypothesize that the continuous removal of THIQ intermediates by their irreversible methylation drives the equilibrium of imine reduction from DHIQs to THIQs.With the encouraging results of N-methylation, we turned to constitution of the artificial biosynthetic pathway in E. coli. To achieve the highest transformation efficiency, each step was evaluated individually in the E. coli system. We first evaluated the effect of temperature and IPTG concentration on the IRED reaction. E. coli stains containing F190M–W191F and GDH (cloned into pET28a and pACYCDuet, respectively) were incubated at 25, 30 or 37 °C and supplemented with 1a and different concentrations of IPTG when their OD600 reached 0.7. The biotransformation results show that lower IPTG dosage (20 μM) is better than a high IPTG dosage (Fig. S8A†) with conversion reaching only 70% and 40% with 50 μM and 100 μM IPTG, respectively, after 3 days. We found that temperature also had a significant impact on the conversion. The biotransformation efficiency at 30 °C is 5 and 10 times higher than that at 25 °C and 37 °C, respectively (after 3 days) (Fig. S8B†). The optimal conditions for the IRED–GDH combination appear to be 20 μM IPTG and 30 °C. Next the conversion of the other four substrates was evaluated under these optimized conditions (F190M–W191F was replaced by F190L–W191F for 2a and 3a). Although 4a and 5a can be efficiently converted into their reduced products (100%), 1a–3a exhibit poor-to-medium conversion (6.5% to 50%) (Fig. S1C†). These ratios are much lower than the conversion ratios of the in vitro enzymatic transformation, suggesting that the 1-phenyl substrates 1a–3a face difficulty in permeating the cell membrane.
Next, the N-methyl transfer step was evaluated. Gratifyingly, all the amine substrates, 1b–5b, are completely converted into their corresponding final products under the same optimal conditions as for IRED–GDH by E. coli containing CNMT (Fig. S5B†). As methylation by CNMT appeared to improve the activity of imine reduction, we co-expressed all three enzymes in a single cell (NMT and GDH were cloned in pACYCDuet and IR45 in pET28a), forming a minimal biosynthetic pathway for the synthesis of THIQAs. As expected 1a, 4a and 5a (50 mg L−1) can be completely converted into their corresponding alkaloids within one to three days (Table 3 and Fig. 2). By extraction with ethyl acetate, pure 1, 4 and 5 can be readily obtained in yields of 95%, 98% and 95%, respectively. Conversion of 2a and 3a also significantly improved (89% and 51% respectively, Fig. S7C†), compared to the conversion by IRED–GDH (6.5% and 9.3%, Fig. S1C†).
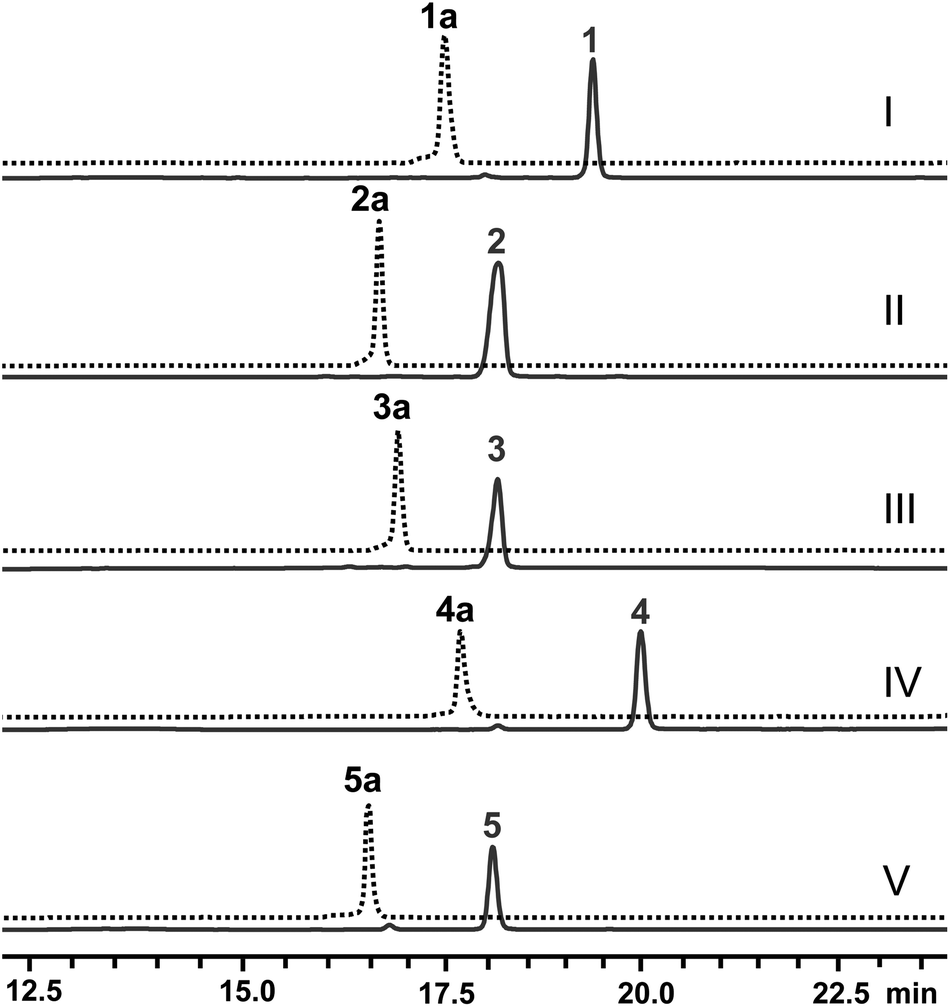 | ||
| Fig. 2 Production of THIQAs 1–5 in the IRED-based biosynthetic systems. The retention time of the precursors 1a–5a is indicated by the dashed lines. Samples from the biosynthetic systems are indicated by the solid lines in dark blue. (I) Biosynthesis of 1 from 1a by E. coli with F190M–W191F + GDH + CNMT; (II) biosynthesis of 2 from 2a by the crude enzymes F190L–W191F + GDH and E. coli with CNMT; (III) biosynthesis of 3 from 3a by the crude enzymes F190L–W191F + GDH and E. coli with CNMT; (IV) biosynthesis of 4 from 4a by E. coli with F190M–W191F + GDH + CNMT; (V) biosynthesis of 5 from 5a by E. coli with F190M–W191F + GDH + CNMT. | ||
In order to overcome the barrier of membrane permeabilization and further improve the conversion of 2a and 3a, we combined enzymatic and whole-cell biotransformation into a single pot reaction. F190L–W191F and GDH were first overexpressed in a single E. coli strain and then lysed by sonication. The crude enzymes were directly added to the E. coli–CNMT culture when the OD600 reached 0.7. At this time, IPTG, DHIQ substrates and NADP (1.1‰ w/v) were added simultaneously to initiate the CNMT expression and cascade biotransformation. To our delight, both 2a and 3a (50 mg L−1) were completely converted into their corresponding THIQAs (Table 3 and Fig. 2). The isolated yields of 2 and 3 are 93% and 96%, respectively. Therefore, by systematically improving enzyme activity and optimizing biocatalytic conditions, we successfully implemented the IRED-based route to biosynthesize two types of plant THIQAs.
Discussion
Lack of knowledge of key biosynthetic genes and poor expression/low reactivity of plant biosynthetic enzymes make reconstitution of THIQA biosynthetic pathways in microbes very challenging. Currently, the biosynthetic enzymes, including the key Pictet–Spengler synthase, that assemble PIAs remain unknown. As such, reconstitution of PIA biosynthesis in microbes is yet to be realized. Although the biosynthesis of many BIAs is well understood and elegant examples of their production in microbes have been reported, the poor performance of NCS O-MT and P450 enzymes results in overall low yields.3 Efforts to bypass such bottleneck enzymes by employing modified precursors are also limited by the narrow substrate scope of NCSs.9 IREDs, owing to their distinct catalytic mechanism, are not restricted by the same limitations as NCSs. Therefore, bottleneck enzymes such as O-MTs and P450 hydroxylases can be omitted and replaced by readily available synthetic DHIQ precursors. In conjunction with NMT and/or additional other modification enzymes, this IRED-based approach has been shown to be very powerful in the biosynthesis of plant THIQAs.The major obstacle for application of this approach is the generally poor tolerance of IREDs to bulky substituents at C1 of DHIQs. IR45 is a steric-hindrance tolerating IRED identified in our previous study.10a Due to their more rigid conformations, 1-phenyl 6,7-dimethoxyl-DHIQ substrates are more sterically hindered than the 1-benzyl 6,7-dimethoxyl-DHIQs, and wild-type IR45 showed no activity on the most sterically hindered substrates 2a and 3a. To address this issue, we engineered IR45 to significantly expand its substrate scope. Instead of engineering the site of direct steric repulsion, we chose to alter the other side of the binding pocket. This strategy was very effective for IR45 and may prove to be useful for engineering other enzymes where certain protein interactions nearby the substrate are critical for maintaining catalytic activity or conformation.
The resultant mutant W191F–F190 efficiently and stereo-selectively (ee > 99%) converted the very sterically hindered substrates (2a and 3a) and the other three substrates (1a, 4a and 5a) into THIQs. The other mutant F190M–W191F showed significantly improved activity toward 1a, 4a and 5a. As these substrates are more sterically hindered than most other natural PIAs and BIAs,1,2 these IR45 mutants are potentially useful for the synthesis of many other THIQAs. Practical application of these enzymes can be backed up by the recently designed strategy for scaling up IRED transformation.19 In addition to the IRED, we also found that the CNMT has a sufficiently relaxed substrate specificity to accept very bulky THIQ substrates. Therefore, the broad substrate specificity of both the IR45 mutants and CNMT has provided a solid basis for the application of this IRED-based biosynthetic strategy.
Although most natural THIQAs contain S-configuration at the C1 position, it is still interesting to generate R-type THIQAs using this strategy. However, our attempt to produce R-1b–5b using the most broad-selective R-type imine reductase IR2 (ref. 10a) was not successful. Like IR45, IR2 shows S-specificity to convert 1a, 4a and 5a into the corresponding S-THIQs (2a and 3a are inactive, data not shown). The reverse stereo-specificity might be due to the occurrence of the 6,7- and 3′,4′-substituent groups in the THIQ scaffold. Currently, protein engineering of IR2 is in progress to reverse its stereo-specificity.
Finally, to accommodate the different physical properties of PIAs and BIAs, we developed two types of biocatalytic approaches for their synthesis. BIAs can be biosynthesized (100% yield) from their DHIQ precursors using the recombinant E. coli strain expressing the artificial biosynthetic pathway. Due to the poor membrane-permeability of their DHIQ precursors, PIAs 2 and 3 were not produced by a traditional whole-cell system in sufficiently high yields. To overcome this challenge and avoid using the expensive S-adenosylmethionine (SAM) cofactor in the N-methylation step, we conceived a novel biocatalytic approach combining the enzymatic transformation of IRED–GDH and the whole-cell transformation of CNMT in one pot. This chimeric system is as efficient as the cell-based system (produces PIAs in 100% yield) and will be useful for biosynthesis of other products with similar problems.
Conclusions
We have developed an IRED-based strategy to successfully biosynthesize five pharmaceutically valuable PIAs and BIAs. Notably, the three PIAs' natural biosynthetic pathways have not been elucidated. Through protein engineering, we significantly expanded the substrate specificity of the IRED IR45. The two resultant mutants F190L–W191F and F190M–W191F can efficiently convert bulky 1-aryl-6,7-dimethoxy-DHIQ substrates into (S)-THIQs. These two IREDs, highly tolerant of steric hindrance, will also be useful for the synthesis of many natural and synthetic THIQs. The N-methylation enzyme (CNMT) was also able to convert highly sterically hindered substrates. We also developed an efficient and cost-effective enzyme/whole-cell chimeric biosynthetic system to overcome the barrier for biotransformation of membrane-impermeable substrates. Our work demonstrates that this IRED-based biosynthetic approach is efficient (100% yield) and flexible for the production of plant THIQAs. By the addition of other modification enzymes, e.g. P450 enzymes, this minimal biosynthetic pathway can be further extended to the biosynthesis of other complex THIQAs.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful to Prof. Wenbo Liu for the help in chiral analysis. This work was supported by the National Key R&D Program of China (2018YFC1706200) and NSFC grant (31800048, 31570057, and 31770063).Notes and references
- (a) W. Liu, S. Liu, R. Jin, H. Guo and J. Zhao, Org. Chem. Front., 2015, 2, 288–299 RSC; (b) M. Chrzanowska, A. Grajewska and M. D. Rozwadowska, Chem. Rev., 2016, 116, 12369–12465 CrossRef CAS PubMed.
- (a) M. Perez, Z. Wu, M. Scalone, T. Ayad and V. Ratovelomanana-Vidal, Eur. J. Org. Chem., 2015, 6503–6514 CrossRef CAS; (b) I. P. Singh and P. Shah, Expert Opin. Ther. Pat., 2017, 27, 17–36 CrossRef CAS PubMed.
- L. Narcross, E. Fossati, L. Bourgeois, J. E. Dueber and V. J. J. Martin, Trends Biotechnol., 2016, 34, 228–241 CrossRef CAS PubMed.
- (a) R. Stadler, T. M. Kutchan and M. H. Zenk, Phytochemistry, 1989, 28, 1083–1086 CrossRef CAS; (b) W. De-Eknamkul, N. Suttipanta and T. M. Kutchan, Phytochemistry, 2000, 55, 177–181 CrossRef CAS PubMed.
- (a) N. Samanani, D. K. Liscombe and P. J. Facchini, Plant J., 2004, 40, 302–313 CrossRef CAS; (b) H. Minami, E. Dubouzet, K. Iwasa and F. Sato, J. Biol. Chem., 2007, 282, 6274–6282 CrossRef CAS PubMed.
- (a) H. Minami, J.-S. Kim, N. Ikezawa, T. Takemura, T. Katayama, H. Kumagai and F. Sato, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7393–7398 CrossRef CAS PubMed; (b) K. M. Hawkins and C. D. Smolke, Nat. Chem. Biol., 2008, 4, 564 CrossRef CAS PubMed; (c) A. Nakagawa, H. Minami, J.-S. Kim, T. Koyanagi, T. Katayama, F. Sato and H. Kumagai, Nat. Commun., 2011, 2, 326 CrossRef PubMed; (d) K. Thodey, S. Galanie and C. D. Smolke, Nat. Chem. Biol., 2014, 10, 837 CrossRef CAS PubMed; (e) E. Fossati, A. Ekins, L. Narcross, Y. Zhu, J.-P. Falgueyret, G. A. W. Beaudoin, P. J. Facchini and V. J. J. Martin, Nat. Commun., 2014, 5, 3283 CrossRef; (f) S. Galanie, K. Thodey, I. J. Trenchard, M. Filsinger Interrante and C. D. Smolke, Science, 2015, 349, 1095–1100 CrossRef CAS; (g) W. C. DeLoache, Z. N. Russ, L. Narcross, A. M. Gonzales, V. J. J. Martin and J. E. Dueber, Nat. Chem. Biol., 2015, 11, 465 CrossRef CAS; (h) E. Fossati, L. Narcross, A. Ekins, J.-P. Falgueyret and V. J. J. Martin, PLoS One, 2015, 10, e0124459 CrossRef; (i) Y. Li and C. D. Smolke, Nat. Commun., 2016, 7, 12137 CrossRef CAS; (j) A. Nakagawa, E. Matsumura, T. Koyanagi, T. Katayama, N. Kawano, K. Yoshimatsu, K. Yamamoto, H. Kumagai, F. Sato and H. Minami, Nat. Commun., 2016, 7, 10390 CrossRef CAS; (k) L. Narcross, L. Bourgeois, E. Fossati, E. Burton and V. J. J. Martin, ACS Synth. Biol., 2016, 5, 1505–1518 CrossRef CAS; (l) Y. Li, S. Li, K. Thodey, I. Trenchard, A. Cravens and C. D. Smolke, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E3922–E3931 CrossRef CAS; (m) C. J. Vavricka, T. Yoshida, Y. Kuriya, S. Takahashi, T. Ogawa, F. Ono, K. Agari, H. Kiyota, J. Li, J. Ishii, K. Tsuge, H. Minami, M. Araki, T. Hasunuma and A. Kondo, Nat. Commun., 2019, 10, 2015 CrossRef.
- S. Li, Y. Li and C. D. Smolke, Nat. Chem., 2018, 10, 395–404 CrossRef CAS.
- (a) F. Yesilirmak and Z. Sayers, Int. J. Plant Genomics, 2009, 2009, 296482 CrossRef; (b) I. J. Trenchard, M. S. Siddiqui, K. Thodey and C. D. Smolke, Metab. Eng., 2015, 31, 74–83 CrossRef CAS PubMed.
- (a) B. M. Ruff, S. Bräse and S. E. O'Connor, Tetrahedron Lett., 2012, 53, 1071–1074 CrossRef CAS; (b) T. Pesnot, M. C. Gershater, J. M. Ward and H. C. Hailes, Adv. Synth. Catal., 2012, 354, 2997–3008 CrossRef CAS; (c) M. Nishihachijo, Y. Hirai, S. Kawano, A. Nishiyama, H. Minami, T. Katayama, Y. Yasohara, F. Sato and H. Kumagai, Biosci., Biotechnol., Biochem., 2014, 78, 701–707 CrossRef CAS; (d) B. R. Lichman, M. C. Gershater, E. D. Lamming, T. Pesnot, A. Sula, N. H. Keep, H. C. Hailes and J. M. Ward, FEBS J., 2015, 282, 1137–1151 CrossRef CAS; (e) A. Bonamore, L. Calisti, A. Calcaterra, O. H. Ismail, M. Gargano, I. D'Acquarica, B. Botta, A. Boffi and A. Macone, ChemistrySelect, 2016, 1, 1525–1528 CrossRef CAS; (f) B. R. Lichman, J. Zhao, H. C. Hailes and J. M. Ward, Nat. Commun., 2017, 8, 14883 CrossRef CAS; (g) V. Erdmann, B. R. Lichman, J. Zhao, R. C. Simon, W. Kroutil, J. M. Ward, H. C. Hailes and D. Rother, Angew. Chem., Int. Ed., 2017, 56, 12503–12507 CrossRef CAS; (h) J. Zhao, B. R. Lichman, J. M. Ward and H. C. Hailes, Chem. Commun., 2018, 54, 1323–1326 RSC; (i) L. Y. P. Luk, S. Bunn, D. K. Liscombe, P. J. Facchini and M. E. Tanner, Biochemistry, 2007, 46, 10153–10161 CrossRef CAS; (j) A. Ilari, S. Franceschini, A. Bonamore, F. Arenghi, B. Botta, A. Macone, A. Pasquo, L. Bellucci and A. Boffi, J. Biol. Chem., 2009, 284, 897–904 CrossRef CAS; (k) Y. Wang, N. Tappertzhofen, D. Mendez-Sanchez, M. Bawn, B. Lyu, J. M. Ward and H. C. Hailes, Angew. Chem., Int. Ed., 2019, 58, 10120–10125 CrossRef CAS.
- (a) J. Zhu, H. Tan, L. Yang, Z. Dai, L. Zhu, H. Ma, Z. Deng, Z. Tian and X. Qu, ACS Catal., 2017, 7, 7003–7007 CrossRef CAS; (b) H. Li, P. Tian, J.-H. Xu and G.-W. Zheng, Org. Lett., 2017, 19, 3151–3154 CrossRef CAS.
- (a) A. Brossi and S. Teitel, Helv. Chim. Acta, 1971, 54, 1564–1571 CrossRef CAS; (b) K. Leander, B. Lüning and E. Ruusa, Acta Chem. Scand., 1969, 23, 244–248 CrossRef CAS.
- D. L. Minor, S. D. Wyrick, P. S. Charifson, V. J. Watts, D. E. Nichols and R. B. Mailman, J. Med. Chem., 1994, 37, 4317–4328 CrossRef CAS.
- M. Rodríguez-Mata, A. Frank, E. Wells, F. Leipold, N. J. Turner, S. Hart, J. P. Turkenburg and G. Grogan, ChemBioChem, 2013, 14, 1372–1379 CrossRef.
- D. Mujahidin and S. Doye, Eur. J. Org. Chem., 2005, 2005, 2689–2693 CrossRef.
- E. Anakabe, L. Carrillo, D. Badía, J. L. Vicario and M. Villegas, Synthesis, 2004, 2004, 1093–1101 Search PubMed.
- L. Chang, J. M. Hagel and P. J. Facchini, Plant Physiol., 2015, 169, 1127–1140 CrossRef.
- (a) K.-B. Choi, T. Morishige, N. Shitan, K. Yazaki and F. Sato, J. Biol. Chem., 2002, 277, 830–835 CrossRef CAS PubMed; (b) K.-B. Choi, T. Morishige and F. Sato, Phytochemistry, 2001, 56, 649–655 CrossRef CAS.
- M. R. Bennett, M. L. Thompson, S. A. Shepherd, M. S. Dunstan, A. J. Herbert, D. R. M. Smith, V. A. Cronin, B. R. K. Menon, C. Levy and J. Micklefield, Angew. Chem., Int. Ed., 2018, 57, 10600–10604 CrossRef CAS.
- A. Bornadel, S. Bisagni, A. Pushpanath, S. L. Montgomery, N. J. Turner and B. Dominguez, Org. Process Res. Dev., 2019, 236, 1262–1268 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03773j |
| This journal is © The Royal Society of Chemistry 2020 |