
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAutomatic retrosynthetic route planning using template-free models†
Kangjie
Lin‡
 a,
Youjun
Xu‡
a,
Jianfeng
Pei
*b and
Luhua
Lai
*ab
a,
Youjun
Xu‡
a,
Jianfeng
Pei
*b and
Luhua
Lai
*ab
aBNLMS, Peking-Tsinghua Center for Life Sciences at the College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, PR China. E-mail: lhlai@pku.edu.cn
bCenter for Quantitative Biology, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing, 100871, PR China. E-mail: jfpei@pku.edu.cn
First published on 3rd March 2020
Abstract
Retrosynthetic route planning can be considered a rule-based reasoning procedure. The possibilities for each transformation are generated based on collected reaction rules, and then potential reaction routes are recommended by various optimization algorithms. Although there has been much progress in computer-assisted retrosynthetic route planning and reaction prediction, fully data-driven automatic retrosynthetic route planning remains challenging. Here we present a template-free approach that is independent of reaction templates, rules, or atom mapping, to implement automatic retrosynthetic route planning. We treated each reaction prediction task as a data-driven sequence-to-sequence problem using the multi-head attention-based Transformer architecture, which has demonstrated power in machine translation tasks. Using reactions from the United States patent literature, our end-to-end models naturally incorporate the global chemical environments of molecules and achieve remarkable performance in top-1 predictive accuracy (63.0%, with the reaction class provided) and top-1 molecular validity (99.6%) in one-step retrosynthetic tasks. Inspired by the success rate of the one-step reaction prediction, we further carried out iterative, multi-step retrosynthetic route planning for four case products, which was successful. We then constructed an automatic data-driven end-to-end retrosynthetic route planning system (AutoSynRoute) using Monte Carlo tree search with a heuristic scoring function. AutoSynRoute successfully reproduced published synthesis routes for the four case products. The end-to-end model for reaction task prediction can be easily extended to larger or customer-requested reaction databases. Our study presents an important step in realizing automatic retrosynthetic route planning.
Introduction
Organic synthesis has a history spanning over 190 years since the synthesis of urea by Friedrich Wöhler in 1828, but remains a rate-limiting step for the discovery of novel medicines and materials.1 One of the critical steps for efficient and environmentally friendly synthesis of valuable molecules lies in well-designed and feasible retrosynthetic routes. Retrosynthetic analysis, first used by Robert Robinson in tropinone synthesis2 and then formalized by E. J. Corey,3 is a fundamental technique that organic chemists use to design target molecules. However, the synthesis route of a molecule is usually diverse, especially for complex compounds like natural products. Historically, synthesis route planning has largely relied on the knowledge of experienced chemists.Since the 1960s, computer-aided retrosynthetic analysis tools have attracted much attention, with the earliest retrosynthesis program likely being the early Logic and Heuristics Applied to Synthetic Analysis (LHASA) work of E. J. Corey.4 Computer-aided synthesis planning has been well-reviewed over the past years.5–11 According to a recent review,11 computer-aided retrosynthetic route planning strategies can be clustered into two main categories: template-based and template-free methods. Template-based methods, including the LHASA software,4,12 have been applied since the philosophy of retrosynthetic analysis was put forward by E. J. Corey. These methods can also be categorized as using either a manual encoding approach or an automated extraction approach. Synthia (formerly Chematica), one of the most well-known, expert-encoded, template-based retrosynthetic analysis tools, is a commercial program developed by Grzybowski and co-workers.9,13–18 This tool uses a manually collected knowledge database containing about 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hand-encoded reaction transformation rules.18 Based on human knowledge of organic synthesis and the encoding of organic rules over a period of more than 15 years, Synthia has been validated experimentally as an efficient toolkit for complex products recently.16 However, it would not be practical to manually collect all the knowledge of organic synthesis considering the exponential growth rate of the number of published reactions.19 Another straightforward strategy for a template-based method, the ReactionPredictor from Baldi's group,20–22 is based on mechanistic views. This method considers the reactions between reactants as electron sinks and sources, and ranks the interactions using approximate molecular orbitals. Although these approaches are logical and interpretable for chemists, the manual encoding of mechanistic rules cannot be avoided and the mechanisms outside the knowledge database cannot be predicted.
000 hand-encoded reaction transformation rules.18 Based on human knowledge of organic synthesis and the encoding of organic rules over a period of more than 15 years, Synthia has been validated experimentally as an efficient toolkit for complex products recently.16 However, it would not be practical to manually collect all the knowledge of organic synthesis considering the exponential growth rate of the number of published reactions.19 Another straightforward strategy for a template-based method, the ReactionPredictor from Baldi's group,20–22 is based on mechanistic views. This method considers the reactions between reactants as electron sinks and sources, and ranks the interactions using approximate molecular orbitals. Although these approaches are logical and interpretable for chemists, the manual encoding of mechanistic rules cannot be avoided and the mechanisms outside the knowledge database cannot be predicted.
In addition to manual rules, automated reaction templates have been extracted by several groups. Based on the algorithms described by Law et al.23 and Bogevig et al.,24 Segler and Waller employed a neural network to score templates and perform retrosynthesis and reaction prediction.25,26 Coupling this method with Monte Carlo Tree Search (MCTS), they built a novel method for synthetic pathway planning.19 Later, Coley et al.27 used automatically extracted templates to perform retrosynthesis analysis based on molecular similarity, where they considered the similarity of both products and reactants to score and rank the templates. Recently, Baylon and coworkers28 applied a multiscale approach based on deep highway networks (DHN) and reaction rule classification for retrosynthetic reaction prediction. Their approach achieved better performance than other previous methods based on automated extraction of reaction templates. However, there are two unavoidable limitations when using automatically extracted reaction templates. First, there is an inevitable trade-off between generalization and specificity in template-based methods. Second, current template extraction algorithms consider reaction centers and their neighboring atoms, but not the global chemical environment of molecules. Moreover, mapping the atoms between products and reactants remains a nontrivial problem for all template-based methods.29
Recently, template-free models have emerged as a promising strategy to predict reactions and retrosynthetic transformations. With the pioneering work using neural networks to generate SMILES30 by Aspuru-Guzik et al.31 and Segler et al.,32 sequence-to-sequence (seq2seq) models have been gradually applied as an important template-free model in reaction outcome prediction and retrosynthetic analysis. The first template-free model in retrosynthetic analysis was proposed by Liu and co-workers,29 who used a seq2seq model to predict SMILES30 strings for reactants of a single product. They used a neural network architecture that involves bidirectional long short-term memory (LSTM) cells with an additive attention mechanism. The seq2seq model performed comparably to its template-based baseline (37.4% versus 35.4% in top-1 accuracy). However, the invalidity rate of the top-10 predicted SMILES strings was greater than 20%, which restricts its potential in further synthetic pathway planning.
In 2017, Vaswani et al.33 proposed a multi-head attention-based Transformer model in machine translation tasks that achieves state-of-the-art performance. Later, two studies used this model to predict the reaction outcome and reactants for single-step retrosynthetic analysis.34,35 Herein, we present a novel template-free strategy for automatic retrosynthetic route planning. The workflow is depicted in Fig. 1. We first trained an end-to-end model for single-step retrosynthetic task prediction using the Transformer architecture on the reactions from the United States patent literature. Our best model achieved a top-1 prediction accuracy of 63.0% using USPTO_MIT (without chiral species) with reaction classification, which exceeds that of the previous similarity-based27 or LSTM-based seq2seq models.29 This model generated fewer invalidity errors of SMILES (99.6% validity rate) compared to the previously reported seq2seq model. When applied recursively, our model successfully performed multi-step retrosynthetic route planning for four case products. It should be noted that none of the products or intermediates appears in the training dataset. We further developed an automatic data-driven end-to-end retrosynthetic route planning system (AutoSynRoute) using MCTS with a heuristic scoring function. AutoSynRoute successfully reproduced the published pathways for the four case products, demonstrating its potential for retrosynthetic pathway planning.
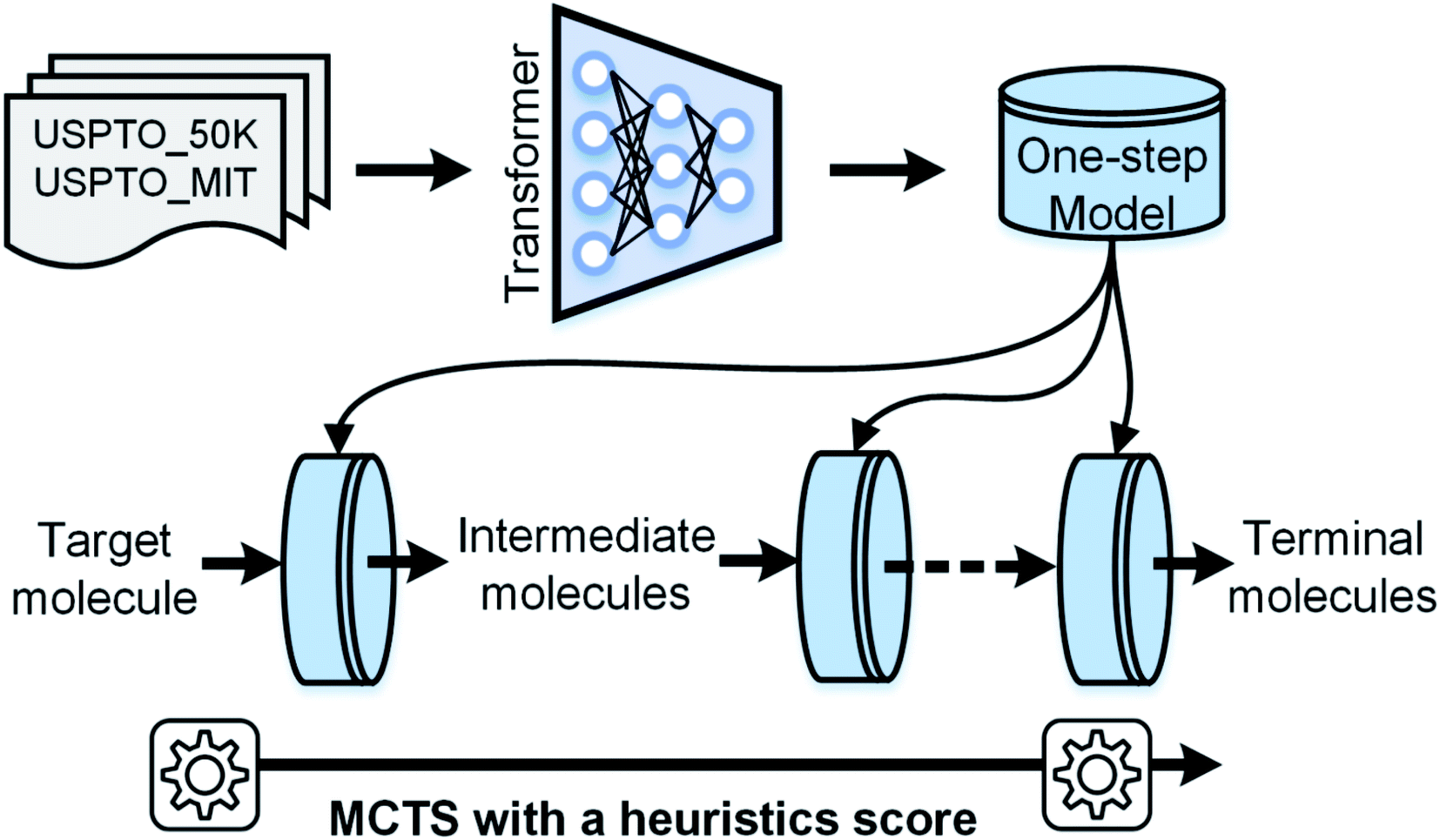 | ||
| Fig. 1 The workflow of AutoSynRoute. The Transformer architecture was used to develop our one-step model based on two datasets. A target molecule was transformed into simpler intermediate molecules using a one-step model. By repeating this operation, terminal molecules can be obtained. An automatic searching system was constructed for retrosynthesis route planning using MCTS with a heuristic score. | ||
Methods
Cadeddu et al.36 described retrosynthesis as natural language processing and termed this idea “chemical linguistics.” Similarly, retrosynthetic analysis can also be treated as a machine translation problem, where the SMILES strings are considered to be sentences and each token or character is treated as a word. In translation, each sentence has several different representations. Similarly, each product SMILES string can be “translated” to several different reactant SMILES strings, consistent with different disconnections in retrosynthetic analysis. Our seq2seq approach was based on the Transformer architecture, which represents one of the state-of-the-art techniques in neural machine translation. Unlike previous LSTM-based seq2seq models, this architecture was solely based on self-attention mechanisms, which have two main advantages: they can significantly improve the efficiency of the training time using parallelizable computation, and they allow the encoder and decoder to peek at different tokens simultaneously, thereby enabling effective computing of long-range dependent sequences and contributing to the production of high-validity SMILES strings. The Transformer architecture is depicted in Fig. 2 and the detailed description of the model can be found in the ESI.†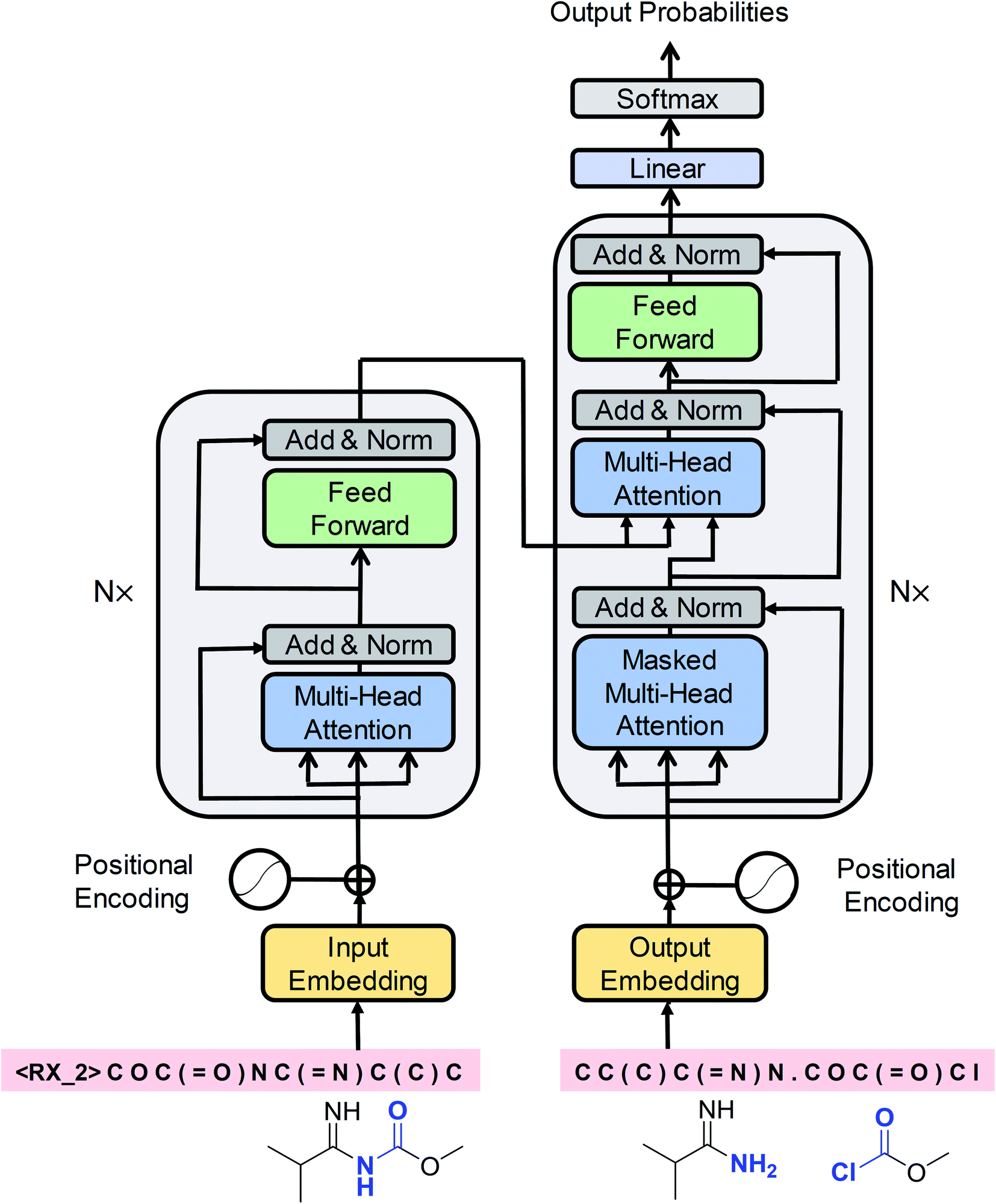 | ||
| Fig. 2 A schematic diagram of the Transformer architecture. Redrawn from ref. 33. | ||
Datasets and data preprocessing
We used two datasets to develop our single-step retrosynthetic prediction models. We first trained our model using a common benchmark dataset with ca. 50000 reactions (USPTO_50K) extracted from the United States patent literature, which was previously used by Liu et al.29 and Coley et al.27 The reaction classes in the dataset were labeled by Schneider and co-workers37 as described in Table 1. Based on the study by Liu et al.,29 we used a 90%/10% training/testing split, and the validation set was randomly sampled from training sets (10%). To develop a more powerful model, we also used a much larger dataset called USPTO_MIT38 from the USPTO,39 with preprocessed training, validation, and testing sets of 424573, 42457 (randomly sampled from training sets), and 38648 reactions, respectively.
| Reaction class | Reaction name | Fraction of USPTO_50k (%) | Fraction of USPTO_MIT (%) |
|---|---|---|---|
| 1 | Heteroatom alkylation and arylation | 30.3 | 29.9 |
| 2 | Acylation and related processes | 23.8 | 24.9 |
| 3 | C–C bond formation | 11.3 | 13.4 |
| 4 | Heterocycle formation | 1.8 | 0.7 |
| 5 | Protections | 1.3 | 0.3 |
| 6 | Deprotections | 16.5 | 14.1 |
| 7 | Reductions | 9.2 | 9.4 |
| 8 | Oxidations | 1.6 | 2.0 |
| 9 | Functional group interconversion (FGI) | 3.7 | 5.0 |
| 10 | Functional group addition (FGA) | 0.5 | 0.2 |
Inspired by Schneider et al.,37,40 the original USPTO_MIT dataset was preprocessed to extract the reactants and products of each reaction. We classified the reactions using a machine learning method based on reaction fingerprints and agent features. Moreover, we tried both token- and character-based methods to tokenize the SMILES strings as model inputs. The details of the reaction classification algorithm and results and the difference between token- and character-based preprocessing are described in the ESI.†
Monte Carlo tree search
To implement automatic retrosynthetic route search, MCTS41 is used to create a search tree where each node corresponds to a set of molecules (shown in Fig. 3). Nodes with terminal molecules (starting materials) are called terminal nodes. Starting with the root node (a target molecule), the search tree grows gradually by iterating four steps, including selection, expansion, simulation, and backpropagation. Each intermediate node has a score of upper confidence bound (UCB)42 indicating how promising it is to explore this subtree. The selection step chooses a node with the maximum UCB, which is subsequently expanded into children nodes generated by our automatic retrosynthetic pathway planning model. For the rollout in the simulation step, paths from the expanded node to terminal nodes are built by a customized approach. The softmax value of a heuristic score (see eqn (1)) of each node offers the prior probability of sampling in one rollout step. The larger the value, the more likely it will be sampled. This approach is considered helpful for better and faster searching than uniformly random rollout. A node at t-1 has a partial retrosynthesis pathway (s1, …, st-1)corresponding to the path from the root to this node. Based on the node st-1, our approach can be used to compute the distribution of the next node st. Sampling from this distribution, the pathway is elongated by one step. Our method repeats elongation until the terminal node occurs. After finishing elongation, the defined reward (the detailed description of the reward can be found in the ESI†) of the generated pathway is used to propagate backward and update the UCB scores of traversed nodes during the backpropagation process. Please see ref. 43 for details about MCTS.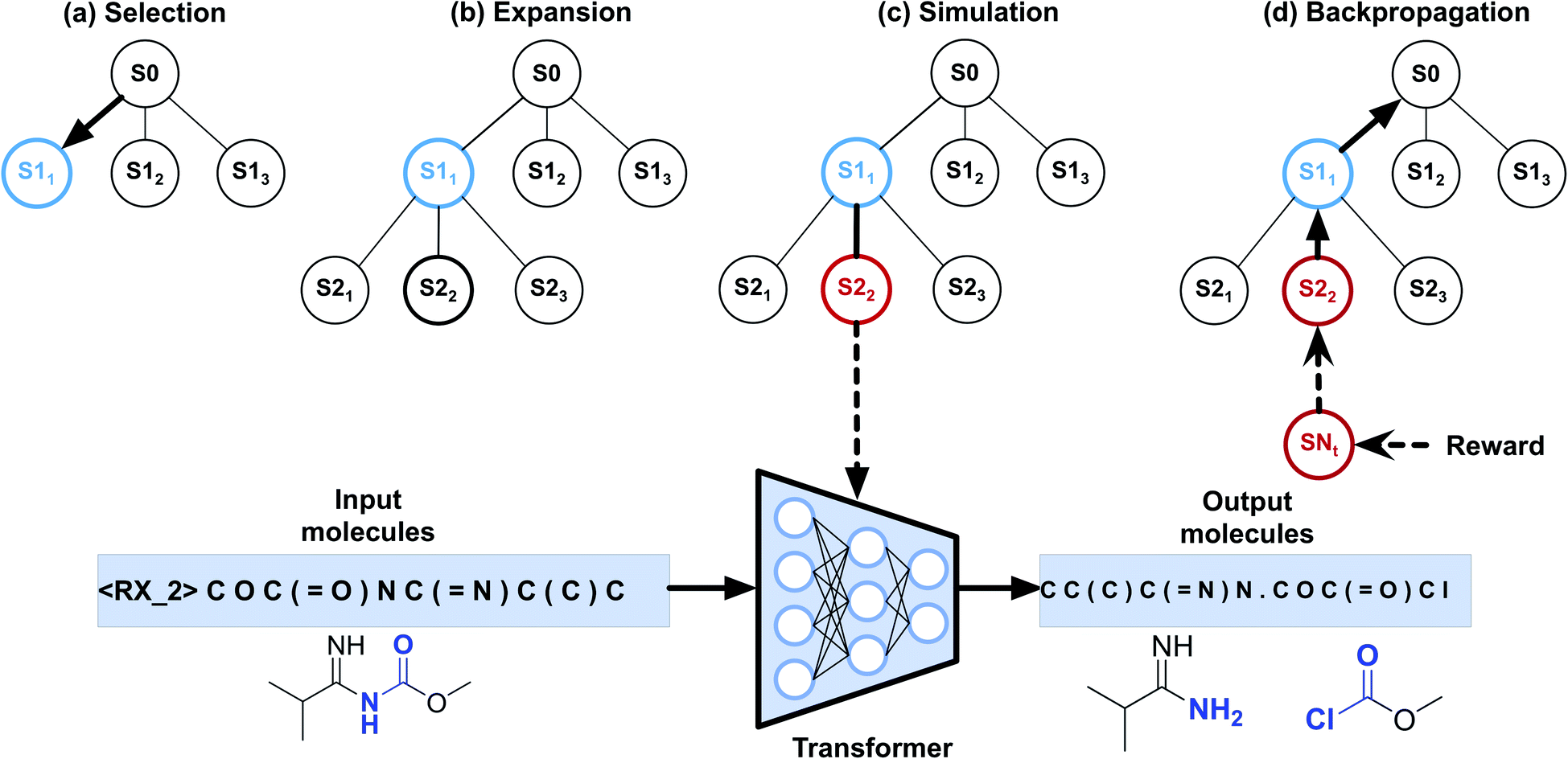 | ||
| Fig. 3 Monte Carlo tree search for retrosynthetic pathway search. (a) Selection step. The search tree is traversed from the root to a leaf by choosing the child with the largest UCB score. (b) Expansion step. Children nodes are created by sampling from the Transformer model. (c) Simulation step. Paths to terminal nodes are created by the rollout procedure using the model with the distribution of a heuristic score function. (d) Backpropagation step. Rewards of the terminal node are computed for updating UCB scores of the upstream nodes. | ||
The source code is available online at http://https://github.com/PKUMDL-AI/AutoSynRoute. All program scripts were written in Python (version 3.6), and the open source RDKit (version 2018.09.02)44 was used for reaction preprocessing and SMILES validation. Our seq2seq model was built with TensorFlow (version 1.12.0),45 and the details of key hyperparameter settings of our models are available in the ESI.†
Results
Single-step evaluation
As summarized in Table 2, our Transformer-based model achieved the best top-1 accuracies of 54.6% and 63.0% for USPTO_50K and USPTO_MIT datasets with additional reaction classes, respectively. With prior reaction class information, the top-1 prediction accuracy of our model is much better than that of the LSTM-based seq2seq model proposed by Liu et al.29 and also higher than that of the similarity-based model by Coley et al.27 As shown in Table 3, when the reaction classes are not provided, the top-1 accuracy of our model is still higher than those of the other methods. The results of Liu et al.'s and Segler et al.'s models are implemented by us using Liu et al.‘s public code and Coley's reproduced code (https://github.com/connorcoley/retrotemp), respectively. The reproduction details of the two baseline experiments can be found in the ESI.† The template-based methods proposed by Baylon et al.28 are also competitive, but currently we cannot make a direct comparison with their approaches because their model and test data are unavailable to us.| top-n accuracy (%), n= | ||||
|---|---|---|---|---|
| Model (dataset) | 1 | 3 | 5 | 10 |
| a Key: “+class” means that reaction class information is added to the model; “+token” means that token-based preprocessing is applied; “+char” means that char-based preprocessing is applied. b The results are implemented by us using Liu et al.'s public code. | ||||
| Liu et al. template +class (USPTO_50K)29,b | 35.4 | 52.3 | 59.1 | 65.1 |
| Liu et al. LSTM +class (USPTO_50K)29,b | 37.4 | 52.4 | 57.0 | 61.7 |
| Coley et al. similarity +class (USPTO_50K)27 | 52.9 | 73.8 | 81.2 | 88.1 |
| Our Transformer +token +class (USPTO_50K) | 54.3 | 74.1 | 79.2 | 84.4 |
| Our Transformer +char +class (USPTO_50K) | 54.6 | 74.8 | 80.2 | 84.9 |
| Liu et al. LSTM +class (USPTO_MIT)29,b | 56.1 | 69.9 | 73.6 | 77.3 |
| Our Transformer +char +class (USPTO_MIT) | 63.0 | 79.2 | 83.4 | 86.8 |
| top-n accuracy (%), n= | ||||
|---|---|---|---|---|
| Model (dataset) | 1 | 3 | 5 | 10 |
| a Key: “+token” means that token-based preprocessing is applied; “+char” means that char-based preprocessing is applied. b The results are implemented by us using Liu et al.'s public code. c The results are implemented by us using Coley's reproduced code (https://github.com/connorcoley/retrotemp). | ||||
| Liu et al. LSTM (USPTO_50K)29,b | 28.3 | 42.8 | 47.3 | 52.8 |
| Liu et al. LSTM (USPTO_MIT)29,b | 46.9 | 61.6 | 66.3 | 70.8 |
| Coley et al. Similarity (USPTO_50K)27 | 37.3 | 54.7 | 63.3 | 74.1 |
| Segler–Coley-retrained (USPTO_50K)25,c | 38.7 | 56.2 | 62.2 | 69.2 |
| Segler–Coley-retrained (USPTO_MIT)25,c | 47.8 | 67.6 | 74.1 | 80.2 |
| Karpov et al. Transformer (USPTO_50K)35 | 42.7 | 63.9 | 69.8 | — |
| Our Transformer +token (USPTO_50K) | 42.0 | 64.0 | 71.3 | 77.6 |
| Our Transformer +char (USPTO_50K) | 43.1 | 64.6 | 71.8 | 78.7 |
| Our Transformer +char (USPTO_MIT) | 54.1 | 71.8 | 76.9 | 81.8 |
The ratio of invalid SMILES strings produced by our model is much lower than that of the previous LSTM-based model, which means that our model has a powerful ability to capture the grammar of SMILES representations. As shown in Table 4, the top-10 invalidity error of our model is 12.6%, which is close to the top-1 invalidity error of Liu's model. When we trained our model on the large-volume USPTO_MIT dataset, the top-1 accuracy increased to 63.0%, which shows the generality ability of our model by increasing the chemical knowledge base. Meanwhile, the error rate of SMILES strings decreases to 0.4% in the top-1 prediction.
| Invalid SMILES rate (%) | ||||
|---|---|---|---|---|
| Model (dataset) | 1 | 3 | 5 | 10 |
| a Key: “+class” means that reaction class information is added to the model; “+token” means that token-based preprocessing is applied; “+char” means that char-based preprocessing is applied. | ||||
| Liu et al. LSTM +class (USPTO_50K)29 | 12.2 | 15.3 | 18.4 | 22 |
| Our Transformer +token (USPTO_50K) | 2.2 | 3.7 | 4.8 | 7.8 |
| Our Transformer +token +class (USPTO_50K) | 2.3 | 4.9 | 7.0 | 12.1 |
| Our Transformer +char (USPTO_50K) | 2.1 | 3.5 | 4.7 | 8.3 |
| Our Transformer +char +class (USPTO_50K) | 2.4 | 4.4 | 6.4 | 12.6 |
| Our Transformer +char +class (USPTO_MIT) | 0.4 | 1.5 | 2.9 | 8.6 |
A comparison of the top-10 accuracies across all classes of our model with those of the previous studies on USPTO_50K and USPTO_MIT datasets is shown in Table S1.† The performance of our model was much better than that of the seq2seq model of Liu et al.29 across all reaction categories. However, our model performed just slightly better or comparably to the similarity-based model in reaction categories 3, 7 and 9.
As shown in Fig. 4, we used the top-5 retrosynthetic disconnections of a compound in a test set as an example to analyze the specificity and generality of our model. We chose a compound in class 1 as an example, in which the ground truth prediction ranks first and the other predicted reactions are also chemically plausible. The results show that our model is able to give reasonably diverse disconnections and the top-5 disconnections comply with the reaction class of heteroatom alkylation. Additional results regarding single-step retrosynthetic disconnections within each reaction class can be found in the ESI (Fig. S1–S10).†
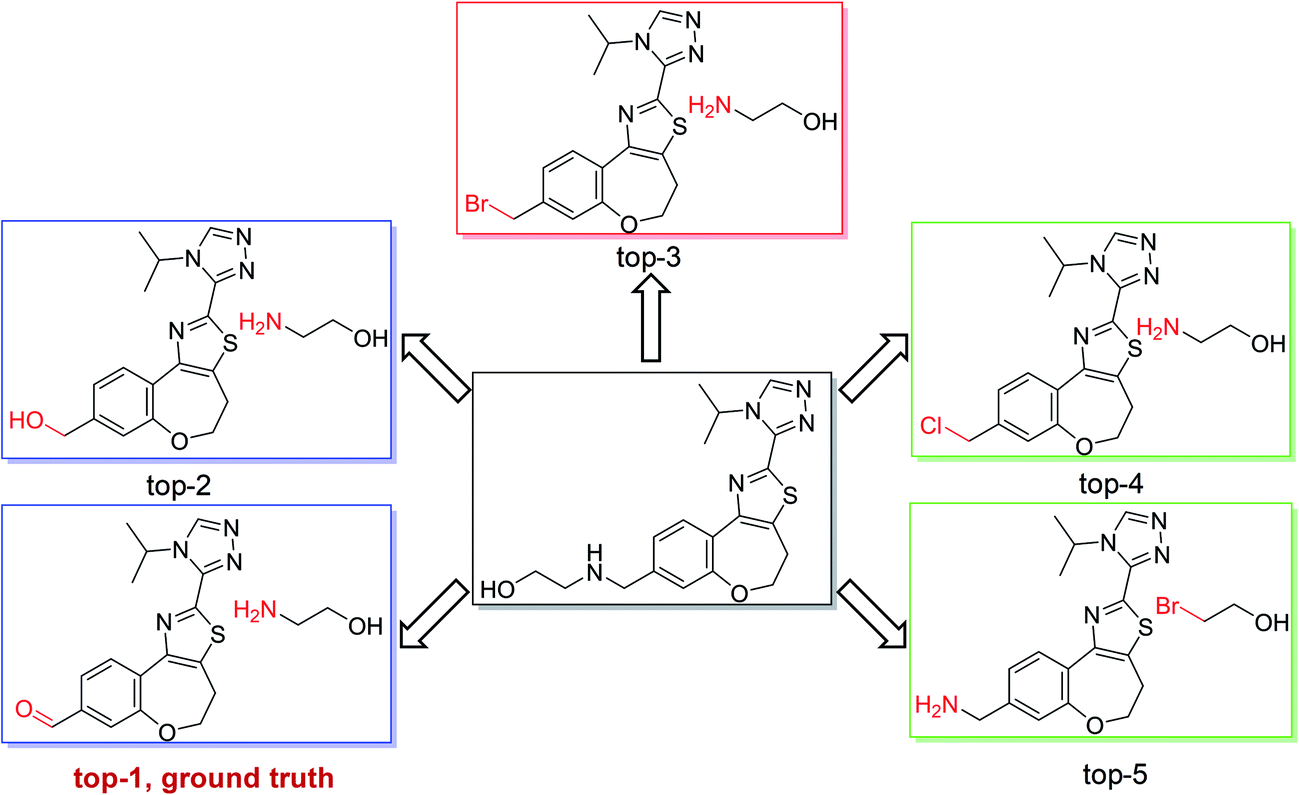 | ||
| Fig. 4 Top-5 retrosynthetic predictions of an example reaction with class 1. The model successfully proposes the recorded reactants with rank 1, corresponding to a heteroatom alkylation. Other suggestions among the top-5 predictions are also chemically reasonable. | ||
Iterative multi-step pathway generation
As the prediction accuracy of our model is quite high (even higher than that of similarity-based methods), we also examined the potential of our model in recursive generation of candidate reactants. We chose four target compounds as examples, including the antiseizure drug Rufinamide,46 a novel allosteric activator for glutathione peroxidase 4 (GPX4),47 and two representative compounds used by other retrosynthetic programs.16,19 By enumerating different reaction classes, we sought to confirm that our model could successfully reproduce the published reaction pathways of the four compounds. The input structures (products or intermediates) of the four examples do not appear in our training set of either the USPTO_50K or USPTO_MIT datasets.For the first example of retrosynthesis pathway planning, Rufinamide (shown in Fig. 5a), the reported first step is the formation of an amide bond, ranking first in reaction class 9 (functional group interconversion). The subsequent step is also found to rank top-1 in class 4 (heterocycle formation), consistent with the mechanistic view. This is followed by another functional group interconversion (FGI) step, the final step, predicted precisely as top-1 in class 9. It is worth mentioning that different reaction classes may have the same disconnections and thus result in the same reactants. For example, the third step of the aforementioned route also ranks first in class 1 (heteroatom alkylation), which is also plausible.
 | ||
| Fig. 5 Iterative multi-step pathway generation. The routes are constructed by iteratively applying single-step retrosynthetic methodology to (a) Rufinamide, (b) an antagonist of the interaction between WDR5 and MLL1, from the example of Grzybowski et al.,16 (c) an allosteric activator for GPX4, and (d) an intermediate of a drug candidate from the example of Segler et al.19 The suggested disconnections are consistent with published pathways. The number before the “.” indicates the reaction class, and the number after the “.” indicates the ranking in the top-10 prediction. | ||
As shown in Fig. 5b, the second example comes from the previous work of Grzybowski et al.,16 which was the retrosynthesis pathway planning of an antagonist of the interaction between WD repeat-containing protein 5 (WDR5) and mixed-lineage leukemia 1 (MLL1).48 Our model could recover the route suggested by the commercial program Synthia. The first step is a FGI predicted as top-2 by our model. The next step is a common amide formation. The final step is a C–C bond formation, which was also predicted by our model as top-1 with the correct reaction class.
The aforementioned two routes are predicted by our model trained on the USPTO_50K dataset. However, another two more challenging routes cannot be completely predicted due to less coverage of chemical space. Remarkably, using the USPTO_MIT dataset (without stereochemistry information), our trained model could completely reproduce the following two routes in our top-10 predictions, suggesting the importance of training on enlarging coverage of the chemical knowledge space.
The third example is the retrosynthesis pathway planning of the GPX4 activator compound, as depicted in Fig. 5c. The published first step ranks first in class 6 (deprotection). The second step could be regarded as acylation and related processes and it is predicted correctly as top-8 by our model. The ground truth of the third step ranks top-1 in class 9, followed by a deprotection step in top-3, and a final acylation and related processes in top-1.
The fourth example, described in Fig. 5d, is the retrosynthesis pathway planning of an intermediate of a drug candidate from the example of Segler and co-workers.19 The first, second, and third steps can be easily reproduced by our model as top-1 or top-2 with the right class. The fourth step is a common functional group addition, followed by an uncommon reduction of a carbonyl group. After the final step of heteroatom alkylation, our model was shown to reproduce the steps predicted by the former template-based method.
Automatic retrosynthetic pathway planning
As shown above, when considering the top 10 prediction of each of the 10 reaction classes, 100 candidate reactants for a target will be predicted in one step. A recursive application in a four-step pathway will produce 100000000 candidate pathways assuming all of the output SMILES strings are valid. To make our model applicable for retrosynthetic pathway planning, we needed to achieve efficient automatic pathway searching and ranking. We used a MCTS algorithm combined with a heuristic scoring function to achieve this purpose. Our heuristic scoring function was inspired by Synthia's Chemical Scoring Function (CSF).9 We considered the Scoremodel produced by our model (representing the decoding log probability from the beam search), the changed SMILES length from the target to the reactants, and the changed number of rings from the target to the reactants. To scale the heuristic scoring function in a comparable range, we presented the scoring function in the formula| Scorestep = a × exp(Scoremodel) − (b × RINGSchanged + SMILESchanged) | (1) |
To make the model applicable, we also needed to define the terminal nodes or reactants, which means the commercially available molecules. We used a dataset containing 84807 building blocks from a chemical supplier (Sigma Aldrich), obtained from the ZINC15 database (http://zinc15.docking.org/) and 17182 molecules from the USPTO_MIT database. The data were used as reactants at least five times as terminal nodes (a building block database of 93563 molecules after removing redundant ones) for searching. Users can also use any specific building block database as a terminal reactant database.
Using our automatic retrosynthetic pathway planning strategy, most of the aforementioned steps in the four examples can be found and ranked in top-10 except for the fourth step of example 3 (ranks top-12) and fifth step of example 4 (ranks top-25). The overall pathway ranking results of the four examples can be found in the ESI (Fig. S11–S14†). Though our heuristic scoring function is simple, these results are impressive. We demonstrated the potential ability of our template-free model to plan the automatic retrosynthetic pathway in a new way other than by using current template-based methods.
Discussion
Advantages and disadvantages of our seq2seq models
As described previously, our models are template-free and free of atom mapping. In addition, our models can learn the global chemical environments of molecules naturally, unlike other template-based methods. However, our seq2seq models still have some problems related to dataset and SMILES representations. In addition to having less coverage of the chemical reaction space, the USPTO dataset does not contain reaction yield information for reactions, which is useful to discriminate whether the predicted pathways are efficient. Because our models were trained on USPTO datasets, their prediction accuracies are currently limited by these problems. A commonly known challenge of using the SMILES or reaction SMARTS format is the poor performance when dealing with stereochemistry and tautomers. Like other template-based methods, our models still have difficulty tackling reactions containing chirality. In fact, our models are able to handle reactants or products with simple chirality, as long as we include reactions containing chirality. However, language models operating on SMILES strings may have trouble learning to meaningfully interpret stereochemistry. Furthermore, because our models do not contain any information about reaction conditions, they are currently unable to deal with asymmetric synthesis, most of which relies on asymmetric catalysts. Meanwhile, tautomers, though chemically equivalent in different molecular structures, are regarded as different inputs and outputs in our model because current SMILES grammar is sequence sensitive. This problem is also common in template-based models, as described by Segler et al.19 Embedding stereochemistry and tautomerization into SMILES representation is a future direction to be explored.Evaluation of different pathways
Retrosynthetic programs can predict thousands of different pathways. However, picking a suitable pathway from the predictions is not easy. Medicinal chemists may want a pathway expanding structure–activity relationship exploration. Organic chemists, especially those working on total synthesis of natural products, may have preferences for the more efficient and greener pathways. The choices of process chemists may be influenced by the cost of starting materials and avoidance of toxic and dangerous molecules. It is difficult to find a pathway that fulfills all these requirements. The heuristic metric proposed by Synthia seems to be a reasonable strategy. This metric has two scoring functions: the CSF and Reaction Scoring Function. Another potential strategy is to use the SCScore metric proposed by Coley and co-workers.49 In general, a comprehensive scoring function will be related to the cost of building blocks, the yield of each step, the avoidance of toxic compounds and functional group incompatibility, the length of the pathway, etc. The design of a perfect pathway scoring function is still an unsolved problem in the community.Evaluation of different models
Evaluation of a retrosynthetic analysis approach is also difficult due to the lack of benchmark metrics. The strategy applied by Segler et al.19 is a reasonable one. They invited professional organic chemists to vote on the predicted and ground truth pathways. If the chemists do not show preference for the ground truth pathway, it means that the predicted one is also reasonable. However, this assessment is difficult to standardize. Certainly, validation in a wet lab is the most reliable way to validate a model. As most chemists are interested in the synthesis of novel complex compounds or finding efficient alternative pathways for valuable molecules, validation of these kinds of compounds with wet experiments should be considered. For example, the cooperation between MilliporeSigma and Grzybowski et al.16 resulted in the efficient syntheses of eight diverse and medicinally relevant targets, demonstrating the reliability of Synthia to the chemistry community.Conclusions
We developed an automatic data-driven retrosynthetic route planning system (AutoSynRoute), which includes retrosynthesis task prediction using a Transformer-based seq2seq model and MCTS with heuristic scoring for route planning. AutoSynRoute can be applied step-by-step and iteratively with user inputs. To demonstrate its application, we predicted the top-10 disconnections for each of ten reaction classes and reproduced the published retrosynthetic pathways for four examples. To further demonstrate the power of AutoSynRoute, we successfully used it to perform automatic retrosynthetic route planning for the above four examples. Unlike other template-based methods, which either rely on experts' laborious work or simple, contextless rule-based systems, our approach is fully end-to-end and naturally incorporates the global molecular context of the reaction species. We demonstrated that a template-free approach can be used to perform automatic retrosynthetic route planning and reproduce the published synthesis routes of valuable compounds. As mentioned by Coley et al., a complete retrosynthetic program should be made up of five components:11 a library containing the disconnection rules, a recursive application engine that generates candidate reactants for target compounds, a building block database containing available compounds to act as terminal nodes, a strategy to guide the retrosynthetic search, and a scoring function for the single-step or pathway. Our approach includes all of these components as described herein.Our approach can be further developed with larger and more diverse chemical knowledge bases for training. Currently, the information regarding reaction conditions like catalysts, solvents, and reagents is missing because of the database used. These conditions can be introduced in the future by using more comprehensive datasets, like the Reaxys database or in-house data. Future work will also tackle problems like SMILES′ poor representations of stereochemistry and tautomerization. Finally, we envision that automatic retrosynthetic route planning will play more important roles in real-world automated synthesis of molecules50 and in de novo molecular design.51
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the National Science and Technology Major Project “Key New Drug Creation and Manufacturing Program”, China (2018ZX09711002), the National Natural Science Foundation of China (21673010 and 21633001), and the Ministry of Science and Technology of China (2016YFA0502303). Computational analysis was performed on the High Performance Computing Platform of the Peking-Tsinghua Center for Life Sciences and the High-performance Computing Platform of Peking University. We thank Dr Weilin Zhang and Wenhao Gao for helpful discussions.References
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS
.
- R. Robinson, J. Chem. Soc., Trans., 1917, 111, 762–768 RSC
- E. J. Corey, Angew. Chem., Int. Ed. Engl., 1991, 30, 455–465 CrossRef
- E. J. Corey and W. T. Wipke, Science, 1969, 166, 178 CrossRef CAS
- M. A. Ott and J. H. Noordik, Recl. Trav. Chim. Pays-Bas, 1992, 111, 239–246 CrossRef CAS
- M. H. Todd, Chem. Soc. Rev., 2005, 34, 247–266 RSC
- A. Cook, A. P. Johnson, J. Law, M. Mirzazadeh, O. Ravitz and A. Simon, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 79–107 CAS
- W. A. Warr, Mol. Inf., 2014, 33, 469–476 CrossRef CAS PubMed
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef PubMed
- F. Feng, L. Lai and J. Pei, Front. Chem., 2018, 6, 199 CrossRef PubMed
- C. W. Coley, W. H. Green and K. F. Jensen, Acc. Chem. Res., 2018, 51, 1281–1289 CrossRef CAS PubMed
- E. J. Corey, A. K. Long and S. D. Rubenstein, Science, 1985, 228, 408 CrossRef CAS PubMed
- K. J. M. Bishop, R. Klajn and B. A. Grzybowski, Angew. Chem., 2006, 118, 5474–5480 CrossRef
- B. A. Grzybowski, K. J. M. Bishop, B. Kowalczyk and C. E. Wilmer, Nat. Chem., 2009, 1, 31 CrossRef CAS PubMed
- M. Kowalik, C. M. Gothard, A. M. Drews, N. A. Gothard, A. Weckiewicz, P. E. Fuller, B. A. Grzybowski and K. J. M. Bishop, Angew. Chem., Int. Ed., 2012, 51, 7928–7932 CrossRef CAS PubMed
- T. Klucznik, B. Mikulak-Klucznik, M. P. McCormack, H. Lima, S. Szymkuć, M. Bhowmick, K. Molga, Y. Zhou, L. Rickershauser, E. P. Gajewska, A. Toutchkine, P. Dittwald, M. P. Startek, G. J. Kirkovits, R. Roszak, A. Adamski, B. Sieredzińska, M. Mrksich, S. L. J. Trice and B. A. Grzybowski, Chem, 2018, 4, 522–532 CAS
- T. Badowski, K. Molga and B. A. Grzybowski, Chem. Sci., 2019, 10, 4640–4651 RSC
- K. Molga, P. Dittwald and B. A. Grzybowski, Chem, 2019, 5, 460–473 CAS
- M. H. S. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604 CrossRef CAS PubMed
- M. A. Kayala, C.-A. Azencott, J. H. Chen and P. Baldi, J. Chem. Inf. Model., 2011, 51, 2209–2222 CrossRef CAS PubMed
- M. A. Kayala and P. Baldi, J. Chem. Inf. Model., 2012, 52, 2526–2540 CrossRef CAS PubMed
- D. Fooshee, A. Mood, E. Gutman, M. Tavakoli, G. Urban, F. Liu, N. Huynh, D. Van Vranken and P. Baldi, Mol. Syst. Des. Eng., 2018, 3, 442–452 RSC
- J. Law, Z. Zsoldos, A. Simon, D. Reid, Y. Liu, S. Y. Khew, A. P. Johnson, S. Major, R. A. Wade and H. Y. Ando, J. Chem. Inf. Model., 2009, 49, 593–602 CrossRef CAS PubMed
- A. Bøgevig, H.-J. Federsel, F. Huerta, M. G. Hutchings, H. Kraut, T. Langer, P. Löw, C. Oppawsky, T. Rein and H. Saller, Org. Process Res. Dev., 2015, 19, 357–368 CrossRef
- M. H. S. Segler and M. P. Waller, Chem.–Eur. J., 2017, 23, 5966–5971 CrossRef CAS PubMed
- M. H. S. Segler and M. P. Waller, Chem.–Eur. J., 2017, 23, 6118–6128 CrossRef CAS PubMed
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2017, 3, 1237–1245 CrossRef CAS PubMed
- J. L. Baylon, N. A. Cilfone, J. R. Gulcher and T. W. Chittenden, J. Chem. Inf. Model., 2019, 59, 673–688 CrossRef CAS PubMed
- B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, ACS Cent. Sci., 2017, 3, 1103–1113 CrossRef CAS PubMed
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed
- M. H. S. Segler, T. Kogej, C. Tyrchan and M. P. Waller, ACS Cent. Sci., 2018, 4, 120–131 CrossRef CAS PubMed
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, Ł. Kaiser and I. Polosukhin, arXiv:1706.03762, 2016.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. Bekas and A. A. Lee, arXiv:1811.02633, 2018.
- K. Pavel, G. Guillaume and T. Igor, ChemRxiv, 2019, 8058464 Search PubMed
- A. Cadeddu, E. K. Wylie, J. Jurczak, M. Wampler-Doty and B. A. Grzybowski, Angew. Chem., Int. Ed., 2014, 53, 8108–8112 CrossRef CAS PubMed
- N. Schneider, N. Stiefl and G. A. Landrum, J. Chem. Inf. Model., 2016, 56, 2336–2346 CrossRef CAS PubMed
- C. W. Coley, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jensen, Chem. Sci., 2019, 10, 370–377 RSC
-
D. M. Lowe, PhD thesis, University of Cambridge, 2012 Search PubMed
- N. Schneider, D. M. Lowe, R. A. Sayle and G. A. Landrum, J. Chem. Inf. Model., 2015, 55, 39–53 CrossRef CAS PubMed
-
R. Coulom, in Computers and Games, Springer Berlin Heidelberg, 2007, pp. 72–83 Search PubMed
- C. B. Browne, E. Powley, D. Whitehouse, S. M. Lucas, P. I. Cowling, P. Rohlfshagen, S. Tavener, D. Perez, S. Samothrakis and S. Colton, IEEE Transactions on Computational Intelligence and AI in Games, 2012, 4, 1–43 Search PubMed
- T. M. Dieb, S. Ju, K. Yoshizoe, Z. Hou, J. Shiomi and K. Tsuda, Sci. Technol. Adv. Mater., 2017, 18, 498–503 CrossRef PubMed
-
G. Landrum, RDKit: Open-source cheminformatics, 2006, http://www.rdkit.org Search PubMed
- M. Abadi, A. Agarwal, P. Barham, E. Brevdo, Z. Chen, C. Citro, G. S. Corrado, A. Davis, J. Dean and M. Devin, arXiv:1603.04467, 2015.
- R. D. Padmaja and K. Chanda, Org. Process Res. Dev., 2018, 22, 457–466 CrossRef CAS
- C. Li, X. Deng, W. Zhang, X. Xie, M. Conrad, Y. Liu, J. P. F. Angeli and L. Lai, J. Med. Chem., 2019, 62, 266–275 CrossRef CAS PubMed
- M. Getlik, D. Smil, C. Zepeda-Velázquez, Y. Bolshan, G. Poda, H. Wu, A. Dong, E. Kuznetsova, R. Marcellus, G. Senisterra, L. Dombrovski, T. Hajian, T. Kiyota, M. Schapira, C. H. Arrowsmith, P. J. Brown, M. Vedadi and R. Al-awar, J. Med. Chem., 2016, 59, 2478–2496 CrossRef CAS PubMed
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, J. Chem. Inf. Model., 2018, 58, 252–261 CrossRef CAS PubMed
- A.-C. Bédard, A. Adamo, K. C. Aroh, M. G. Russell, A. A. Bedermann, J. Torosian, B. Yue, K. F. Jensen and T. F. Jamison, Science, 2018, 361, 1220 CrossRef PubMed
- Y. Xu, K. Lin, S. Wang, L. Wang, C. Cai, C. Song, L. Lai and J. Pei, Future Med. Chem., 2019, 11, 567–597 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03666k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |