
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterization of reaction enthalpy and kinetics in a microscale flow platform†
Agnieszka
Ładosz‡
 ,
Christina
Kuhnle‡
and
Klavs F.
Jensen
*
,
Christina
Kuhnle‡
and
Klavs F.
Jensen
*
Department of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: kfjensen@mit.edu
First published on 30th September 2020
Abstract
We report an isothermal flow calorimeter for characterization of reaction enthalpy and kinetics. The platform consists of a thermoelectric element and a glass–silicon microreactor to measure heat flux and an inline IR spectrometer to monitor reaction conversion. The thermally insulated assembly is calibrated with a thin film heater placed between the microreactor and the thermoelectric element. Without any reconfiguration of hardware, the setup can also be used to efficiently characterize reaction kinetics in transient flow experiments. We tested the calorimeter with hydrolysis of acetic anhydride as a model reaction. We determined the exothermic reaction enthalpy and the endothermic heat of mixing of the reagent to be −63 ± 3.0 kJ mol−1 and +8.8 ± 2.1 kJ mol−1 respectively, in good agreement with literature values and theoretical predictions. Following calorimetry studies, we investigated reaction kinetics by applying carefully controlled residence time ramps at four different temperatures, and we obtained kinetic rate constants of 0.129 min−1 up to 0.522 min−1 for temperatures between 20 °C and 56.3 °C, also fitting well with data reported in the literature.
Introduction
In this manuscript we describe a microfluidic platform for characterization of enthalpy and kinetics of chemical reactions. Especially for highly exothermic processes, accurate enthalpy and kinetics data are essential to find safe and optimal operating conditions. This is usually achieved via batch experimentation, often requiring large volumes of reactants. To address this problem, we envisioned application of microreactors to realize a continuous flow platform for calorimetric and kinetic measurements.Microfluidic devices offer excellent heat transfer due to high surface-to-volume ratio,1 enabling investigation of reactions at elevated reactant concentrations when compared to standard equipment, which in turn means that lower overall reagent volumes suffice to achieve a measurable heat output. Processes run in continuous fashion are easier to control than batch reactions, which further improves process safety, facilitates automation and enables use of otherwise hazardous reagents, especially important when investigating highly exothermic reactions.2–4 Development of in-flow analytical equipment (e.g. Mettler Toledo ReactIR with flow cells) opens up new possibilities for reaction characterization when coupled with microfluidic devices.5 Several studies have already demonstrated such advantageous coupling of analytical and microfluidic technologies, including e.g. a high-throughput oscillatory droplet reactor to probe reaction space for optimal conditions;6–8 and a self-optimising automated flow API synthesis with an at-line HPLC for sample analysis.9
Given clear advantages of flow units over their batch counterparts, first applications of microfluidic devices in reaction characterization have been reported. In calorimetry, early implementations include channels with integrated thin film thermopiles10–12 and microstructured devices coupled with commercially available calorimeters.13,14 Since then various techniques have been utilized to measure heat released in microchannels: heat flux measurement utilizing thermoelectric effect,10–12,15–22 energy balance based on temperature measurements with thermocouples23 or infrared cameras,24–26 or a combination of several techniques.27
Depending on the applied method, reaction conversion is measured either directly in the microreactor by colorimetry,20,21,24 estimated from the heat released at varied flow rates,15,17–20,26 or by analysing samples off-line.28 An interesting example of how microfluidic technology can be used to collect data otherwise difficult to measure experimentally is the spatially-resolved calorimeter developed at Fraunhofer Institute for Chemical Technology.15,17,18 By combining a microstructured device with an array of thermoelectric elements, the group created a tool to follow the progress of chemical reactions along the channel, enabling thermokinetic characterization of chemical processes.18 Similar approach was used by Reichmann et al. to evaluate the extent of mixing.20,21 Zhang et al. realized spatially resolved calorimetric measurements using an infrared camera and stainless steel tubing as a microreactor.25,26 By adjusting flow rates to obtain full or partial conversion, both reaction enthalpy as well as kinetics were measured within the same device at the fraction of time normally needed in conventional equipment.25,26 Recently, a meso-scale flow reactor with integrated temperature sensors was used to obtain a spatially-resolved temperature profile in an exothermic organolithium reaction, supporting subsequent scale-up of the process.29
Traditionally, reaction kinetics is investigated in a series of batch experiments at varied reaction times and temperatures. Direct adaptation of kinetic experiments to flow is hindered by long wait times to reach steady state in flow reactors, as a rule of thumb usually assumed equal to three to four residence times. This results in long and material-intensive experimentation, contrary of what is widely regarded as benefits of microfluidic technology. To overcome this issue, Moore & Jensen pioneered the use of transient flow experiments, in which a precisely controlled residence time or temperature ramp is combined with inline IR analysis to quickly obtain reliable kinetic data.30,31 Since then the method has been extended to simultaneous residence time and temperature ramps,32 to linear and non-linear ramps,33 and combined with non-invasive Raman measurements at different locations along the reactor,34 further maximizing experimental output. It has been also successfully applied with a more time-consuming HPLC analysis.35–37 In terms of generated data, the method can be applied to discriminate between various kinetic models,32,38 but as discussed by Waldron et al., who compared the output of the transient flow experiments with data obtained from DoE or model-based DoE campaigns, the precision of estimated kinetic parameters depends on the ramp parameters selected by the user.36,37 An alternative approach to use a step change in flow instead of a ramp was developed by Mozharov et al., who studied a Knoevenagel condensation with an inline Raman probe.39 This so-called ‘push-out’ method results in a short residence time ramp between the two steady states at low and high flow rate and is especially useful if application of a full ramp is constrained by the experimental setup. This technique was successfully applied in a commercially available meso-scale Vapourtec system, albeit with some modifications,40 and used together with a specially-constructed HPLC ‘sample parking’ interface for an extensive characterization of a photochemical cycloaddition.41
In our work, we have combined a microreactor with an inline IR unit and a thermoelectric element to develop a safe and versatile platform for reaction characterization. The unit can be operated either in calorimetry mode, where residence time is kept constant and heat signal and conversion are recorded simultaneously to evaluate reaction enthalpy, on in transient flow mode to gather data for kinetic modeling. In this manuscript, we present the details of the calorimeter setup and showcase its application on hydrolysis of acetic anhydride.
Method
Setup design
Our microcalorimeter comprises a glass–silicon microreactor and a thermoelectric element (TE), sandwiched between a heating block to maintain constant reaction temperature and a thick glass cover for insulation and visual access (see Fig. 1). Whenever there is a temperature difference between its two sides, the thermoelectric element (Tellurex) generates a voltage which is recorded continuously and recalculated into heat output using a calibration curve. A thin polyimide film heater (Omega Engineering) is placed between the reactor and the TE for the calibration of the latter.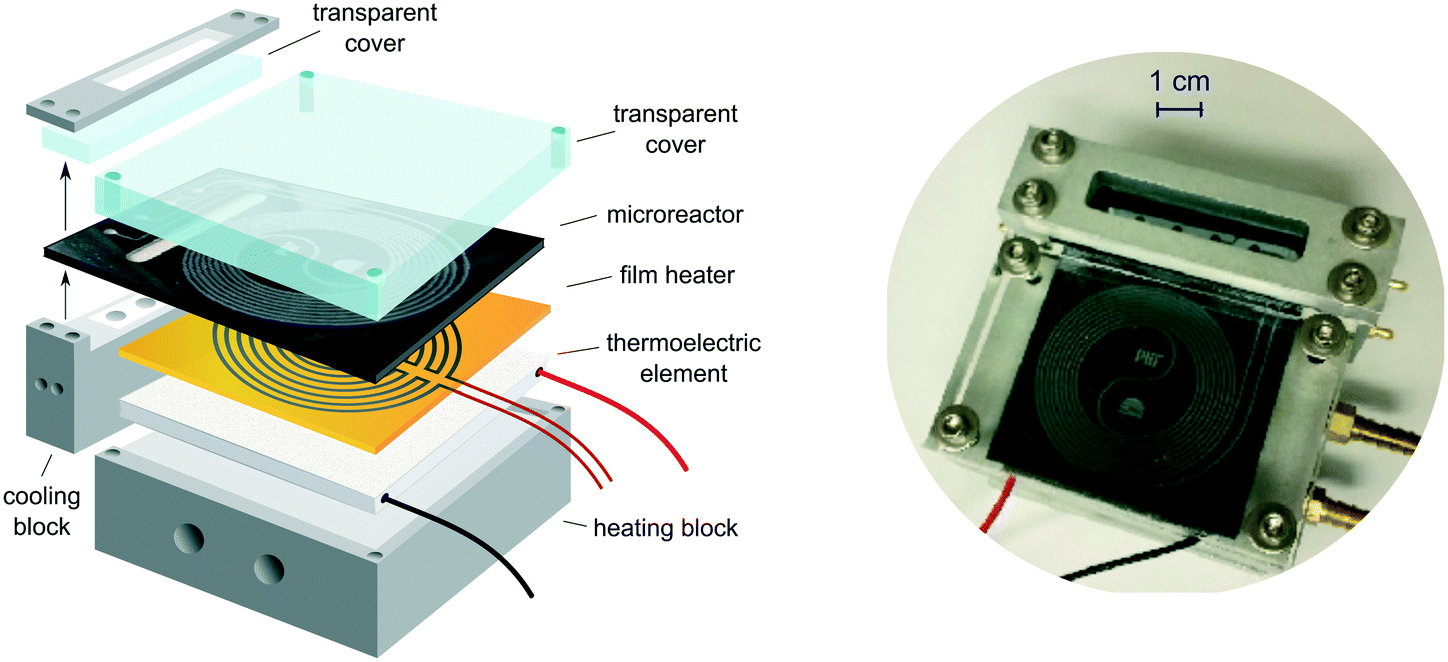 | ||
| Fig. 1 Microcalorimeter assembly. Two separate aluminum blocks enable control of temperatures in the inlet/outlet zone and reaction zone. | ||
The in-house designed glass–silicon microreactor (manufactured by Little Things Factory) consists of an inlet/outlet section and a spiral reaction section, separated by an elliptical recess in the silicon to minimize the heat transfer between the two zones (see Fig. 2).32 In the inlet section, the two reactant inlets merge into a T-mixer shortly before entering the spiral reaction zone. The third inlet gives the option of quenching the reaction as it joins the reaction stream directly after the reaction zone. The entire stream then leaves the reactor through the outlet opening. Most of the channels on the chip have a square cross section of 500 × 500 μm, giving the total volume of the reaction zone of about 180 μl. The side inlet of the T-mixer is narrower, only 200 μm, to form smaller droplets if two-phase flow is being investigated. The width of the channel increases to 800 μm after the quench stream joins the outlet of the reaction zone. The inlet and reaction zones are attached to two separate aluminum blocks which can be cooled or heated with recirculating water. The recess in the silicon enables simultaneous heating of the reaction zone and cooling of the inlet section. This combination of independently cooled and heated sections on the chip allowed us to reach temperatures as low as 10 °C in the cooling zone, while maintaining the reaction zone at around 60 °C. In the future, further improvements for the temperature control could be made by replacing recirculating water heaters with sensitive electrical heaters/coolers. The cooling block also houses a Teflon insert with channels for the inlets and outlets, installed to protect the aluminum block from corrosive reagents (Fig. S1 in ESI†). The Teflon insert is a compromise between safe handling of acids and a good thermal control. The temperature on chip is monitored with three thermocouples attached to the silicon surface of the chip. The whole assembly is insulated to reduce heat losses and environmental influences.
 | ||
| Fig. 2 Experimental setup. | ||
Three syringe pumps (Harvard Apparatus) control the flow of reactants through PFA tubing into the reactor (Fig. 2). Reaction conversion is continuously monitored with an inline FTIR spectrometer – a Mettler Toledo ReactIR iC 15 unit – equipped with a DS micro flow cell with an integrated silicon attenuated-total-reflection (ATR) sensor. The outlet tubing connecting the reactor to the IR is also cooled to provide additional quench and ensure that measured concentrations correspond to the conditions at the end of the reaction zone. A 40 psi back-pressure regulator is placed after the ReactIR to stabilize the flow through the flow cell. The entire setup is controlled with LabView, IR data is recorded and processed with iC Quant software (Mettler Toledo).
Thermoelectric element
The enthalpy of reaction is calculated using the heat flow measured with the thermoelectic element and the known amount of the consumed reagent ṅreacted: | (1) |
Measurement of the heat output of the reaction occurs via the thermoelectric element thanks to the Seebeck effect. It describes the generation of an electric potential when different temperatures prevail at two sides of a TE. This temperature difference creates an electromotive force and consequently an open circuit voltage, the ‘Seebeck voltage’.42 The Seebeck voltage is stable for a constant temperature difference ΔT and is given by:
| ΔU = SΔT = S(Thot − Tcold) | (2) |
 | (3) |
The resulting heat flow ![[q with combining dot above]](https://www.rsc.org/images/entities/i_char_0071_0307.gif) TE through the TE depends on material properties along with the temperature difference ΔT:
TE through the TE depends on material properties along with the temperature difference ΔT:
 | (4) |
 | (5) |
To determine the value of α, the calorimeter is calibrated using a thin film heater (Omega Engineering Inc., resistance of 32 Ω) attached to the bottom of the chip; it imitates the heat release of an exothermic reaction, allowing to determine the dependency between the heat transferred and voltage output of the TE. We assumed the presence of the thin film (approximately 200 μm thick) and its potential influence on heat flux measured during a reaction to be negligible. For the calibration, the temperatures of the two aluminum blocks are set to match the final reaction conditions and water is pumped through the reactor at the flow rate equal to the total flow rate during an actual calorimetry experiment. Once a stable TE signal is reached, the power output of the heater is increased stepwise and the output of the TE is measured (Fig. 3). Power increments of 20 mW from 0 to 200 mW, held for 10 minutes each, were found to give sufficient range and accuracy in the calibration. When plotting the power output of the power supply against the measured voltage, one obtains a linear graph with the slope equal to the value of α (Fig. 3).
| heater = α(ΔU − ΔU0) | (6) |
 | ||
| Fig. 3 Calibration of the TE: to determine the value of alpha, the power output generated by the power source is correlated with the voltage generated by the TE. The offset voltage ΔU0, marked in the second pane as the voltage at zero power input, is subtracted before linear regression. The slope of the line equals alpha. | ||
Heat of mixing
Mixing of the reagents may also release or consume heat; the heat of mixing is usually measured together with the reaction enthalpy. Decoupling the two values is difficult to realize experimentally, because the substrates will start reacting as soon as they are mixed. We have estimated the heat of mixing indirectly by conducting experiments at various reactant conversions. We utilize the fact that the heat of mixing equals the heat released at 0% conversion, when the educts are mixed but have not reacted yet. In the presence of significant heat of mixing, eqn (1) can be rewritten as: | (7) |
Reaction kinetics
For kinetic experiments in flow, we follow the residence time ramp method developed by Moore & Jensen.30 It uses the fact that a flow reactor displaying plug flow behaviour can be approximated as a series of batch reactors.43 Thus by carefully controlling the residence time in the unit, a series of batch kinetic experiments can be replaced by one batch-like flow experiment, saving both time and reagents.30 Large deviations from plug flow can occur in laminar flow devices if the radial concentration gradients caused by the parabolic flow profile are not eliminated by diffusion. Owing to the small channel width, microdevices we tested show small deviations from plug flow and therefore can be used for batch-like kinetics experimentation.44In the experiment, the residence time is ramped at a constant rate, as described with the following equation:
 | (8) |
Residence time experienced by each fluid element arriving at the measurement point is given by eqn (9), which includes a correction due to the dead volume between the exit of the reaction zone and the detector:
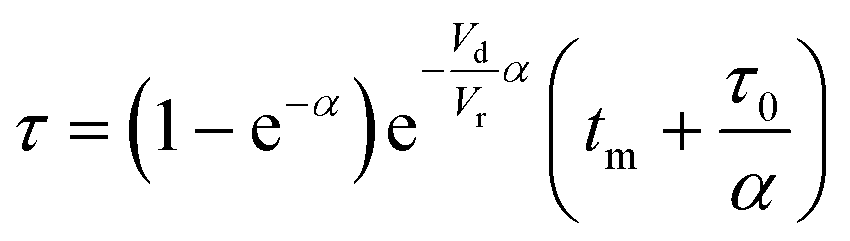 | (9) |
Experimental procedure
Before starting an experiment, the entire system is flushed with the reagents to remove air. Once the temperature in both aluminum blocks is set, the system requires 30–60 min for the TE signal to stabilize. For a calorimetry experiment, first a baseline signal is collected: the flow of one of the reagents and the quench (if used) is started; the flow rate of the reagent is set to the total flow rate of all reactants. After the voltage signal of the TE has been stable for at least 15 minutes the reaction is started by setting the flow of both reagents to the planned flow rates. The TE signal increases immediately once the reactants come into contact, see Fig. 4. Once it reaches a stable value, usually after 2–3 min from the start of the reagent flow for the flow rates investigated here, it is recorded for another 15 minutes. Upon completion of the measurement, the entire system is cleaned by flushing with an appropriate solvent. | ||
| Fig. 4 Example of the recorded voltage from the TE, corrected with the baseline value. After collection of the baseline signal, flow of reactants is started. Voltage increases immediately to the value ΔU, corresponding to the heat released during the reaction. | ||
For a kinetic investigation, the flow of reagents is set to obtain a constant initial residence time τ0; steady state is reached once a stable signal is measured with the ReactIR. At this point the experiment time is reset to 0 and the residence time ramp is started. Upon completion, the system is washed with an appropriate solvent. To collect more information, the residence time ramp can be repeated at different temperatures.
Results
Hydrolysis of acetic anhydride
We chose the hydrolysis of acetic anhydride to acetic acid as the model reaction to test the calorimeter (Fig. 5). It often serves as a model reaction for calorimetry and has been thoroughly studied in literature, with the enthalpy and kinetics well known.45–50 The synthesis can be performed as a single- or two-phase reaction as Ac2O is partially miscible with water. To ensure a single-phase reaction the miscibility can be improved by increasing the reaction temperature and by adding AcOH to the starting solution. Hydrochloric acid can be used as a catalyst for the reaction. | ||
| Fig. 5 Hydrolysis of acetic anhydride. | ||
To ensure that the reaction follows a first-order kinetics, we conducted the experiments under excess of water with a flow rate ratio of Ac2O solution to water of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6.5. To improve the solubility of acetic anhydride in water 25% acetic acid was added to the Ac2O starting solution.
:6.5. To improve the solubility of acetic anhydride in water 25% acetic acid was added to the Ac2O starting solution.
For the calorimetric measurements, we set the temperature in the reaction zone to 60 °C. Outlet of the reactor was kept at 10 °C to quench the reaction. The heat release during the hydrolysis of Ac2O may be significantly influenced by the heat of mixing. Therefore to determine both the reaction enthalpy as well as the heat of mixing we run the hydrolysis at a range of conversions: we varied the flow rate of Ac2O starting solution from 80 to 5 μL min−1, corresponding to total flow rate between 600 and 37.5 μL min−1, residence times in the reaction zone from 0.3 min to 4.7 min, and Ac2O conversion between 16% and 100%. In each experiment the baseline signal of the thermoelectric was collected with water flow only, as we found that addition of other components had a negligible influence on the sensible heat (see ESI†). Subsequently the flow of Ac2O was switched on to start the reaction.
The results of these experiments are shown in Fig. 6. We performed ordinary least squares linear regression using R (R Project) to calculate regression coefficients and associated uncertainties. Uncertainties in the heat of mixing and reaction enthalpy are calculated from 95% confidence intervals in the estimated regression coefficients; uncertainty in the total enthalpy at 100% conversion is obtained from 95% prediction interval for this value (see ESI†). The total heat released equals to −54.2 ± 3.9 kJ mol−1, with the heat of mixing of approximately +8.8 ± 2.1 kJ mol−1, indicating endothermic mixing process within the applied conditions. The enthalpy of the reaction was thus determined to be −63 ± 3.0 kJ mol−1, in good agreement with the calculated value of −58.2 kJ mol−1 (obtained from Hess law, see ESI† for details). Enthalpies reported in literature range between −57 and −65 kJ mol−1 and in most cases refer to combined reaction enthalpy and the heat of mixing, and reactions performed in presence of HCl.45,47–50 Studies separating the two values are scarce, but indicate a change from exothermal to endothermal mixing as the temperature increases. Zogg et al. (2003) report heat of mixing values increasing from −3 to +3 kJ mol−1 at 25 °C and 55 °C respectively in a batch reaction catalysed by 0.1 M HCl.45 Becker and Walisch (1965) determined heat of mixing of −4.25 kJ mol−1 at room temperature, also in presence of 0.1 M HCl.47 Fritzler et al. (2014) observed endothermic mixing at 60 °C but did not measure the heat consumed.51 While our result for the heat of mixing may not be directly comparable to those values due to differences in the composition of the starting mixture, the endothermal heat of mixing we measured fits into the described trend. The excellent agreement of the total enthalpy with literature values together with low standard deviation demonstrate the accuracy and reliability of the system we developed (Table 1).
| Reaction enthalpy kJ mol−1 | Heat of mixing kJ mol−1 | |
|---|---|---|
| Experimental data | −63 ± 3.0 | +8.8 ± 2.1 |
| Temperature | 60 °C | 60 °C |
| Literature45,47 | Between −57 and −65 | Between −4.25 and +3 |
| Temperature | Between 0 °C and 55 °C | Between 25 °C and 55 °C |
| Theoretical value | −58.2 |
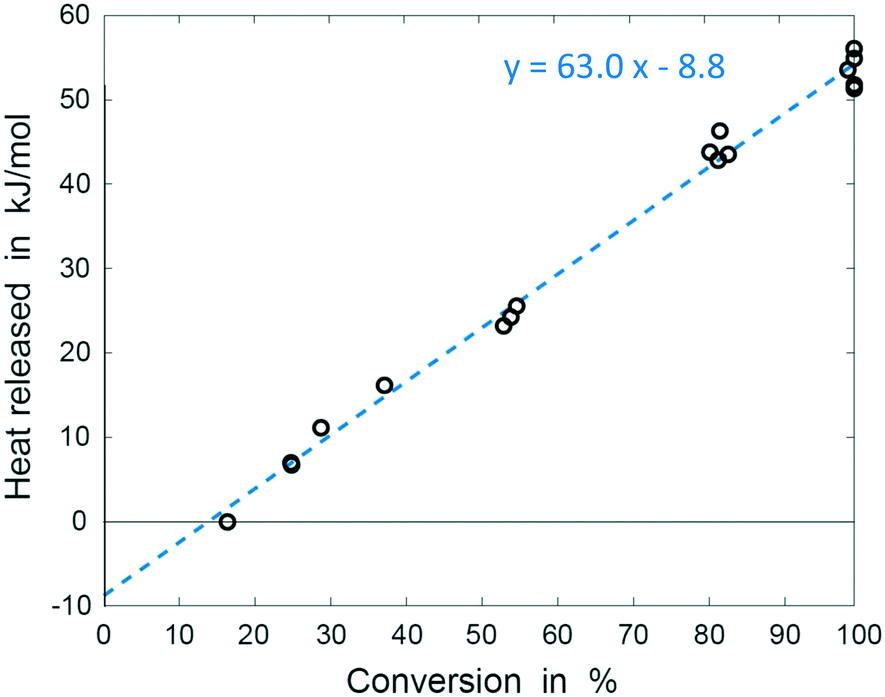 | ||
| Fig. 6 Plot of heat release against conversion. Linear regression parameters represent reaction enthalpy (slope) and the heat of mixing (intercept). | ||
For the kinetic experiments, we applied a residence time ramp with α = 0.4 and the total flow rate ranging from 375 down to 37.5 μL min−1, corresponding to the residence time of 0.48 to 4.8 min. The same ramp was repeated at four different temperatures in the reaction zone, as listed in Table 2. With the large excess of water the reaction follows a pseudo-first order kinetics, Fig. 7.
| T °C | k min−1 |
|---|---|
| 20 | 0.129 |
| 29.1 | 0.173 |
| 43.2 | 0.274 |
| 56.3 | 0.522 |
 | ||
| Fig. 7 Residence time ramps at constant temperature: experimental data (circles) and fitted model (lines). | ||
Obtained reaction rate constants (Table 2) are in good agreement with data available from literature for hydrolysis in absence of hydrochloric acid: Mitzner et al. measured 0.122 and 0.225 min−1 at temperature of 20 °C and 30 °C respectively, while kinetic parameters estimated by Glasser & Williams result in k = 0.165 min−1 at 30 °C.52,53 Values at higher temperatures are also within the ranges reported in literature, see Fig. S5† for a full comparison. Relatively large variability in the reaction rate constant reported in literature is most likely due to differences in starting conditions, especially concentration of the initial mixture which could influence the reaction rate.
Accuracy of the calorimeter
The signal-to-noise-ratio of the thermoelectric element is an indicator of the influence of the surroundings on the calorimeter. There is a trade-off between the accessibility of the reactor and the noise in the signal due to the environmental influences on the TE. A compromise was found by insulating the reactor assembly with an exception of the transparent reactor cover, and placing the entire system in a closed box with a window. We measured the signal from a TE placed inside and outside of the box; signal-to-noise ratio was significantly improved for the insulated TE. The remaining error of the thermoelectric was found to be approximately 11 mW, corresponding to approximately 0.3 mV uncertainty in the voltage output of the TE.Conclusion
We developed a robust and versatile microfluidic platform for fast investigations of reaction enthalpy and kinetics. Reaction enthalpy is evaluated in steady state isothermal experiments by using a thermoelectric element and a microreactor, sandwiched between a heater and transparent cover for visual access. An inline IR spectrometer is used to quantify reaction conversion. The same setup can be used for transient flow experiments to obtain kinetic data without any hardware manipulation, reducing reagent consumption and experiment duration. We successfully tested the microfluidic calorimeter on hydrolysis of acetic anhydride as a model reaction, achieving good agreement with literature for both reaction enthalpy and kinetics. Calorimetry measurements revealed enthalpy of the reaction of −63.0 ± 3 kJ mol−1 and the heat of mixing of +8.8 ± 2.1 kJ mol−1 at 60 °C. We measured the reaction rate constant in four residence time ramp experiments, each at different temperature, obtaining reaction rate constant values between 0.129 up to 0.522 min−1 for corresponding temperatures from 20 °C to 56.3 °C. Good agreement with literature demonstrates how a single microfluidic setup can be used to generate thermodynamic as well as kinetic data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Novartis MIT Center for Continuous Manufacturing supported part of this effort. A. Ł. gratefully acknowledges the Swiss National Science Foundation for financial support (grant number P2EZP2_175152).References
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS.
- K. S. Elvira, X. Casadevall i Solvas, R. C. R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef CAS.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- C. F. Carter, H. Lange, S. V. Ley, I. R. Baxendale, B. Wittkamp, J. G. Goode and N. L. Gaunt, Org. Process Res. Dev., 2010, 14, 393–404 CrossRef CAS.
- M. Abolhasani and K. F. Jensen, Lab Chip, 2016, 16, 2775–2784 RSC.
- Y.-J. Hwang, C. W. Coley, M. Abolhasani, A. L. Marzinzik, G. Koch, C. Spanka, H. Lehmann and K. F. Jensen, Chem. Commun., 2017, 53, 6649–6652 RSC.
- L. M. Baumgartner, C. W. Coley, B. J. Reizman, K. W. Gao and K. F. Jensen, React. Chem. Eng., 2018, 3, 301–311 RSC.
- N. Holmes, G. R. Akien, A. J. Blacker, R. L. Woodward, R. E. Meadows and R. A. Bourne, React. Chem. Eng., 2016, 1, 366–371 RSC.
- J. M. Köhler and M. Zieren, Fresenius' J. Anal. Chem., 1997, 358, 683–686 CrossRef.
- J. M. Köhler and M. Zieren, Thermochim. Acta, 1998, 310, 25–35 CrossRef.
- J. Lerchner, A. Wolf, R. Hüttl and G. Wolf, Chem. Eng. J., 2004, 101, 187–194 CrossRef CAS.
- M. A. Schneider, T. Maeder, P. Ryser and F. Stoessel, Chem. Eng. J., 2004, 101, 241–250 CrossRef CAS.
- M. A. Schneider and F. Stoessel, Chem. Eng. J., 2005, 115, 73–83 CrossRef CAS.
- J. Antes, D. Schifferdecker, S. Loebbecke and H. Krause, Chem. Ing. Tech., 2005, 77, 994–996 CrossRef.
- J. Lerchner, A. Wolf, G. Wolf, V. Baier, E. Kessler, M. Nietzsch and M. Krügel, Thermochim. Acta, 2006, 445, 144–150 CrossRef CAS.
- J. Antes, M. Gegenheimer, H. Krause, S. Löbbecke, R. Wirker and A. Knorr, Chem. Ing. Tech., 2008, 80, 1270 CrossRef CAS.
- J. Antes, M. Gegenheimer, S. Löbbecke and H. Krause, Reaction calorimetry in microreactors: fast reaction screening and process design, San Diego, 2008, p. 3 Search PubMed.
- C. Hany, H. Lebrun, C. Pradere, J. Toutain and J.-C. Batsale, Chem. Eng. J., 2010, 160, 814–822 CrossRef CAS.
- F. Reichmann, S. Millhoff, Y. Jirmann and N. Kockmann, Chem. Eng. Technol., 2017, 40, 2144–2154 CrossRef CAS.
- F. Reichmann, K. Vennemann, T. A. Frede and N. Kockmann, Chem. Ing. Tech., 2019, 91, 622–631 CrossRef CAS.
- M. C. Maier, M. Leitner, C. O. Kappe and H. Gruber-Woelfler, React. Chem. Eng., 2020, 5, 1410–1420 RSC.
- K. Wang, Y. C. Lu, H. W. Shao and G. S. Luo, AIChE J., 2010, 56, 1045–1052 CAS.
- M. Romano, C. Pradere, F. Sarrazin, J. Toutain and J. C. Batsale, Chem. Eng. J., 2015, 273, 325–332 CrossRef CAS.
- Y. Zhang, N. E. Benes and R. G. H. Lammertink, Chem. Eng. J., 2016, 284, 1342–1347 CrossRef CAS.
- C. Zhang, J. Zhang and G. Luo, J. Flow Chem., 2020, 10, 219–226 CrossRef CAS.
- C. Pradere, C. Hany, J. Toutain and J.-C. Batsale, Exp. Heat Transfer, 2009, 23, 44–62 CrossRef.
- G. Glotz, D. J. Knoechel, P. Podmore, H. Gruber-Woelfler and C. O. Kappe, Org. Process Res. Dev., 2017, 21, 763–770 CrossRef CAS.
- F. Mortzfeld, J. Polenk, B. Guélat, F. Venturoni, B. Schenkel and P. Filipponi, Organic Process Research & Development, 2020 Search PubMed.
- J. S. Moore and K. F. Jensen, Angew. Chem., 2014, 126, 480–483 CrossRef.
- J. S. Moore, C. D. Smith and K. F. Jensen, React. Chem. Eng., 2016, 1, 272–279 RSC.
- K. C. Aroh and K. F. Jensen, React. Chem. Eng., 2018, 3, 94–101 RSC.
- B. M. Wyvratt, J. P. McMullen and S. T. Grosser, React. Chem. Eng., 2019, 4, 1637–1645 RSC.
- S. Schwolow, F. Braun, M. Rädle, N. Kockmann and T. Röder, Org. Process Res. Dev., 2015, 19, 1286–1292 CrossRef CAS.
- C. Hone, N. Holmes, G. R. Akien, R. A. Bourne and F. L. Muller, React. Chem. Eng., 2017, 2, 103–108 RSC.
- C. Waldron, A. Pankajakshan, M. Quaglio, E. Cao, F. Galvanin and A. Gavriilidis, React. Chem. Eng., 2019, 4, 1623–1636 RSC.
- C. Waldron, A. Pankajakshan, M. Quaglio, E. Cao, F. Galvanin and A. Gavriilidis, React. Chem. Eng., 2020, 5, 112–123 RSC.
- S. D. Schaber, S. C. Born, K. F. Jensen and P. I. Barton, Org. Process Res. Dev., 2014, 18, 1461–1467 CrossRef CAS.
- S. Mozharov, A. Nordon, D. Littlejohn, C. Wiles, P. Watts, P. Dallin and J. M. Girkin, J. Am. Chem. Soc., 2011, 133, 3601–3608 CrossRef CAS.
- T. Durand, C. Henry, D. Bolien, D. C. Harrowven, S. Bloodworth, X. Franck and R. J. Whitby, React. Chem. Eng., 2016, 1, 82–89 RSC.
- C. P. Haas, S. Biesenroth, S. Buckenmaier, T. van de Goor and U. Tallarek, React. Chem. Eng., 2020, 5, 912–920 RSC.
- S. P. Beeby, Z. Cao and A. Almussallam, in Multidisciplinary Know-How for Smart-Textiles Developers, ed. T. Kirstein, Woodhead Publishing, 2013, pp. 306–328 Search PubMed.
- O. Levenspiel, Chemical Reaction Engineering, Wiley, 1999 Search PubMed.
- K. D. Nagy, B. Shen, T. F. Jamison and K. F. Jensen, Org. Process Res. Dev., 2012, 16, 976–981 CrossRef CAS.
- A. Zogg, U. Fischer and K. Hungerbühler, Ind. Eng. Chem. Res., 2003, 42, 767–776 CrossRef CAS.
- A. Zogg, U. Fischer and K. Hungerbühler, Chemom. Intell. Lab. Syst., 2004, 71, 165–176 CrossRef CAS.
- F. Becker and W. Walisch, Z. Phys. Chem., 1965, 46, 279–293 CrossRef CAS.
- T. L. Smith, J. Phys. Chem., 1955, 59, 385–388 CrossRef CAS.
- J. B. Conn, G. B. Kistiakowsky, R. M. Roberts and E. A. Smith, J. Am. Chem. Soc., 1942, 64, 1747–1752 CrossRef CAS.
- W. Köhler, O. Riedel and H. Scherer, Chem. Ing. Tech., 1973, 45, 1289–1294 CrossRef.
- B. C. Fritzler, S. Dharmavaram, R. T. Hartrim and G. F. Diffendall, Int. J. Chem. Kinet., 2014, 46, 151–160 CrossRef CAS.
- R. Mitzner and F. Lemke, Z. Chem., 1985, 25, 406–407 CrossRef CAS.
- D. Glasser and D. F. Williams, Ind. Eng. Chem. Fundam., 1971, 10, 516–519 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the experimental setup, experimental data and data analysis. See DOI: 10.1039/d0re00304b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |