
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous one-flow multi-step synthesis of active pharmaceutical ingredients
Victor R. L. J.
Bloemendal
 a,
Mathilde A. C. H.
Janssen
a,
Jan C. M.
van Hest
*b and
Floris P. J. T.
Rutjes
*a
a,
Mathilde A. C. H.
Janssen
a,
Jan C. M.
van Hest
*b and
Floris P. J. T.
Rutjes
*a
aInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: Floris.Rutjes@ru.nl
bBio-organic chemistry, Eindhoven University of Technology, P.O. Box 513 (STO 3.31), 5600 MB Eindhoven, The Netherlands. E-mail: J.C.M.v.Hest@tue.nl
First published on 26th May 2020
Abstract
During the past two decades, continuous flow chemistry has been developed into a mature field as shown by numerous examples in which complex molecules are synthesised. In this review we discuss recent one-flow multistep syntheses to produce active pharmaceutical ingredients (APIs) including challenges and solutions that have been encountered.
 Victor R. L. J. Bloemendal | Victor R. L. J. Bloemendal received his MSc in Chemistry from Radboud University in 2016, during which he focused on the synthesis of bio-orthogonally functionalised carbohydrates. Currently, he is a PhD-candidate working on a collaboration between Eindhoven University of Technology and Radboud University. As researcher on the EU-FETOPEN One-Flow project, his goal is to develop one-flow cascade reactions for the synthesis of cannabinoids. |
 Mathilde A. C. H. Janssen | Mathilde A. C. H. Janssen is a Master's student in Chemistry at Radboud University. During her Master's thesis she performed research on carbohydrate inhibitors. More recently, she did a second Master's project at Acerta Pharma B.V. (Oss, The Netherlands, member of the AstraZeneca group) were she is working on the development of covalent small molecule inhibitors in the oncology field. |
 Jan C. M. van Hest | Jan C. M. van Hest obtained his PhD from Eindhoven University of Technology in 1996 in macro-organic chemistry with Prof. E.W. Meijer. He worked as a postdoc with Prof. D. A. Tirrell on protein engineering. In 2000 he was appointed full professor in Bio-organic chemistry at Radboud University. As of September 2016 he holds the chair of Bio-organic Chemistry at Eindhoven University of Technology. He is recipient of an ERC Advanced grant and member of the Royal Netherlands Academy of Arts and Sciences. The group's focus is to develop well-defined compartments for nanomedicine and artificial cell research. |
 Floris P. J. T. Rutjes | Floris P. J. T. Rutjes received his PhD from the University of Amsterdam in 1993 with Profs. W. N. Speckamp and H. Hiemstra and conducted postdoctoral research in the group of Prof. K. C. Nicolaou at The Scripps Research Institute, La Jolla, USA. In 1995 he was appointed assistant professor in Amsterdam, and in 1999 he became full professor in organic synthesis at Radboud University. Awards include the Gold Medal of the Royal Netherlands Chemical Society (KNCV, 2002), the AstraZeneca award for research in organic chemistry (2003), and Most Entrepreneurial Scientist of the Netherlands (2008). He is currently director of the Institute for Molecules and Materials at Radboud University and President-Elect of the European Chemical Society (EuChemS). |
Introduction
The synthesis of complex biologically and pharmacologically relevant molecules traditionally proceeds through multistep batchwise reactions. This not only concerns small-scale syntheses in academic labs, but also large scale processes in the fine chemical and pharmaceutical industry.1 In the past two decades, however, there has been a steep increase in the development and use of continuous flow reactions in organic synthesis. This initially concerned single reactions, but in the past few years involved more and more multistep sequences in which consecutive reactions carried out in a so-called one-flow system lead to a complete synthesis of functional small molecules.2,3Prompted by the societal wish to be able to locally and rapidly produce important medicines on demand, it is of particular interest to synthesise active pharmaceutical ingredients (APIs) in one-flow systems.4,5 It is envisaged that in such an approach by using relatively small and mobile dedicated equipment, cartridges of suitable reagents and solvents, vital drugs can be produced in a straightforward manner and in reasonable amounts due to the continuous character of the process. This is also considered a safe approach owing to the reproducibility, efficient heat transfer and small dimensions of the actual reaction vessels.6,7 Additionally, in a one-flow process workup and intermediate purification steps are excluded leading to faster production and more environmentally benign processes.8 Inspired by our EU FETOPEN ONE-FLOW project,9 in which one-flow approaches are being developed for the synthesis of pharmaceutically relevant small molecules, this review highlights various recently published one-flow syntheses of APIs. It does not only include the synthetic steps, but also addresses challenges and incompatibilities that were encountered by combining multiple steps into a single flow system. In this review, we distinguish between one-flow systems that consist of fully continuous flow operations, and interrupted one-flow systems, meaning approaches in which at least one intermediate isolation/collection step is included (Fig. 1). One-flow systems, in particular set-ups that are not interrupted by intermediate phase switches or purification steps, have the challenge that incompatibilities between reagents in subsequent steps may occur, that solvents optimal for one step, may be detrimental for a second one, side-products may be carried along etc. In addition, with long continuous flow lines high back-pressures may be created, which will hamper the throughput and may give even rise to leakages.10 Microfluidic behaviour is also heavily influenced by pressure, and might require (static) mixers to be included.11 Conceptually, however, a one-flow system is very attractive and in principle leads to a straightforward flow scheme. Interrupted one-flow systems, on the other hand, offer opportunities to install more rigorous phase switches, or even work-up steps, in which salts, excess reagents or even side-products may be removed, and incompatibilities in the system can be avoided. Although beneficial for the overall process, these interruptions contribute to complexity of the system.
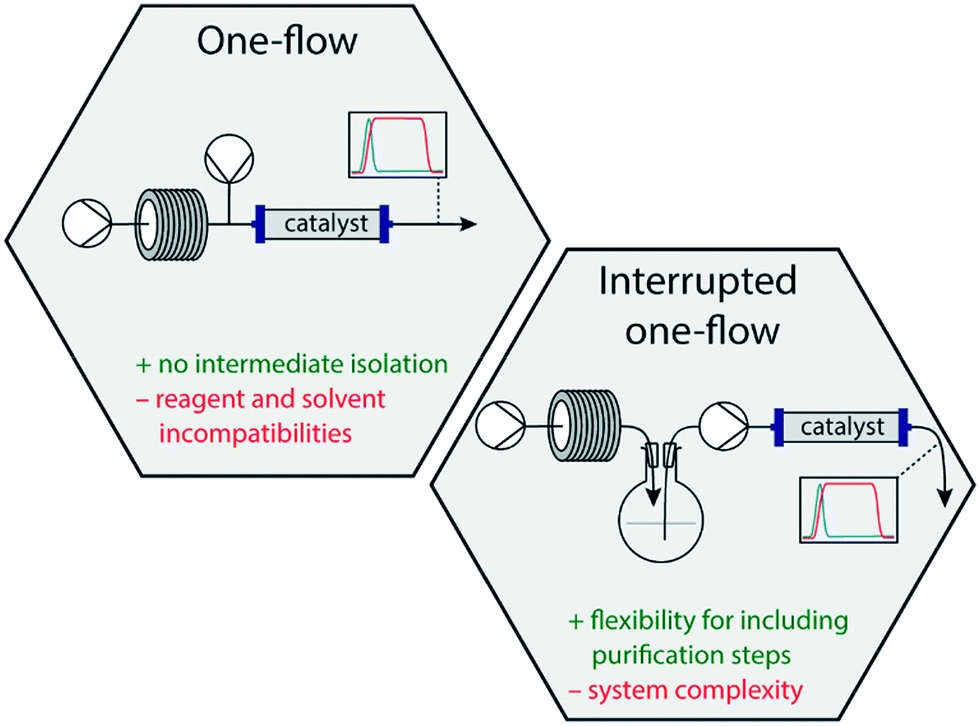 | ||
| Fig. 1 Schematic overview over the different types of flow reactions. | ||
One-flow systems
We will start with highlighting several successful approaches to synthesise APIs in one-flow systems in the last few years, including addressing some of the drawbacks.Pioneering work in the scientific field of continuous flow chemistry has been carried out by the Yoshida group. In various publications, they have demonstrated the possibility to synthesise API precursors in flow through multiple chemical transformations in unusually short reaction times (<10 s).12,13 This unique strategy, termed flash chemistry, allows building-up molecules by extremely fast quenching of highly reactive intermediates, which can only be realised under flow conditions. Not unimportantly, typically these reactions involving strong bases and organometallic reagents are carried out at low temperatures, but again, due to the immediate quenching, these reactions proceed in high yields even at room temperature. A challenge in this approach is, however, that while using these organometallic reagents and reactive intermediates the slightest leakage or supersaturation may lead to salt-formation and hence clogging of the entire system. One of the prime examples published by Yoshida is the synthesis of TAC-101 (4) in a one-flow system, combining six chemical transformations in a single continuous system (Fig. 2). The term flash chemistry is appropriate as transformation of tribromide 1 into TAC-101 (4) was achieved in approximately 13 s.
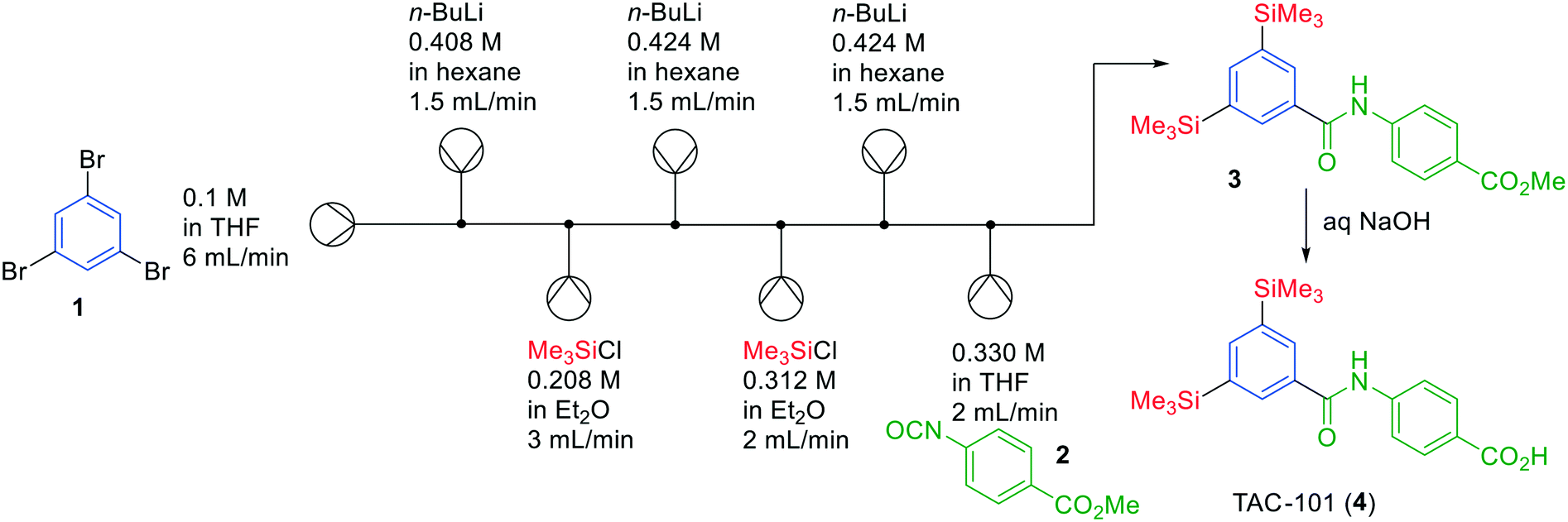 | ||
| Fig. 2 Synthesis of TAC-101 (4) by Yoshida and co-workers using a flash chemistry one-flow approach. Adapted with permission from RSC Adv., 2011, 1, 758–760. Copyright 2011 Royal Society of Chemistry.12 | ||
Ley and co-workers reported the synthesis of imatinib (8) and described a multistep flow process using in-line purification strategies.14 The system contained a crucial in-line solvent switch to limit the amount of human operations and increase the overall efficiency of the synthesis. Recently, Jamison and co-workers went a step further by realising a one-flow synthesis of imatinib (8) without a solvent switch.15 The API itself and several analogues were prepared in three chemical transformations which were optimised separately in flow and combined in a final single setup (Fig. 3). One of the key-steps in the synthesis was the Pd-catalysed amidation in a near-homogeneous solvent system. This system was formed by vigorous mixing of dioxane and an alkaline solution in a cross-mixer with a small inner diameter (ID 0.02 inch). The authors postulated that creating aqueous microdroplets by vigorous mixing would increase interfacial contact and enhance the conversion. Alternatively, a packed-bed mixing strategy appeared unsuccessful and resulted in aggregation on the outlet frit of the reactor.16
 | ||
| Fig. 3 Synthesis of imatinib (8) by Jamison and co-workers making use of a one-flow system. Adapted with permission from Org. Lett., 2019, 21, 6112–6116. Copyright 2019 American Chemical Society.15 | ||
The synthesis of imatinib (8) commenced with the hydration of nitrile 5, mediated by a stoichiometric amount of base at high temperature. The next step was the Pd-catalysed Buchwald–Hartwig amidation of aryl halide 6 with the hydrated intermediate using BrettPhos Pd G4. The integration of this final step was challenging because of the low solubility of 2-aminopyrimidine 7. To circumvent this problem, the authors premixed the Pd-catalyst and K3PO4 prior to the addition of 7. Also, they utilised the aforementioned near-homogenous mixture of 1,4-dioxane and water to increase interfacial contact and allow for a fast reaction between the base K3PO4 and the substrate. Additionally, the system was diluted by 25%, and a final iPrOH hydration module was implemented to decrease product precipitation after leaving the final reactor. This one-flow approach led to a moderate yield of 58% in the preparation of imatinib (8).
Several synthetic approaches have been employed in the synthesis of ACE inhibitors, which are used to treat hypertension.17 Jamison and co-workers developed a three-step continuous flow system, in which eight different ACE inhibitors, including quinapril (11), imidapril, and perindopril were synthesised (Fig. 4).18 The synthesis of quinapril (11) started with homophenylalanine derivative 9, which was coupled to amino ester 10. The authors initially focused on amide bond formation in flow by activating the carboxylic acid into the corresponding acid chloride. This led to precipitation, causing the authors to employ other amide formation strategies. Thus, the carboxylate was activated with N,N-carbonyldiimidazole (CDI) leading to the corresponding protected quinapril precursor in 92% yield. By adding a static mixer to the system, the yield of the amidation was increased, most likely due to a better conversion in the activated ester. In the subsequent coupling with amino ester 10 also diketopiperazine (DKP) impurities were observed, which could be effectively circumvented by varying the temperature: significant DKP formation was observed above 50 °C, while only amide coupling occurred at temperatures below 50 °C. Next, TFA deprotection of the tert-butyl group was incorporated in the flow system to generate the final product. In this way, the API quinapril (11) was obtained in a total yield of 86% within an overall reaction time of 175 min. Notably, the designed system was successfully used to synthesise other ACE inhibitors as well by varying coupling partners, showing the versatility of the system. Finally, the scalability of the system was tested by the larger scale formation of ACE-inhibitor enalapril. In this case, the flow rate was doubled compared to the original rate, giving rise to a yield of 86% for the scaled reaction.
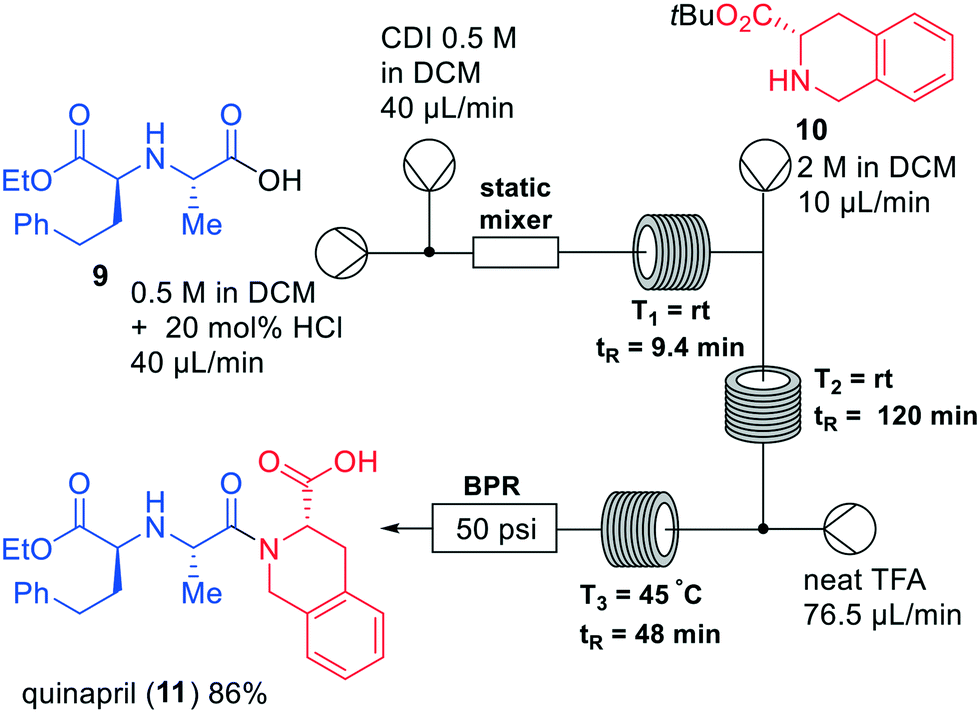 | ||
| Fig. 4 Synthesis of quinapril (11) by Jamison and co-workers making use of a one-flow system. Adapted with permission from Chem. – Eur. J., 2019, 25, 14527–14531. Copyright 2019 Wiley-VCH.18 | ||
Jamison and coworkers were also interested in reducing safety risks involving organic azides as they are highly energetic substances. The authors reported the synthesis of rufinamide (14), in which as key step a [3 + 2] Huisgen cycloaddition reaction was described for the preparation of a 1,2,3-triazole (Fig. 5).19 Rufinamide (14) was previously described as an antiepileptic drug for the treatment of neurogenic pain. It is conventionally prepared in batch,20 albeit that extensive comparisons with flow approaches have been made using life cycle assessment.21,22 Most of the synthetic routes relied on the isolation of the benzyl azide intermediate, which was circumvented in a continuous system. Firstly, the transformation of difluorobenzyl bromide (12) to the corresponding azide was investigated in flow. Gratifyingly, full conversion to the benzyl azide intermediate was obtained in DMSO in reaction times below 1 minute, supposedly because of efficient mixing in the reactor. Next, Jamison and coworkers examined the amidation of methyl propiolate (13) with ammonium hydroxide. This reaction is prone to polymerisation and was performed in flow in short reaction times without additional heating. Using 4 equivalents of ammonium hydroxide, and 5 min average residence time at room temperature, more than 95% conversion into the propiolamide was observed.
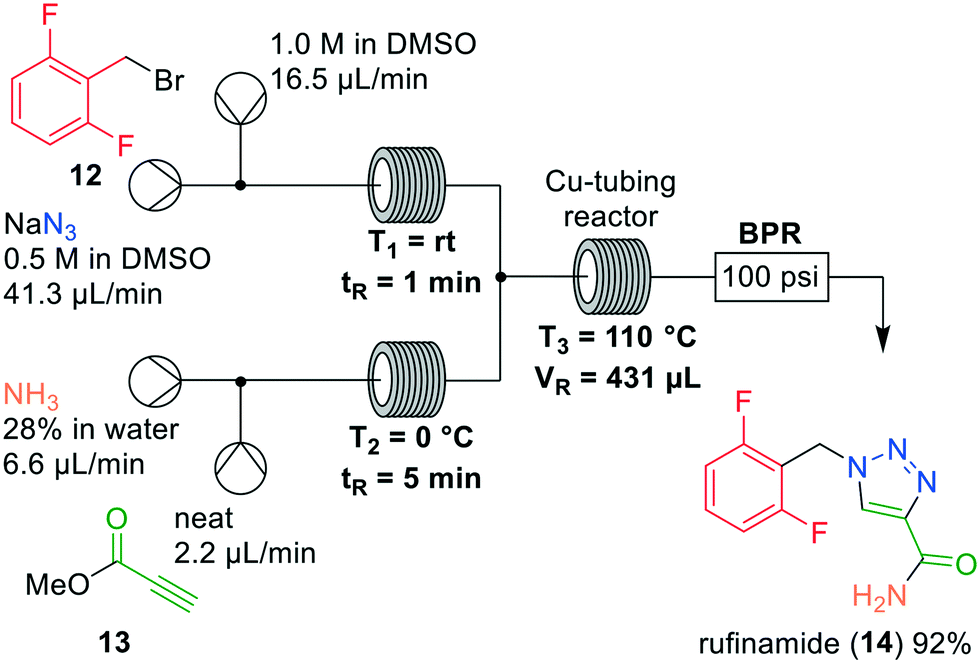 | ||
| Fig. 5 Synthesis of rufinamide (14) by Jamison and co-workers making use of a one-flow system. Adapted with permission from Org. Process Res. Dev., 2014, 18, 1567–1570. Copyright 2014 American Chemical Society.19 | ||
The challenge was combining both intermediates to conduct the [3 + 2] Huisgen cycloaddition in a next reactor. Mixing both reactor outlets did not cause any solid formation, but showed <10% conversions in perfluoroalkoxy (PFA) tubing. Inspired by Bogdan and Sach,23 the authors then used copper tubing and elevated temperatures to observe 82% conversion into rufinamide (14). Notably, there was occasional ammonia gas formation in the reactor so that a 100 psi BPR was implemented in the system. In order to further improve the yield, the temperature was lowered to 110 °C which gave 98% conversion, and 92% isolated yield. This short and elegant approach significantly reduced safety hazards because the azide intermediate was not accumulated, but in near-quantitative amounts immediately converted into the product rufinamide (14).
Recently, Gilmore and co-workers demonstrated a novel automated synthesis of rufinamide.24 They described a novel radial system, in which the reaction parameters can be changed by applying smart system loops via a digital LabVIEW interface. The system consists of various components which can be individually controlled (Fig. 6). Firstly, pressurised reagent feedstocks can be selected using multi-position valves and loaded in the connected syringes (reagent delivery system (RDS)). The syringe loaded reagents can be transferred to a sample loop and connected to a mass flow controller (MFC). To initiate the reaction, the MFC pumps the sample loops to the central switching station (CSS), which will feed the combined streams to a heated PFA reactor, photoreactor or in-line NMR system, after which a FlowIR continuously analyses the outlet stream. Depending on the analysis, the product can flow back to the RDS, stored in the standby module (SM) sample loop, or collected in the collection vessel (CV).
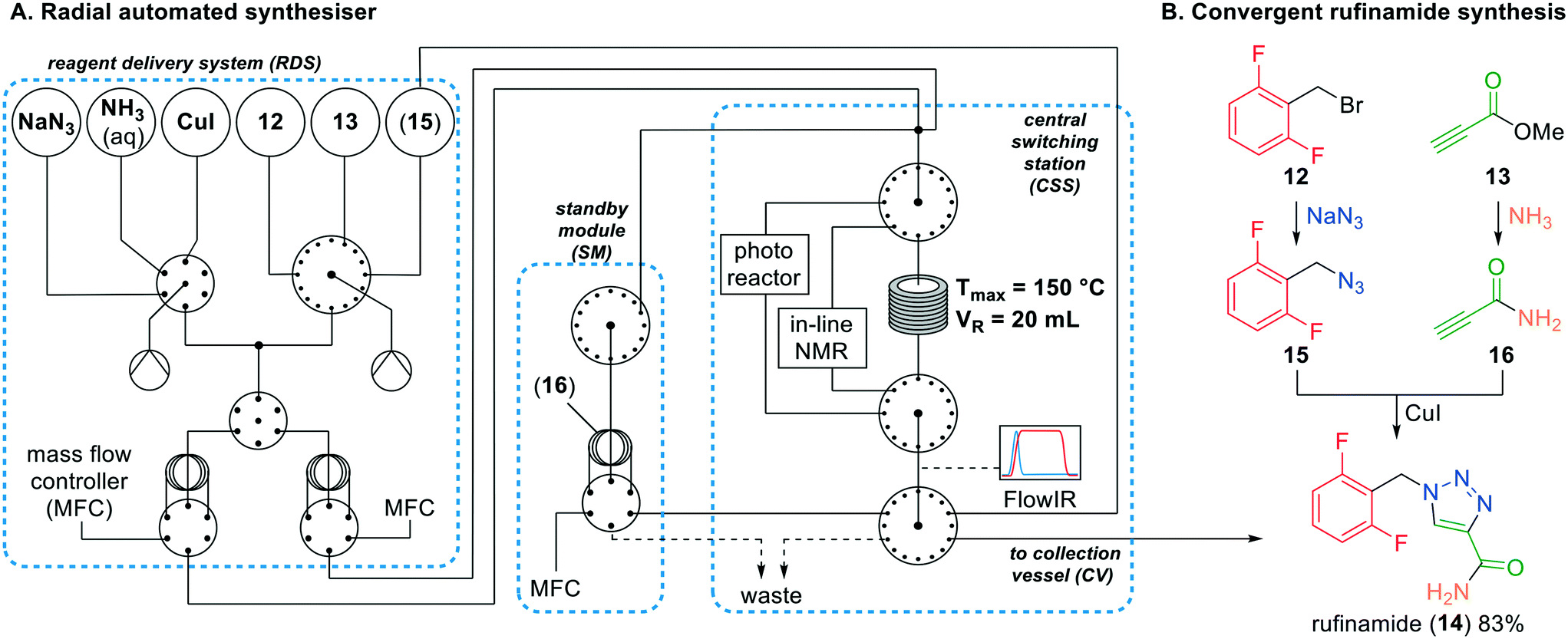 | ||
| Fig. 6 A. The simplified automated radial synthesiser; B. the convergent radial synthesis of rufinamide (14) by Gilmore and co-workers; dotted boxes: system components. Adapted with permission from Nature, 2020, 579, 379–384. Copyright 2020 Springer Nature.24 | ||
Following this methodology, the authors synthesised rufinamide in a convergent and linear sequence. Firstly, difluorobenzyl bromide (12) and sodium azide were selected in the RDS, loaded in the sample loops and fed to the CSS reactor at 40 °C. The formed difluorobenzyl azide (15) was recycled back to an empty vessel in the RDS. Secondly, methyl propiolate (13) was amidated using aqueous NH3 to obtain propiolamide (16) which was stored in the SM sample loop. Finally, difluorobenzyl azide (15), propiolamide (16) and copper iodide were selected and fed to the CSS reactor to afford rufinamide (14) in 83% conversion.
Additionally, the radial system was used for synthesising a library of rufinamide derivatives by adding more reagents to the RDS. This is in our view the first time that such a modular system is created that allows to readily adjust reaction parameters, optimise reaction conditions and use the same system for the production of compound libraries. The authors were convinced that the radial automatic system could be used as well for other processes, and did so by successfully using the photoreactor for nickel-mediated photoredox-catalysed cross-couplings. This would lead to more entries in the rufinamide library. Without question, the automated platform is highly effective for the screening and optimisation of multistep flow reactions. Not unimportantly, the system was created from commercially available equipment and hardware and the required software was freely accessible.
(−)-Oseltamivir (21) is a drug used for the treatment of influenza types A and B by acting as a neuraminidase inhibitor. The drug is included in the WHO-list of prequalified medicinal products, and different synthetic routes have been developed.25 A batch one-pot synthesis of (−)-oseltamivir (21) was described in 2013 by Hayashi and co-workers.26 No evaporation and no solvent switch were needed during this synthesis and therefore they postulated that a new one-flow approach for its synthesis may be realised.27 However, the original one-pot reaction took 57 h to reach completion, which implied that reduction of the reaction time was required. This was first attempted in batch, leading to 170 and 60 min reaction times, by going from conventional to microwave heating.
Next, the authors synthesised (−)-oseltamivir (21) in flow in five consecutive reactions (Fig. 7). Firstly, an asymmetric Michael reaction of aldehyde 16 with nitroalkene 15 was performed. This reaction was catalysed by Schreiner's thiourea (17) in combination with (S)-prolinol derivative 18. The main challenge was the solubility of nitroalkene 15, which dissolves in polar solvents leading, however, to epimerisation to the undesired syn-configuration of the resulting adduct. Toluene and chlorobenzene were examined on their ability to dissolve the nitroalkene without affecting the stereochemistry. In the end, toluene exhibited the best results to afford aldehyde 19. A second Michael reaction with phosphoryl acrylate 20 was conducted in the next reactor, after which an intramolecular Horner–Wadsworth–Emmons reaction was performed. This cyclisation predominantly afforded the undesired (5R)-isomer. In the third reactor, protonation of the potassium nitronate by TMSCl in EtOH was performed, whereby HCl was formed in situ. Subsequent desilylation with TBAF gave epimerisation to provide a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereoisomers at the 5-position. Lastly, the nitro group was reduced using zinc and TMSCl to afford (−)-oseltamivir (21). Zinc was packed in a column with Celite to prevent precipitation of material in the tubing. In the end, (−)-oseltamivir (21) was formed in an overall reaction time of 310 min in a yield of 13%.
:1 mixture of diastereoisomers at the 5-position. Lastly, the nitro group was reduced using zinc and TMSCl to afford (−)-oseltamivir (21). Zinc was packed in a column with Celite to prevent precipitation of material in the tubing. In the end, (−)-oseltamivir (21) was formed in an overall reaction time of 310 min in a yield of 13%.
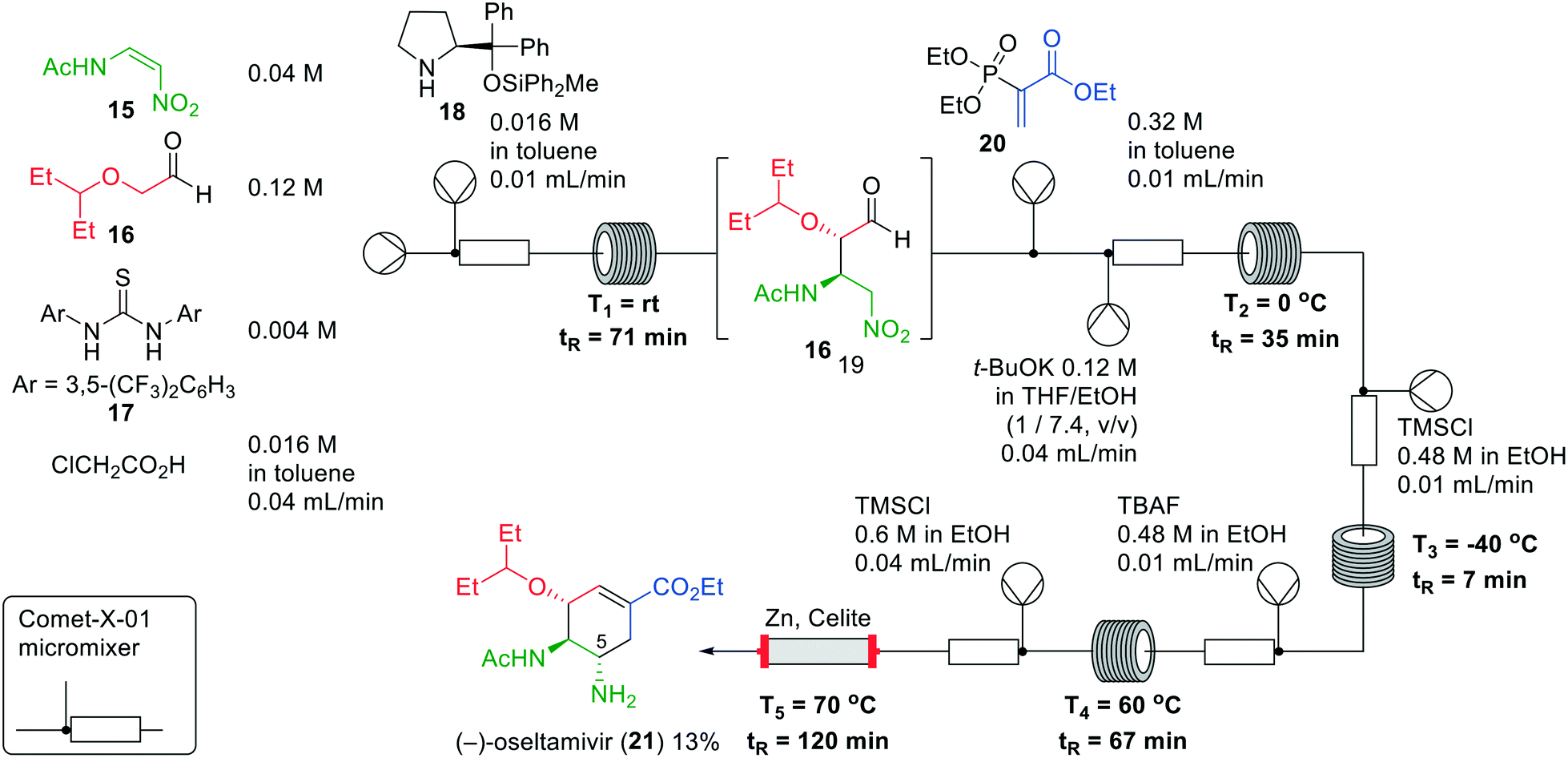 | ||
| Fig. 7 The synthesis of (−)-oseltamivir (21) by Hayashi and co-workers making use of a one-flow system. Adapted with permission from Synthesis, 2017, 424–428. Copyright 2017 Georg Thieme Verlag KG.27 | ||
Even though Hayashi and co-workers successfully developed a continuous flow synthesis of (−)-oseltamivir (21), the overall yield of the reaction was rather low and the zinc Celite reactor appeared only stable for approximately 5 h. The use of the column, which requires replacement during this process, renders this system somewhat more laborious and not a fully continuous system in practice.
In the last decade, various API flow syntheses have been published by the group of Jamison in which continuous flow extractions have been incorporated.28,29 Using a variety of inventive chemistries, he demonstrated inline purifications in which excess reagents and side products were removed to optimise yields of reaction products. An example is the formation of atropine (25) in flow, which was initially published in 2015 providing the target compound in 8% yield (Fig. 8).30 A drawback of this flow synthesis was the instability of intermediate products, which lowered the overall yield. Therefore, Jamison and co-workers revisited the synthesis in 2017 and optimised several steps, resulting in a continuous flow process with an overall yield of 22%.31 The early flow system had three different in-line liquid–liquid separations, while the revised system only had one. In the more recent one-flow system first esterification took place between tropinol 22 and acid chloride 23. Subsequently, an aldol reaction with aqueous formaldehyde (24) was performed to create the desired product atropine (25), which was isolated after in-line liquid–liquid separation.
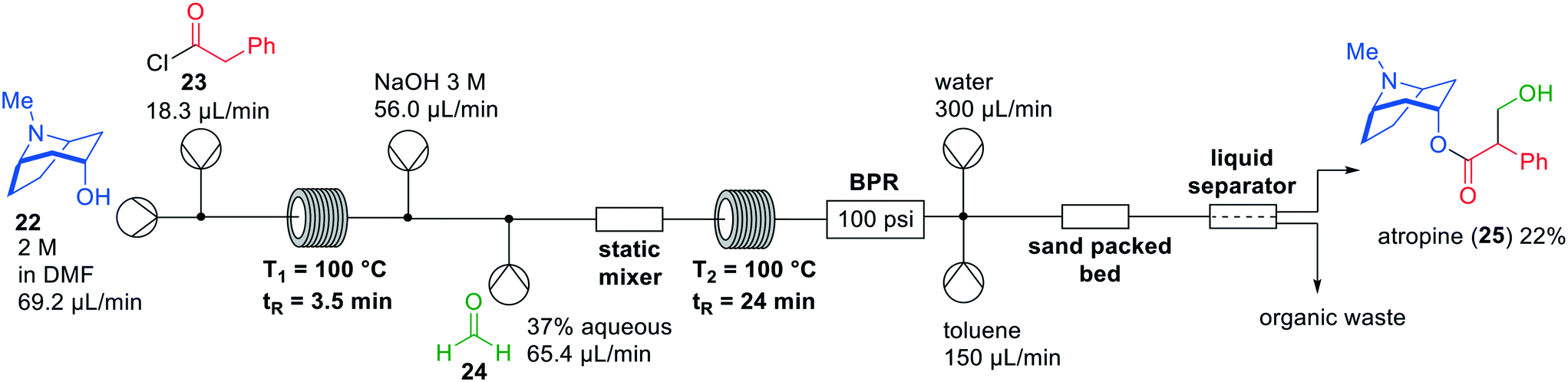 | ||
| Fig. 8 Synthesis of atropine (25) by Jamison and co-workers making use of a one-flow system. Adapted with permission from Bioorg. Med. Chem., 2017, 25, 6233–6241. Copyright 2017 Elsevier.30,31 | ||
Lomustine (33) is a widely used anticancer agent, of which the synthesis involves carcinogenic compounds. Due to new regulations regarding the handling of such types of compounds, production costs have increased and the availability of lomustine has dropped.32 To overcome these challenges, Thompson and co-workers investigated a continuous one-flow system to synthesise lomustine, which involves two chemical steps and an in-line extraction (Fig. 9).33 Firstly, the two reactions were optimised in glass microreactors and then implemented in PFA tubing. The first step was a carbamoylation of cyclohexylamine (31) with 1-chloro-2-isocyanato-ethane (30) and triethylamine (TEA), which was optimised regarding solvent, temperature, residence time and stoichiometry of the reagents. Most of the solvents that were examined led to clogging, except for THF which was then used in the system.
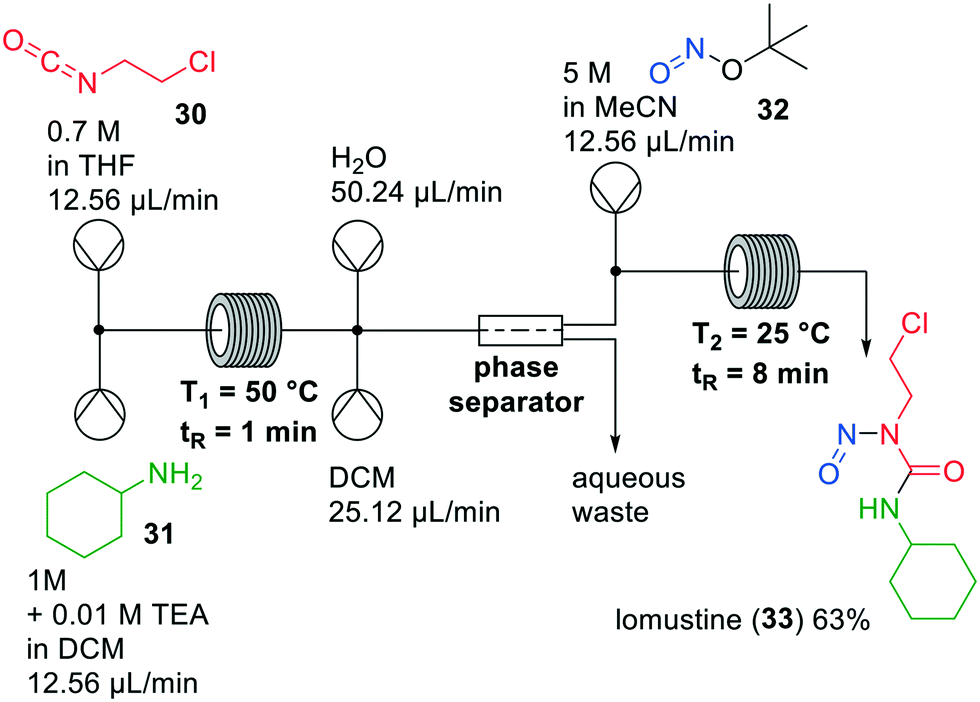 | ||
| Fig. 9 Synthesis of lomustine (33) by Thompson and co-workers making use of a one-flow system. Adapted with permission from Org. Process Res. Dev., 2019, 23, 334–341. Copyright 2019 American Chemical Society.33 | ||
The subsequent nitrosation was initially carried out with sodium nitrite in formic acid at temperatures below 0 °C to avoid sodium nitrite decomposition. Under optimised conditions the isolated yield of the reaction was 74%. It could, however, be increased to 91% when tert-butyl nitrite (TBN, 32) was used instead of sodium nitrite. Combining the carbamoylation and nitration in a continuous flow set up led to a low yield, because excess TEA reacted with TBN, hampering the nitrosation. Therefore, an extraction step to remove TEA was added after the first reaction to prevent the undesired side reaction from happening. With this one-flow system, lomustine (33) was obtained in an overall yield of 63%.
Grignard reactions are commonly used for the construction of carbon–carbon bonds and show exothermic behaviour which can be dangerous in large-scale batch processes. The use of Grignard reagents in flow can be beneficial because of the high control of reaction conditions, facile heat transport and small effective reaction volume.6,34 A recent example was published by Kiil and co-workers, who synthesised melitracen (36) in a one-flow system.35 Kiil hypothesised that the seven unit operations required in batch could be decreased by combining a hydrolysis and dehydration step, and removing a phase separation (Fig. 10).
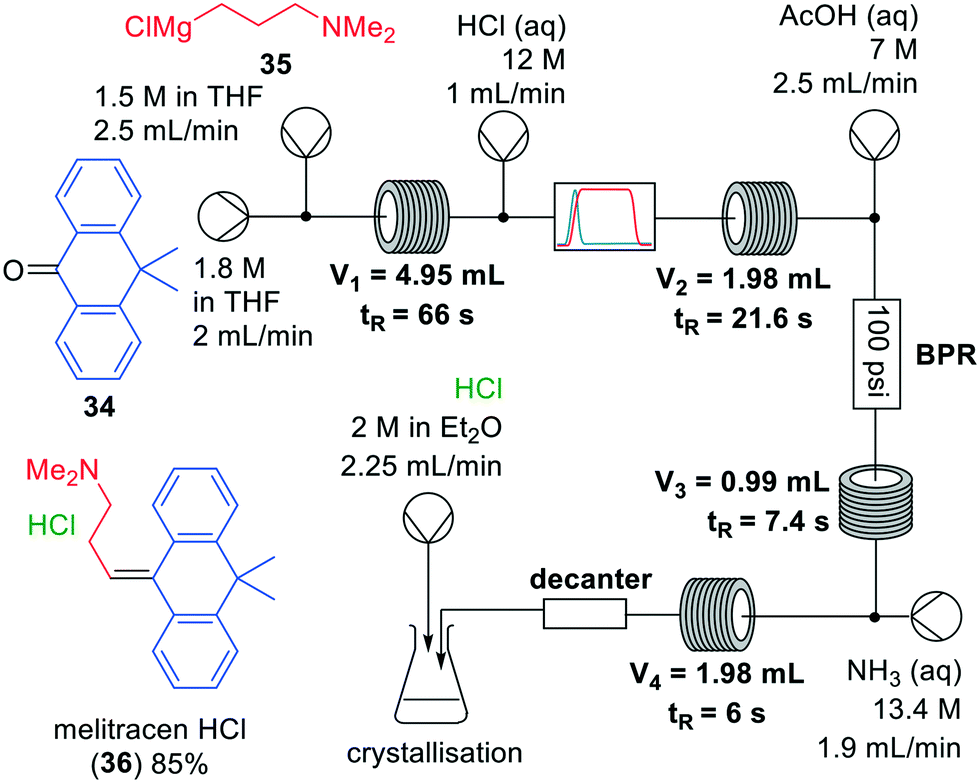 | ||
| Fig. 10 Synthesis of melitracen HCl-(36) by Kiil and co-workers making use of a one-flow system. Adapted with permission from Org. Process Res. Dev., 2018, 22, 228–235. Copyright 2018 American Chemical Society.35 | ||
The investigation commenced with finding a suitable solvent for the Grignard reaction in which starting materials 34, 35 and intermediate products would dissolve. After having identified THF as the most suitable option, the next challenge was to find an acid that could induce both hydrolysis and dehydration in a single step. Hydrochloric acid was able to perform both transformations, however, precipitation was observed. Thus, hydrochloric acid molarities ranging from 1–12 M were tested. However, while even at the lowest molarity precipitation was observed, it also appeared that below 6 M the dehydration reaction did not proceed. Since the precipitation could not be prevented, a molarity of 12 M was eventually used. The individually optimised transformations were then combined in a one-flow continuous system. Most troublesome was that addition of HCl to the reaction mixture led to an exothermic reaction and boiling of the solvent. Therefore, a back-pressure regulator was employed so that melitracen (36) could be successfully synthesised as its HCl-salt in approximately 85% yield.
Interrupted one-flow systems
The subsequent addition of multiple reagents into a single continuous flow system obviously requires a high level of compatibility between the different reagents themselves, the remains of reagents and the solvent involved.3,4 An additional complication is the build-up of a high back-pressure, which sometimes can be beneficial for transformations as with concerted reactions,36 oxidations with gaseous reagents37 or superheating solvents,38 but also may give rise to equipment failure. By interrupting the system, e.g. through introducing a wash step and storage of an intermediate product, or by applying a solvent switch, it is possible to effectively reduce the system pressure and alleviate equipment stress.In one-flow systems it can also be challenging to combine fast and slower transformations. To circumvent incompatibilities in reaction rates, one could opt to increase temperature or the residence time in case of slow transformations and vice versa for faster reactions. Again, interrupting the system by collecting an intermediate in a storage vessel, will allow applying differences in flow rates.
Next to technical drawbacks, one-flow systems can give rise to chemical incompatibilities in sequential transformations. Leaching of heterogeneous catalysts can lead to poisoning of other reagents39 or damaging downstream reactors,40 which is undesired. A solution to such chemical incompatibilities can involve interrupting the system by using inline extraction or purification to remove excess reagents. Alternatively, interruptions involving solvent switches and side-product removal with concomitant collection of the intermediate may be installed.19,41,42
A general challenge in flow chemistry is the undesired precipitation of either substrates, products, reagents or salts. As an alternative to solvent switches or washing steps to avoid precipitation, Kulkarni and co-workers recently described a novel flow reactor which can efficiently handle solid particles in suspension-like solutions.43 A cost-effective and readily applicable method to the aforementioned problem may again invoke interrupting the system. In case transformations that produce solid particles (e.g.Fig. 10, HCl-salt formation and subsequent precipitation) cannot be applied in flow, interrupting the one-flow process using a storage vessel or intermediate purification will solve the problem. In the sequel, we will discuss several API flow syntheses in which such interruptions have been incorporated.
An interrupted one-flow approach was published by Kobayashi et al. in 2015, focussing on the formation of both the (R)- and (S)-rolipram (29) enantiomers (Fig. 11).44 Immobilised heterogeneous catalysts were used for the synthesis of the products. Starting from the commercially available aldehyde 26 and nitromethane, the final product was formed as a single enantiomer via four catalytic transformations. In the first transformation, a nitroalkene was formed through a Henry reaction catalysed by Si-immobilised ammonia. The authors observed that the system became more stable by adding CaCl2 to the column, leading to higher yields. Various nitroalkenes were synthesised in this way showing the versatility of the method. Additionally, a column containing 4 Å molecular sieves was installed to dry the solvent. Again, this column was not essential, but provided higher yields. Then, an asymmetric 1,4-addition of dimethyl malonate (27) catalysed by immobilised PS-(S)-pybox-CaCl2 took place, followed by hydrogenation over a Pd-catalyst to amine 28.45 The latter step, which includes H2-degassing, is an interruption to enable purification of the intermediate product, which proceeds through salt removal by flushing over Amberlyst 15Dry and collection of amine 28. A Celite column was included in the system to enable mixing of the intermediate and the solvent for the decarboxylation and lactam formation using a Si-immobilised carboxylic acid to obtain (S)-rolipram (29) in a yield of 50% and 96% ee. By using the opposite enantiomer of the Pybox ligand, (R)-rolipram was obtained in comparable yield and enantioselectivity.
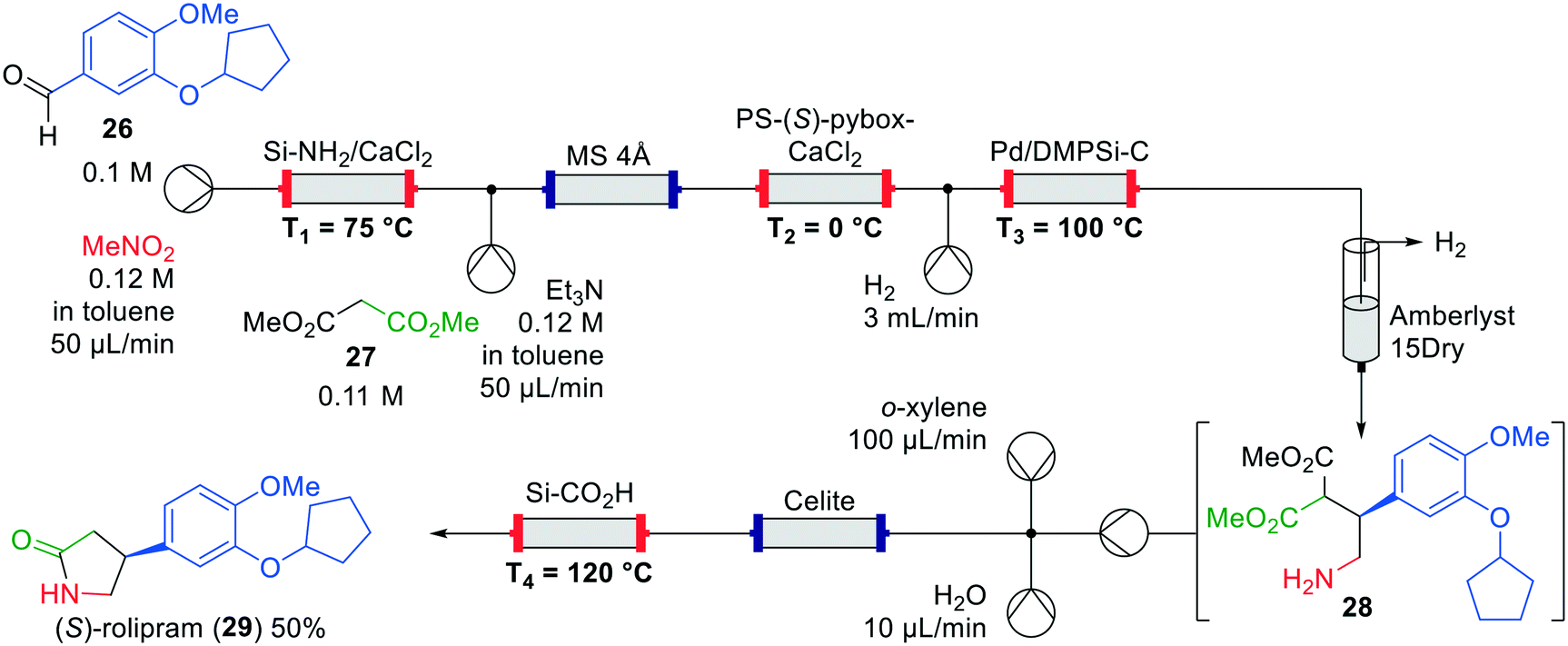 | ||
| Fig. 11 Synthesis of (S)-rolipram (29) by Kobayashi and co-workers making use of an interrupted one-flow system. Adapted with permission from Nature, 2015, 520, 329–332. Copyright 2015 Springer Nature.44 | ||
Kulkarni and co-workers described a one-flow synthesis of ivacaftor (39) involving an ozonolysis reaction as a key-step (Fig. 12).46 Initially, the synthesis was developed in batch after which every step was separately optimised in flow and combined in a one-flow system. The key-step ozonolysis in flow appeared particularly challenging since known reagents such as PPh3, NaBH4, H2O2 and P(OEt)3 to convert the intermediate ozonide were not successful. In the approach of Kulkarni the use of these reagents was avoided, which would lead to a more facile purification step. The synthesis started with the flow ozonolysis of indole 37, after which the carbonyl oxide intermediate was captured in situ followed by a reduction to intermediate 38. The reaction was completed within 2 seconds and gave an overall throughput of 84 grams per day in a 30 mL reactor after optimisation. After the ozonolysis, a cyclisation was performed in DMF/DMA of which the reaction time of 12 h was reduced to less than 60 min by increasing the concentration. Lastly, ivacaftor (39) was obtained by methoxide-mediated cleavage of the carbonate. The overall yield was 60% for the three steps, resulting in a production of 7.2 gram of ivacaftor (39) per day.
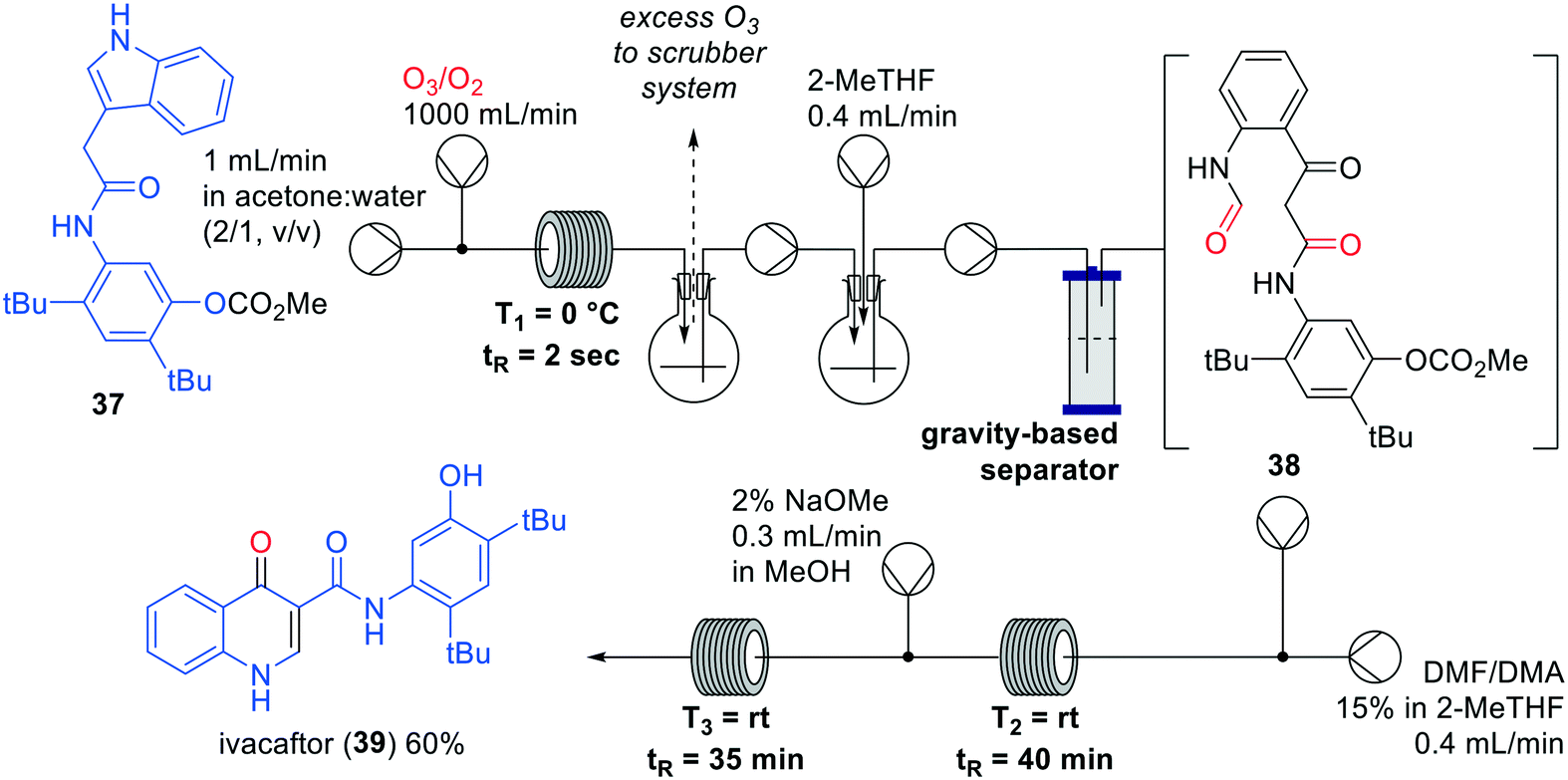 | ||
| Fig. 12 Synthesis of Ivacaftor (39) by Kulkarni and co-workers making use of an interrupted one-flow system. Adapted with permission from React. Chem. Eng., 2018, 3, 520–526. Copyright 2018 Royal Society of Chemistry.46 | ||
Flibanserin (44) has been developed as a treatment for depression, but eventually approved as treatment for female hypoactive sexual desire disorder. In literature several batch syntheses have been described with yields ranging from 32–51%.47–49 Greiner and co-workers developed an interrupted one-flow system for the synthesis of flibanserin (44) with a minimal number of manual unit operations.50 Previous existing batch protocols to flibanserin included protecting group introduction and removal as separate steps which was not considered feasible.49 Alternatively, reported N-alkylations were challenging and required forcing reaction conditions with insoluble bases.47 The authors therefore devised a novel synthetic route for the synthesis of flibanserin in flow (Fig. 13).
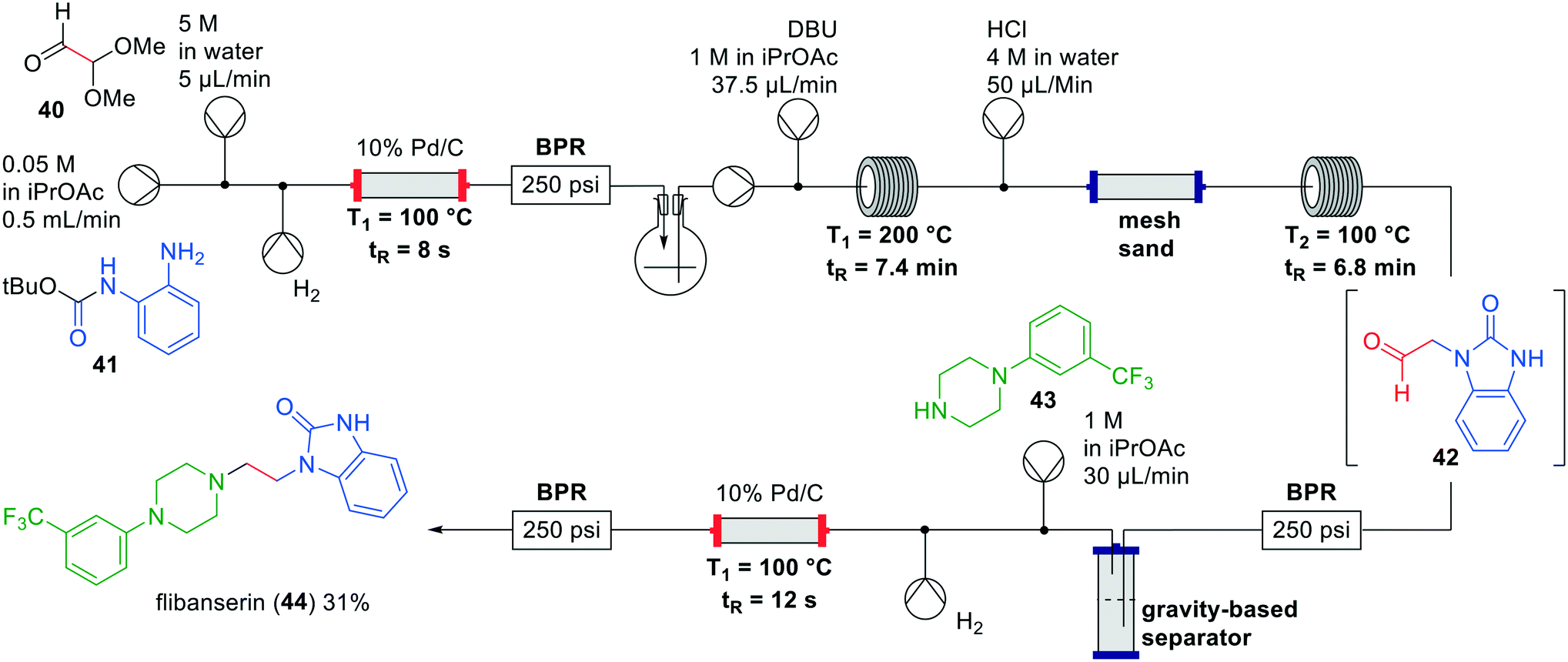 | ||
| Fig. 13 Synthesis of flibanserin (44) by Greiner and co-workers making use of an interrupted one-flow system. Adapted with permission from React. Chem. Eng., 2019, 4, 652–657. Copyright 2019 Royal Society of Chemistry.50 | ||
The authors started by investigating suitable solvents for the reagents, intermediates and catalyst, which led to isopropyl acetate as the preferred solvent. Additionally, polar compounds formed in the process such as tert-butanol and methanol acted as co-solvents assisting in dissolving some of the intermediates involved. As in most one-flow processes, the individual steps were optimised first and afterwards combined in a single system. Starting from aniline 41 a reductive amination was performed in a continuous flow hydrogenation reactor at 100 °C. The product mixture was collected in a flask for degassing and pumped into the next section. The authors then investigated the ring-closure to the corresponding benzimidazolone, which took place with DBU at 200 °C as opposed to NaH and KOtBu that were used in batch experiments, but gave solubility problems in flow. HCl-mediated acetal hydrolysis showed the best results albeit that initially the desired product 42 was present in both layers after the gravity based extraction. In order to optimise this conversion, the ratio between the aqueous and organic phase was made 1:10 respectively to maximise the material in the organic phase, and mixed using an inert packed column with mesh sand. The final reductive amination with piperazine 43 was again performed in a continuous flow hydrogenation reactor at 100 °C. Combining all these steps in this interrupted one-flow system led to the synthesis of flibanserin (44) in a yield of 31% and a residence time of approximately 20 min.
Jamison and co-workers developed a seven-step one-flow system for the synthesis of linezolid (50), a drug that is used as a last line of defence against multi-drug resistant gram-positive bacteria.51 This seven-step synthesis works in a fully continuous manner without solvent exchanges, but utilises a gravity-based liquid–liquid separator.52 The existing batch synthesis of linezolid (50) is rather time consuming,53 but with Jamison's investigations a fast protecting group free flow synthesis of linezolid has become available (Fig. 14).52 Starting from (+)-epichlorohydrin (45) a Ritter-type reaction was performed with acetonitrile using BF3·OEt2 as the Lewis acid. Several side reactions were observed, however, which was overcome by changing to the corresponding dibutyl etherate. It was hypothesised that the reactivity of BF3·OBu2 is slightly diminished by the sterically more demanding butyl groups, thereby preventing the side reactions from occurring. Next, isopropanol was added to the system to trap the nitrilium ion and quench the Lewis to the corresponding solubilised boric ester. The desired epoxide 48 was obtained after the addition of lithium tert-butoxide in THF and 1,2-dichloroethane, by efficient intramolecular substitution of the chloride. The addition of 1,2-dichloroethane was necessary to solubilise the formed lithium chloride and avoid precipitation. Having established the successful construction of epoxide 48, the synthesis of the second building block, aniline 49, was investigated.
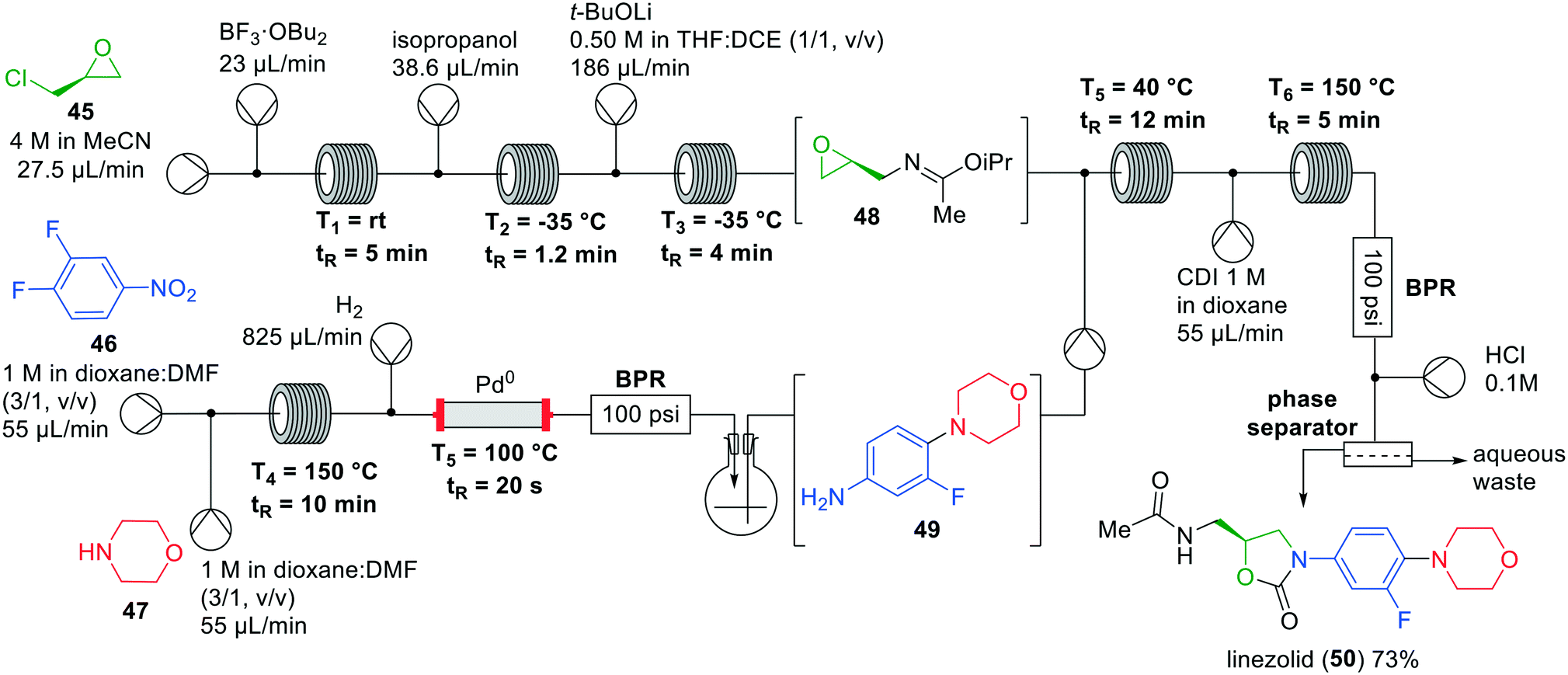 | ||
| Fig. 14 Synthesis of linezolid (50) by Jamison and co-workers making use of an interrupted one-flow system. Adapted with permission from Angew. Chem., Int. Ed., 2019, 58, 7678–7681. Copyright 2019 Wiley-VCH.52 | ||
The aniline building block was formed by nucleophilic aromatic substitution of morpholine (47) with nitrobenzene 46, followed by hydrogenation to the aniline. To remove the excess of H2 gas the reaction mixture was transferred into a flask, which was directly pumped into the flow system for coupling with epoxide 48. Nucleophilic epoxide opening, followed by reaction with CDI and subsequent cyclisation at 150 °C provided the oxazolidinone ring. Finally, acidic hydrolysis of the imino ether and aqueous extraction provided linezolid (50) in an isolated overall yield of 73%.
Conclusion and outlook
A vast number of examples in literature have demonstrated that continuous flow chemistry is a relevant approach to synthesise APIs.4 This review has listed a series of one-flow and interrupted one-flow approaches, aimed at providing insights in the challenges that researchers face when trying to establish a one-flow multistep synthesis system.The advantages of continuous flow for the synthesis of APIs compared to batch chemistry are numerous.54 Flow chemistry allows for a large variety of chemical conversions and specific reaction conditions tailored to the reactants to optimise the synthesis.55,56 Reaction time, mixing, temperature, stoichiometry, concentration, (side-)product solubility, pressure control and continuous operating modes are a few of the benefits which are widely used in the pharmaceutical industry.57
Currently, however, numerous biologically relevant molecules are still prepared in a batch fashion, as most of organic chemistry is still focused on this conventional methodology. Batch chemistry is more straightforward to execute and requires readily available multi-purpose flasks and reactors and is hence the primary choice for the synthetic organic chemist. Also chemistry education still heavily relies on traditional batchwise transformations.58 Inversely, flow chemistry approaches are typically not considered by chemists because of the higher level of complexity of the equipment and lack of hands-on experience.59 The transition from batch to flow chemistry remains a challenge, not only in chemical education, but also in academic and industrial labs. It is a process that requires training, choosing the right equipment, and investments in dedicated flow machinery.60 We firmly feel though that eventually the transition into flow chemistry will result in more safe and efficient laboratory and manufacturing practices.
Notwithstanding, flow chemistry does not solve all shortcomings in the conventional synthesis of pharmaceuticals. Chemical transformations which utilise gaseous reagents or show formation of gaseous (side-)products, like hydride reductions, increase overall pressure in flow systems.10 Moreover, by combining multiple reactors in one continuous setup, the small dimensions of microchannels in the flow system add additional pressure complications.3 While higher pressure can have a positive effect on various chemical reactions, it does put serious stress on all reaction equipment in the flow system. One could opt for more high-pressure equipment, but this can be a costly measure.60 Fortunately, interrupted one-flow chemistry solves these problems by release of pressure through isolation of an intermediate in a batch-like fashion. The reactants can then be concentrated in vacuo or used directly for the following chemical conversion using a peristaltic flow pump.
Additionally, the formation of products with low solubility, like salts, pose a challenge to flow chemistry. As highlighted in this review, the search for solvents which circumvent precipitation of (side-)products is often accompanied with solvent-catalyst incompatibility. A direct solution can be found in the implementation of a continuous phase separator, which allows for purification and removal of unwanted (aqueous) side products. This equipment, however, is not commonly employed, and again has technical limitations concerning lifetime of the membrane separator, cost, and incompatibility with various solvents and reagents.31,33,50,52 Alternatively, continuous gravity-based phase separation techniques can circumvent these problems.61 Finally, solvent-catalyst incompatibilities can be overcome using solvent switching techniques.62 This introduces some complexity in the flow system, but its feasibility was recently demonstrated by Ley and co-workers.63,64
In conclusion, we have distinguished between interrupted and non-interrupted one-flow approaches for the synthesis of APIs. Moreover, we demonstrated the limitations of the reported flow syntheses, and compared it with earlier conducted batch chemistry approaches. We envision that the preparation of biologically relevant molecules will shift from batch to (interrupted) one-flow chemistry, of which the latter is unmatched in tailored reaction conditions and precision optimisations. Though we feel that the shift to this revolutionary chemistry is only beginning, we are excited to contribute to this next generation organic synthesis.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We kindly acknowledge funding by the H2020-FETOPEN-2016-2017 programme of the European Commission (Grant agreement number: 737266-ONE FLOW).Notes and references
- D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680 RSC.
- S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chem., Int. Ed., 2015, 54, 3449–3464 CrossRef CAS PubMed.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194–1219 CrossRef CAS PubMed.
- R. Gérardy, N. Emmanuel, T. Toupy, V. E. Kassin, N. N. Tshibalonza, M. Schmitz and J. C. M. Monbaliu, Eur. J. Org. Chem., 2018, 2018, 2301–2351 Search PubMed.
- M. Movsisyan, E. Delbeke, J. Berton, C. Battilocchio, S. Ley and C. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- P. D. Morse, R. L. Beingessner and T. F. Jamison, Isr. J. Chem., 2017, 57, 218–227 CrossRef CAS.
- L. Rogers and K. F. Jensen, Green Chem., 2019, 21, 3481–3498 RSC.
- R. Mechoulam, P. Braun and Y. Gaoni, J. Am. Chem. Soc., 1967, 89, 4552–4554 CrossRef CAS PubMed.
- C. J. Mallia and I. R. Baxendale, Org. Process Res. Dev., 2016, 20, 327–360 CrossRef CAS.
- M. Berton, J. M. de Souza, I. Abdiaj, D. T. McQuade and D. R. Snead, J. Flow Chem., 2020, 10, 73–92 Search PubMed.
- A. Nagaki, K. Imai, H. Kim and J.-i. Yoshida, RSC Adv., 2011, 1, 758–760 RSC.
- J.-i. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896–9904 RSC.
- M. D. Hopkin, I. R. Baxendale and S. V. Ley, Chem. Commun., 2010, 46, 2450–2452 RSC.
- W. C. Fu and T. F. Jamison, Org. Lett., 2019, 21, 6112–6116 CrossRef CAS PubMed.
- J. C. Yang, D. Niu, B. P. Karsten, F. Lima and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 2531–2535 CrossRef CAS PubMed.
- N. R. Poulter, D. Prabhakaran and M. Caulfield, Lancet, 2015, 386, 801–812 Search PubMed.
- C. P. Breen and T. F. Jamison, Chem. – Eur. J., 2019, 25, 14527–14531 CrossRef CAS PubMed.
- P. Zhang, M. G. Russell and T. F. Jamison, Org. Process Res. Dev., 2014, 18, 1567–1570 CrossRef CAS.
- L. Sorbera, P. Leeson, X. Rabasseda and J. Castaner, Drugs Future, 2000, 25, 1145–1149 CrossRef CAS.
- D. Ott, S. Borukhova and V. Hessel, Green Chem., 2016, 18, 1096–1116 RSC.
- S. Borukhova, T. Noël, B. Metten, E. de Vos and V. Hessel, ChemSusChem, 2013, 6, 2220–2225 CrossRef CAS PubMed.
- A. R. Bogdan and N. W. Sach, Adv. Synth. Catal., 2009, 351, 849–854 CrossRef CAS.
- S. Chatterjee, M. Guidi, P. H. Seeberger and K. Gilmore, Nature, 2020, 579, 379–384 CrossRef CAS PubMed.
- J. Magano, Chem. Rev., 2009, 109, 4398–4438 CrossRef CAS PubMed.
- T. Mukaiyama, H. Ishikawa, H. Koshino and Y. Hayashi, Chem. – Eur. J., 2013, 19, 17789–17800 Search PubMed.
- S. Ogasawara and Y. Hayashi, Synthesis, 2017, 49, 424–428 CAS.
- D. R. Snead and T. F. Jamison, Chem. Sci., 2013, 4, 2822–2827 RSC.
- J. Britton and T. F. Jamison, Angew. Chem., Int. Ed., 2017, 56, 8823–8827 CrossRef CAS PubMed.
- C. Dai, D. R. Snead, P. Zhang and T. F. Jamison, J. Flow Chem., 2015, 5, 133–138 Search PubMed.
- A.-C. Bédard, A. R. Longstreet, J. Britton, Y. Wang, H. Moriguchi, R. W. Hicklin, W. H. Green and T. F. Jamison, Bioorg. Med. Chem., 2017, 25, 6233–6241 CrossRef PubMed.
- T. Chakkath, S. Lavergne, T. M. Fan, D. Bunick and L. Dirikolu, Vet. Sci., 2015, 2, 52–68 CrossRef.
- Z. Jaman, T. J. Sobreira, A. Mufti, C. R. Ferreira, R. G. Cooks and D. H. Thompson, Org. Process Res. Dev., 2019, 23, 334–341 CrossRef CAS.
- M. J. Pedersen, S. Born, U. Neuenschwander, T. Skovby, M. J. Mealy, S. Kiil, K. Dam-Johansen and K. F. Jensen, Ind. Eng. Chem. Res., 2018, 57, 4859–4866 CrossRef CAS.
- M. J. Pedersen, T. Skovby, M. J. Mealy, K. Dam-Johansen and S. Kiil, Org. Process Res. Dev., 2018, 22, 228–235 CrossRef CAS.
- I. Nekkaa, M. Palkó, I. M. Mándity and F. Fülöp, Beilstein J. Org. Chem., 2018, 14, 318–324 Search PubMed.
- C. A. Hone and C. O. Kappe, in Accounts on Sustainable Flow Chemistry, ed. T. Noël and R. Luque, Springer International Publishing, Cham, 2020, pp. 67–110, DOI:10.1007/978-3-030-36572-1_3.
- R. Labes, C. Battilocchio, C. Mateos, G. R. Cumming, O. de Frutos, J. A. Rincón, K. Binder and S. V. Ley, Org. Process Res. Dev., 2017, 21, 1419–1422 CrossRef CAS.
- K. Masuda, T. Ichitsuka, N. Koumura, K. Sato and S. Kobayashi, Tetrahedron, 2018, 74, 1705–1730 CrossRef CAS.
- N. Cherkasov, A. O. Ibhadon and E. V. Rebrov, Lab Chip, 2015, 15, 1952–1960 Search PubMed.
- D. R. Snead and T. F. Jamison, Angew. Chem., Int. Ed., 2015, 54, 983–987 CrossRef CAS PubMed.
- H. Lin, C. Dai, T. F. Jamison and K. F. Jensen, Angew. Chem., Int. Ed., 2017, 56, 8870–8873 CrossRef CAS PubMed.
- M. K. Sharma, A. Suru, A. Joshi and A. A. Kulkarni, Ind. Eng. Chem. Res., 2020 DOI:10.1021/acs.iecr.9b06864.
- T. Tsubogo, H. Oyamada and S. Kobayashi, Nature, 2015, 520, 329–332 CrossRef CAS PubMed.
- T. Tsubogo, Y. Yamashita and S. Kobayashi, Chem. – Eur. J., 2012, 18, 13624–13628 CrossRef CAS.
- N. Vasudevan, M. K. Sharma, D. S. Reddy and A. A. Kulkarni, React. Chem. Eng., 2018, 3, 520–526 RSC.
- R. D. Shingare, A. S. Kulkarni, R. L. Sutar and D. S. Reddy, ACS Omega, 2017, 2, 5137–5141 CrossRef CAS PubMed.
- G. Bietti, F. Borsini, M. Turconi, E. Giraldo and M. Bignotti, United States Pat., US5576318A, 1991 Search PubMed.
- F. Yang, C. Wu, Z. Li, G. Tian, J. Wu, F. Zhu, J. Zhang, Y. He and J. Shen, Org. Process Res. Dev., 2016, 20, 1576–1580 Search PubMed.
- P. Bana, Á. Szigetvári, J. Kóti, J. Éles and I. Greiner, React. Chem. Eng., 2019, 4, 652–657 RSC.
- D. Clemett and A. Markham, Drugs, 2000, 59, 815–827 CrossRef CAS.
- M. G. Russell and T. F. Jamison, Angew. Chem., Int. Ed., 2019, 58, 7678–7681 CrossRef CAS.
- W. R. Perrault, B. A. Pearlman, D. B. Godrej, A. Jeganathan, K. Yamagata, J. J. Chen, C. V. Lu, P. M. Herrinton, R. C. Gadwood and L. Chan, Org. Process Res. Dev., 2003, 7, 533–546 CrossRef CAS.
- I. R. Baxendale, J. Chem. Technol. Biotechnol., 2013, 88, 519–552 Search PubMed.
- M. K. Sharma, R. B. Acharya, C. A. Shukla and A. A. Kulkarni, Beilstein J. Org. Chem., 2018, 14, 1917–1936 CrossRef CAS PubMed.
- I. R. Baxendale, L. Brocken and C. J. Mallia, Green Process. Synth., 2013, 2, 211–230 CAS.
- E. Palao and J. Alcazar, in Flow Chemistry: Integrated Approaches for Practical Applications, ed. Luis, S. V and E. Garcia-Verdugo, The Royal Society of Chemistry, 2019, pp. 86–128 Search PubMed.
- D. Blanco-Ania and F. P. J. T. Rutjes, J. Flow Chem., 2017, 7, 157–158 CrossRef CAS.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS.
- J. H. Bannock, S. H. Krishnadasan, M. Heeney and J. C. de Mello, Mater. Horiz., 2014, 1, 373–378 Search PubMed.
- D. E. Fitzpatrick, T. Maujean, A. C. Evans and S. V. Ley, Angew. Chem., Int. Ed., 2018, 57, 15128–15132 CrossRef CAS PubMed.
- P. Gruber, M. P. C. Marques, B. O'Sullivan, F. Baganz, R. Wohlgemuth and N. Szita, Biotechnol. J., 2017, 12, 1700030 CrossRef PubMed.
- C. Battilocchio, B. J. Deadman, N. Nikbin, M. O. Kitching, I. R. Baxendale and S. V. Ley, Chem. – Eur. J., 2013, 19, 7917–7930 Search PubMed.
- B. J. Deadman, C. Battilocchio, E. Sliwinski and S. V. Ley, Green Chem., 2013, 15, 2050–2055 RSC.
| This journal is © The Royal Society of Chemistry 2020 |