DOI:
10.1039/C9RE00467J
(Communication)
React. Chem. Eng., 2020,
5, 263-269
A chemo-enzymatic tandem reaction in a mixture of deep eutectic solvent and water in continuous flow†
Received
5th December 2019
, Accepted 3rd January 2020
First published on 3rd January 2020
Abstract
The combination of metal- and biocatalysis is a challenging but forward-looking topic in synthetic chemistry. The unique selectivity of enzymes paired with the broad range of applications of chemical catalysts enables an undreamed-of number of novel processes. Herein, we describe the application of immobilized phenolic acid decarboxylase (PAD) for the decarboxylation of para-coumaric acid and subsequent Pd-catalyzed Heck cross-coupling with an aryl halide in a fully integrated two-step continuous flow process to synthesize (E)-4-hydroxy-stilbene. The application of a choline chloride-based deep eutectic solvent (DES) proved to be crucial to overcome the problem of solvent compatibility and enabled an increase in substrate concentration (from 5 mM in buffer to 20 mM in DES) as well as a process with a homogeneous starting solution. The two-step process was successfully operated for more than 16 h in continuous flow and full conversion was achieved. The results underline the usefulness of DES to overcome compatibility problems in tandem-catalytic processes. The system benefits from its simplicity due to increased substrate solubility, the possibility to conduct both reactions at their optimal temperatures and the elimination of isolating the reaction intermediate, which is prone to polymerization.
Introduction
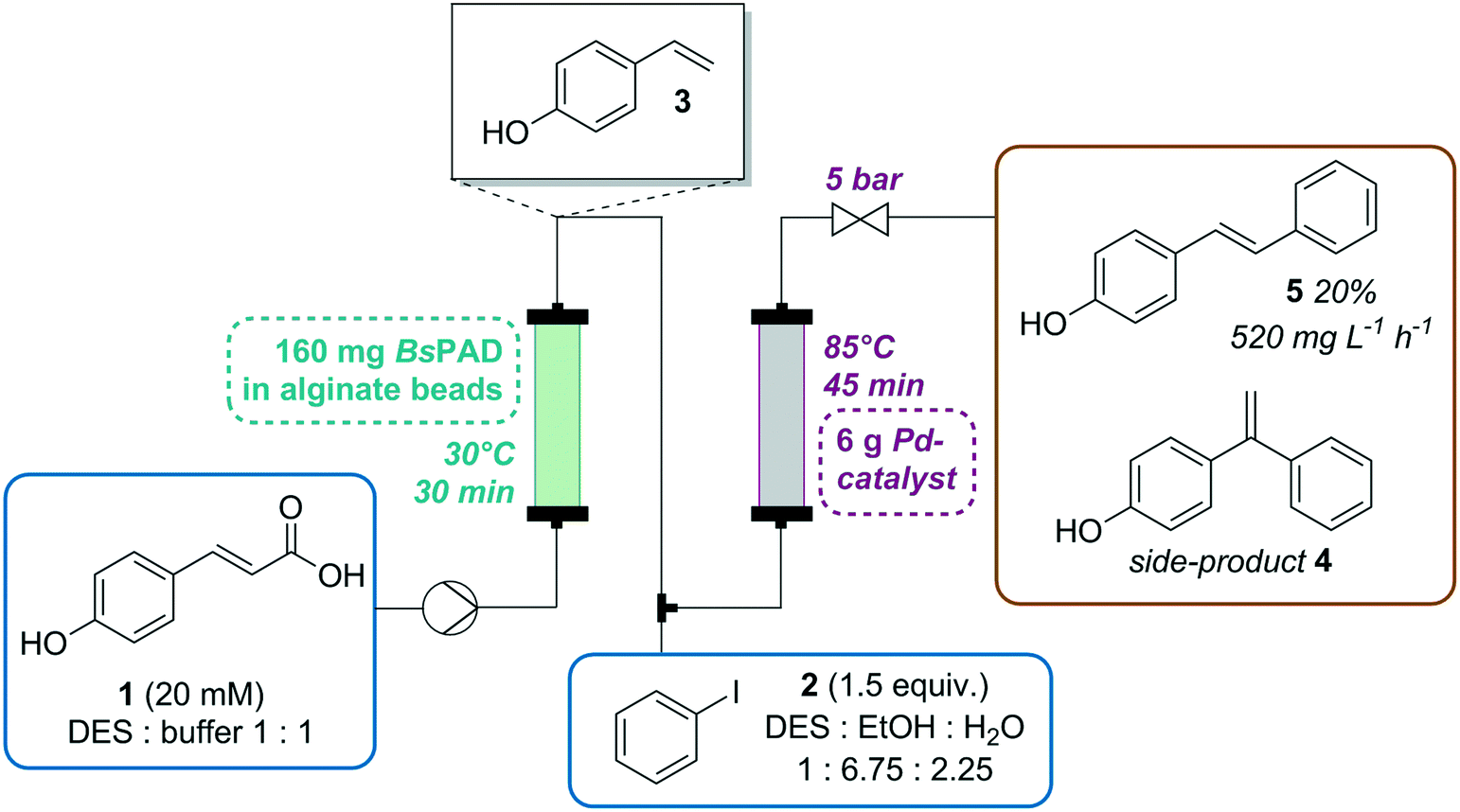
Chemo-enzymatic one-pot syntheses have been attracting increasing attention of researchers in the past decade.1–7 These tandem reactions offer the best of both worlds, the versatility of traditional chemical catalysis is replenished with the unbeatable selectivity and mild reaction conditions of enzymes. Nevertheless, besides numerous advantages these promising systems also pose several challenges. One obstacle is the compatibility of the considerable differences of ideal reaction conditions for the individual process steps.4,8 Especially, the choice of an appropriate solvent is crucial. Most enzymes show optimal activity in aqueous systems. However, low substrate solubility and the hydrolysis sensitivity of many chemo-catalysts are crucial arguments against the use of neat buffer systems. Alternative solvents, such as deep eutectic solvents (DESs) can help to overcome this obstacle of solvent compatibility.9–13 DESs are mixtures of two or more components which have a lower melting point than the individual compounds at a certain mixing ratio.14 In recent years these solvents have gained attention due to their simple preparation and tuneable properties. With lower intrinsic toxicity than ionic liquids, DESs have emerged as alternative solutions for issues such as low substrate solubility, enzyme activity and stability. Besides theses advantages, DESs also bring along some drawbacks including occasional toxicity and high viscosity, depending on the starting materials. The latter is a challenge in the scale-up of processes, but can be tackeled by mixing with water.15–18 Up to 50% of water the characteristics of DESs is preserved. In mixtures containing a lower fraction of DES (in the second step in the process described in this work, after adding ethanol and water) their properties are comparable with salt solutions.19 This good miscibility with water becomes an issue, when the environmental compatibility is discussed. Therefore, a well thought-out process and post-process treatment of DESs and it mixtures with water is required in order to get DES into industrial application.20,21 Nevertheless, an impressive series of chemical22,23 and enzymatic24–26 reactions and also flow applications27–29 were successfully conducted in DESs. We have recently shown the feasibility to combine the enzymatic decarboxylation of hydroxycinnamic acids catalyzed by phenolic acid decarboxylase from Bacillus subtilis (BsPAD) in water and Ru-catalyzed homo-coupling in organic solvents in a one-pot reaction for the synthesis of a series of bio-based stilbene derivatives.30 In order to expand the scope of the decarboxylation reaction towards the synthesis of asymmetric stilbenes, we envisioned a combination of enzymatic decarboxylation with the Heck reaction.31 The most prominent derivatives of stilbenes are resveratrol and its analogues. Due to their conjugated double bond system, many stilbenes possess anti-oxidant, anti-inflammatory, antidiabetic and antiaging properties32,33 and are under investigation as active pharmaceutical ingredients (APIs) for cancer preventive drugs.34 For the synthesis of these important compounds, encapsulation proved to be a practical approach to achieve spatial and temporal separation of the enzymatic in aqueous environment and the metal-catalyzed step in organic solvent.30 Yet, biphasic systems are complex and difficult to scale. Therefore, the goal of this work is a continuous flow process using a solvent compatible for both catalysts and allowing high substrate loadings. Continuous flow synthesis yields a number of benefits over batch operation. Besides consistent product quality and reduced reaction time, the elimination of separation of the catalyst from the reaction solution is a major advantage.35 Furthermore, splitting the two reaction steps into separate continuous flow reactors in a row made it possible to conduct the reaction at optimal reaction temperature for both catalysts (30 °C for enzymatic reaction, 85 °C for Heck reaction). Yet, the limited solubility of hydroxycinnamic acids in buffer of a few mM and the need to add additional 30% ethanol for the Heck reaction would require to operate with very diluted substrate solutions in order to prevent reactor clogging. Recent findings showed that PAD is highly active in DES.36 Experiments on interactions of the used DES and the biocatalyst were not coducted so far and are not scope of this work. A series of recently investigated DES-protein systems can be found in literature.37 Encouraged by the fact that PAD is highly active in DES, we envisioned that this would be the appropriate solution for the transformation of the one-pot reactions to an integrated multi-step continuous flow process. Furthermore, the combination of both reactions as cascade in flow lower the risks associated to the spontaneous polymerization of the intermediate hydroxystyrene. While conventional flow chemistry,38–43 also including C–C coupling reactions,39,44–47 and continuous biocatalysis48–50 are currently in the focus of many researchers, examples for their successful combination are rare, which can also be attributed by the difficulties to find suitable reaction media.51 In this work we present a fully integrated two-step flow setup consisting of an enzymatic decarboxylation with a phenolic acid decarboxylase from Bacillus subtilis (BsPAD), followed by a Heck reaction catalyzed by a Pd-substituted Ce–Sn-oxide as shown in Scheme 1. For the proof of concept of the synthesis of asymmetric stilbenes, we chose the synthesis of (E)-4-hydroxy stilbene 5 from para-coumaric acid 1 and iodobenzene 2 because of the low price and good availability of these substrates.
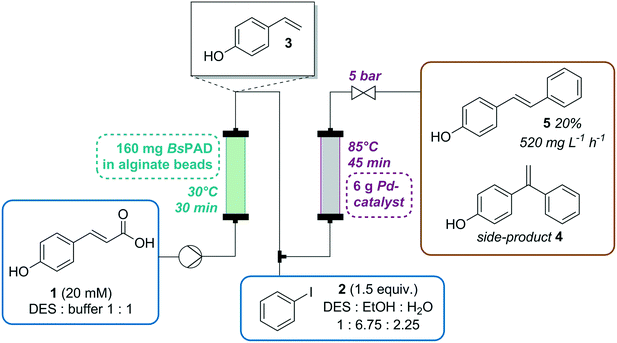 |
| | Scheme 1 General reaction scheme for enzymatic decarboxylation with a phenolic acid decarboxylase from Bacillus subtilis (BsPAD) yielding 4-ethenylphenol 3, which serves as substrate for a Pd-catalyzed cross-coupling reaction with aryl halide 2, resulting in (E)-4-hydroxy-stilbene 5 and the side product (para-hydroxy-1,1-diphenylethylene 4). | |
Results and discussion
Decarboxylation in batch
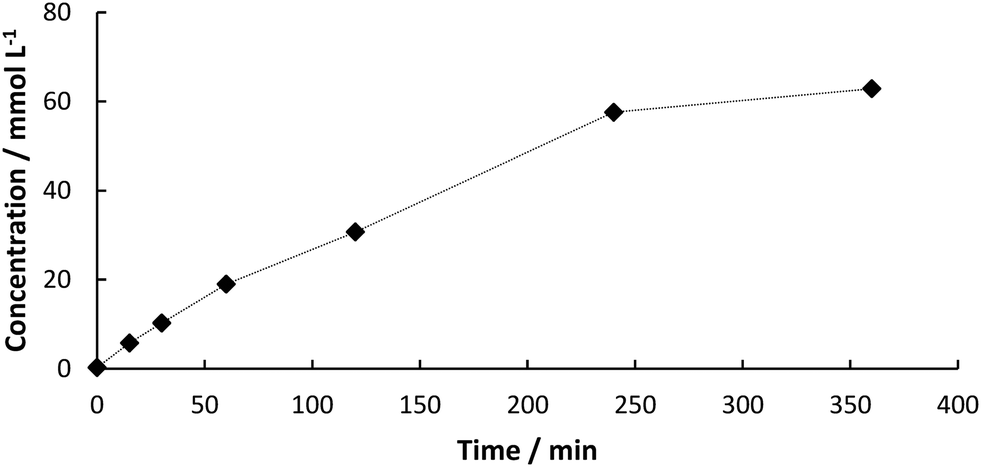
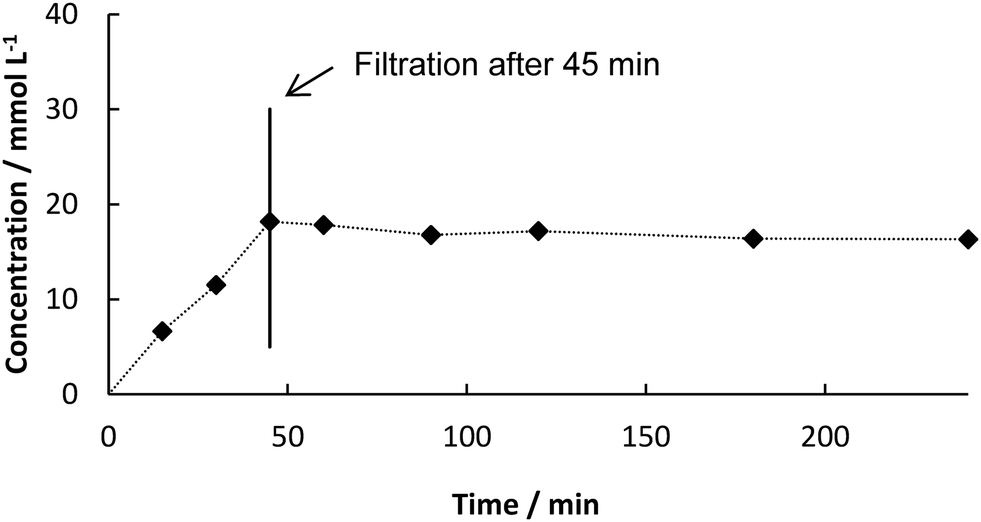
We first started with batch experiments to investigate the compatibility of the systems. We recently showed the excellent compatibility of BsPAD with DESs.36 Choline chloride (ChCl)-based eutectic mixtures were tested neat and in dilution with potassium phosphate buffer (50 mM, pH 6.0) for the decarboxylation of a variety of phenolic acid derivatives. Our results proved that not only the free enzyme, but also immobilized PAD shows excellent activity in deep eutectic solvents. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) dilution of ChCl/glycerol 1:2 (mol/mol) with buffer gave the best results for PAD. Mixing DES with buffer has not only the advantage of increased enzyme activity and enhanced CO2 release from the system, but also decreases the viscosity of the solvent system enabling its application in continuous flow.29 With up to 50% water the properties of the DES are predominant and substrate solubility is significantly increased.19 Therefore, this solvent system was chosen for the first step in our two-step synthesis. As the final process requires a heterogeneous biocatalyst, PAD cell-free extract (CFE) was immobilized in 2% alginate beads and tested in the DES:buffer mixture. The formation of the decarboxylation product 3 proceeded linearly (Fig. 1) and full conversion of 1 was reached after 6 h (70 mM) and a yield of 3 of 90% was determined by HPLC. In order to test the heterogeneity of the biocatalyst, a “hot”-filtration test was conducted.52Fig. 2 shows the formation of 3 in the reaction mixture until the beads were filtered off after 45 min reaction time, which stopped the reaction, thus indicating that no active enzyme leaches from the alginate beads under these reaction conditions (details in ESI†). Since the hot filtration test does not prove heterogeneity of a catalyst as a standaline method, further investigations regarding enzyme leaching were required in the flow experiment.
:1 (v/v) dilution of ChCl/glycerol 1:2 (mol/mol) with buffer gave the best results for PAD. Mixing DES with buffer has not only the advantage of increased enzyme activity and enhanced CO2 release from the system, but also decreases the viscosity of the solvent system enabling its application in continuous flow.29 With up to 50% water the properties of the DES are predominant and substrate solubility is significantly increased.19 Therefore, this solvent system was chosen for the first step in our two-step synthesis. As the final process requires a heterogeneous biocatalyst, PAD cell-free extract (CFE) was immobilized in 2% alginate beads and tested in the DES:buffer mixture. The formation of the decarboxylation product 3 proceeded linearly (Fig. 1) and full conversion of 1 was reached after 6 h (70 mM) and a yield of 3 of 90% was determined by HPLC. In order to test the heterogeneity of the biocatalyst, a “hot”-filtration test was conducted.52Fig. 2 shows the formation of 3 in the reaction mixture until the beads were filtered off after 45 min reaction time, which stopped the reaction, thus indicating that no active enzyme leaches from the alginate beads under these reaction conditions (details in ESI†). Since the hot filtration test does not prove heterogeneity of a catalyst as a standaline method, further investigations regarding enzyme leaching were required in the flow experiment.
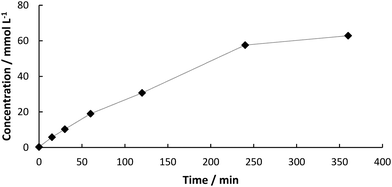 |
| | Fig. 1 Concentration of 3 from the decarboxylation of 1 (0.7 mmol) using BsPAD immobilized in alginate beads (2% (w/v) Na-alginate and 38.3 mg BsPAD (CFE) in potassium phosphate buffer (1 mL, 50 mM, pH 6.0), cross-linking was induced by 2% (w/v) BaCl2 solution), 10 mL solvent (DES:buffer 1:1 (v/v)), 30 °C. | |
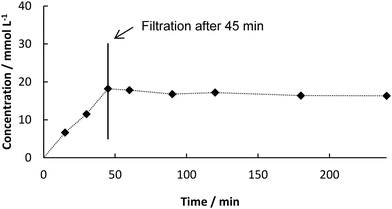 |
| | Fig. 2 Concentration of 3 from decarboxylation of 1 using BsPAD immobilized in alginate beads (2% (w/v) Na-alginate and 38.3 mg BsPAD (CFE) in 1 mL potassium phosphate buffer (50 mM, pH 6.0)), 100 mM substrate in 10 mL solvent (DES:buffer 1:1 (v/v)), 30 °C, catalyst beads filtered off after 45 min. | |
Heck coupling in batch
For the second step, the Heck coupling of 3 with 2, an in-house developed heterogeneous Pd-catalyst45,53‡ (Pd-substituted cerium–tin-oxide with the molecular formula Sn0.79Ce0.20Pd0.01O2−δ) was successfully tested for its activity in the novel solvent system. Full conversion of 3 was reached within 30 min (Fig. 3) with a mixture of DES:buffer 1:1 (v/v) (from the first step in the continuous application) plus ethanol:water 1:1 (v/v) containing substrate 2 and the required base, K2CO3, (both in 1.5 mol-eq. with respect to 3).
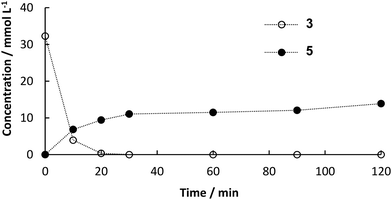 |
| | Fig. 3 Concentration of 3 (0.7 mmol) and 5 in a batch Heck coupling with 2 (1.5 mol-eq.) in the presence of a base (K2CO3, 1.5 mol-eq.) using Sn0.79Ce0.20Pd0.01O2−δ as heterogeneous catalyst (0.05 mol%) in 20 mL solvent (DES:buffer:ethanol:water 1:1:1:1 (v/v/v/v)), 85 °C. | |
Due to the similar reactivity of both sp2-atoms in the terminal olefin group, formation of two isomers (Scheme 1) is expected during the Heck reaction, that can either be separated or applied as antioxidant mixture. Unfortunately, there is a number of potential side-reactions, such as homo-coupling of 2 or 3, subsequent Heck reaction on the double-bond of the product or polymerization of 3. These reactions led to a low isolated yield of the desired product 5 of 35% in the batch reaction (see ESI†).
Combined decarboxylation and Heck coupling in batch
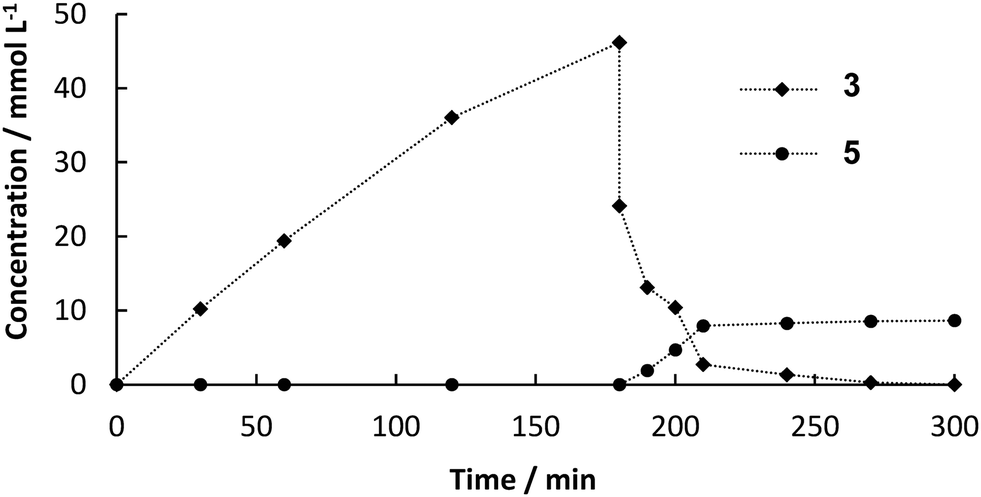
A one-pot reaction was conducted to investigate potential negative impact of the first step on the second one (Fig. 4). A suspension of 1 (70 mM) was stirred together with 80 mg immobilized PAD alginate beads in DES:buffer 1:1 (v/v). After 180 min, the enzyme was filtered off and solvent, substrates and catalyst for the second step were added. Within 2 h all previously formed 3 was converted. Therefore, we can conclude that remaining 1, which was not converted in the first step, does not negatively affect the performance of the catalyst in the second step and does also not serve as substrate leading to unwanted side-products. It was a very important finding that components from the immobilized CFE do not interfere with the Heck-reaction, which is a crucial requirement for a continuous approach.
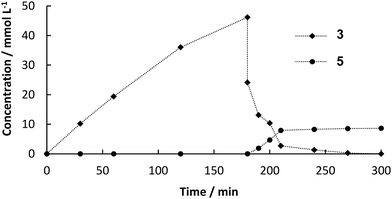 |
| | Fig. 4 Product formation over time in one-pot cascade reaction of 1 (0.7 mmol, 70 mM) in 10 mL DES:buffer 1:1 (v/v), 30 °C, 80 mg BsPAD immobilized in 2% (w/v) alginate beads. Beads were filtered off after 180 min and add 10 mL ethanol:water 1:1 (v/v) containing 2 and K2CO3 (1.05 mmol, 1.5 mol-eq. to 1), 25.2 mg (0.05 mol% Pd loading) of Pd-catalyst, 85 °C. | |
Individual steps in flow
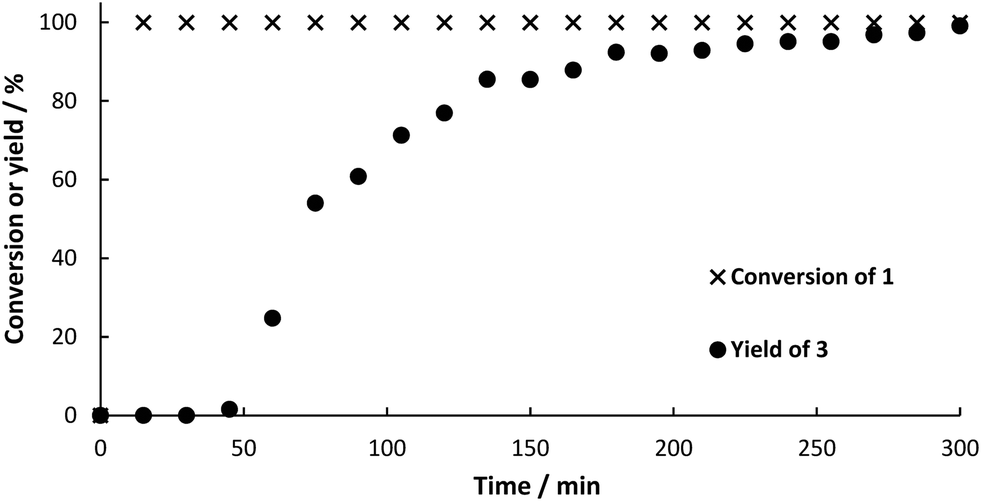
Before both reactions were combined in a two-step flow process, each reaction was tested in continuous mode. Immobilization of PAD in alginate beads with an average diameter of 2 mm proved to be a straightforward method using cell-free extract, thus avoiding any steps for enzyme purification. The beads were packed into two stainless steel columns (40 mm × 8 mm i.d.) connected in a series with a total volume of 4 mL. This “packed bed reactor” was heated to 30 °C in a water bath. The reactor was filled and flushed with solvent (DES:buffer 1:1 (v/v)) for 1 h before switching to the feed solution containing 1 (20 mM). This lower concentration was chosen to guarantee complete solubility of the substrate (substrate crystals need to be avoided in order to prevent clogging of the capillaries in the continuous setup). The feed stock was pumped with a syringe pump equipped with a 20 mL stainless steel syringe at a flow rate of 45.5 μL min−1 resulting in a residence time of 75 min in the whole set-up, of which 30 min was the residence time within the reactor. Product formation was followed by taking samples of the product stream in 15 min intervals (Fig. 5). The progress of this step could be followed visually as CO2 bubbles left the reactor as soon a full conversion of the substrate was achieved and no back pressure regulator was used. Details on the flow set-up are available in ESI.† In order to investigate the heterogeneity of the immobilized biocatalyst, samples were taken and not quenched, but additional substrate was added. No further product formation was observed, indicating that there is no active enzyme leaching into to product stream.
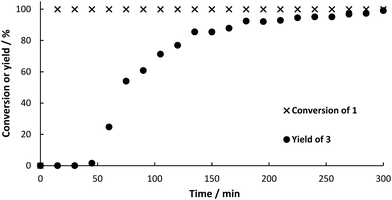 |
| | Fig. 5 Conversion‡ of 1 and yield‡ (determined by HPLC) of 3 for the continuous flow synthesis. 160 mg BsPAD immobilized in alginate beads (4 mL beads volume), 20 mM substrate in DES:buffer 1:1 (v/v), 30 °C, flow rate: 45.5 μL min−1. | |
Continuous Heck coupling was conducted by packing the catalyst powder (2 g, Sn0.79Ce0.20Pd0.01O2−δ) in a stainless steel column (40 mm × 8 mm i.d.).§ This packed bed reactor was heated to 85 °C in a water bath. The reactor was flushed with solvent (aqueous solution of DES:buffer:ethanol:water 1:1:1:1 (v/v/v/v)) for 1 h before switching to the feed stock, which contained 3 (10 mM, simulating the product stream from the first step) as well as 1.5 mol-eq. of 2 and K2CO3, respectively, dissolved in DES:buffer:ethanol:water 1:1:1:1 (v/v/v/v). The feed solution was pumped with a syringe pump equipped with a 20 mL stainless steel syringe at a flow rate of 91 μL min−1. Samples were taken from the product stream in 15 min intervals and analysed with HPLC. After a start-up time of 200 min a approximate steady-state with an average conversion of 90% of 3 was reached. The residence time within the reactor was determined to be 45 min. More information can be found in the ESI.†
Combined decarboxylation and Heck coupling in flow
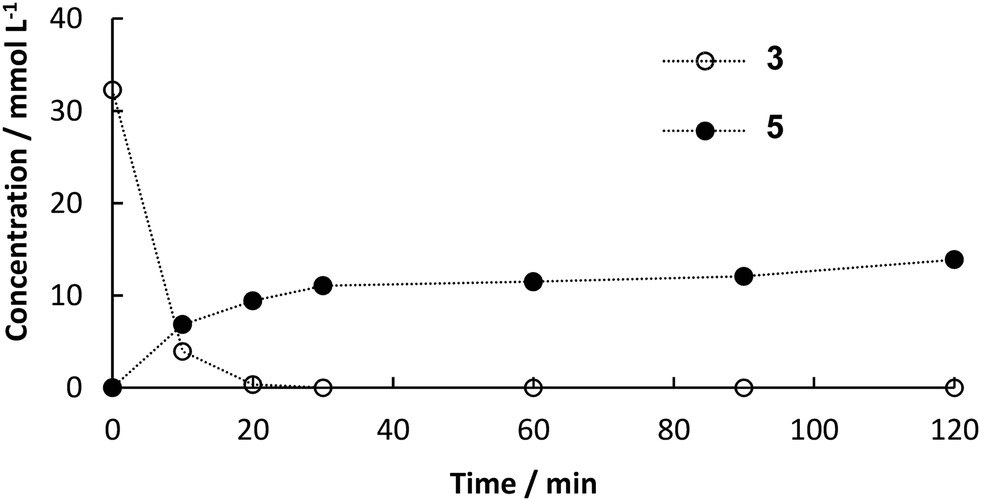
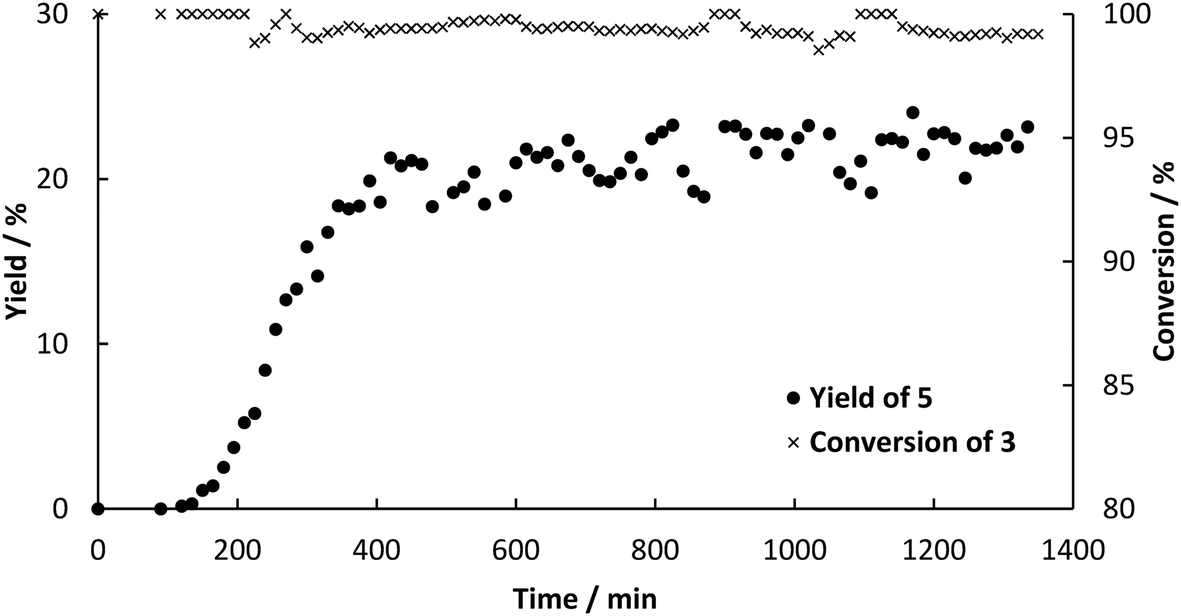
Since the outcome of both continuous processes was satisfactory for us, the combination of them was the next goal. For the first step, all the preparations were similar to the single step continuous flow. To improve the performance of the second step, some changes needed to be done. In order to achieve complete dissolution of the substrates, the solvent composition of the feed stock for the second step was adapted to DES:ethanol:H2O 1:6.75:2.25. Therefore, the final solvent composition for the Heck reaction was 30 vol% DES, 25 vol% buffer, 34 vol% ethanol and 11 vol% water. Furthermore, the amount of catalyst was increased. Instead of a column of 40 mm in length a column with the dimensions 120 mm × 8 mm i.d. was used (6 g of catalyst). The flow rate was set to 45.5 μL min−1 for both pumps (90 μL min−1 in sum). Samples of 100 μL were collected every 15 min at the outlet of the reactor. As soon as decarboxylation product was detected at the outlet of the a column packed with PAD, both steps were connected. CO2 initially leaving the decarboxylation in bubbles, were not observed after connecting the reactors, because the pressure increased to 5 bar and CO2 dissolved in the reaciton solvent. In a first attempt, some leaching of the enzyme led to channel clogging in the pre-heating tube before the Pd-packed reactor after 11 h (see Fig. S3 in ESI†). Blocking of the capillary reduced the supply of intermediate 3 for the second column, which led to increased formation of biphenyl by homo-coupling of the excess 2. Leaching could easily be overcome by a thorough overnight flushing of loosely bound enzyme from the surface prior to the continuous experiment. In addition to that, a filter (HPLC pre-column filter) was installed after the pre-heating channel, catching the denaturized enzyme. The filter could easily be changed and hence the continuous process could proceed for 16 h in a steady-state with full conversion of both 1 in the first step and 3 in the second step yielding an average of 20% of desired product 5 (Fig. 6). The yield in the batch process was higher than in the continuous setup, because after full conversion we observed isomerization of 4 to the more stable and desired product 5. The ratio of both products was determined to be 3:1 (mol/mol) and shift towards 5 with increasing reaction time. However, the rate of isomerization is too small to be considered as a way to synthesize 5. The product could easily be isolated by evaporation of ethanol followed by extraction in MTBE. The residence time in the reactors was determined to be 30 min for the decarboxylation and 45 min for the Heck coupling resulting in a space–time yield of 4.8 g L−1 h−1 for decarboxylation and 0.52 g L−1 h−1 for Heck coupling. Since constant feed of 3 could be achieved this time, side-product formation due to homo coupling of 2 could be limited to a maximum of 0.8 mM (see Fig. S4 ESI†). Other potential side reactions are polymerization of 3 resulting in high converstion, but not leading to any detectable product.
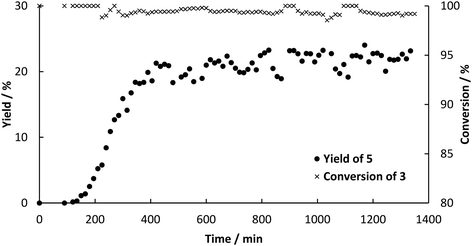 |
| | Fig. 6 Yield‡ (determined by HPLC) of 5 and conversion‡ of 3 over time for the continuous synthesis by enzymatic decarboxylation by BsPAD and subsequent Pd-catalyzed Heck reaction using Sn0.79Ce0.20Pd0.01O2−δ to couple 2 and 3. | |
Conclusion
In conclusion, we were able to develop a fully integrated flow process combining an enzymatic decarboxylation by encapsulation of the enzyme and subsequent Heck coupling via a heterogeneous Pd-catalyst. To our knowledge, this is the first example of a chemo-enzymatic cascade in flow using a non-conventional solvent system of DES with water. The outstanding potential of DES to overcome obstacles like substrate solubility and solvent compatibility is a major step towards broad application of continuous flow chemo-enzymatic reactions. In comparison to the one-pot setup, continuous flow requires a reduction in substrate concentration. However, long-term usage of catalysts in continuous flow is economically and ecologically more favourable. Admittedly, the complex chemistry of the chosen reaction leads to a number of side reactions and thus a low yield of the desired product. Nevertheless, due to the elimination of the isolation of the highly reactive intermediate, the use of bio-based, cheap substrates and the reuse of the expensive Pd-catalyst, this proof-of-concept can be the first step on the path to establish flow reactions in aqueous DES mixtures.
In near future further substrates will be tested for their potential to serve as bio-based substrates for the decarboxylation in order to extend the scope of this setup. Furthermore, experiments regarding catalyst and enzyme activity, stability and heterogeneity are planned. Additionally, optimization of process parameters and product ratio are currently under investigation.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- E. Liardo, N. Ríos-Lombardía, F. Morís, F. Rebolledo and J. González-Sabín, Hybrid Organo- and Biocatalytic Process for the Asymmetric Transformation of Alcohols into Amines in Aqueous Medium, ACS Catal., 2017, 7, 4768–4774, DOI:10.1021/acscatal.7b01543.
- N. Ríos-Lombardía, J. García-Álvarez and J. González-Sabín, One-Pot Combination of Metal- and Bio-Catalysis in Water for the Synthesis of Chiral Molecules, Catalysts, 2018, 8, 75, DOI:10.3390/catal8020075.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Opportunities and challenges for combining chemo- and biocatalysis, Nat. Catal., 2018, 1, 12–22, DOI:10.1038/s41929-017-0010-4.
- S. Schmidt, K. Castiglione and R. Kourist, Overcoming the Incompatibility Challenge in Chemoenzymatic and Multi-Catalytic Cascade Reactions, Chem. – Eur. J., 2018, 24, 1755–1768, DOI:10.1002/chem.201703353.
- J. Enoki, C. Mügge, D. Tischler, K. Miyamoto and R. Kourist, Chemoenzymatic Cascade Synthesis of Optically Pure Alkanoic Acids by Using Engineered Arylmalonate Decarboxylase Variants, Chemistry, 2019, 25, 5071–5076, DOI:10.1002/chem.201806339.
- T. Himiyama, M. Waki, Y. Maegawa and S. Inagaki, Cooperative Catalysis of an Alcohol Dehydrogenase and Rhodium-Modified Periodic Mesoporous Organosilica, Angew. Chem., Int. Ed., 2019, 58(27), 9150–9154, DOI:10.1002/anie.201904116.
- H. Gröger and W. Hummel, Combining the 'two worlds' of chemocatalysis and biocatalysis towards multi-step one-pot processes in aqueous media, Curr. Opin. Chem. Biol., 2014, 19, 171–179, DOI:10.1016/j.cbpa.2014.03.002.
- J. M. Sperl, J. M. Carsten, J.-K. Guterl, P. Lommes and V. Sieber, Reaction Design for the Compartmented Combination of Heterogeneous and Enzyme Catalysis, ACS Catal., 2016, 6, 6329–6334, DOI:10.1021/acscatal.6b01276.
- J. Paris, N. Ríos-Lombardía, F. Morís, H. Gröger and J. González-Sabín, Novel Insights into the Combination of Metal- and Biocatalysis, ChemCatChem, 2018, 10, 4417–4423, DOI:10.1002/cctc.201800768.
- L. Cicco, N. Ríos-Lombardía, M. J. Rodríguez-Álvarez, F. Morís, F. M. Perna, V. Capriati, J. García-Álvarez and J. González-Sabín, Programming cascade reactions interfacing biocatalysis with transition-metal catalysis in Deep Eutectic Solvents as biorenewable reaction media, Green Chem., 2018, 20, 3468–3475, 10.1039/C8GC00861B.
- V. Gotor-Fernández and C. E. Paul, Deep eutectic solvents for redox biocatalysis, J. Biotechnol., 2019, 293, 24–35, DOI:10.1016/j.jbiotec.2018.12.018.
- N. Guajardo, C. R. Müller, R. Schrebler, C. Carlesi and P. Domínguez de María, Deep Eutectic Solvents for Organocatalysis, Biotransformations, and Multistep Organocatalyst/Enzyme Combinations, ChemCatChem, 2016, 8, 1020–1027, DOI:10.1002/cctc.201501133.
- Z. Maugeri and P. Domínguez de María, Whole-Cell Biocatalysis in Deep-Eutectic-Solvents/Aqueous Mixtures, ChemCatChem, 2014, 6, 1535–1537, DOI:10.1002/cctc.201400077.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Deep eutectic solvents (DESs) and their applications, Chem. Rev., 2014, 114, 11060–11082, DOI:10.1021/cr300162p.
- E. E. L. Tanner, K. M. Piston, H. Ma, K. N. Ibsen, S. Nangia and S. Mitragotri, The Influence of Water on Choline-Based Ionic Liquids, ACS Biomater. Sci. Eng., 2019, 5, 3645–3653, DOI:10.1021/acsbiomaterials.9b00243.
- C. Ma, A. Laaksonen, C. Liu, X. Lu and X. Ji, The peculiar effect of water on ionic liquids and deep eutectic solvents, Chem. Soc. Rev., 2018, 47, 8685–8720, 10.1039/c8cs00325d.
- M. Kuddushi, G. S. Nangala, S. Rajput, S. P. Ijardar and N. I. Malek, Understanding the peculiar effect of water on the physicochemical properties of choline chloride based deep eutectic solvents theoretically and experimentally, J. Mol. Liq., 2019, 278, 607–615, DOI:10.1016/j.molliq.2019.01.053.
- F. Gabriele, M. Chiarini, R. Germani, M. Tiecco and N. Spreti, Effect of water addition on choline chloride/glycol deep eutectic solvents, J. Mol. Liq., 2019, 291, 111301, DOI:10.1016/j.molliq.2019.111301.
- O. S. Hammond, D. T. Bowron and K. J. Edler, The Effect of Water upon Deep Eutectic Solvent Nanostructure, Angew. Chem., Int. Ed., 2017, 56, 9782–9785, DOI:10.1002/anie.201702486.
- P. Xu, G.-W. Zheng, M.-H. Zong, N. Li and W.-Y. Lou, Recent progress on deep eutectic solvents in biocatalysis, Bioresour. Bioprocess., 2017, 4, 34, DOI:10.1186/s40643-017-0165-5.
- A. Paiva, A. A. Matias and A. R. C. Duarte, How do we drive deep eutectic systems towards an industrial reality?, Curr. Opin. Green Sustain. Chem., 2018, 11, 81–85, DOI:10.1016/j.cogsc.2018.05.010.
- D. A. Alonso, A. Baeza, R. Chinchilla, G. Guillena, I. M. Pastor and D. J. Ramón, Deep Eutectic Solvents, Eur. J. Org. Chem., 2016, 2016, 612–632, DOI:10.1002/ejoc.201501197.
- X. Marset, A. Khoshnood, L. Sotorríos, E. Gómez-Bengoa, D. A. Alonso and D. J. Ramón, Deep Eutectic Solvent Compatible Metallic Catalysts, ChemCatChem, 2017, 9, 1269–1275, DOI:10.1002/cctc.201601544.
- Z.-L. Huang, B.-P. Wu, Q. Wen, T.-X. Yang and Z. Yang, Deep eutectic solvents can be viable enzyme activators and stabilizers, J. Chem. Technol. Biotechnol., 2014, 89, 1975–1981, DOI:10.1002/jctb.4285.
- I. Wazeer, M. Hayyan and M. K. Hadj-Kali, Deep eutectic solvents, J. Chem. Technol. Biotechnol., 2018, 93, 945–958, DOI:10.1002/jctb.5491.
- I. Juneidi, M. Hayyan and M. A. Hashim, Intensification of biotransformations using deep eutectic solvents, Process Biochem., 2018, 66, 33–60, DOI:10.1016/j.procbio.2017.12.003.
- X. Liu, X.-Y. Meng, Y. Xu, T. Dong, D.-Y. Zhang, H.-X. Guan, Y. Zhuang and J. Wang, Enzymatic synthesis of 1-caffeoylglycerol with deep eutectic solvent under continuous microflow conditions, Biochem. Eng. J., 2019, 142, 41–49, DOI:10.1016/j.bej.2018.11.007.
- N. Guajardo, R. A. Schrebler and P. Domínguez de María, From batch to fed-batch and to continuous packed-bed reactors, Bioresour. Technol., 2019, 273, 320–325, DOI:10.1016/j.biortech.2018.11.026.
- N. Guajardo, P. Domínguez de María, K. Ahumada, R. A. Schrebler, R. Ramírez-Tagle, F. A. Crespo and C. Carlesi, Water as Cosolvent, ChemCatChem, 2017, 9, 1393–1396, DOI:10.1002/cctc.201601575.
- Á. Gómez Baraibar, D. Reichert, C. Mügge, S. Seger, H. Gröger and R. Kourist, A One-Pot Cascade Reaction Combining an Encapsulated Decarboxylase with a Metathesis Catalyst for the Synthesis of Bio-Based Antioxidants, Angew. Chem., Int. Ed., 2016, 55, 14823–14827, DOI:10.1002/anie.201607777.
- S. E. Payer, K. Faber and S. M. Glueck, Non-Oxidative Enzymatic (De)Carboxylation of (Hetero)Aromatics and Acrylic Acid Derivatives, Adv. Synth. Catal., 2019, 361, 2402–2420, DOI:10.1002/adsc.201900275.
- W. Nawaz, Z. Zhou, S. Deng, X. Ma, X. Ma, C. Li and X. Shu, Therapeutic Versatility of Resveratrol Derivatives, Nutrients, 2017, 9(11), 1188–1214, DOI:10.3390/nu9111188.
- H.-Y. Tsai, C.-T. Ho and Y.-K. Chen, Biological actions and molecular effects of resveratrol, pterostilbene, and 3′-hydroxypterostilbene, J. Food Drug Anal., 2017, 25, 134–147, DOI:10.1016/j.jfda.2016.07.004.
- M. Savio, D. Ferraro, C. Maccario, R. Vaccarone, L. D. Jensen, F. Corana, B. Mannucci, L. Bianchi, Y. Cao and L. A. Stivala, Resveratrol analogue 4,4′-dihydroxy-trans-stilbene potently inhibits cancer invasion and metastasis, Sci. Rep., 2016, 6, 19973, DOI:10.1038/srep19973.
- D. Webb and T. F. Jamison, Continuous flow multi-step organic synthesis, Chem. Sci., 2010, 1, 675, 10.1039/c0sc00381f.
- A. K. Schweiger, N. Ríos-Lombardía, C. K. Winkler, S. Schmidt, F. Morís, W. Kroutil, J. González-Sabín and R. Kourist, Using Deep Eutectic Solvents to Overcome Limited Substrate Solubility in the Enzymatic Decarboxylation of Bio-Based Phenolic Acids, ACS Sustainable Chem. Eng., 2019, 7, 16364–16370, DOI:10.1021/acssuschemeng.9b03455.
- T. El Achkar, S. Fourmentin and H. Greige-Gerges, Deep eutectic solvents, J. Mol. Liq., 2019, 288, 111028, DOI:10.1016/j.molliq.2019.111028.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, The Hitchhiker's Guide to Flow Chemistry, Chem. Rev., 2017, 117, 11796–11893, DOI:10.1021/acs.chemrev.7b00183.
- T. Noël and S. L. Buchwald, Cross-coupling in flow, Chem. Soc. Rev., 2011, 40, 5010–5029, 10.1039/c1cs15075h.
- B. Gutmann, D. Cantillo and C. O. Kappe, Continuous-Flow Technology—A Tool for the Safe Manufacturing of Active Pharmaceutical Ingredients, Angew. Chem., Int. Ed., 2015, 54, 6688–6728, DOI:10.1002/anie.201409318.
- R. Porta, M. Benaglia and A. Puglisi, Flow Chemistry, Org. Process Res. Dev., 2016, 20, 2–25, DOI:10.1021/acs.oprd.5b00325.
- M. Baumann and I. R. Baxendale, The synthesis of active pharmaceutical ingredients (APIs) using continuous flow chemistry, Beilstein J. Org. Chem., 2015, 11, 1194–1219, DOI:10.3762/bjoc.11.134.
- J. Britton and C. L. Raston, Multi-step continuous-flow synthesis, Chem. Soc. Rev., 2017, 46, 1250–1271, 10.1039/c6cs00830e.
- P. L. Lau, R. W. K. Allen and P. Styring, Continuous-flow Heck synthesis of 4-methoxybiphenyl and methyl 4-methoxycinnamate in supercritical carbon dioxide expanded solvent solutions, Beilstein J. Org. Chem., 2013, 9, 2886–2897, DOI:10.3762/bjoc.9.325.
- G. J. Lichtenegger, M. Maier, J. G. Khinast and H. Gruber-Wölfler, Continuous Suzuki—Miyaura reactions with novel Ce—Sn—Pd oxides and integrated crystallization as continuous downstream protocol, J. Flow Chem., 2016, 6, 244–251, DOI:10.1556/1846.2016.00021.
- N. Nikbin, M. Ladlow and S. V. Ley, Continuous Flow Ligand-Free Heck Reactions Using Monolithic Pd [0] Nanoparticles, Org. Process Res. Dev., 2007, 11, 458–462, DOI:10.1021/op7000436.
- K. Hiebler, S. Soritz, K. Gavric, S. Birrer, M. C. Maier, B. Grabner and H. Gruber-Woelfler, Multistep synthesis of a valsartan precursor in continuous flow, J. Flow Chem., 2019, 116, 1074, DOI:10.1007/s41981-019-00044-x.
- J. Britton, S. Majumdar and G. A. Weiss, Continuous flow biocatalysis, Chem. Soc. Rev., 2018, 47, 5891–5918, 10.1039/c7cs00906b.
- N. Guajardo and P. Domínguez de María, Continuous Biocatalysis in Environmentally-Friendly Media, ChemCatChem, 2019, 11, 3128–3137, DOI:10.1002/cctc.201900773.
- Y. Zhu, Q. Chen, L. Shao, Y. Jia and X. Zhang, Microfluidic immobilized enzyme reactors for continuous biocatalysis, React. Chem. Eng., 2019, 36, 73, 10.1039/C9RE00217K.
- C. A. Denard, H. Huang, M. J. Bartlett, L. Lu, Y. Tan, H. Zhao and J. F. Hartwig, Cooperative tandem catalysis by an organometallic complex and a metalloenzyme, Angew. Chem., Int. Ed., 2014, 53, 465–469, DOI:10.1002/anie.201305778.
- G. J. Lichtenegger and H. Gruber-Woelfler, Strategies to develop leaching-free heterogeneous catalysts, Chim. Oggi, 2015, 33, 12–19 CAS.
- G. J. Lichtenegger, M. Maier, M. Hackl, J. G. Khinast, W. Gössler, T. Griesser, V. S. P. Kumar, H. Gruber-Woelfler and P. A. Deshpande, Suzuki-Miyaura coupling reactions using novel metal oxide supported ionic palladium catalysts, Mol. Catal., 2017, 426, 39–51, DOI:10.1016/j.molcata.2016.10.033.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9re00467j |
| ‡ A(limiting) + B → P; yield Y [%] = (cP,t/cA,0) × 100; conversion X [%] = (cA,0 − cA,t)/cA,0 × 100, yield and conversion determined by HPLC after calibration. |
| § Leaching of the inorganic catalyst was not investigated in this work, but will be topic of future investigations. Leaching data of the catalyst under similar conditions can be found in ref. 45 and 47. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Anna K.
Schweiger
*a,
Anna K.
Schweiger