
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining retrosynthesis and mixed-integer optimization for minimizing the chemical inventory needed to realize a WHO essential medicines list†
Hanyu
Gao
 a,
Connor W.
Coley
a,
Thomas J.
Struble
a,
Linyan
Li
b,
Yujie
Qian
c,
William H.
Green
a and
Klavs F.
Jensen
*a
a,
Connor W.
Coley
a,
Thomas J.
Struble
a,
Linyan
Li
b,
Yujie
Qian
c,
William H.
Green
a and
Klavs F.
Jensen
*a
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: kfjensen@mit.edu
bHarvard T.H. Chan School of Public Health, 677 Huntington Ave, Boston, MA 02115, USA
cMIT Computer Science & Artificial Intelligence Laboratory, 32 Vassar St, Cambridge, MA 02139, USA
First published on 2nd January 2020
Abstract
The access to essential medicines remains a problem in many low-income countries for logistic and expiration limits, among other factors. Enabling flexible replenishment and easier supply chain management by on demand manufacturing from stored starting materials provides a solution to this challenge. Recent developments in computer-aided chemical synthesis planning have benefited from machine learning in different aspects. In this manuscript, we use those techniques to perform a combined analysis of a WHO essential medicines list to identify synthetic routes that minimize chemical inventory that would be required to synthesize the all the active pharmaceutical ingredients. We use a synthesis planning tool to perform retrosynthetic analyses for 99 targets and solve a mixed-integer programming problem to select a combination of pathways that uses the minimal number of chemicals. This work demonstrates the technical feasibility of reducing storage of active pharmaceutical ingredients to a minimal inventory of starting materials.
Introduction
The WHO Model List of Essential Medicines (EML), created in 1977, contains medicines that are needed to satisfy the highest priority healthcare needs and that should be available in sufficient quantities at all times.1 However, access to essential medicines is still problematic for about one-third of the world's population.2 Prices for some medicines make them unaffordable in less developed areas,3 and the fragility of global supply chains and manufacturing has led to frequent and extended drug shortages.4 No single solution can currently address this complicated problem, though there have been approaches proposed from policy perspective to improve access, including forming public-private partnerships, developing appropriate pricing models, and intellectual property access strategies.5The expiration of stockpiles of pharmaceuticals is one significant contributor to drug shortages,6 in particular in low-income countries, where drug donations can include drugs that are either expired or close to expiration.7 Another reason for expired stockpiles is poor forecasts of future demands8 or stockpiling for emergencies that fortunately do not occur. Drug expiration often results from the final formulated drug having a much shorter storage life than the chemical starting materials. Thus, it is of potential interest to identify the minimum number of starting materials needed to produce a desired set of medicines, and store the starting materials instead of the final drug products. Recent advances in computer-aided synthesis enable fast planning of large numbers of possible pathways to individual targets.9–14 Tools have also been developed to evaluate proposed pathways and identify which pathway suggestions are likely to be feasible.15–23 This progress makes it possible to combine synthesis planning for multiple targets. Molga et al. performed retrosynthetic analysis for multiple targets on the same graph and penalized new reaction types that appeared during the search, to promote the use of common intermediates.36 In this application, we explicitly consider the criteria of maximizing the overlap in the chemicals needed to realize a set of molecules. An approach of different syntheses sharing chemicals would have the advantage of allowing more flexible replenishment since starting materials tend to have more sources and be easier to obtain than the final drug products. Additionally, minimizing the total number of chemicals also promotes sharing common reaction conditions such as solvents and catalysts. It then becomes more likely to have reactions that share the same or similar conditions and thus the possibility of using common reactors, which facilitates modular process development. On the other hand, minimizing the number of chemicals may lead to pathways that are suboptimal for each molecular target individually. It increases the risk of choosing more practically challenging reactions or longer synthetic pathways, which are both undesirable.
We develop a framework to discover and select an optimal combination of pathways for molecules in the WHO essential medicines list accessible by chemical synthesis (more specific criteria in the Methods section). A machine learning based retrosynthesis platform24 is used to find candidate synthetic routes for all the targets, and then a multi-objective mixed-integer programming problem is solved25 to minimize the number of chemicals used in all the syntheses (including those used as solvents, reagents and catalysts), while maximizing the perceived chemical feasibility of the pathways. It is worth noting that this is a hypothetical study focusing on the conceptual feasibility of the problem. Building such a system for producing the WHO EML requires many other considerations, including the quantity and demand for different drugs, the expertise for conducting chemical synthesis and developing chemical processes, and cost associated with separation and purification of the product.
Methods
Dataset processing
The WHO essential medicines list has more than 600 entries categorized based on their therapeutic area. Some drugs appear multiple times because they are effective for different diseases. After consolidating these duplicates, 420 entries remained. We then used the NIH chemical identifier resolver tool (https://cactus.nci.nih.gov/chemical/structure) to resolve the chemical names to SMILES notation,26 which produced 330 molecular targets. The unresolved names were predominantly biomolecules or vaccines that did not have SMILES representation. Additional filters further narrowed the list down according to the Lipinski's rule of 5, which is commonly used in the pharmaceutical industry for coarsely examining “drug likeliness” of a molecule. The list was restricted by the following criteria:1) Molecular weight not greater than 500;
2) No fewer than 10 carbon atoms;
3) No greater than 5 hydrogen donors;
4) No greater than 10 hydrogen acceptors;
5) Estimated octanol–water partition coefficient, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P ≤ 5.
P ≤ 5.
6) No more than three chiral centers.
The final list of 127 targets used in our analysis is provided in the Table S1,† along with the full list of the 420 molecules and information about what filters caused the exclusion of particular targets.
Retrosynthesis analysis
Retrosynthetic analysis was performed for these 127 targets. Retrosynthetic templates were recursively applied to the target molecule (and the generated intermediates) until stopping criteria (defined as molecules that are smaller than 10 carbon atoms, three nitrogen atoms and five oxygen atoms in this context) were met, or specified maximal search depth (10) or time (100 seconds) were exceeded. A maximum of 10000 trees were returned for each target. We use a template-based upper confidence bound tree search (UCB) framework similar to the work by Segler et al.12 The methodology for retrosynthesis is the same as described in reference27 (with some parameters set as above). Here we provide brief recapitulations of some key elements in the methodology, and the readers are encouraged to refer to ref. 27 and the open source implementation at https://github.com/connorcoley/ASKCOS for more details. Note that we did not explicitly search known reactions. The first reason is that known reactions/routes may not be able to provide the benefit of sharing chemicals and minimizing the chemical inventory, e.g. published reactions might report only one out of many equivalent leaving groups that could provide the desired reactivity. Second, we would like the framework to generalize to new molecules, for which literature lookup would not suffice.
When using the in-scope filter in the search, the threshold for filtering out a reaction suggestion was when its score is below 0.75. In practice, this value helped achieve a good balance between filtering out false negative reactions and including false positive reactions, leading to a good number of reasonable pathways from which to choose.
000.
For the stopping criteria (or how to determine whether a chemical is a “starting material”), we defined a molecule that has no more than 10 carbon atoms, 3 nitrogen atoms and 5 oxygen atoms as a terminal node in the tree search. Different criteria could be chosen for the retrosynthetic analysis. Specifically, we did not use the price of the chemicals (which seems intuitive) for a few reasons: a) many of these essential medicines, including the intermediates in the syntheses, are likely to be identified as commercially available at a cheap price, and b) there lacks a complete catalog that has accurate price estimates for all commonly used chemicals. In the meantime, these small molecules as defined here are likely to be obtained in a sufficiently affordable manner.
Pathway evaluation
The pathway search time for each target was set to 100 seconds (parallelized over 16 cores). After the synthetic pathways were identified, reaction conditions (including up to one catalyst, two solvents and two reagents) were suggested for every reaction, using the exact model described in reference.23 The reaction conditions were then combined with the reactants to predict the scores for all potential outcomes of the reaction, following reference.19 If the intended product was within the list of potential outcomes, the forward evaluation score of the intended product was used as an indication of the likelihood of success of the reaction. Otherwise the score was zero. The top-five reaction conditions were tested, and the one with the highest forward evaluation score was taken as the final condition for the reaction. Pathways that had two or more steps that had less than 0.05 forward evaluation scores (which we refer to as low-score steps) were eliminated from the pool of pathways. Then we set the probability of all remaining low-score steps to 0.05 as a way to tolerate one low-score step in a pathway. Later, in the Analysis with stricter quality requirements section, we explored the scenario where all low-score steps were excluded from the analysis. The overall pathway score is calculated as the cumulative probability of all the reaction steps, and we define the penalty for a pathway as the negative natural logarithm of the pathway score.Optimization of pathway selection
The starting materials and all the conditions (chemicals used as solvents/reagents/catalysts) were extracted from the found pathways. The problem was defined as minimizing the total number of chemicals used in all the syntheses, while maximizing the likelihood of success for all the pathways. Aligning with the goal of driving the retrosynthesis towards simpler starting materials, the chemicals were weighted by their synthetic complexity scores (SCScore), as developed by Coley et al.29 The SCScore for solvents/reagents/catalysts were set to 1, which is the lowest synthetic complexity score based on this definition. By doing this, in case there were multiple solution with the same number of chemicals, the solution with simpler chemicals would be preferred. The mathematical formulation of the model is as follows: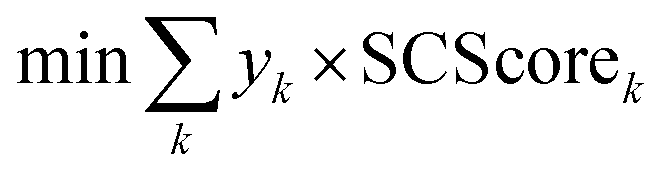 | (1) |
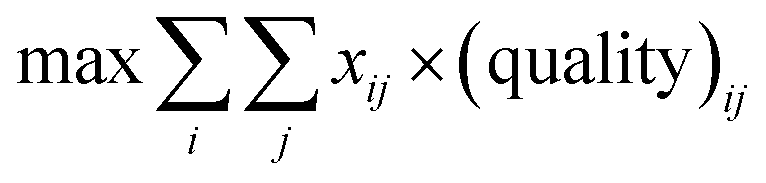 | (2) |
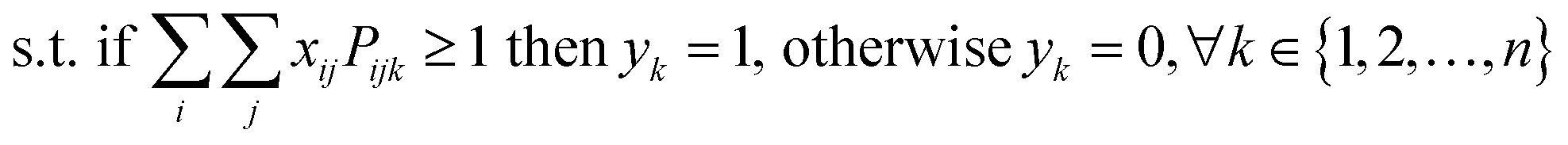 | (3) |
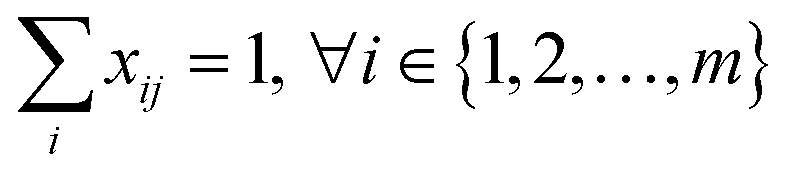 | (4) |
| xij, yk ∈ {0, 1} | (5) |
As a common strategy to solve a multi-objective optimization problem, we used a weighting parameter, λ, to balance between the two objectives. The first constraint also needed reformulation to a pure mathematical form. The final mixed-integer multi-objective optimization is presented as follows:
 | (6) |
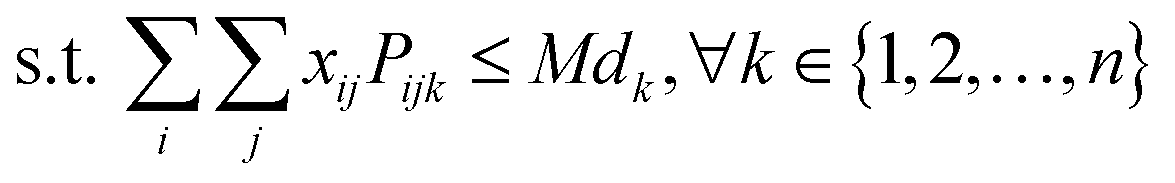 | (7) |
| y ≥ 1 − M(1 − dk) | (8) |
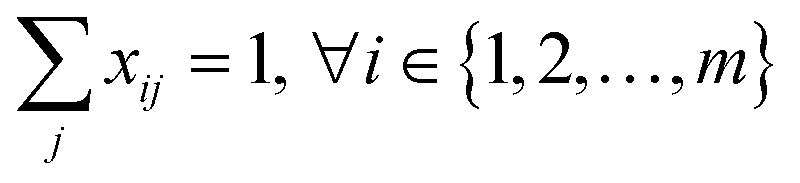 | (9) |
| xij, yk, dk ∈ {0, 1} | (10) |
The optimization problem is solved using the CPLEX solver with the Python API.25 Note that the optimization problem is linear, so there is no need to supply an initial guess and it is guaranteed that the solution found is global optimal regardless of the starting point.
We changed the value of λ to test the tradeoff between the two objectives. The value of λ varied from 0 to 1. They represented two extremes. With λ being one, solving the optimization problem was essentially trying to find the minimal number of chemicals required without considering the feasibility of the pathways. λ being zero indicated that we were trying to find pathways for individual targets that had the maximum score without considerations of the overlap of starting materials.
Results and discussion
Retrosynthesis analysis
The whole process for the 127 targets including retrosynthesis analysis, condition recommendation, and forward evaluation required 75 hours on a workstation with a dual processor Intel® Xeon(R) CPU E6-2690@2.9GHz processors and 128G RAM. Synthetic pathways were found for 112 out of 127 targets using the settings described in the Methods section; the total number of unique pathways found was 492860, where each target was limited to at most 10000 pathways. Fig. S1† lists the drug molecules for which no pathway was returned in the specified amount of time under our strict expansion criteria (see Methods). While these molecules revealed some limitations of the retrosynthesis capability originating from the limitation of available reaction data, in the meantime the success rate of the retrosynthesis analysis was high (88.2%), meaning pathways were found for a majority of the targets in this set.
As described in the Methods section, we used machine learning models to predict the top-five most plausible sets of conditions23 and to assess the probability that the desired product is the major outcome given the reactants and each of the five sets of reaction conditions.19 The condition set that yields the highest reaction evaluation score was retained as the final condition for each reaction. The pathways with more than one low-probability (<0.05) step are filtered out, and targets that include more than one molecules are also excluded at this stage, resulting in 193597 pathways for 99 targets. These remaining pathways involved 3340 chemicals used as starting materials and 570 chemicals used in the reaction conditions (i.e., as solvents, catalysts or reagents).
Optimal selection of pathways
If the pathways with the highest scores were chosen for each target, the 99 targets required 236 starting materials and 135 additional chemicals (reagents, catalysts, solvents); we refer to this as the “separate analysis”. By solving the optimization problem to minimize the number of chemicals while maximizing the sum of pathway scores, the number of starting materials required for these 99 targets was 187, and the number of additional reagents, catalysts, and solvents was 99; this is the “combined analysis”. This represented an approximate reduction in the total number of chemicals by 25%. Note that “combined analysis” and “separate analysis” essentially represent solving the optimization problem using two different λ values: 0.5 and 0.When comparing the chemicals used in the above two scenarios, it was found that 106 chemicals were present in both scenarios. 130 chemicals were used only in the separate analysis and 81 chemicals were used only in the combined analysis. As exemplified in Fig. 1, a starting material was shared by different targets in the combined analysis but was used for fewer targets in the separate analysis. In the combined analysis, 1-bromo-4-chlorobenzene was shared by three different targets (pyrimethamine, halopericol and chloropromazine), whereas in the separate analysis only halopericol used it as a starting material (Fig. 1A). Di-tert-butyl dicarbonate was shared by three different targets as a protecting agent in the combined analysis, while only one target used it in the separate analysis (Fig. 1B). Dimethylamine was another example that was used by more syntheses in the combined analysis than in the separate analysis (4 vs. 2, Fig. 1C). The full list pathways for all the targets in the combined and separate analyses are in the ESI† (Fig. S2 and S3). We manually examined the first fifty targets in both analyses, and pointed out potential problematic reactions. Common issues include missing reaction conditions (e.g., solvents and bases), or ambiguous selectivity. These topics are further discussed later in the Limitations section.
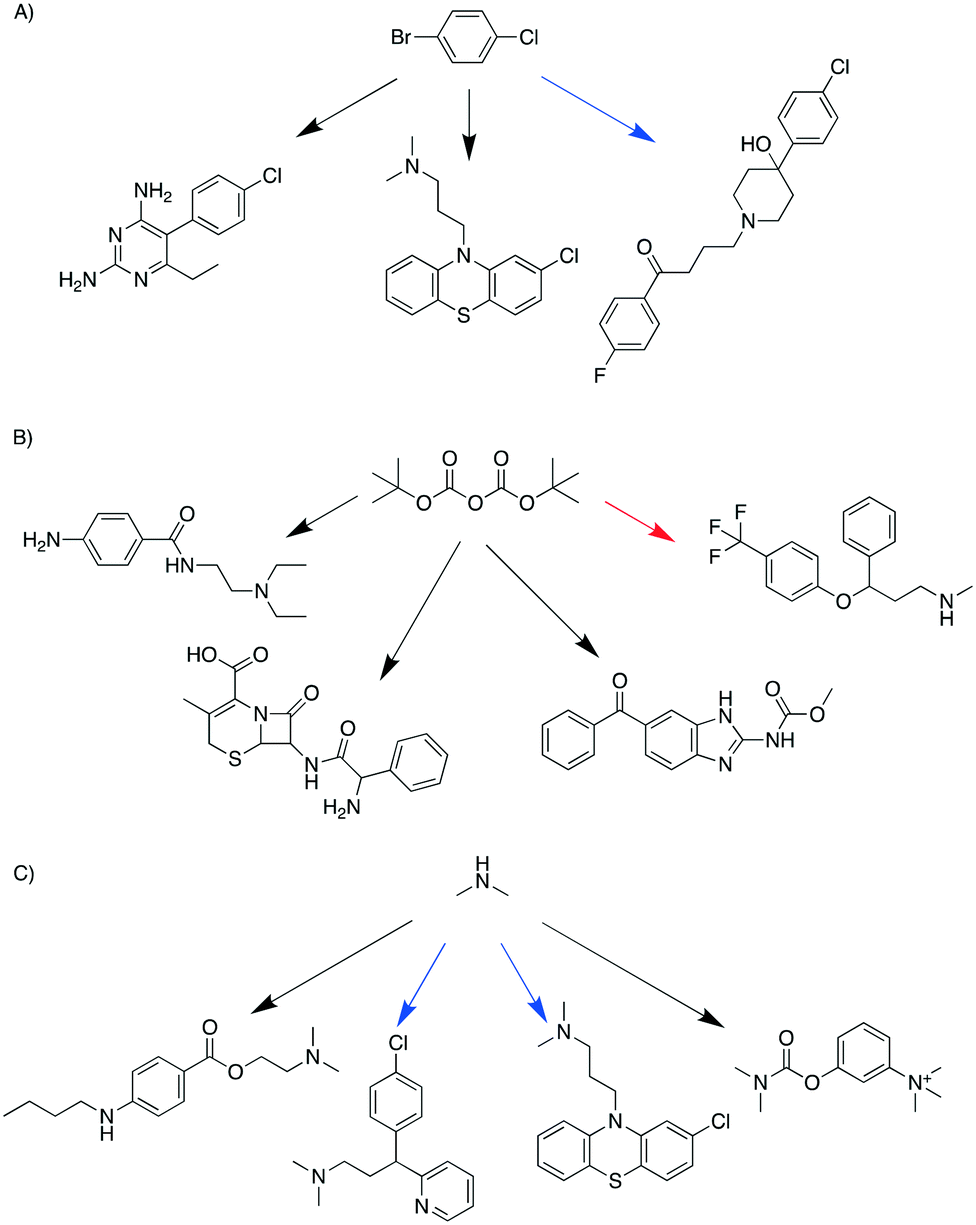 | ||
| Fig. 1 Examples of chemicals that were shared by more targets in the combined analysis compared to the separate analysis. A) 1-Bromo-4-chlorobenzene; B) di-tert-butyl dicarbonate; C) dimethylamine. The arrows in black represent that the starting material was used for the target only in the combined analysis but not the separate analysis. The arrows in blue represent that the starting materials was used for the target both in the combined and separate analysis. The arrows in red represent that the starting material was used for the target only in the separate analysis but not the combined analysis. | ||
The entire list of chemicals along with the number of times each chemical was used by all the syntheses are provided in ESI† (Fig. S4, S5 and S6). The chemicals were generally shared by more syntheses in the combined analysis, with the most frequent chemical (methanol) used for 10 different targets. We also found that, by visual inspection, in the separate analysis, chemicals that are small variations of each other are used in different synthesis, which indicates some redundancy, and in the combined analysis, we found fewer similarly-structured chemicals.
To further examine how the reduction in the number of chemicals was achieved, we counted the number of chemicals used in each synthesis for both the combined analysis and separate analysis. Then when we calculated the average number of chemicals used in each synthesis. Interestingly enough, the combined analysis and separate analysis were very close (8.28 vs. 8.41). This indicated that the combined analysis was not mainly selecting syntheses that used fewer chemicals, but identifying pathways that had overlap in the chemicals they used.
For some pairs of targets, we found that their syntheses shared more common chemicals in the combined analysis. For example, the combined analysis recommended performing the syntheses of both propyliodone and probenecid in one step (Fig. 2). Whereas the targets were structurally different, the analysis find commonalities in their syntheses: both used 1-bromopropane to introduce the propyl group and both reactions happened in dimethylformamide (DMF). The recommended pathways in the separate analysis syntheses did not share any common chemicals. The scores given in the combined analysis, although slightly lower than the separate analysis, still indicated that the reactions were likely to occur.
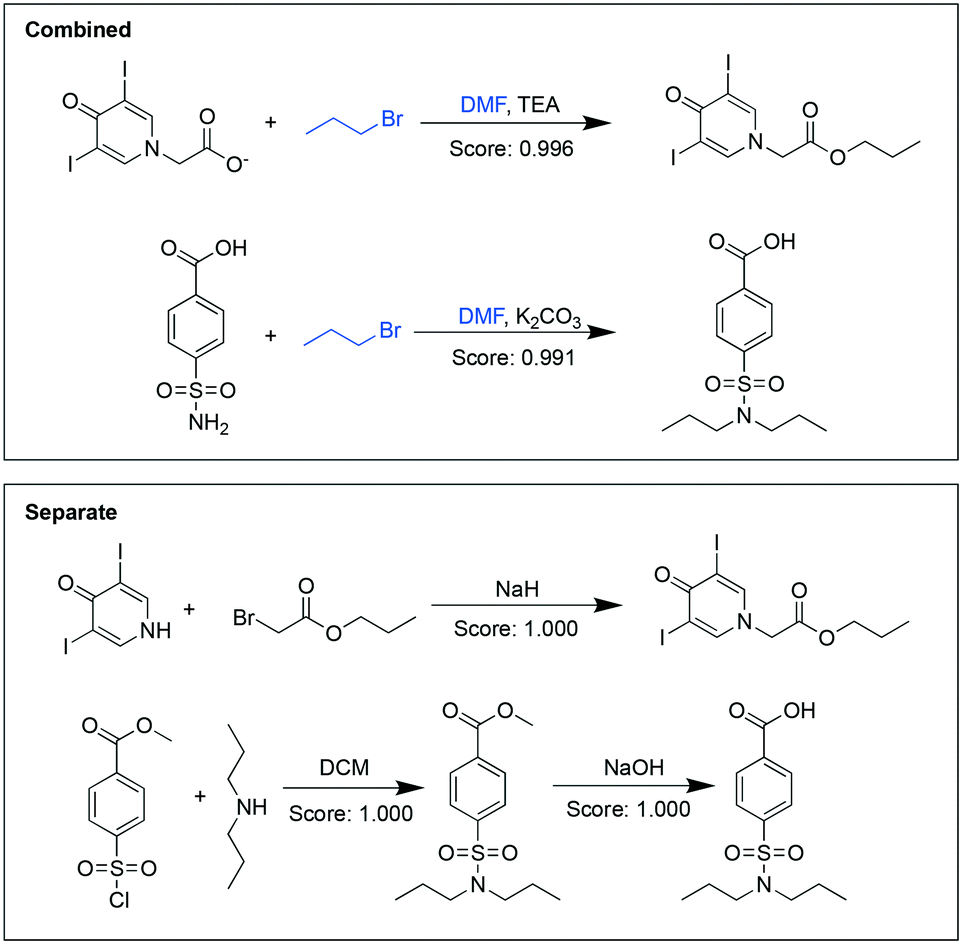 | ||
| Fig. 2 Synthetic pathways suggested for propyliodone and probenecid in the combined analysis (upper panel) and separate analysis (lower panel). Chemicals highlighted in blue were shared between the two target molecules. The last reaction in the combined analysis for probenecid likely represents a multi-step reaction recorded as an overall transformation. | ||
For probenecid, the combined analysis proposed alkylation of a sulfonamide in the presence of a carboxylic acid using sodium hydroxide. However, multiple evidence in the literature has shown reactivity in the reverse order – the carboxylic acid was more reactive than the sulfonamide.29,30 To ameliorate the side reactivity, either the acid was protected,31 or it was alkylated and later hydrolyzed back to the acid (in the reaction workup).32 It is worth noting here that the model-suggested reactions are not necessarily single step operations. There might be some human bias in the practice of recording reactions in the database – some workup steps might be omitted and only the desired overall transformation is recorded. For this reaction (and likely many other similar reactions), alkylation of the sulfonamide is the desired outcome over alkylation of the carboxylic acid, but due to cross-reactivity in basic conditions, a workup is sometimes used to hydrolyze the resulting ester back to the carboxylic acid. However, the recorded reaction (in most datasets) does not typically indicate a separate workup step and the carboxylic acid would appear unchanged. This example illustrates that the previously developed data-driven models used to make condition and reactivity predictions (before solving the optimization problem) can have bias based on reaction datasets.
Another example target pair shown here is neostigmine and tetracaine (Fig. 3). In the combined analysis, the synthesis of neostigmine and the second step for tetracaine synthesis uses a common substrate (dimethylamine) and the same solvent (benzene). While in the separate analysis, for both targets pathways with higher perceived likelihood were chosen, but at the expense of not having any overlapping starting materials or reaction conditions.
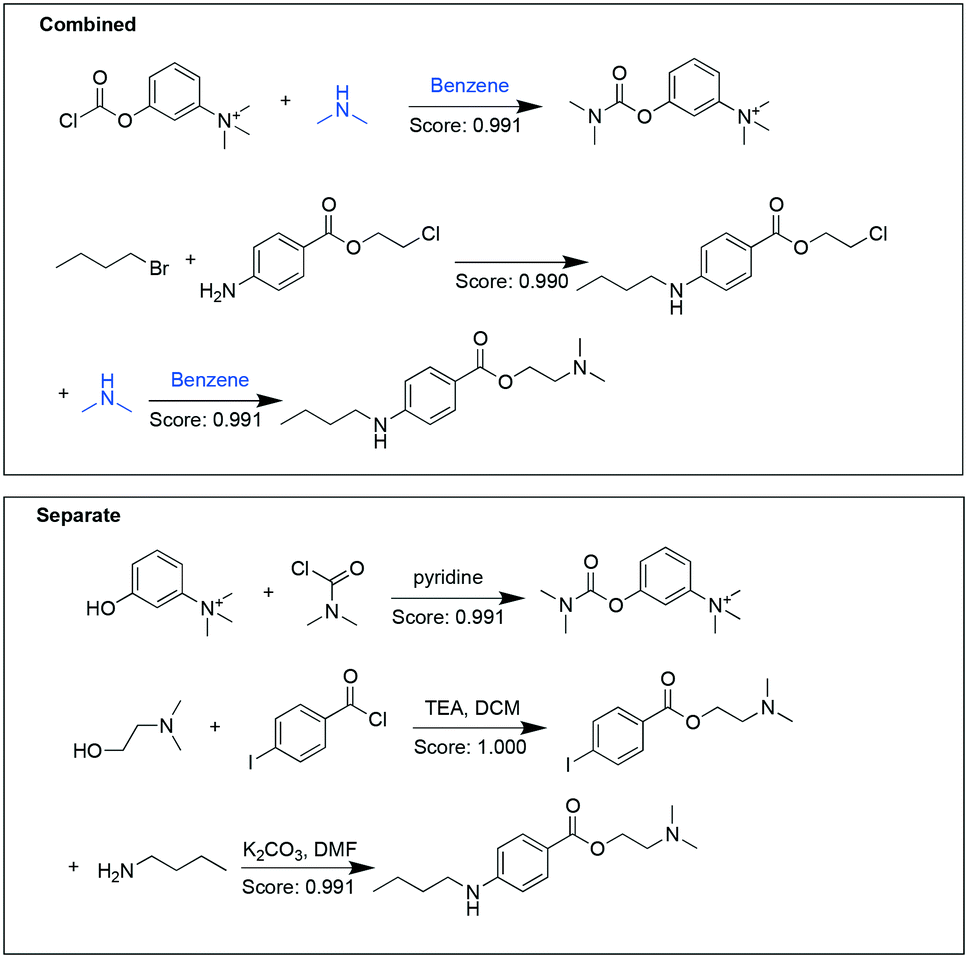 | ||
| Fig. 3 Synthetic pathways suggested for neostigmine and tetracaine in the combined analysis (upper panel) and separate analysis (lower panel). Chemicals highlighted in blue were shared between the two target molecules. | ||
It was, however, not universal to have improved pairwise similarity for all target pairs. Fig. S7† visualizes the pairwise similarity between the syntheses of different targets using a heatmap. The similarity was calculated as the ratio of the number of overlapping chemicals to the total number of unique chemical used in the two syntheses. Mathematically this is referred to as the Tanimoto similarity, and it is a common metric to calculate similarity for molecular fingerprints.33 We calculated this value for each target pair to create the heatmap. It was not clear that the combined analysis had improved pairwise similarity over the separate analysis, and when we calculated the difference, on average the pairwise similarity was only 0.0006 higher in the combined analysis than the separate analysis, which was not a significant margin.
It turned out that for many chemicals, they did not specifically overlap with another single target, but shared chemicals with several other targets. Although some targets needed more starting materials in the combined analysis than the separate analysis, many of the starting materials were shared with other targets, so that they would not add additional requirements to the overall chemical inventory. Fig. 4 and 5 show the entire pathways for synthesizing procainamide and ephedrine, together with how the starting materials can be shared with other targets (only one example target is shown for a starting material; there can be other targets that also share the starting material). In the combined analysis, both syntheses were longer and used more chemicals than in the separate analysis, but more of the starting materials were shared with some other targets (procainamide 5/5 and ephedrine 2/3).
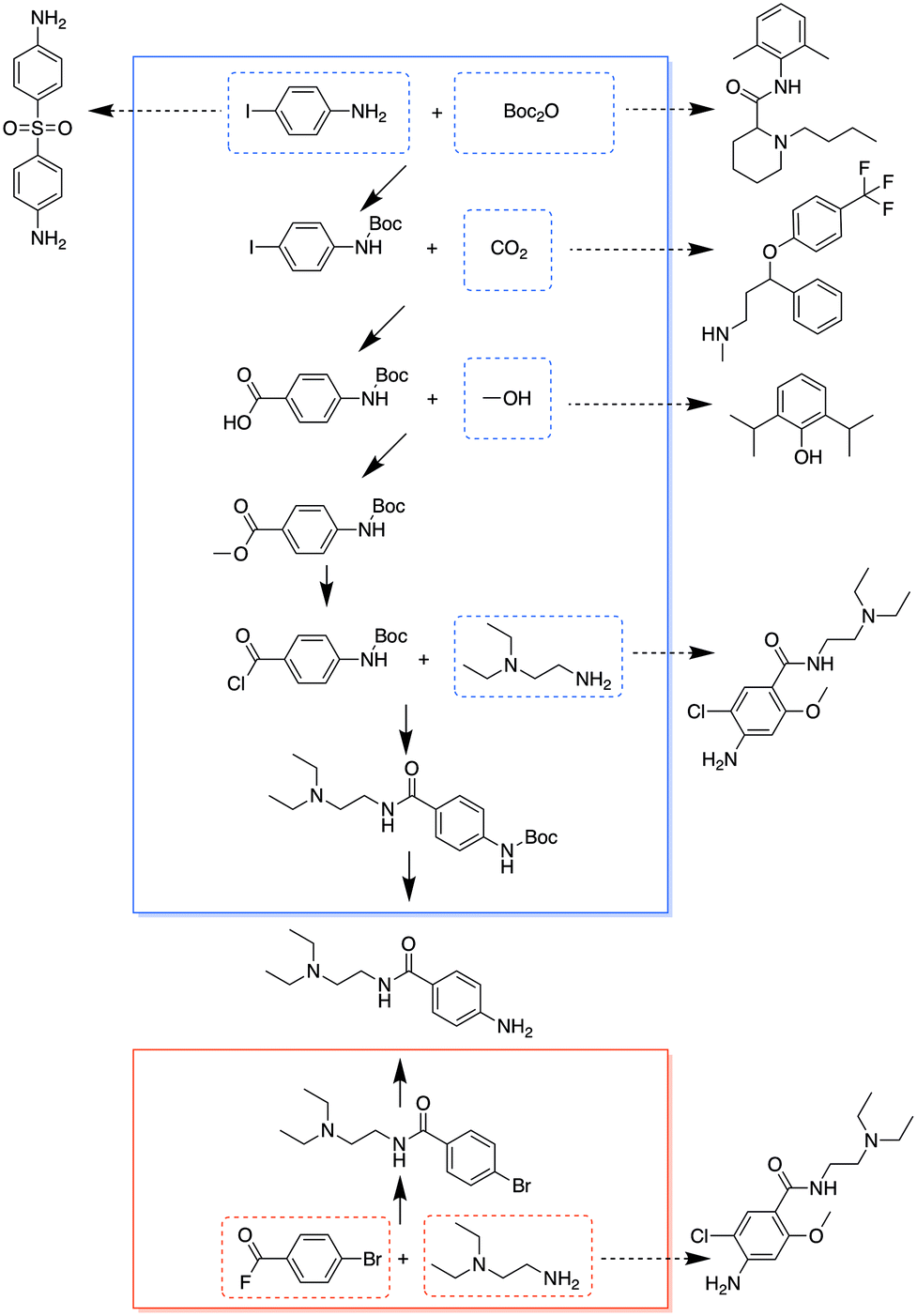 | ||
| Fig. 4 Comparison of syntheses of procainamide in the combined analysis (blue box) and the separate analysis (red box). The chemicals in dashed boxes are starting materials used in the syntheses, some with dashed arrows pointing to another target which this starting material can be used for. | ||
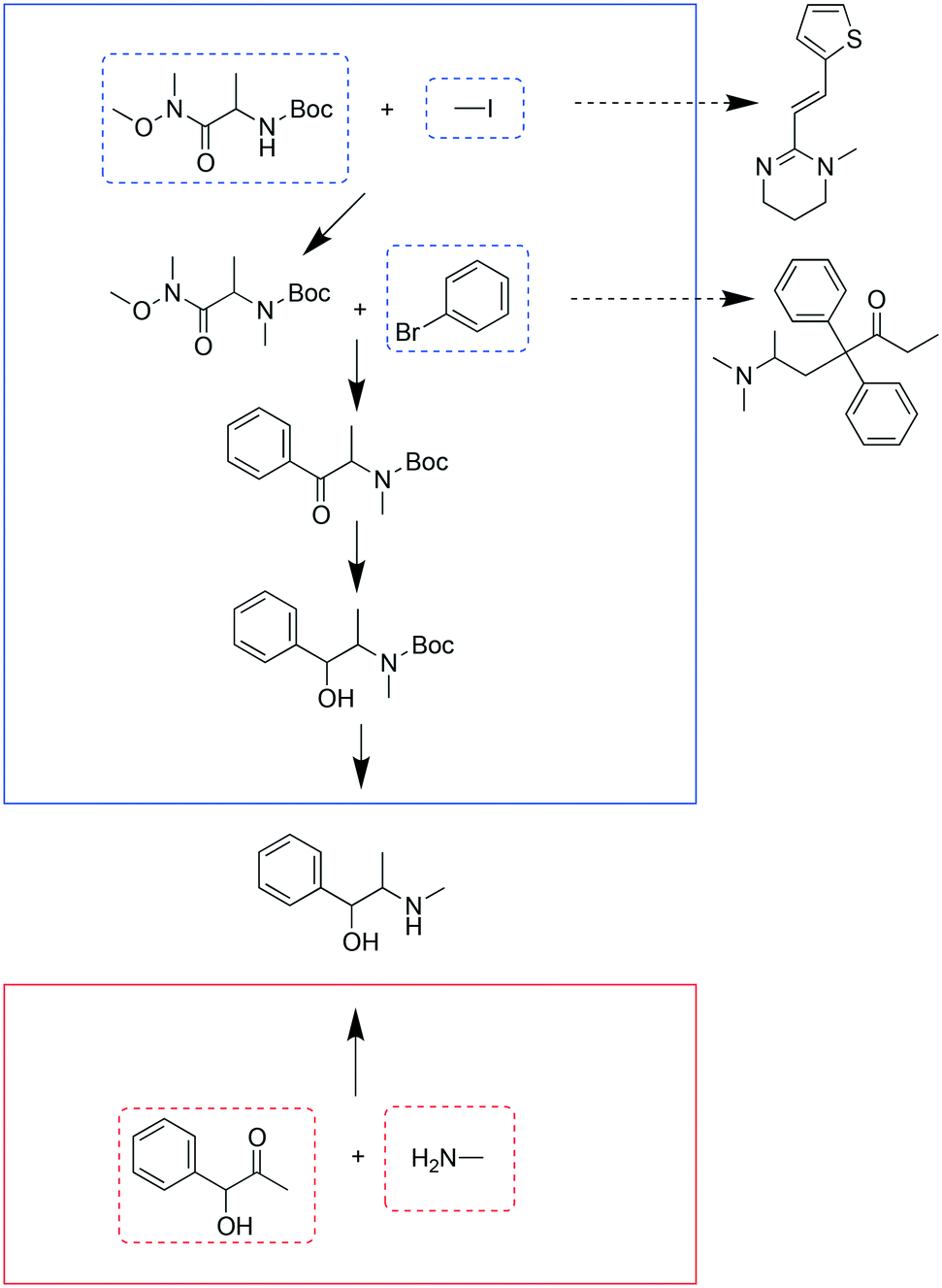 | ||
| Fig. 5 Comparison of syntheses of ephedrine in the combined analysis (blue box) and the separate analysis (red box). The chemicals in dashed boxes are starting materials used in the syntheses, some with dashed arrows pointing to another target which this starting material can be used for. | ||
We also explored the tradeoff between the two objectives, minimizing the number of chemicals and minimizing the penalty of the pathways, by adjusting the weighting parameter, λ, between the two goals (eqn. (6)). The results are shown as Pareto frontiers in Fig. 6, with λ taking values from an increasing sequence from right to left: 0, 0.01, 0.1, 0.25, 0.33, 0.50, 1. The points on the Pareto curve represent the states from which one objective cannot be improved without worsening another objective. As shown in Fig. 6, reducing the total number of chemicals leads to decreased average scores of the pathways. Emphasizing the minimum number of chemicals (λ = 1) without regard to pathway feasibility led to 175 starting materials and an average pathway score of 0.230. At the other extreme (λ = 0), when pathways with the highest scores were chosen for every individual target, the total number of chemicals needed was 236, more than 30% higher than the other extreme, and the average pathway score was 0.663. The sharp change in the Pareto frontiers on the right hand side of Fig. 6 revealed that the number of chemicals decreased significantly with only a small decrease in the average pathway score. When λ = 0.1, the number of starting materials was 199 with an average pathway score of 0.648. Decreasing λ to 0.5 reduced the number of starting materials to 187 and the average pathway score to 0.566. The sharp curvature at the right end of the figure shows that reductions of the number of chemicals can be achieved without significantly sacrificing the predicted quality of the pathways.
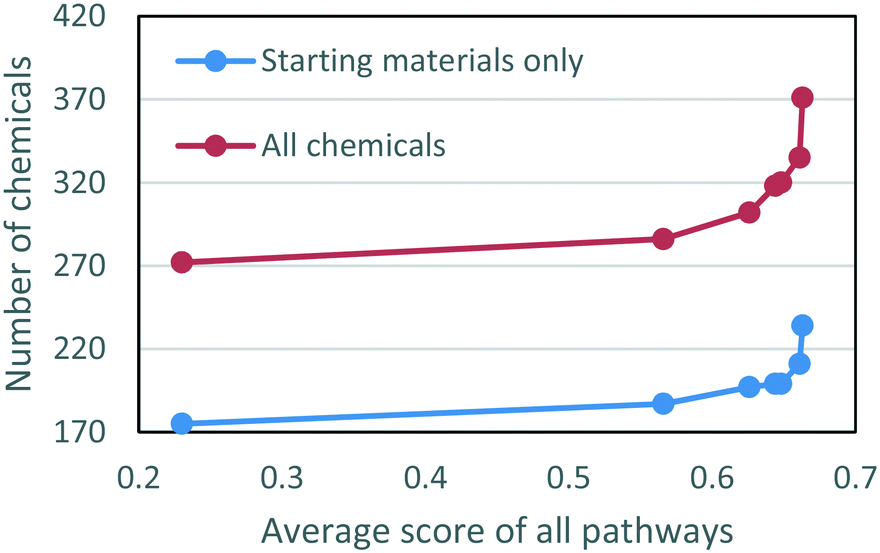 | ||
| Fig. 6 Pareto frontiers showing the tradeoff between the number of chemicals used in all syntheses and the average score of the pathways. The λ values corresponding to the points from right to left are 0, 0.01, 0.1, 0.25, 0.33 0.50, 1, respectively. | ||
Analysis with stricter quality requirements
In the current scheme we allowed each pathway to have at most one low-probability step, which allows some reactions that were rejected by the reaction evaluation model to be included (i.e. predicted score less than 0.05). This was because some low-scoring reactions could be possible which exposes limitations in the models rather than fundamental reactivity issues.In Fig. 7, we show several representative examples with different types of problems. Fig. 7a and b can be classified as false negative predictions by the reaction evaluation model, as literature precedence of the exact or very similar reactions can be found.34,35 These reactions usually involve many bond changes or even multiple reaction steps (usually recorded in the database as a single step), which can be out of scope for the reaction evaluation model. If we can expand the scope of the reaction evaluation model, or break down the retrosynthetic templates to reflect the multiple reaction steps separately, these reactions should be predicted viable. Fig. 7c–e are examples where the primary issue was reaction conditions – an atom source is missing so the reaction is chemically nonsensical. Fig. 7c only predicted a solvent and base as reagent, and most importantly there is no methylating agent. Fig. 7d does not provide a carbon source for the epoxidation. Fig. 7e missed water which is needed for a workup step and also supply the oxygen. These reactions cannot be possible as they are presented, but if the condition recommendation model can be improved to propose the necessary conditions, these reactions could become viable suggestions. For example, if methyl iodide is supplied as an additional reagent, the score of the reaction in Fig. 7d increases to 0.41, which can make it favorable than the current scheme presented. Fig. 7f and g represent examples where there were fundamental reactivity concerns. These are the true negative reactions that we would like to be eliminated from the optimization. With further improvement of the underlying models used before the optimization, using the reaction score as a filter will help us more exclusively filter out reactions that have fundamental reactivity concerns.
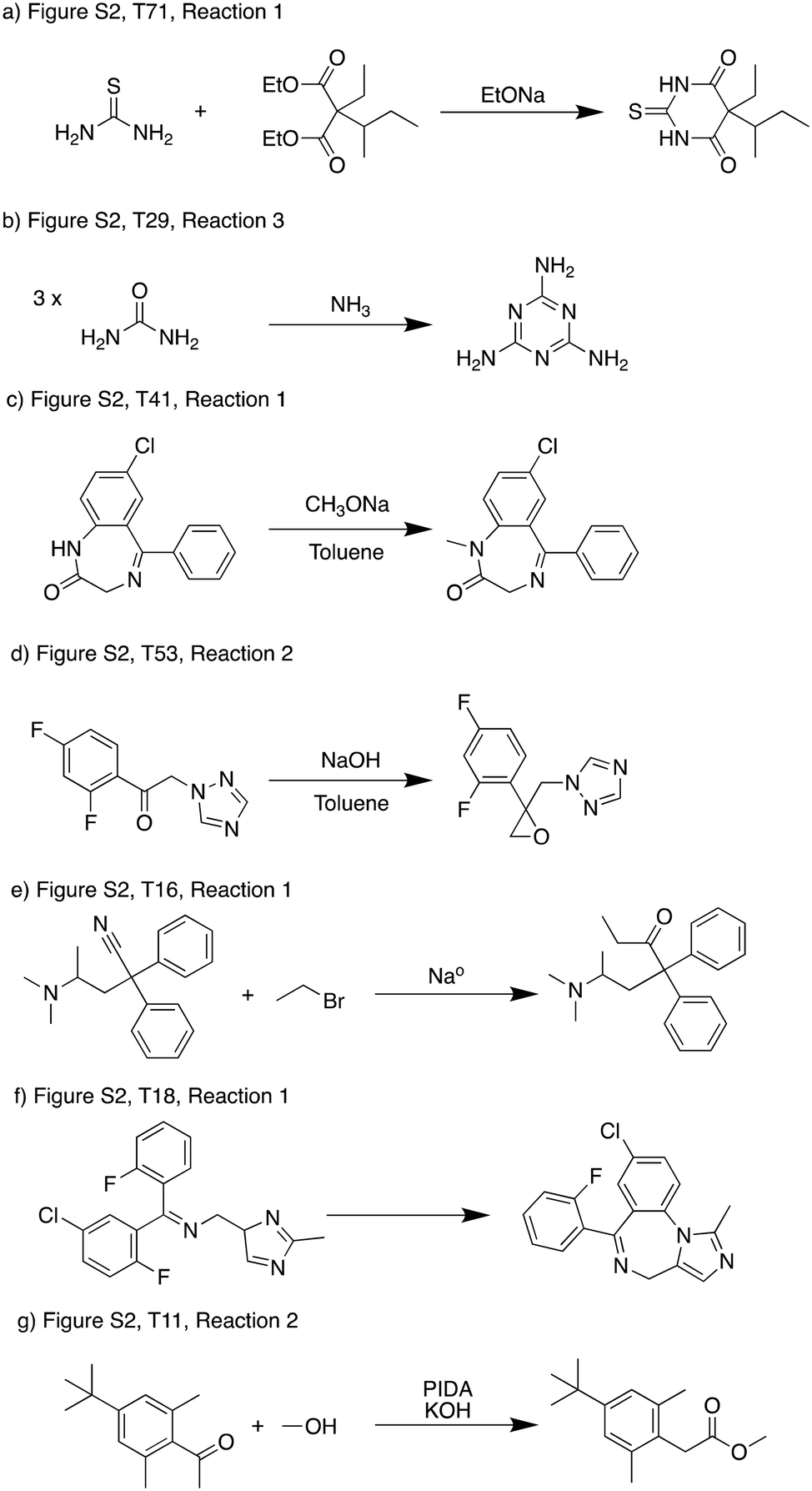 | ||
| Fig. 7 Examples of low scoring reactions. a) and b) Represent cases where the reaction evaluation model was not able to recognize feasible reactions.34,35 c)–e) Represent cases where a necessary reagent was missing that was supposed to supply atoms to the product. f) and g) Represent cases where reactivity was problematic. | ||
We highlighted all low probability steps in Fig. S2 and S3† to make the pathways more clear to readers. As we explained, these are not straightforward one-step operations, and their inclusion can be attributed to limitations of the retrosynthetic model, condition recommendation model or reaction evaluation model.
In addition, in order to present a more realistic collection of pathways, we performed an analysis in which we filtered out all pathways that have low-probability reactions. This results in 54204 pathways for 78 targets. We ran the same combined (λ = 0.5) and separate analysis (λ = 0) and the total number of chemicals for these two cases were 250 and 301. The reduction is smaller (17%) since there are fewer options to choose from, but it still demonstrates the benefit of planning synthesis in a combined manner for multiple targets. The full pathways for this sensitivity analysis are shown in Fig. S8 (combined analysis) and Fig. S9† (separate analysis). As computer-aided synthesis planning programs improve, we expect that the number of targets with pathways found and the quality of proposed pathways will improve; our optimization framework for multi-target planning demonstrates the value even now.
Limitations
As mentioned in the Introduction section, this method only addresses the initial question of what would be the minimal inventory needed to synthesize the WHO EML, which is the first step to establish an on-demand synthesis system. Other practical aspects, including experimental validation, process development, policy approval, are beyond the scope of this work. Nevertheless, this work explores the conceptual feasibility to develop integrated syntheses for the WHO EML, which can drive further developments.We understand the current model-suggested pathways are not perfect and improvements in multiple aspects can be made. In the ESI† we manually examined the first 50 pathways and made notes about reactions that might be problematic.
A large fraction of cases can be attributed to missing reaction conditions, like solvents and bases (e.g. Fig. S2:† T8, T10-2; Fig. S3:† T7, T8, T10-3). This is largely related to the quality of the data used to train the condition recommendation model. Some common solvents/reagents are omitted in the database (due to human error in data entry); a typical example is that water is frequently missing when the model recommends a reagent like NaOH or HCl. This needs to be addressed in future improvement of data and model for the condition recommendation model.
Some reactions seem to have ambiguous selectivity (e.g. Fig. S2 and S3:† T0–1, T14). While the reaction evaluation score should reflect this aspect, the reactivity/selectivity can only be confirmed via experiments. Models that are more focused and robust on selectivity prediction are currently under development that can improve the confidence in this task.
Sometimes the pathways seem redundant and circular (e.g. Fig. S2 and S3:† T5). This either requires improving the tree retrieval procedure, or the suggestions can be avoided by changing the optimization objective to favor shorter pathways.
As mentioned in the previous section, a more serious flaw is that some reactions are missing an necessary reagent that contributes atoms (e.g. T41 in Fig. S2 and S3†). The alkylating agent is recorded as a reactant in some cases and reagents in others in the training dataset. This type of error occurs when the retrosynthetic template omits the alkylating agent in the reactant but the condition recommendation model fails to predict it as a reagent. Solving this issue requires cleaning the data to fix the role of the alkylating agent.
These are also common issues with the rest of reactions shown Fig. S2 and S3,† and will need to be addressed in future improvements of the dataset quality and model performance. However, these issues appear similarly in both the combined and separate analysis, which indicates that the optimization method can stay useful with improved tools for retrosynthetic analysis.
We are also limited by retrosynthetic analysis settings, as we set the maximal number of trees for a target and maximal depth of the trees. Also, we chose only one set of condition for every reaction, which limits the ability to further reduce the number of solvents, reagents, etc. These can be solved by improved optimization algorithms to optimize on a graph instead of enumerate distinct tree options.
Conclusion
In this work we developed a workflow to perform combined synthesis planning and predict the minimal chemical inventory needed for multiple small molecule targets in the WHO essential medicines list. By performing a multi-objective optimization on the selection of pathways, the number of chemicals used was reduced by 20–30%. A considerable reduction (∼20%) in the chemical inventory could be achieved without substantially decreasing the quality of proposed pathways to 99 targets on the WHO list. Even when using stricter requirements of pathway quality, the combined analysis achieved a 17% reduction in the number of unique chemicals required to synthesize the subset of 78 targets. Examples demonstrated that the reduction was enabled by sharing chemicals between different syntheses, either increasing pairwise similarity or sharing with multiple other targets. This work evaluated a hypothetical solution to the problem of drug expiration by designing synthetic routes that start from simpler starting materials that tend to have longer shelf-lives, and minimizing the inventory of starting materials and reaction conditions that need to be stored. The methodology used in this work is sufficiently general that it can be applied to solve other multi-compound synthesis tasks as well. This work can be enriched in the future with improvement in the capacity of the retrosynthetic analysis, and developing other metrics for pathway evaluation to design pathways based on more versatile criteria.The code for running the retrosynthetic analysis and optimization is available at https://github.com/Coughy1991/Combined_synthesis_planning.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Machine Learning for Pharmaceutical Discovery and Synthesis Consortium and the DARPA Make-It program under contract ARO W911NF-16-2-0023. We thank Elsevier for the permission to use the Reaxys API. We also thank IBM for the CPLEX academic license.References
- The World Heath Organization, WHO Model Lists of Essential Medicines, https://www.who.int/medicines/publications/essentialmedicines/en/, Accessed on June 08, 2019 Search PubMed.
- The Secretary General, The Right to Health, http://www.who.int/medicines/areas/human_rights/A63_263.pdf, Accessed on June 08, 2019 Search PubMed.
- A. M. Hill, M. J. Barber and D. Gotham, BMJ Glob. Health, 2018, 3, e000571 CrossRef PubMed.
- A. Gray and H. R. Manasse, Bull. W. H. O., 2012, 90, 158 CrossRef PubMed.
- H. Stevens and I. Huys, Front. Med., 2017, 4, 218 CrossRef.
- P. F. Kamba, M. E. Ireeta, S. Balikuna and B. Kaggwa, Bull. W. H. O., 2017, 95, 594–598 CrossRef.
- E. Schouten, BMJ, 1995, 311, 684 CrossRef CAS PubMed.
- J. K. Nakyanzi, F. E. Kitutu, H. Oria and P. F. Kamba, Bull. W. H. O., 2010, 88, 154–158 CrossRef PubMed.
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2017, 3, 1237–1245 CrossRef CAS PubMed.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef.
- M. H. S. Segler and M. P. Waller, Chem. – Eur. J., 2017, 23, 5966–5971 CrossRef CAS.
- M. H. S. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604 CrossRef CAS.
- B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, ACS Cent. Sci., 2017, 3, 1103–1113 CrossRef CAS PubMed.
- A. Bøgevig, H.-J. Federsel, F. Huerta, M. G. Hutchings, H. Kraut, T. Langer, P. Löw, C. Oppawsky, T. Rein and H. Saller, Org. Process Res. Dev., 2015, 19, 357–368 CrossRef.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. Bekas and A. A. Lee, 2018, arXiv:1811.02633 [physics.chem-ph].
- P. Schwaller, T. Gaudin, D. Lanyi, C. Bekas and T. Laino, Chem. Sci., 2018, 9, 6091–6098 RSC.
- M. A. Kayala and P. Baldi, J. Chem. Inf. Model., 2012, 52, 2526–2540 CrossRef CAS PubMed.
- C. W. Coley, R. Barzilay, T. S. Jaakkola, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2017, 3, 434–443 CrossRef CAS PubMed.
- C. W. Coley, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jensen, Chem. Sci., 2019, 10, 370–377 RSC.
- D. Fooshee, A. Mood, E. Gutman, M. Tavakoli, G. Urban, F. Liu, N. Huynh, D. Van Vranken and P. Baldi, Mol. Syst. Des. Eng., 2018, 3, 442–452 RSC.
- J. Nam and J. Kim, 2016, arXiv:1612.09529 [cs.LG].
- J. N. Wei, D. Duvenaud and A. Aspuru-Guzik, ACS Cent. Sci., 2016, 2, 725–732 CrossRef CAS PubMed.
- H. Gao, T. J. Struble, C. W. Coley, Y. Wang, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2018, 4, 1465–1476 CrossRef CAS PubMed.
- ASKCOS, http://askcos.mit.edu, Accessed on June 08, 2019.
- IBM Co., IBM ILOG CPLEX, Accessed on June 08, 2019 Search PubMed.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- C. W. Coley, D. A. Thomas, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamison and K. F. Jensen, Science, 2019, 365, eaax1566 CrossRef CAS PubMed.
- Reaxys, https://new.reaxys.com/, Accessed on June 08, 2019.
- F. Mincione, F. Benedini, S. Biondi, A. Cecchi, C. Temperini, G. Formicola, I. Pacileo, A. Scozzafava, E. Masini and C. T. Supuran, Bioorg. Med. Chem. Lett., 2011, 21, 3216–3221 CrossRef CAS.
- N. Yamakawa, K. Suzuki, Y. Yamashita, T. Katsu, K. Hanaya, M. Shoji, T. Sugai and T. Mizushima, Bioorg. Med. Chem., 2014, 22, 2529–2534 CrossRef CAS.
- F. Caturla, J.-M. Jiménez, N. Godessart, M. Amat, A. Cárdenas, L. Soca, J. Beleta, H. Ryder and M. I. Crespo, J. Med. Chem., 2004, 47, 3874–3886 CrossRef CAS PubMed.
- S. Wanaski, S. Collins, J. Kincaid and G. Dodson, WO Pat., WO2013059648, 2013 Search PubMed.
- D. Bajusz, A. Rácz and K. Héberger, Aust. J. Chem., 2015, 7, 20 Search PubMed.
- M. Tang, J. Ding, R. Zhang and Q. Yu, CN Pat., CN102558071, 2012 Search PubMed.
- W. Zhu, A. Nasier, M. Song, X. Liu and Q. Wang, CN Pat., CN102295615, 2011 Search PubMed.
- K. Molga, P. Dittwald and B. A. Grzybowski, Chem. Sci., 2019, 10, 9219–9232 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9re00348g |
| This journal is © The Royal Society of Chemistry 2020 |