
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and structural characterization of CO2-soluble oxidizers [Bu4N]BrO3 and [Bu4N]ClO3 and their dissolution in cosolvent-modified CO2 for reservoir applications†
Katherine L. Hull *a,
Desmond E. Schippera and
Allen G. Oliverb
*a,
Desmond E. Schippera and
Allen G. Oliverb
aAramco Services Company: Aramco Research Center – Houston, 16300 Park Row, Houston, TX 77084, USA. E-mail: katherine.hull@aramcoamericas.com
bThe Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, USA
First published on 21st December 2020
Abstract
CO2 utilization in upsteam oil and gas applications requires CO2-soluble additives such as polymers, surfactants, and other components. Here we report the facile synthesis of CO2-soluble oxidizers composed of judiciously selected organic cations paired with oxidizing anions. [Bu4N]BrO3 and [Bu4N]ClO3 are prepared using a double displacement synthetic strategy, whereby the crystalline product is readily obtained in high yield and structurally characterized using single-crystal X-ray diffraction. The facility of the approach is demonstrated through the preparation of several additional alkylammonium bromate compounds. Static solubility studies using a high-pressure cell with viewing windows showed that tetrabutylammonium compounds could be solubilized using cosolvent-modified CO2. Using 4 mol% ethanol as cosolvent, >3 mM [Bu4N]BrO3 could be dissolved in CO2, while ∼0.75 mM [Bu4N]ClO3 could be dissolved in the same solvent system. The solubility properties of [Bu4N]BrO3 along with its thermal stability up to ∼200 °C suggest that it is a promising oilfield oxidizer that can be utilized in subterranean CO2 applications.
Introduction
As the global demand for reducing CO2 emissions continues to rise, the need for better methods to sequester, utilize, and convert CO2 also continues to increase. Injecting CO2 in subterranean rock formations is an important technology under development that can ultimately store large quantities of CO2.1 Meanwhile CO2 utilization in the energy sector for enhanced oil recovery (EOR)2,3 or shale fracturing operations4,5 may lead to some CO2 uptake while also providing a means of reducing valuable groundwater consumption. Hydraulic fracturing in shale reservoirs with extended reach horizontal drilling and multistage fracturing can consume tens of millions of gallons of water in a single well. Using CO2 for fracturing, however, is very challenging due to its low viscosity and poor dissolving power for the hydrophilic fracturing fluid additives that have been developed over the decades. Advances in nonfluorinated CO2-soluble polymers and surfactants suggests that CO2-based hydraulic fracturing is attainable.6 However no examples of complementary CO2-soluble oxidizers have been reported. These components are vitally important in fracturing fluid systems for breaking polymer chains to reduce viscosity at a specified time during the well operation and to aid in clean-up. Typical oxidizing salts used in aqueous fracturing fluids have negligible solubilities in liquid or supercritical CO2. Therefore, preparing oxidizers with solubility and reactivity properties suitable for use in CO2 media are needed.We turned our attention to bromate-based oxidizing salts, which have seen consistent application in subterranean stimulation fluids.7,8 Their usage is partially due to the convenient overlap in the temperature range over which they are active toward relevant reductants and the typical bottom hole static temperature of reservoirs. Bromate-containing fluids injected from the surface into rock formations experience an inherent delay before activation due to the time required to heat the fluid to reservoir temperatures and thereby activate the bromate. In situ pH changes can also be used to alter the reactivity of bromate after a specified amount of time. Not only does the thermal stability of bromate lend to its usefulness in subsurface applications but also its high electron per mole oxidizing capacity (6e− to form the reduced species Br−) if all oxidizing equivalents are utilized.9 Other common oxidizers used in these applications such as persulfate, chlorite, perborate, and peroxide are able to receive as low as 2e− per mole and/or have limited thermal stability. In some cases, many of these oxidizers also produce reduced species with the potential to generate scale, namely divalent salt precipitate, which may block hydrocarbon flow in pore spaces or propped fractures.
We recently demonstrated the use of two different bromate-based fluid systems for upstream well stimulation applications. The first fluid is composed of ammonium salts and sodium bromate where the ammonium is oxidized to generate mineral or organic acids depending upon the counteranion.10 A second fluid system incorporating bromate was designed to fracture unconventional shale source rock by oxidizing the ductile polymeric organic matter dispersed on the fracture faces.11 Both water-based stimulation fluids capture the effects of delayed oxidizer activation using natural reservoir heating as well as the high oxidizing power of bromate. Laboratory studies indicate that Br− ion, the fully reduced species, is the ultimate fate of the bromate system in the presence of hydrocarbon-bearing formation rocks such as carbonate and shale.
In order to bring the advantages of bromate oxidizer to CO2-based systems, a synthetic method was developed to replace the metal cation of a bromate salt with an organic cation to increase the overall hydrophobicity of the molecule. This approach is similar to the hydrophobic ion pairing (HIP) strategy that has been employed for drug delivery and protein solubilization in organic media where a charged hydrophilic molecule of interest is paired with a hydrophobic ion of opposite charge.12–16 The bromate anion electrostatically coupled to an organic cation could potentially be solubilized in CO2. In general, CO2 does not solubilize compounds that are high molecular weight, polar, or ionic to a significant extent. This solubility aspect handicaps the use of CO2 as a solvent in most industrial applications for these types of compounds. However, it has been demonstrated that CO2 can be modified with small amounts of polar cosolvent in order to drastically enhance the solubility of polar solutes and could potentially aid the solubility of ionic solutes given the proper choice of cation and anion. Recent studies on the solubility of light hydrocarbons in CO2 demonstrated notable improvements when cosolvent was added to the fluid.17,18 The current application requires that only small amounts of oxidizing salts be dissolved in CO2. For subterranean applications where oxidizers are utilized in aqueous fluids, concentrations are typically in the range of a few pounds per thousand gallons of fluid (<0.1 wt%).19,20 The solubility of alkali salts of bromates in liquid or supercritical CO2 is negligible, so enhancing the solubility through cation modification is highly desirable.
Here we report the preparation, structural characterization, and solubilization of bromate-based ionic compounds with hydrophobic character. A few structurally characterized examples of ionic compounds composed of bromate and organic cation exist, though the molecules were designed to be rich in reactive nitrogen sites for use as solid propellants.21 We envisioned stable molecules where organic components are relatively inert to oxidation, such that the bromate would target an external source. Chlorate analogues were also prepared in order to demonstrate the utility of the general synthetic method for other oxidizing ionic liquids. Finally, the stability of the prepared compounds was investigated through thermogravimetric analysis, and their solubility in CO2 was examined.
Experimental
Chemical reagents and materials
All chemicals and solvents used in the preparation of the compounds described herein were of reagent grade and used without further purification (Table S1†). (WARNING: This series of reactions should be handled with extreme caution.)General procedure to prepare compounds 1–8
The general strategy for the synthesis of alkylammonium bromates and chlorates is through a double-displacement reaction of [RxN4−x]2SO4 with Ba(XO3)2 to yield two equivalents of [RxN4−x]XO3 and one equivalent of BaSO4 (R = alkyl; X = Br, Cl). Experimental preparations of [Bu4N]BrO3 (1), [Bu4N]ClO3 (2), [Bu3NH]BrO3 (5), [Bu2NH2]BrO3 (6), [BuNH3]BrO3 (7) (Bu = Butyl), and [Me4N]BrO3 (8) were carried out using this double displacement technique. The sulfate salts of [Bu3NH]+, [Bu2NH2]+, and [BuNH3]+ salts were prepared by the reaction of the respective amine (Bu3N, Bu2NH, and BuNH2, respectively) with sulfuric acid. The synthesis of bis(triphenylphosphine)iminium, [PPN]+, bromate (3) and chlorate (4) was performed by reacting [PPN]Cl and alkali salts of XO3− where X = Br, Cl. Detailed synthetic methods for each of compounds 1–8 along with their yields and characterization are provided in the ESI.†X-ray crystallography
Single crystal X-ray diffraction analysis was performed on 1–4 using either a Bruker PHOTON-II or Bruker APEX-II diffractometer. Details of the measurements are provided in the ESI.†CO2 solubility tests
The solid sample was placed in a 450 mL high-pressure autoclave composed of corrosion-resistant Hastelloy metal alloy with see-through windows on two sides of the vessel as shown in Fig. S4.† Typical concentrations of solute ranged from 1.5–5 mM (e.g., 0.10 g [Bu4N]BrO3 in ∼170 g liquid CO2). Optionally, cosolvent was also added along with the solid oxidizer with the concentration of cosolvent ranging from 1–5 mol% relative to the amount of CO2. After combining solute and cosolvent, the autoclave was sealed and charged with liquid CO2 to a specified mass. Most experiments were conducted with 170 g of liquid CO2 such that the top level of the liquid was viewable through the see-through window. A propeller was used to stir the CO2-cosolvent-oxidizer mixture while monitoring through the window to determine whether the oxidizer is soluble, slightly soluble, or insoluble.Thermogravimetric analysis
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements were performed on TA Instruments SDT Q600 Simultaneous DSC-TGA. The samples were placed in a tared alumina ceramic crucible and were submitted to a non-oxidative (nitrogen) atmosphere under a flow rate of 100 mL min−1 under standard temperature and pressure conditions. They were heated from ambient temperature to 1000 °C at a rate of 10 °C min−1. For each experiment, approximately 10–15 mg of solid material was placed in the crucible of the thermobalance.Results and discussion
Overview
Common cations utilized in ionic liquids were considered for their compatibility with bromate and chlorate oxidizing anions such as tetraphenylphosphonium [Ph4P]+, imidazolium [Im]+, pyridinium [Pyr]+, pyrrolidinium [Pyrr]+, and tetraalkylammonium [R4N]+ as shown in Scheme 1. The most closely reported, related structure in the literature is that of guanidinium bromate.22 No structures have been reported of these common ionic liquid cations with bromate or chlorate. [Pyr]+, [Pyrr]+, and [Im]+ were each considered as potential cations for pairing with bromate. The relative accessibility of the oxidizable N atoms suggested that the compounds, if formed, may not be stable. A systematic study was performed and will later be discussed to assess the stability of bromate in the presence of N with variable degree of substitution. A second consideration regarding cation selection is the relative cost as this is a significant factor for contemplating scalability for oilfield application. In this respect, [Pyr]+, [Pyrr]+, and [Im]+ are much more expensive than the alternative options.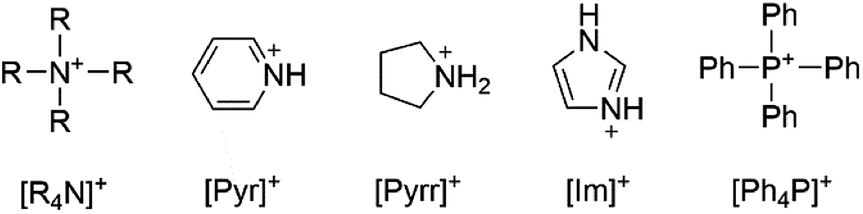 | ||
| Scheme 1 Cations commonly used in ionic liquids. | ||
Tetraalkylammonium and tetraalkylphosphonium cations are frequently used in ionic liquids due to their high chemical and thermal stability.23 The sterically protected N means that the cations have low susceptibility to oxidation. Tetraalkylphosphonium cations are less preferred for oilfield chemistry applications as any degradation of the cation to form phosphate ions could be detrimental to the reservoir environment where divalent scaling can plug pore throats or fractures. By contrast, tetraalkylammonium salts are already used in the oilfield for clay stabilization24 or biocidal purposes, so scaling new products for field application may be accelerated. These salts are routinely added to fluids for subterranean application in clay-bearing shales where there is strong potential for water swelling25 and hence destabilization of the formation surface. Therefore, adding the new tetraalkylammonium bromate/chlorate compounds to existing aqueous or CO2-based fluid formulations offers a dual benefit.
Literature precedent exists for dissolving CO2 in alkylammonium-based ionic liquids. For example, tributylmethylammonium methylsulfate was demonstrated to dissolve CO2 and did so even to a greater extent than the imidazolium analogue 1-butyl-3-methyl-imidazolium methylsulfate.26 Similarly, a tetraalkylammonium-based poly(ionic liquid) exhibited particularly high dissolution of CO2, absorbing as much as 77% of its weight in CO2.27 The solubility of alkylammonium salts in CO2 is less well understood. There is precedent for dissolving ionic compounds by fluorinating either the cation or both the cation and anion.28 Fluorinated alkylammonium cations are expected to be moderately soluble, but the scale of the application would prevent widespread adoption due to the cost prohibitive nature of fluorinated compounds. There is some evidence for improving the solubility of salts in CO2 by exchanging alkali cations for tetraalkylammonium cations. The solubility of the tetrabutylammonium salt of diethyldithiocarbamate was 2.7 times more soluble than the sodium salt.29 Though this is an ionic compound, the anion has favorable solubility in CO2 so the oxyhalides of the present application are expected to be more challenging to dissolve.
Alkylated ammonium compounds are frequently used as phase transfer catalysts. Their solubilities and ion pairing attributes in water and organic solvents has been studied thoroughly, though limited information is available about their solubilities in CO2. In water, tetraalkylated ammonium cations pair more readily with larger halide anions based on the cation–anion radial distribution function predicted by molecular dynamics.30 Tetramethylammonium behaves as a harder cation, though the softer structures of tetraethylammonium and tetrapropylammonium allow some water to penetrate.30 Tetramethylammonium salts are known to have very low solubility in chloroform,31 so they are unlikely to dissolve in CO2. Though the solubility of similar salts increases with the length of the alkyl chain, there may be a reverse at high chain lengths.31 Tetrahexylammonium ions appear to create large pockets where solvent is excluded and large anions can be housed.32 Pairing an oxidizing anion with an alkylammonium cations with long alkyl chains may ultimately hinder the anion's reactivity with substrates.
Tetrabutylammonium may serve as an ideal intermediate cation size with both sufficient hydrophobicity to encourage solubility in CO2 while also allowing reactivity between the anion and substrate. Tetrabutylammonium cations are one of the most ubiquitous cations used for phase transfer catalysis and their structures, ion–ion interactions, and transport across liquid/liquid interfaces have been studied in detail.33,34 Their salts have good solubility in both aqueous and organic phases and are a good target for a subterranean application where miscibility with reservoir fluids including both water and hydrocarbons is desirable. Tetrabutylammonium cations have been used as phase transfer catalysts for oxidation reactions.35 For example, tetrabutylammonium hypochlorite can be generated in situ and transferred to an organic phase where benzyl alcohol is oxidized to aldehyde.36–38 Tetrabutylammonium cation has also been paired with bromite39 or dichromate40 to name a couple more examples. The synthesis of [Bu4N]BrO3 and [Pr4N]BrO3 by bubbling chlorine gas through a solution of the tetraalkylammonium bromide was reported, though the compounds isolated were yellow or orange in color suggesting the formation of the tribromide salts or bromine contamination.41
Synthesis and structural characterization
The synthetic strategy for [Bu4N]BrO3 (1) and [Bu4N]ClO3 (2) is a straightforward double displacement reaction between the partially soluble Ba(BrO3)2 or Ba(ClO3)2 and a commercially available tetraalkylammonium sulfate to form insoluble BaSO4. The formation of 1 and 2 are shown in Scheme 2, though other alkylammonium salts such as tetramethylammonium and trimethylhexadecylammonium bromates were also prepared in like fashion. High yields, facile preparation, and pure crystalline solid isolates are important features of this methodology as compared to ion exchange chromatography42 for preparing tetrabutylammonium salts. A preparation method for pure tetraalkylammonium bromates or chlorates has not previously been reported. | ||
| Scheme 2 Synthesis of tetrabutylammonium bromate/chlorate. | ||
Tetrabutylammonium bromate monohydrate crystallizes as colorless block-like crystals from an aqueous solution. The asymmetric unit consists solely of the ammonium cation, bromate anion and water of crystallization. The water and bromate anion form a hydrogen-bonded chain parallel to the crystallographic b-axis (Fig. 1) and is the dominant interaction within this compound. Tetrabutylammonium chlorate (2) also crystallizes as colorless block-like crystals. There are two crystallographically independent, yet chemically identical molecules of the salt in the asymmetric unit (Fig. 2). The two independent molecules of the [Bu4N]ClO3 differ from each other in that one molecule has a minor disorder component of the butyl chains in the cation and the position of the chlorine atom in the chlorate anion. This disorder has been omitted from the figures.
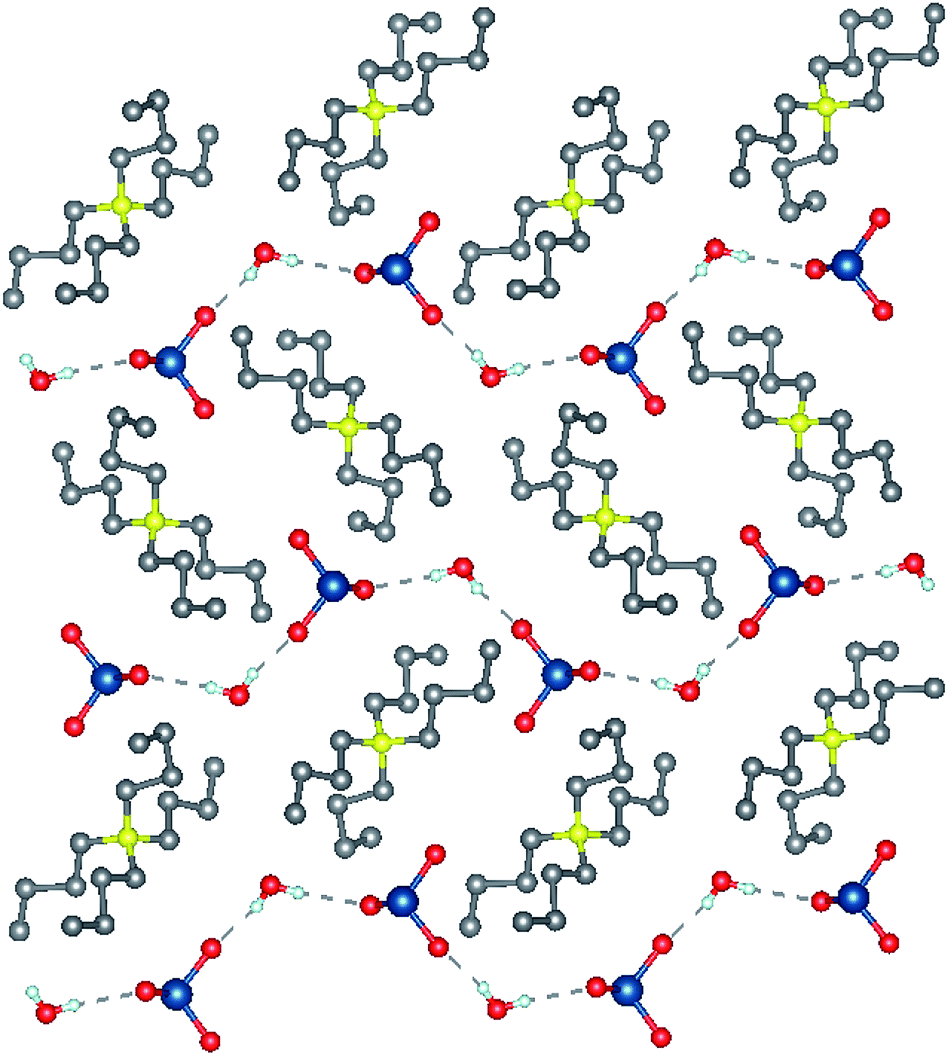 | ||
| Fig. 1 Crystal structures [Bu4N]BrO3 viewed along the a-axis. Selected hydrogen atoms have been removed for clarity. Gray = C, yellow = N, red = O, blue = Br, white = H. | ||
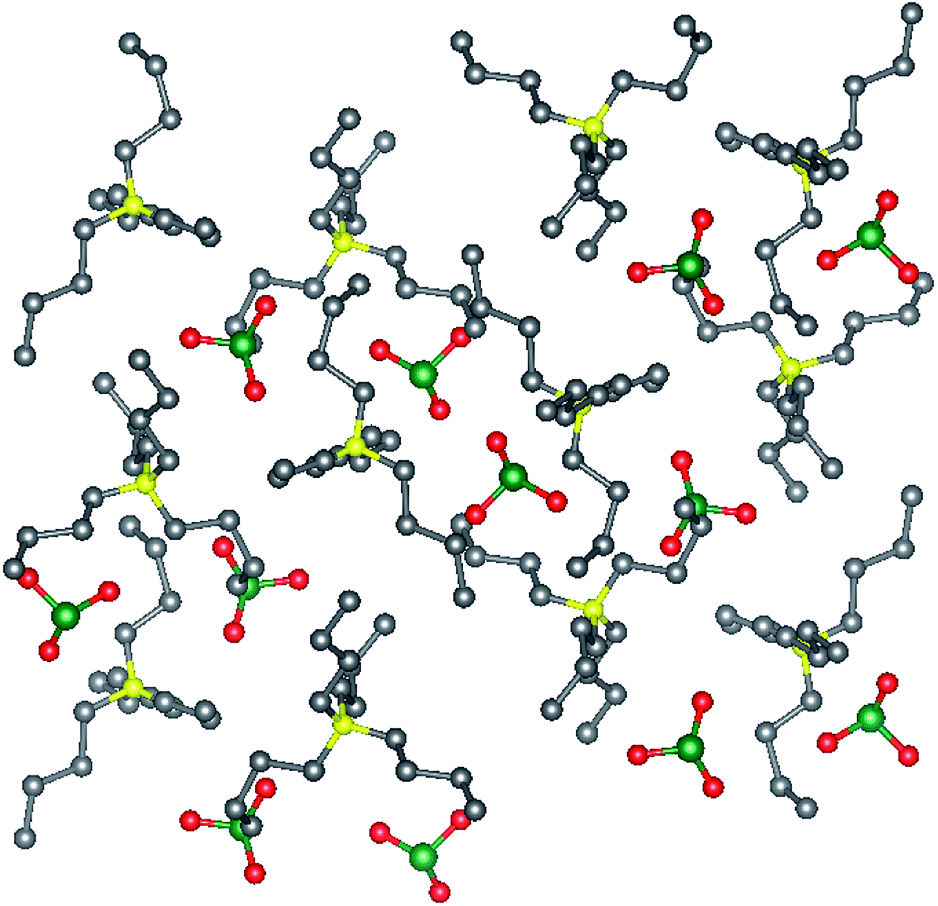 | ||
| Fig. 2 Crystal structures [Bu4N]ClO3 viewed along the a-axis. Selected hydrogen atoms have been removed for clarity. Gray = C, yellow = N, red = O, green = Cl, white = H. | ||
[PPN]+ has an established history over 50 years as a convenient counterion to stabilize air-sensitive anions such as metal carbonyls.43–45 Its bulky nature, sterically protected N atom, low overall susceptibility to oxidation, and crystallization ease made it an attractive candidate for establishing a general preparation method for bromates and chlorates paired with organic cations. The differing solubility properties of [PPN]+ salts relative to tetrabutylammonium salts led to different synthetic procedures. [PPN]+ is readily available commercially as the chloride salt with low solubility at room temperature and high solubility upon heating to 100 °C, lending itself to ready isolation of solid product. Upon combining [PPN]Cl and NaBrO3 and heating to 100 °C for 15 minutes, a liquid formed at the bottom of the tube, then crystallized upon cooling to yield [PPN]BrO3 (3). Meanwhile, the identical reaction performed between [PPN]Cl and KClO3 resulted in solid [PPN]ClO3 (4) precipitation at 100 °C. Both products were easily redissolved in water and recrystallized. The reactions are summarized in Scheme S1.†
Thermal stability
The general synthetic procedure was also applied to butylammonium bromate compounds with successively fewer butyl substituents to demonstrate the utility of the approach. Compounds 5–7 were readily synthesized by protonating the respective amine with sulfuric acid to form the alkylammonium salt. A double displacement reaction between the alkylammonium sulfate and Ba(BrO3)2 was then performed to yield the alkylammonium bromate series. Scheme S2† illustrates the reaction sequence. The stability of the resulting compounds changed sharply as the number of butyl substituents on the alkylammonium cation was reduced. Dibutyl- and tributylammonium bromates were each isolated as liquids and are stable during manipulation. Monobutylammonium bromate was isolated as a clean solid that was stable during manipulation and storage for several days. However, after approximately 10 days, the material decomposed with rapid gas evolution. Compounds 1–2 have been observed to be stable indefinitely (>6 months) while stored in a sealed container.The reactivity and thermal decomposition of 1 follows a straightforward sequence as shown in Fig. 3. Heating the solid at 10 °C per minute resulted in a gradual mass loss of 7–8% while heating to nearly 200 °C, consistent with the complete evaporation of water from the solid. When the temperature reaches 192 °C, a large exotherm is generated accompanied by abrupt and complete mass loss. Interestingly, 1 has a notably lower melting point (54 °C) than other tetrabutylammonium salt derivatives as shown in Table S3.† This observation can be rationalized by the hydrate crystal structure, where bromate-water linkages form throughout the lattice. The low melting point and high decomposition temperature means that this compound has a large stable liquid temperature range of ∼138 °C. This may open additional applications for this chemistry such as gas hydrate removal. The melting point depression through hydrate or other lattice modification may also provide an approach for ionic liquid-based CO2 capture technologies that are often limited due to their high melting points.
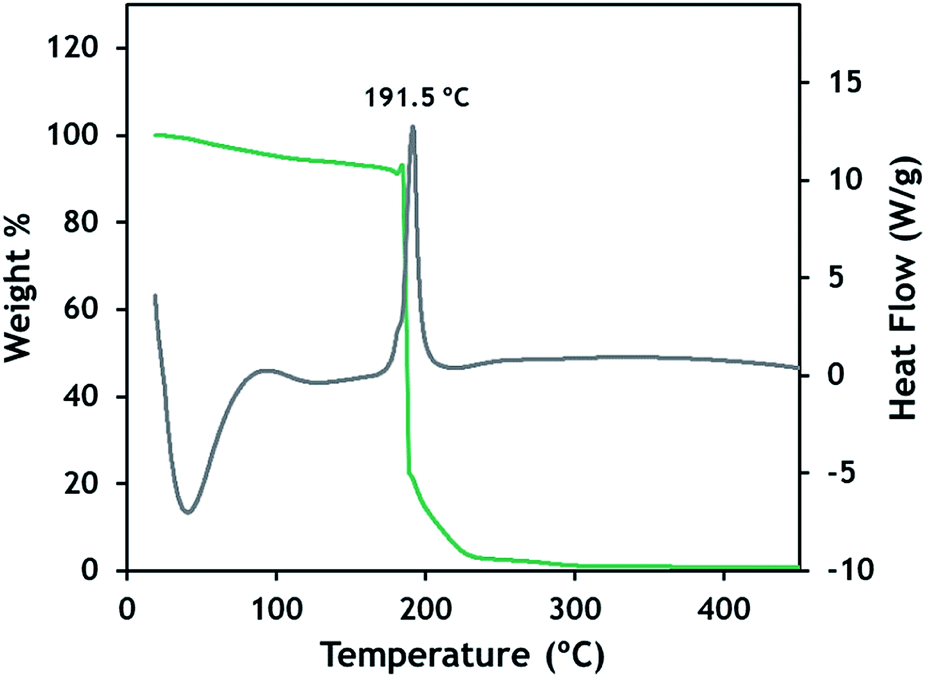 | ||
| Fig. 3 TGA-DSC of 1 while heating from room temperature to 400 °C. | ||
CO2 solubility
In order to test the solubility of the newly prepared 1 in CO2, an autoclave with see-through windows as shown in Fig. S4† was used in order to visualize the dissolution process and determine whether each solute could dissolve in the CO2. A static view cell apparatus has previously been reported for other CO2 solubility studies.46 Addition of liquid CO2 to either 1 or 2 in the see-through autoclave (1.5 mM) resulted in opaque fluid indicating that the solids were either insoluble or sparingly soluble. 2 separates as a white solid while 1 separates from the mixture as an oily liquid, consistent with its low melting point (54 °C) under atmospheric conditions. A limited number of polar solutes have been directly dissolved in CO2, but adding a small amount of polar cosolvent has been shown to enhance the solubility significantly by changing the thermodynamic properties of the system.47,48 For example, aspirin solubility in CO2 was enhanced five times with acetone cosolvent and 14 times with methanol or ethanol cosolvent.49 Typical cosolvents used with CO2 to enhance the solubility of various solutes include ketones, alkanes, and alcohols.47 Alcohols can act as hydrogen-bonding acceptors and donors, while ketones are only able to act as hydrogen-bonding acceptors. Alkanes are nonpolar and can only interact with solutes molecules via van der Waals interactions. Cosolvents are typically added up to 5 mol% though in some cases reach 10 mol%.50Beyond pharmaceutical examples such as aspirin, naproxen,51,52 and acetaminophen,53 many other examples exist for solubilizing polar compounds in CO2 through polar cosolvent addition. Caffeine can be extracted from green tea using ethyl lactate, ethyl acetate, and ethanol where ethyl lactate showed the strongest effects upon solubilization due to its ability to hydrogen bond through both hydroxyl and carbonyl groups.54 Benzoic acid and salicylic acid solubilities are improved with methanol and ethanol due to hydrogen bonding between the solute and cosolvent.55,56 Acetic acid and methyl acetate are each suitable cosolvents for stearic acid, though acetic acid has a much stronger effect on solubility.57 A combination of cosolvents also aids in the solubilization of stearic acid in CO2, though the effect of binary cosolvent was observed to be intermediate to the two individual cosolvents.58 Perhaps most relevant to the present study is an ionic compound composed of an alkylated ammonium salt. Acid Red 57, an anionic dye, can be ion paired with dodecyltrimethylammonium cation and solubilized in CO2 with 10 mol% methanol as cosolvent.59
Solubility experiments were then conducted on 1 in the presence of cosolvents. A wide range of cosolvents were tested including acetone, ethyl acetate, ethyl lactate, ethanol, and acetic acid. The results of dissolution tests performed with 4 mol% cosolvent in the presence of 1.5 mM 1 solute are shown in Fig. 4. Very little, if any, solute is dissolved in CO2 modified with either acetone or ethyl acetate, even after stirring for a few hours. On the other hand, CO2 modified with either ethyl lactate or ethanol yielded fully homogenous solutions with no visible solute. The positive effect of these two cosolvents is likely attributed to their hydrogen-bond donating capabilities and not to their dipole moments, because ethanol has a low dipole moment relative to both acetone and ethyl acetate.
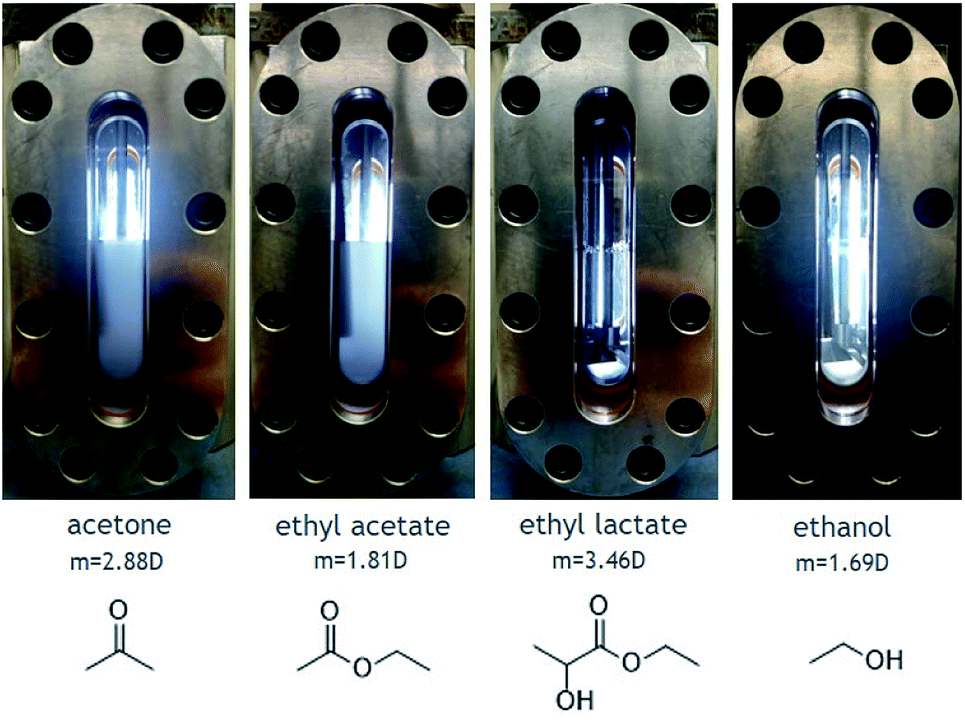 | ||
| Fig. 4 Dissolution tests of 1 (1.5 mM) in carbon dioxide (20 °C, ∼800 psi) with 4 mol% cosolvent. | ||
Increasing the concentration of 1 two-fold to 3 mM while maintaining the concentration of cosolvent distinguished the dissolution effects of ethanol from ethyl lactate. Fig. 5(a) shows the results of the two tests. While 3 mM of 1 with 4 mol% ethyl lactate demonstrated low solubility in CO2, 3 mM 1 with 4 mol% ethanol was fully dissolved in the CO2 solution. However, 4.5 mM of 1 with 4 mol% ethanol exhibited some cloudiness, indicating that the solubility limit is slightly less than that (Fig. S5†). In order to examine the role of the butyl chains in the solubility of this compound, the solubility of [Me4N]BrO3 (8) was also examined. The results of 1.5 mM 8 dispersed in CO2 with 4 mol% of either ethyl lactate or ethanol are shown in Fig. 5(b). As is evident in the images, 8 exhibits low solubility in CO2 in the presence of either cosolvent, indicating that the longer butyl chains provide the requisite hydrophobicity to enhance the solubility of these compounds in nonpolar CO2 solvent. Although 3 and 4 could be readily prepared in high yield and isolated as crystalline solids, their solubility in CO2 was limited in the presence of any of the cosolvents examined.
 | ||
| Fig. 5 Dissolution tests of (a) 1 (3 mM) and (b) 8 (1.5 mM) in carbon dioxide (20 °C, ∼800 psi) with 4 mol% cosolvent. | ||
Compound 2 exhibited much lower solubility than 1 with the various cosolvents. With 4 mol% of ethanol, ethyl lactate, ethyl acetate, or acetone, 1.5 mM of 2 in CO2 is not fully dissolved. Decreasing the concentration of solute to 0.75 mM yielded full solubility in CO2 with 4 mol% ethanol but still a significant undissolved fraction in CO2 modified with 4 mol% ethyl lactate. Meanwhile, the commercially available [Bu4N]ClO4 exhibited even lower solubility than 2, where even 0.75 mM [Bu4N]ClO4 in CO2 modified with 4 mol% ethanol was sparingly soluble. The two solubility tests performed at 0.75 mM are shown in Fig. S6.† By contrast, [Bu4N]Cl exhibited solubility results very similar to 1 where 3 mM was fully dissolved but 4.5 mM is cloudy. The solubility trend of the [Bu4N]+ salt series in CO2 modified with 4 mol% ethanol follows the trend:
| BrO3− ∼ Cl− ≫ ClO3− > ClO4− |
The higher solubility of the bromate salt is consistent with the bromate versus chlorate crystal structures. The bromate ions form a hydrogen-bonded chain with water molecules, while the chlorate salt is anhydrous. It has been shown that bromate exhibits both structure-making and structure-breaking properties in water, where the three oxygen atoms acts kosmotropically while the bromine atom behaves chaotropically.60 In the current system, bromate may interact more favorably with the hydrogen-bond donating solvents than chlorate or perchlorate, yielding higher solubility. Notably, the solubility of [Bu4N]BrO3 achieved (>12 wt%) with ethanol cosolvent significantly exceeds the typical concentrations utilized in the application (∼0.03 wt%).
Conclusions
Oxybromine chemistries continue to be actively explored for their utility in chemical transformations and their presence in aqueous environmental systems. The synthesis of oxidizers composed of bulky organic cations and oxybromine/oxychlorine anions is now reliably demonstrated. The oxidizers are prepared in high yield using a straightforward double displacement reaction method and isolated as crystalline solids whose structures were verified crystallographically. [Bu4N]+ paired with bromate or chlorate is stable upon heating to ∼200 °C, well above subterranean temperature conditions. Dissolution of 1 and 2 in cosolvent-modified CO2 was achieved using ethanol and, to a lesser degree, ethyl lactate, demonstrating the utility of these compounds for potential field application.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Joshua Brothers for assistance with FT-IR measurements and Aditya Dimri for assistance with some of the CO2 solubility experiments.References
- S. Krevor, M. J. Blunt, J. P. M. Trusler and S. D. Simone, in Carbon Capture and Storage, 2019, pp. 238–295 Search PubMed.
- Z. Dai, R. Middleton, H. Viswanathan, J. Fessenden-Rahn, J. Bauman, R. Pawar, S.-Y. Lee and B. McPherson, An Integrated Framework for Optimizing CO2 Sequestration and Enhanced Oil Recovery, Environ. Sci. Technol. Lett., 2014, 1, 49–54 Search PubMed.
- B. Jia, J.-S. Tsau and R. Barati, A review of the current progress of CO2 injection EOR and carbon storage in shale oil reservoirs, Fuel, 2019, 236, 404–427 Search PubMed.
- W. Lin, A. M. Bergquist, K. Mohanty and C. J. Werth, Environmental Impacts of Replacing Slickwater with Low/No-Water Fracturing Fluids for Shale Gas Recovery, ACS Sustainable Chem. Eng., 2018, 6, 7515–7524 Search PubMed.
- R. S. Middleton, J. W. Carey, R. P. Currier, J. D. Hyman, Q. Kang, S. Karra, J. Jiménez-Martínez, M. L. Porter and H. S. Viswanathan, Shale gas and non-aqueous fracturing fluids: Opportunities and challenges for supercritical CO2, Appl. Energy, 2015, 147, 500–509 Search PubMed.
- T. Sarbu, T. Styranec and E. J. Beckman, Non-fluorous polymers with very high solubility in supercritical CO2 down to low pressures, Nature, 2000, 405, 165–168 Search PubMed.
- A. F. Ibrahim, H. A. Nasr-El-Din, A. Rabie, G. Lin, J. Zhou and Q. Qu, A New Friction-Reducing Agent for Slickwater-Fracturing Treatments, SPE Prod. Oper., 2018, 33, 583–595 Search PubMed.
- T. Almubarak, J. H. Ng, H. A. Nasr-El-Din, K. Sokhanvrian and M. AlKhaldi, Dual-Polymer Hydraulic-Fracturing Fluids: A Synergy Between Polysaccharides and Polyacrylamides, SPE J., 2019, 24, 2635–2652 Search PubMed.
- G. Joshi, S. Bhadra, S. Ghosh, M. K. Agrawal, B. Ganguly, S. Adimurthy, P. K. Ghosh and B. C. Ranu, Making Full Use of the Oxidizing Equivalents in Bromate in the Selective Oxidation of Thiols, Sulfides, and Benzylic/Secondary Alcohols into Disulfides, Sulfoxides, and Aldehydes/Ketones, Ind. Eng. Chem. Res., 2010, 49, 1236–1241 Search PubMed.
- K. L. Hull, A. J. Cairns and M. Haq, Bromate Oxidation of Ammonium Salts: In Situ Acid Formation for Reservoir Stimulation, Inorg. Chem., 2019, 58, 3007–3014 Search PubMed.
- K. L. Hull, D. Jacobi and Y. N. Abousleiman, Oxidative Kerogen Degradation: A Potential Approach to Hydraulic Fracturing in Unconventionals, Energy Fuels, 2019, 33, 4758–4766 Search PubMed.
- K. D. Ristroph and R. K. Prud’homme, Hydrophobic ion pairing: encapsulating small molecules, peptides, and proteins into nanocarriers, Nanoscale Adv., 2019, 1, 4207–4237 Search PubMed.
- K. Benaissi, M. Poliakoff and N. R. Thomas, Solubilisation of α-chymotrypsin by hydrophobic ion pairing in fluorous systems and supercritical carbon dioxide and demonstration of efficient enzyme recycling, Green Chem., 2010, 12, 54–59 Search PubMed.
- S. Taguchi, T. Ichikawa, T. Kato and H. Ohno, Nano-biphasic ionic liquid systems composed of hydrophobic phosphonium salts and a hydrophilic ammonium salt, Chem. Commun., 2012, 48, 5271–5273 Search PubMed.
- D. J. Kozuch, K. Ristroph, R. K. Prud’homme and P. G. Debenedetti, Insights into Hydrophobic Ion Pairing from Molecular Simulation and Experiment, ACS Nano, 2020, 14, 6097–6106 Search PubMed.
- T. N. Q. Phan, R. Ismail, B. Le-Vinh, S. Zaichik, F. Laffleur and A. Bernkop-Schnürch, The Effect of Counterions in Hydrophobic Ion Pairs on Oral Bioavailability of Exenatide, ACS Biomater. Sci. Eng., 2020, 6, 5032–5039 Search PubMed.
- H. Gong, X. Qin, S. Shang, C. Zhu, L. Xu, Q. San, Y. Li and M. Dong, Enhanced Shale Oil Recovery by the Huff and Puff Method Using CO2 and Cosolvent Mixed Fluids, Energy Fuels, 2020, 34, 1438–1446 Search PubMed.
- Y. Du, H. Zhang, X. Li, Y. Zhang and S. Yuan, Molecular Dynamics Study on the Suitable Compatibility Conditions of a CO2-Cosolvent-Light Hydrocarbon System by Calculating the Solubility Parameters, Energy Fuels, 2020, 34, 3483–3492 Search PubMed.
- G. A. Al-Muntasheri, A Critical Review of Hydraulic-Fracturing Fluids for Moderate- to Ultralow-Permeability Formations Over the Last Decade, SPE Prod. Oper., 2014, 29, 243–260 Search PubMed.
- R. Barati and J.-T. Liang, A review of fracturing fluid systems used for hydraulic fracturing of oil and gas wells, J. Appl. Polym. Sci., 2014, 131, 40735 Search PubMed.
- M. H. H. Wurzenberger, N. Szimhardt and J. Stierstorfer, Nitrogen-Rich Copper(II) Bromate Complexes: an Exotic Class of Primary Explosives, Inorg. Chem., 2018, 57, 7940–7949 Search PubMed.
- J. Drenth, W. Drenth, A. Vos and E. H. Wiebenga, On the crystal structure of guanidinium bromate, Acta Crystallogr., 1953, 6, 424 Search PubMed.
- M. K. Mishra, S. P. Kelley, J. L. Shamshina, H. Choudhary and R. D. Rogers, Can Melting Point Trends Help Us Develop New Tools To Control the Crystal Packing of Weakly Interacting Ions?, Cryst. Growth Des., 2018, 18, 597–601 Search PubMed.
- Petroleum Engineer's Guide to Oil Field Chemicals and Fluids, ed. J.Fink, 2nd edn, Gulf Professional Publishing, Boston, 2015, pp. 121–145 Search PubMed.
- E. J. M. Hensen and B. Smit, Why Clays Swell, J. Phys. Chem. B, 2002, 106, 12664–12667 Search PubMed.
- M. Ramdin, T. J. H. Vlugt and T. W. de Loos, Solubility of CO2 in the Ionic Liquids [TBMN][MeSO4] and [TBMP][MeSO4], J. Chem. Eng. Data, 2012, 57, 2275–2280 Search PubMed.
- S. Supasitmongkol and P. Styring, High CO2 solubility in ionic liquids and a tetraalkylammonium-based poly(ionic liquid), Energy Environ. Sci., 2010, 3, 1961–1972 Search PubMed.
- A. E. Zweber, M. Wagner, J. DeYoung and R. G. Carbonell, Mechanism of Extreme Ultraviolet Photoresist Development with a Supercritical CO2 Compatible Salt, Langmuir, 2009, 25, 6176–6190 Search PubMed.
- J. Wang and W. D. Marshall, Recovery of Metals from Aqueous Media by Extraction with Supercritical Carbon Dioxide, Anal. Chem., 1994, 66, 1658–1663 Search PubMed.
- J. Heyda, M. Lund, M. Ončák, P. Slavíček and P. Jungwirth, Reversal of Hofmeister Ordering for Pairing of NH4+ vs. Alkylated Ammonium Cations with Halide Anions in Water, J. Phys. Chem. B, 2010, 114, 10843–10852 Search PubMed.
- J. Czapkiewicz, Solubilization of some tetramethylammonium salts and of ethyltrimethylammonium bromide by their homologues in chloroform, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2669–2674 Search PubMed.
- J. A. Faust, L. P. Dempsey and G. M. Nathanson, Surfactant-Promoted Reactions of Cl2 and Br2 with Br– in Glycerol, J. Phys. Chem. B, 2013, 117, 12602–12612 Search PubMed.
- I. Benjamin, Recombination, Dissociation, and Transport of Ion Pairs across the Liquid/Liquid Interface. Implications for Phase Transfer Catalysis, J. Phys. Chem. B, 2013, 117, 4325–4331 Search PubMed.
- D. Bhowmik, N. Malikova, G. Mériguet, O. Bernard, J. Teixeira and P. Turq, Aqueous solutions of tetraalkylammonium halides: ion hydration, dynamics and ion–ion interactions in light of steric effects, Phys. Chem. Chem. Phys., 2014, 16, 13447–13457 Search PubMed.
- N. Kikkawa, T. Ishiyama and A. Morita, Molecular dynamics study of phase transfer catalyst for ion transfer through water–chloroform interface, Chem. Phys. Lett., 2012, 534, 19–22 Search PubMed.
- D. C. M. Albanese, F. Foschi and M. Penso, Sustainable Oxidations under Phase-Transfer Catalysis Conditions, Org. Process Res. Dev., 2016, 20, 129–139 Search PubMed.
- L. M. Bradley, J. M. Lesica and K. B. Albertson, Kinetics of phase-transfer-catalyzed oxidations of p-substituted benzyl alcohols with aqueous hypochlorite, React. Kinet. Catal. Lett., 1997, 62, 3–7 Search PubMed.
- J. S. Do and T. C. Chou, Kinetics of the oxidation of benzyl alcohol by hypochlorite ion in the presence of phase-transfer catalyst, Ind. Eng. Chem. Res., 1990, 29, 1095–1103 Search PubMed.
- D. Pletcher and N. Tomov, The use of electrogenerated hypobromite for the phase transfer catalysed oxidation of benzyl alcohols, J. Appl. Electrochem., 1977, 7, 501–504 Search PubMed.
- K. Bijudas, P. Bashpa and T. D. R. Nair, Kinetic Studies on the Selective Oxidation of Benzyl Alcohols in Organic Medium under Phase Transfer Catalysis, Bull. Chem. React. Eng. Catal., 2014, 9, 142–147 Search PubMed.
- U. Nath, S. S. Das, D. Deb and P. J. Das, Tetra-n-alkylammonium bromates—new and efficient reagents for deoximation, New J. Chem., 2004, 28, 1423–1425 Search PubMed.
- V. D. Bhatt and K. Gohil, Ion exchange synthesis and thermal characteristics of some [N+4444] based ionic liquids, Thermochim. Acta, 2013, 556, 23–29 Search PubMed.
- J. K. Ruff, Chemistry of the dinuclear metal carbonyl anions. I. Insertion reactions, Inorg. Chem., 1967, 6, 2080–2082 Search PubMed.
- J. K. Ruff, Chemistry of the dinuclear carbonyl anions. II. Mixed-metal derivatives, Inorg. Chem., 1968, 7, 1818–1821 Search PubMed.
- J. K. Ruff, Chemistry of the dinuclear carbonyl anions. III. Halogen-bridged anions, Inorg. Chem., 1968, 7, 1821–1825 Search PubMed.
- E. Pérez, A. Cabañas, J. A. R. Renuncio, Y. Sánchez-Vicente and C. Pando, Cosolvent Effect of Methanol and Acetic Acid on Dibenzofuran Solubility in Supercritical Carbon Dioxide, J. Chem. Eng. Data, 2008, 53, 2649–2653 Search PubMed.
- A. I. Frolov and M. G. Kiselev, Prediction of Cosolvent Effect on Solvation Free Energies and Solubilities of Organic Compounds in Supercritical Carbon Dioxide Based on Fully Atomistic Molecular Simulations, J. Phys. Chem. B, 2014, 118, 11769–11780 Search PubMed.
- X. Zhang, B. Han, Z. Hou, J. Zhang, Z. Liu, T. Jiang, J. He and H. Li, Why Do Co-solvents Enhance the Solubility of Solutes in Supercritical Fluids? New Evidence and Opinion, Chem. – Eur. J., 2002, 8, 5107–5111 Search PubMed.
- Z. Huang, Y. C. Chiew, W.-D. Lu and S. Kawi, Solubility of aspirin in supercritical carbon dioxide/alcohol mixtures, Fluid Phase Equilib., 2005, 237, 9–15 Search PubMed.
- J. M. Dobbs, J. M. Wong, R. J. Lahiere and K. P. Johnston, Modification of supercritical fluid phase behavior using polar cosolvents, Ind. Eng. Chem. Res., 1987, 26, 56–65 Search PubMed.
- S. S. T. Ting, D. L. Tomasko, N. R. Foster and S. J. Macnaughton, Solubility of naproxen in supercritical carbon dioxide with and without cosolvents, Ind. Eng. Chem. Res., 1993, 32, 1471–1481 Search PubMed.
- S. S. T. Ting, D. L. Tomasko, N. R. Foster and S. J. Macnaughton, Chemical-physical interpretation of cosolvent effects in supercritical fluids, Ind. Eng. Chem. Res., 1993, 32, 1482–1487 Search PubMed.
- S. Bristow, B. Y. Shekunov and P. York, Solubility Analysis of Drug Compounds in Supercritical Carbon Dioxide Using Static and Dynamic Extraction Systems, Ind. Eng. Chem. Res., 2001, 40, 1732–1739 Search PubMed.
- D. V. Bermejo, E. Ibáñez, G. Reglero and T. Fornari, Effect of cosolvents (ethyl lactate, ethyl acetate and ethanol) on the supercritical CO2 extraction of caffeine from green tea, J. Supercrit. Fluids, 2016, 107, 507–512 Search PubMed.
- J. Ke, S. Jin, B. Han, H. Yan and D. Shen, Hydrogen bonding of some organic acid in supercritical CO2 with polar cosolvents, J. Supercrit. Fluids, 1997, 11, 53–60 Search PubMed.
- Q. Xu, B. Han and H. Yan, Equilibrium Constant and Enthalpy for the Hydrogen Bonding of Acetic Acid with Tetrahydrofuran in Supercritical CO2, J. Phys. Chem. A, 1999, 103, 5240–5245 Search PubMed.
- M. Zhong, B. Han and H. Yan, Solubility of stearic acid in supercritical CO2 with cosolvents, J. Supercrit. Fluids, 1997, 10, 113–118 Search PubMed.
- B. Guan, B. Han and H. Yan, Effect of acetic acid+acetonitrile and ethanol+acetonitrile mixed cosolvents on the solubility of stearic acid in supercritical CO2, Fluid Phase Equilib., 1998, 149, 277–286 Search PubMed.
- A. Özcan and A. S. Özcan, Solubility of an acid dye in supercritical carbon dioxide by ion-pairing with dodecyltrimethylammonium bromide, Fluid Phase Equilib., 2006, 249, 1–5 Search PubMed.
- B. Sharma and A. Chandra, Born–Oppenheimer Molecular Dynamics Simulations of a Bromate Ion in Water Reveal Its Dual Kosmotropic and Chaotropic Behavior, J. Phys. Chem. B, 2018, 122, 2090–2101 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, crystallographic data for compounds 1–2 and 6–7, TGA-DSC, melting points for tetrabutylammonium salts, CO2 solubility test results. CCDC 2039678–2039681. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra09563j |
| This journal is © The Royal Society of Chemistry 2020 |