
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecise synthesis of amphiphilic diblock copolymers consisting of various ionic liquid-type segments and their influence on physical gelation behavior in water†
Daichi Yokota ,
Arihiro Kanazawa and
Sadahito Aoshima*
,
Arihiro Kanazawa and
Sadahito Aoshima*
Department of Macromolecular Science, Graduate School of Science, Osaka University, 1-1 Machikaneyama, Toyonaka, Osaka 560-0043, Japan. E-mail: aoshima@chem.sci.osaka-u.ac.jp; Fax: +81-6-6850-5448; Tel: +81-6-6850-5448
First published on 21st November 2020
Abstract
Appropriately designed amphiphilic diblock vinyl ether (VE) copolymers consisting of an ionic liquid-type segment and a hydrophobic segment were demonstrated to undergo physical gelation in water at extremely low concentrations. The precursor diblock copolymers were synthesized by the living cationic polymerization of 2-chloroethyl VE with a hydrophobic VE through an appropriately designed initiating system such as optimized temperature and catalyst. A relatively high temperature such as 20 °C was important for controlled polymerization of octadecyl VE. Ionic liquid moieties with imidazolium salt structures were subsequently introduced into the side chains of the diblock copolymers via chemical modifications of the 2-chloroethyl groups. The physical gelation behavior of the obtained diblock copolymers was examined in water, with a particular focus on the influence of the hydrophobic VEs, the hydrophilicity of the counteranions and the substituents on the ionic liquid-type segments, and the length of each segment. Based on this systematic investigation, physical gelation at concentrations as low as 0.2 wt% was achieved with diblock copolymers with a suitable balance of these factors.
1 Introduction
The careful design of the hydrophilic and hydrophobic segments of amphiphilic block copolymers is important because their self-assembled structures and macroscopic physical properties depend on the nature of these segments. Amphiphilic block copolymers self-assemble via the hydrophobic interactions among hydrophobic segments in aqueous solutions, and their self-assembled structures depend on the hydrophilicity/hydrophobicity balance of each segment.1,2 For example, copolymers with long hydrophobic segments form large aggregates, such as biomolecular membranes, cylindrical micelles, and vesicles. Moreover, some studies have demonstrated unique changes in the macroscopic physical properties by varying the hydrophobic segments.3–9 An amphiphilic diblock copolymer with a polypeptide chain as the hydrophobic segment exhibited physical gelation behavior in water at an extremely low concentration via the formation of polypeptide-derived aggregated structures, such as β-sheets or α-helices.3 In addition, the self-assembled structures of amphiphilic block copolymers with a stimuli-responsive segment can be altered in response to external stimuli. For example, a diblock copolymer composed of poly(ethylene glycol) and poly(N-isopropylacrylamide) segments, the latter of which exhibits lower critical solution temperature (LCST)-type phase separation behavior in water,4,5 aggregates and dissociates in water upon heating and cooling, respectively.6 The particle size of a block copolymer with carboxy groups as the hydrophilic moieties changes depending on the pH of the aqueous solution.7,8Ionic liquid-type polymers, which have ionic liquid structures in their side chains, show various physical properties due to the high designability of the ionic liquid moieties.10–15 Both experimental and theoretical studies have been conducted to investigate the effects of ionic liquid moieties on the properties of polymers. In particular, the designability of the cation and counteranion structures, which can include imidazolium, ammonium, and phosphonium groups, has facilitated the creation of polymers with various novel functions.16–21 For example, our group16,17 reported that vinyl ether (VE) polymers with pendant imidazolium ionic liquid segments exhibited LCST-type or UCST-type phase-separation behaviors in organic solvents and water, respectively, depending on the substituents on the imidazolium moiety and the hydrophilicity of the counteranions. In addition, Ohno and coworkers20,21 reported polymeric gels composed of ionic liquid-type polymers. The polymeric gels possess a high ion conductivity due to the ionic properties of the ionic liquid-type polymers.
We demonstrated that upper critical solution temperature (UCST)-type amphiphilic diblock copolymers with imidazolium ionic liquid segments exhibit thermosensitive physical gelation behavior at extremely low concentrations in water.22 The precise synthesis of precursor diblock copolymers consisting of 2-chloroethyl VE (CEVE) and isobutyl VE (IBVE) was feasible with an elaborately designed initiating system. The copolymers have MWs that are in agreement with the theoretical values. An aqueous solution of the amphiphilic diblock copolymers prepared by chemical modification and counteranion exchange of these precursor copolymers underwent thermosensitive physical gelation at concentrations of 1 or 0.2 wt%. The formation of large micelles with extended corona chains based on electrostatic repulsion and hydration of the ionic liquid segments was likely responsible for the physical gelation behavior in water at a very low concentration.23–25 Therefore, the elaborate design of the ionic liquid-type amphiphilic block copolymers, such as the structures of the hydrophilic and hydrophobic monomers and the ratio of their chain lengths, are expected to result in remarkable physical gelation behavior, such as gelation at much lower concentrations, adjustments of the thermosensitivity in water, and the formation of tough physical gels.
In this study, amphiphilic diblock copolymers composed of imidazolium-type poly(ionic liquid)s as hydrophilic segments and different hydrophobic poly(VE) segments were synthesized to investigate the effects of both the hydrophilicity of the ionic liquid structures and the hydrophobicity of the hydrophobic segments on the physical gelation behavior (Scheme 1). The hydrophilicity of the counteranions and the substituents on the imidazolium ring influenced the critical gelation concentration and thermosensitivity in water. For example, a copolymer with Cl−, a hydrophilic counteranion, exhibited physical gelation behavior at a lower concentration than that of the polymer with BF4− in water. In addition, amphiphilic diblock copolymers with octadecyl side chains and very long ionic liquid segments (e.g., 800 or 1200 monomer units) exhibited physical gelation behavior at a very low concentration in water. To prepare copolymers with various structures, suitable initiating systems were also investigated with a focus on the appropriate conditions for the block polymerization of octadecyl VE (ODVE) and CEVE.
 | ||
| Scheme 1 Synthesis of amphiphilic diblock copolymers consisting of ionic liquids structures with various substituents and counteranions and different VEs. | ||
2 Experiments
2.1 Materials
IBVE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (TCI; >99%) was washed with 10 wt% aqueous sodium hydroxide solution and then with water, dried overnight over potassium hydroxide (pellets), and distilled twice over calcium hydride. CEVE (TCI; >97%) was washed with 10% aqueous sodium hydroxide solution and then with water, dried overnight over sodium sulfate, and distilled twice under reduced pressure over calcium hydride. ODVE (BASF) was distilled over calcium hydride under reduced pressure. 2-Phenoxyethyl VE (PhOVE) was prepared by the reaction of CEVE with phenol in dimethyl sulfoxide in the presence of sodium hydroxide, and it then was distilled twice over calcium hydride under reduced pressure.26,27 Toluene (Wako; >99.5%) was dried using solvent purification columns (Glass Contour; Solvent Dispensing System). 2,6-Di-tert-butylpyridine (DTBP: Sigma-Aldrich; 97%) was distilled twice over calcium hydride under reduced pressure. 1,4-Dioxane (Wako; 99.5%) was distilled over calcium hydride and then lithium aluminum hydride. Ethyl acetate (Wako; >99.5%) was dried overnight over molecular sieves 3 A and 4 A and distilled twice over calcium hydride. The adduct of IBVE with acetic acid [IBEA; CH3CH(OiBu)OCOCH3] was prepared from the addition reaction of IBVE with acetic acid according to the literature method.28 Et1.5AlCl1.5 (Nippon Aluminum Alkyls; 1.0 M solution in toluene) and SnCl4 (Sigma-Aldrich; 1.0 M solution in heptane) were used without further purification. 1-Methylimidazole (Aldrich; 99%), 1,2-dimethylimidazole (Wako; >97%), 1-ethylimidazole (TCI; >98%), 1-butylimidazole (Aldrich; 98%), sodium chloride (Nacalai Tesque; >99%), sodium tetrafluoroborate (Sigma-Aldrich; 98%), sodium iodide (Wako; >99.5%), sodium hexafluoroantimonate (Sigma-Aldrich; 90%), potassium hexafluorophosphate (Wako; 97%), lithium bis(trifluoromethanesulfonyl)imide (Wako: >98%), and all solvents expect for the polymerization solvent, i.e., toluene, were used without further purification.
(TCI; >99%) was washed with 10 wt% aqueous sodium hydroxide solution and then with water, dried overnight over potassium hydroxide (pellets), and distilled twice over calcium hydride. CEVE (TCI; >97%) was washed with 10% aqueous sodium hydroxide solution and then with water, dried overnight over sodium sulfate, and distilled twice under reduced pressure over calcium hydride. ODVE (BASF) was distilled over calcium hydride under reduced pressure. 2-Phenoxyethyl VE (PhOVE) was prepared by the reaction of CEVE with phenol in dimethyl sulfoxide in the presence of sodium hydroxide, and it then was distilled twice over calcium hydride under reduced pressure.26,27 Toluene (Wako; >99.5%) was dried using solvent purification columns (Glass Contour; Solvent Dispensing System). 2,6-Di-tert-butylpyridine (DTBP: Sigma-Aldrich; 97%) was distilled twice over calcium hydride under reduced pressure. 1,4-Dioxane (Wako; 99.5%) was distilled over calcium hydride and then lithium aluminum hydride. Ethyl acetate (Wako; >99.5%) was dried overnight over molecular sieves 3 A and 4 A and distilled twice over calcium hydride. The adduct of IBVE with acetic acid [IBEA; CH3CH(OiBu)OCOCH3] was prepared from the addition reaction of IBVE with acetic acid according to the literature method.28 Et1.5AlCl1.5 (Nippon Aluminum Alkyls; 1.0 M solution in toluene) and SnCl4 (Sigma-Aldrich; 1.0 M solution in heptane) were used without further purification. 1-Methylimidazole (Aldrich; 99%), 1,2-dimethylimidazole (Wako; >97%), 1-ethylimidazole (TCI; >98%), 1-butylimidazole (Aldrich; 98%), sodium chloride (Nacalai Tesque; >99%), sodium tetrafluoroborate (Sigma-Aldrich; 98%), sodium iodide (Wako; >99.5%), sodium hexafluoroantimonate (Sigma-Aldrich; 90%), potassium hexafluorophosphate (Wako; 97%), lithium bis(trifluoromethanesulfonyl)imide (Wako: >98%), and all solvents expect for the polymerization solvent, i.e., toluene, were used without further purification.
2.2 Synthesis of the precursor block copolymer
Polymerization was carried out under a dry nitrogen atmosphere in a glass tube equipped with a three-way stopcock. The glass tube was baked using a heat gun (Ishizaki; PJ-206A; blow temperature: approximately 450 °C) under dry nitrogen for 10 min before use. A prechilled Et1.5AlCl1.5 solution was added to a prechilled mixture containing IBEA, an added base, a VE monomer (IBVE, PhOVE, or ODVE), DTBP, and toluene at 0 °C (for IBVE and PhOVE) or 20 °C (for ODVE) using dry syringes. The polymerization was initiated by the addition of a prechilled SnCl4 solution at 0 °C (for IBVE and PhOVE) or at 20 °C (for ODVE). The polymerization that was conducted without SnCl4 was initiated by the addition of a prechilled Et1.5AlCl1.5 solution. After a predetermined period, CEVE was added to the mixture solution to start the second stage polymerization. The polymerization was quenched using methanol containing a small amount of an aqueous ammonia solution. The quenched reaction mixture was diluted with dichloromethane and subsequently washed with water to remove the initiator residues. The product polymer was recovered from the organic layer by evaporation of volatiles under reduced pressure and subsequently vacuum-dried for at least 6 h. The monomer conversion was determined by a gravimetric method and 1H NMR analysis of the product.2.3 Substitution of the chlorine atoms in the copolymer with imidazole derivatives
An amphiphilic diblock copolymer was dissolved in a N,N-dimethylformamide (DMF) solution containing an imidazole derivative and NaI (five times the molar amounts of CEVE units).29 The solution was stirred with a magnetic stir bar at 80 °C for 72 h. The reaction mixture was dialyzed against deionized water at least 24 h to remove DMF, the unreacted imidazole, and residual salts. After purification, the polymer solution was dialyzed against deionized water in the presence of arbitrary salts (five times the amounts of CEVE units) at least 24 h to exchange the counteranions, followed by evaporation under reduced pressure. The obtained polymer was vacuum-dried at 60 °C for at least 6 h. The degree of substitution to the imidazolium moieties was determined by 1H NMR analysis.2.4 Anion exchange reaction
Anion exchange was carried out according to the literature.30 The obtained diblock copolymers (0.60 g) were dissolved in water (3.0 mL) and then mixed with sodium, potassium, or lithium salts having various anions (equivalent to 5 imidazolium salt units). For water-insoluble polymers, the precipitate was washed with water to remove excess salts. For the other polymers, the polymer obtained was purified by dialysis against deionized water for at least 24 h, followed by evaporation under reduced pressure. The product was vacuum-dried for at least 6 h. An upfield shift in the signal assignable to the imidazolium ring proton confirmed the anion exchange.2.5 Characterization
The molecular weight distribution (MWD) of polymers in chloroform was measured at 40 °C using gel permeation chromatography (GPC) with polystyrene gel columns [Tosoh TSKgel GMHHR-M × 2 (exclusion limit molecular weight = 4 × 106; bead size = 5 μm; column size = 7.8 mm i.d. × 300 mm; flow rate = 1.0 mL min−1)] connected to a Tosoh DP-8020 pump, a CO-8020 column oven, a UV-8020 ultraviolet detector, and an RI-8020 refractive index detector. The number-average molecular weight (Mn) and polydispersity ratio [weight-average molecular weight/number-average molecular weight (Mw/Mn)] were calculated from the chromatograph with respect to 16 polystyrene standards (Tosoh; Mw = 5.0 × 102 to 1.09 × 106, Mw/Mn < 1.2). 1H NMR spectra were recorded using a JEOL ECA500 (500 MHz) spectrometer at 30 °C. The particle size was measured by dynamic light scattering (DLS; Otsuka Electronics FPAR-1000HG, λ = 632.8 nm, scattering angle = 90°). The dynamic viscoelasticity measurement was performed with the stress-controlled rheometer MCR302 (Anton Paar) using a cone-plate with a diameter of 40 mm and an angle of 4°. For the dynamic viscoelastic measurements, the angular frequency sweep tests were carried out from 0.1 to 100 rad s−1.3 Results and discussion
3.1 Precise synthesis of various precursor copolymers by living cationic copolymerization
Precursor block copolymers consisting of CEVE and IBVE with different IBVE segment lengths were synthesized by living cationic polymerization under the conditions developed in our previous study.22 First, the homopolymerization of IBVE at different monomer concentrations (0.040–0.80 M) was conducted using Et1.5AlCl1.5 and SnCl4 as catalysts in the presence of 1,4-dioxane and DTBP in toluene at 0 °C. Et1.5AlCl1.5 is used to generate an IBVE–HCl adduct from IBEA.31 The polymerizations were completed in approximately 3 min regardless of the amount of IBVE present. After completion of the initial polymerization, CEVE was added. The second-stage polymerization was complete in 1.0–6.0 h. The obtained block copolymers had MWs that were comparable to the theoretical values with very narrow MWDs (Mn = 26–76 × 103, Mw/Mn = 1.03–1.07; entries 1–13, Table 1). In addition, statistical copolymers composed of IBVE and CEVE were synthesized by living cationic copolymerization under the same polymerization conditions. Both monomers were quantitatively consumed in 40 min, and IBVE was preferentially consumed due to its higher reactivity compared to that of CEVE. The obtained copolymers had gradient-type sequences and narrow MWDs (entries 14 and 15).| Entry | Copolymer | Time | Conv (%) | Mn × 10−3 (GPC) | Mw/Mn |
|---|---|---|---|---|---|
| a Polymerization conditions: [CEVE]0 or add = 0.20–4.8 M, [IBEA]0 = 4.0 mM, [Et1.5AlCl1.5]0 = 5.0 mM, [SnCl4]0 = 10 mM, [DTBP]0 = 10 mM, in toluene; for entries 1–13: [IBVE]0 = 0.040–0.80 M, [1,4-dioxane] = 1.2 M, at 0 °C; for entries 14–15: [IBVE]0 = 0.40 or 0.80 M, [1,4-dioxane] = 1.2 M, at 0 °C; for entry 16–22: [ODVE]0 = 0.040–0.20 M, [ethyl acetate] = 1.0 M, at 20 °C; for entry 23: [ODVE]0 = 0.20 M, [ethyl acetate] = 1.0 M, at 20 °C; for entries 24–26: [PhOVE]0 = 0.040–0.40 M, [1,4-dioxane] = 1.2 M, at 0 °C. | |||||
| 1 | IBVE10-b-CEVE400 | 2.5 min + 2.0 h | ∼200 | 40.5 | 1.03 |
| 2 | IBVE20-b-CEVE400 | 2.5 min + 1.0 h | 197 | 42.6 | 1.06 |
| 3 | IBVE20-b-CEVE800 | 2.5 min + 6.0 h | 196 | 76.0 | 1.07 |
| 4 | IBVE30-b-CEVE400 | 2.5 min + 2.0 h | ∼200 | 40.7 | 1.03 |
| 5 | IBVE40-b-CEVE400 | 2.5 min + 2.0 h | ∼200 | 41.9 | 1.04 |
| 6 | IBVE50-b-CEVE400 | 2.5 min + 2.0 h | 197 | 47.1 | 1.06 |
| 7 | IBVE50-b-CEVE800 | 2.5 min + 1.0 h | ∼200 | 68.5 | 1.04 |
| 8 | IBVE100-b-CEVE400 | 3 min + 2.5 h | ∼200 | 49.3 | 1.06 |
| 9 | IBVE100-b-CEVE300 | 2.5 min + 2.5 h | 186 | 33.7 | 1.05 |
| 10 | IBVE200-b-CEVE400 | 2.5 min + 2.5 h | 192 | 59.4 | 1.05 |
| 11 | IBVE200-b-CEVE200 | 2.5 min + 1.0 h | 197 | 39.0 | 1.05 |
| 12 | IBVE200-b-CEVE100 | 2.5 min + 1.0 h | 198 | 30.3 | 1.05 |
| 13 | IBVE200-b-CEVE50 | 2.5 min + 1.0 h | 199 | 26.2 | 1.05 |
| 14 | IBVE100-co-CEVE400 | 40 min | 190 | 27.3 | 1.18 |
| 15 | IBVE200-co-CEVE400 | 40 min | 193 | 50.2 | 1.06 |
| 16 | ODVE10-b-CEVE400 | 30 s + 40 min | 196 | 40.1 | 1.06 |
| 17 | ODVE10-b-CEVE800 | 30 s + 24 h | ∼200 | 57.8 | 1.20 |
| 18 | ODVE10-b-CEVE1200 | 30 s + 24 h | ∼200 | 55.1 | 1.36 |
| 19 | ODVE20-b-CEVE400 | 30 s + 40 min | ∼200 | 43.1 | 1.11 |
| 20 | ODVE20-b-CEVE800 | 30 s + 24 h | 194 | 44.1 | 1.29 |
| 21 | ODVE20-b-CEVE1200 | 30 s + 24 h | 198 | 56.9 | 1.45 |
| 22 | ODVE50-b-CEVE400 | 30 s + 40 min | 193 | 52.1 | 1.07 |
| 23 | ODVE50-co-CEVE400 | 25 min | 199 | 46.3 | 1.05 |
| 24 | PhOVE10-b-CEVE400 | 2.5 min + 2.5 h | ∼200 | 43.7 | 1.04 |
| 25 | PhOVE50b-CEVE400 | 2.5 min + 2.5 h | ∼200 | 48.2 | 1.04 |
| 26 | PhOVE100-b-CEVE400 | 2.5 min + 2.5 h | ∼200 | 56.4 | 1.03 |
To examine the influence of the properties of the hydrophobic segments on the physical gelation behavior, we synthesized block copolymers with ODVE possessing a long alkyl chain that enhances crystallinity32–35 or PhOVE, which has an aromatic ring,26 as the hydrophobic segments. The block copolymerization of ODVE and CEVE was first examined in toluene at 0 °C because side reactions such as the elimination of 2-chloroethanol from the main chain of the CEVE block can occur in the polymerization using only Et1.5AlCl1.5 at 30 °C, as demonstrated in a previous study.22 However, in the polymerization of ODVE using Et1.5AlCl1.5 at 0 °C, monomer conversion plateaued at approximately 40% (Fig. 1A, diamond symbols; the left curves in Fig. 1C). Importantly, the polymerization solution became heterogeneous (or gelled), and the conversion leveled off. This result is most likely due to the aggregation of the poly(ODVE) chains at 0 °C.35 Therefore, the polymerization was conducted at 20 °C, which resulted in a quantitative consumption of ODVE without gelation (circle symbols in Fig. 1A; the middle curves in Fig. 1C). The sequential addition of CEVE led to block copolymerization from the propagating poly(ODVE) chains, yielding a polymer with a relatively narrow MWD. However, 1H NMR analysis of the product revealed that side reactions (dealcoholization) occurred during the polymerization of CEVE (Fig. S1,† 5.3–5.4 and 5.6–5.7 ppm). In addition, the Mn values of the products were lower than the theoretical value (circle symbols in Fig. 2A).
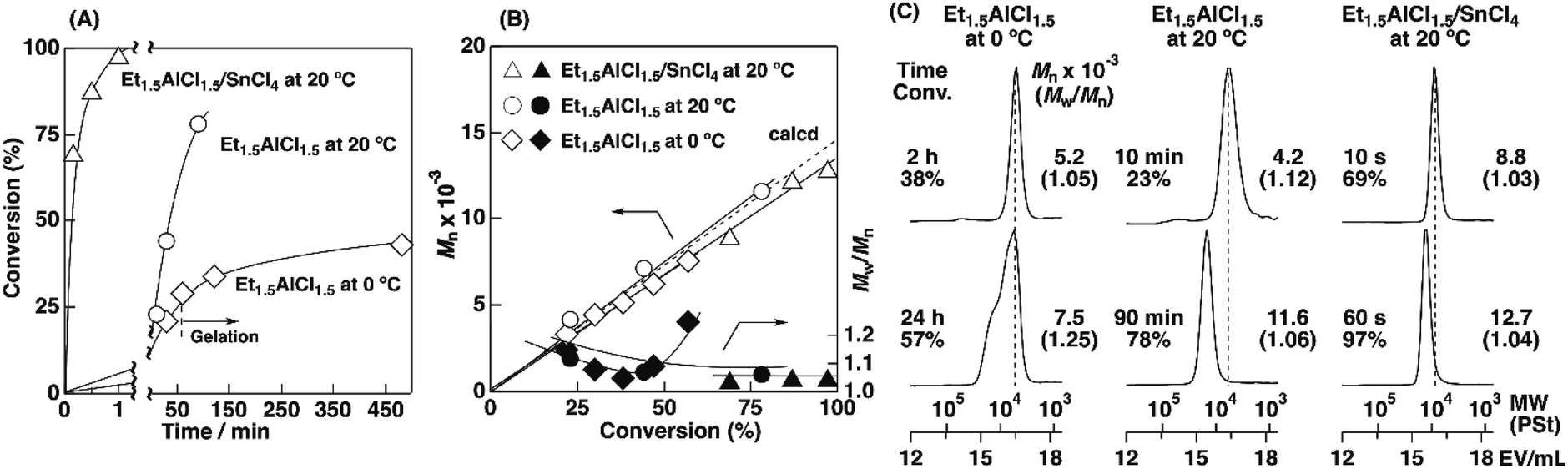 | ||
| Fig. 1 Synthesis of poly(ODVE)s by living cationic polymerization. (A) Time-conversion plots, (B) the Mn (open symbols) and Mw/Mn (fill symbols) values of the products, and (C) MWD curves of the products obtained: [ODVE]0 = 0.20 M, [IBEA]0 = 4.0 mM, [ethyl acetate] = 1.0 M, in toluene; for diamond symbols: [Et1.5AlCl1.5]0 = 20 mM, at 0 °C; for circle symbols: [Et1.5AlCl1.5]0 = 20 mM, at 20 °C; for triangle symbols: [Et1.5AlCl1.5]0 = 5.0 mM, [SnCl4]0 = 10 mM, [DTBP]0 = 10 mM, at 20 °C. | ||
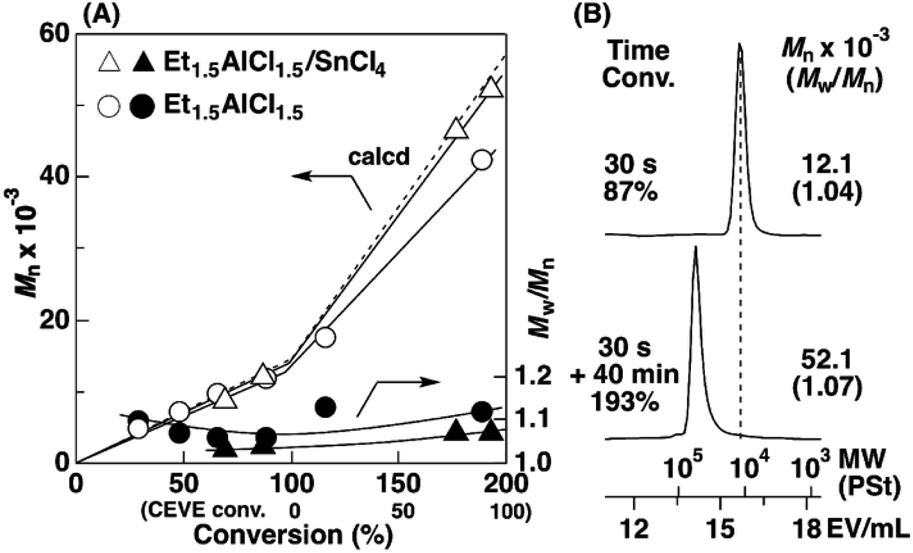 | ||
| Fig. 2 (A) The Mn and Mw/Mn values and (B) MWD curves of the products obtained by the polymerization of ODVE (upper curve in B) and the subsequent block copolymerization with CEVE (lower curve): [ODVE]0 = 0.20 M, [CEVE]add = 1.6 M, [IBEA]0 = 4.0 mM, [ethyl acetate] = 1.0 M, in toluene at 20 °C; for triangle symbols: [Et1.5AlCl1.5]0 = 5.0 mM, [SnCl4]0 = 10 mM, [DTBP]0 = 10 mM (entry 22, Table 1); for circle symbols: [Et1.5AlCl1.5]0 = 20 mM. | ||
The Et1.5AlCl1.5/SnCl4-combined system, which was developed for CEVE polymerization at 0 °C, was efficient for both ODVE polymerization and CEVE polymerization even at a higher temperature (20 °C). The polymerization of ODVE was nearly completed in 60 s, making it more than 100 times faster than the reaction using only Et1.5AlCl1.5 (Fig. 1A, triangle symbols). In addition, the Mn values of the obtained poly(ODVE)s increased in agreement with the theoretical values while maintaining narrow MWDs (Fig. 1B, triangle symbols; the right curves in Fig. 1C). The 1H NMR spectrum of the product did not contain peaks associated with side reactions, indicating that the polymerization proceeded in a living manner (Fig. 3A). For the block copolymer synthesis, the second-stage polymerization of CEVE was nearly complete in approximately 40 min, resulting in a clear shift in the MWD to a higher MW region (Fig. 2B). The MWs of the block copolymers increased in agreement with the theoretical values with very narrow MWDs (triangle symbols in Fig. 2A). Notably, 1H NMR analysis indicated that side reactions (dealcoholization) did not occur during the second-stage polymerization (Fig. 3B), unlike in the polymerization using only Et1.5AlCl1.5 at 20 °C (Fig. S1†). In addition, the products had an ODVE:CEVE molar ratio of 1:8, which is in agreement with the feed ratio of ODVE and CEVE (Fig. 3B). Moreover, diblock copolymers with various segment lengths were obtained by changing the amount of each monomer (ODVEn-b-CEVEm; n = 10–50, m = 400–1200, Mn = 40–57 × 103, Mw/Mn = 1.06–1.45, entries 16–22, Table 1). Statistical copolymers composed of ODVE and CEVE were also successfully synthesized by living cationic copolymerization under the same conditions as those employed for block copolymerization (entry 23, Table 1; Fig. S2†).
 | ||
| Fig. 3 1H NMR spectra of (A) ODVE50 (the upper in Fig. 2B) and (B) ODVE50-b-CEVE400 (entry 22, Table 1; the lower in Fig. 2B) (in CDCl3 at 30 °C; * toluene, ethyl acetate, water, CH2Cl2). | ||
The copolymers of PhOVE and CEVE were synthesized by living cationic polymerization with the Et1.5AlCl1.5/SnCl4-combined system at 0 °C. PhOVE has an aromatic ring in its side chain.26 The copolymers had an Mn comparable to the theoretical value and a narrow MWD (Fig. 4). The 1H NMR spectra of both poly(PhOVE) and PhOVE-b-CEVE showed no peaks associated with side reactions (Fig. S3†). The lengths of each segment of the block copolymer were calculated to be approximately 50 and 400 units for the PhOVE and CEVE segments, respectively, based on 1H NMR analysis of the obtained product. Furthermore, diblock copolymers with various PhOVE segment lengths were obtained by varying the amount of PhOVE (PhOVEn-b-CEVE400; n = 10–100, Mn = 44–56 × 103, Mw/Mn = 1.03–1.04, entries 24–26 in Table 1).
 | ||
| Fig. 4 (A) The Mn and Mw/Mn values and (B) MWD curves of the products obtained by the polymerization of PhOVE (upper) and the subsequent block copolymerization with CEVE (lower): [PhOVE]0 = 0.20 M, [CEVE]add = 1.6 M, [IBEA]0 = 4.0 mM, [Et1.5AlCl1.5]0 = 5.0 mM, [SnCl4]0 = 10 mM, [DTBP]0 = 10 mM, [1,4-dioxane] = 1.2 M in toluene at 0 °C (entry 25, Table 1). | ||
The chlorine atoms in the CEVE segment of these obtained diblock copolymers were substituted with arbitrary imidazole derivative in the presence of NaI in DMF at 80 °C for 72 h.22,29 Subsequently, the diblock copolymers were treated with five molar equivalents of sodium, potassium, or lithium salts having various anions in water for 24 h, which resulted in the exchange of the counteranions. Analysis of 1H NMR confirmed that both the introduction of imidazolium cation and the subsequent counteranion exchange proceeded quantitatively (Fig. S4†).
3.2 Relationship between physical gelation behavior in water and the structures of the amphiphilic diblock copolymers having ionic liquid segments
In this section, the relationship between physical gelation in water and the structures of amphiphilic diblock copolymers with pendant ionic liquid segments was investigated, as shown in Scheme 2. | ||
| Scheme 2 Summary of relationship between physical gelation behavior and structures of amphiphilic diblock copolymers. | ||
To examine the thermosensitivity of the physical gelation behavior of the copolymers with Cl− as the counteranion in water, we conducted DLS measurements with a 0.01 wt% aqueous solution at various temperatures. IBVE50-b-[Me2Im][Cl]400 forms micelles with a diameter of approximately 200 nm at 55 °C (open circle symbols in Fig. 5A). Interestingly, the diameter remained almost constant (ca. 200 nm) as the temperature was decreased from 55 °C to 10 °C (Fig. 5B). In our previous study, we reported that the diameter of IBVE50-b-[Me2Im][BF4]400 decreased from 190 nm at 55 °C to 140 nm at 15 °C in water. This behavior was due to the thermosensitivity of the [Me2Im][BF4] segments.17,22 The aqueous solution of IBVE50-b-[Me2Im][BF4]400 gelled at 25 °C via packing of the large micelles with extended corona chains based on electrostatic repulsion and hydration of the ionic liquid segments.22,36 This result corresponds to the temperature-independent physical gelation of IBVE50-b-[Me2Im][Cl]400 in water (entry 2 in Table 2).
 | ||
| Fig. 5 DLS measurement for 0.01 wt% aqueous solution of IBVE50-b-[Me2Im][Cl]400. (A) The results at 15 and 55 °C and (B) the diameter at different temperature. | ||
The amphiphilic copolymer with Cl− as the counteranion exhibited physical gelation behavior at a very low concentration in water. IBVE50-b-[Me2Im][Cl]400 formed a gel at 0.2 wt% in water (entry 2 in Table 2). However, the critical gelation concentration of the BF4− analogue (IBVE50-b-[Me2Im][BF4]400) was 0.7 wt% (entry 1). The hydrogen-bonding ability of Cl− has been experimentally and theoretically demonstrated to be much higher than that of BF4− in bulk ionic liquids.37–40 Cl−-containing polymers may form micelles with smaller numbers of polymer chains than their BF4− counterparts, and/or the stronger interactions of the Cl−-containing segments with water molecules or other micelles may contribute to micelle packing or percolation at a lower concentration compared to that for the BF4−-containing polymer.
| Entry | IBVE50-b-[R1R2Im][X]400 | Critical gelation concentrationa | Gelation temperatureb | ||
|---|---|---|---|---|---|
| R1 = | R2 = | X− = | |||
| a Status of solution at 50 °C.b Status in 1 wt% aqueous solution. | |||||
| 1 (ref. 22) | Me | Me | BF4 | 0.7 wt% | 25 °C |
| 2 | H | Me | BF4 | 0.2 wt% | Below 0 °C |
| 3 | H | Et | BF4 | 0.8 wt% | 12 °C |
| 4 | H | Bu | BF4 | — | Insoluble |
| 5 | Me | Me | Cl | 0.2 wt% | Temperature-independent |
| 6 | H | Me | Cl | 0.2 wt% | Temperature-independent |
| 7 | H | Et | Cl | 0.4 wt% | Temperature-independent |
| 8 | H | Bu | Cl | 0.6 wt% | Temperature-independent |
The length of the alkyl group of the imidazolium ring influenced the physical gelation behavior of the amphiphilic diblock copolymers in water. A 1 wt% aqueous solution of IBVE50-b-[MeIm][BF4]400, which has a shorter alkyl group than the other copolymers, formed physical gel. The solution remained in the gel state even at 0 °C (entry 2 in Table 3), unlike IBVE50-b-[EtIm][BF4]400 (entry 3). Since the UCST-type phase separation temperature of poly([MeIm]) (5 °C) is lower than that of poly([EtIm]) (8 °C),17 the difference in the gelation temperature is most likely related to the difference in the phase separation temperature. IBVE50-b-[BuIm][BF4]400, which has longer alkyl groups than the other copolymers, was insoluble in water over the whole concentration and temperature range examined (entry 4). The solubility and gelation behavior of the amphiphilic diblock copolymers in water were dependent on the structures of the ionic liquid-containing poly(VE) segments.17
The amphiphilic diblock copolymers possessing Cl− as counteranions rather than BF4− exhibited physical gelation behavior at a lower concentration in water regardless of the substituents on the imidazolium moieties (entries 5–8 in Table 3). Even a diblock copolymer with [BuIm][Cl] segments, which was insoluble in water before ion exchange from BF4− (entry 4), exhibited physical gelation behavior in water (entry 8). Interestingly, the critical concentration varied depending on the substituents on the imidazolium ring. For example, the concentrations of IBVE50-b-[MeIm][Cl]400, IBVE50-b-[EtIm][Cl]400, and IBVE50-b-[BuIm][Cl]400 were 0.2, 0.4, and 0.6 wt%, respectively. These results indicate that the substituents on the imidazolium ring may be related to the micelle packing behavior at a low concentration in water.
The substituents of the hydrophobic segments also influenced the physical gelation behavior of the amphiphilic diblock copolymers in water. A 0.6 wt% aqueous solution of the amphiphilic diblock copolymers with PhOVE segments exhibited physical gelation behavior (entries 3 and 4 in Table 4). Amphiphilic diblock copolymers with ODVE segments bearing long alkyl side chains that enhance crystallinity also form gels in water at 0.6 wt% (entry 5 in Table 4, Fig. S5†). The ODVE segment length sufficient for physical gelation behavior in water was 10 units due to the high hydrophobicity of the side chain. These copolymers (entries 3–5) also exhibited UCST-type thermosensitive physical gelation behavior derived from the thermosensitivity of the [Me2Im][BF4] segments as in the case of the copolymers with IBVE segments (entries 1 and 2). To examine the influence of the substituents and chain lengths of the hydrophobic segments on the viscoelastic behavior of the physical gels, we performed dynamic viscoelasticity measurements of the aqueous solutions containing the amphiphilic diblock copolymers with ODVE or PhOVE chains as the hydrophobic segments. The storage modulus (G′) of the copolymers was almost constant over a wide range of frequencies and was higher than the loss modulus (G′′), and both were frequency-independent, which is consistent with the behavior of a typical physical gel (Fig. 6 and S6†). In addition, the G′ of the copolymers with PhOVE segments increased as the PhOVE segment length increased (Fig. S6†).
| Entry | Hydrophobic VE | Unit (n) | Conc. (wt%) | |||
|---|---|---|---|---|---|---|
| 1 | 0.8 | 0.6 | 0.4 | |||
| a Determined using the test-tube inversion method at 50 °C. | ||||||
| 1 | IBVE | 50 | Gel | Gel | Sol | — |
| 2 | IBVE | 100 | Gel | Sol | — | — |
| 3 | PhOVE | 50 | Gel | Gel | Gel | Sol |
| 4 | PhOVE | 100 | Gel | Gel | Gel | Sol |
| 5 | ODVE | 10 | Gel | Gel | Gel | Sol |
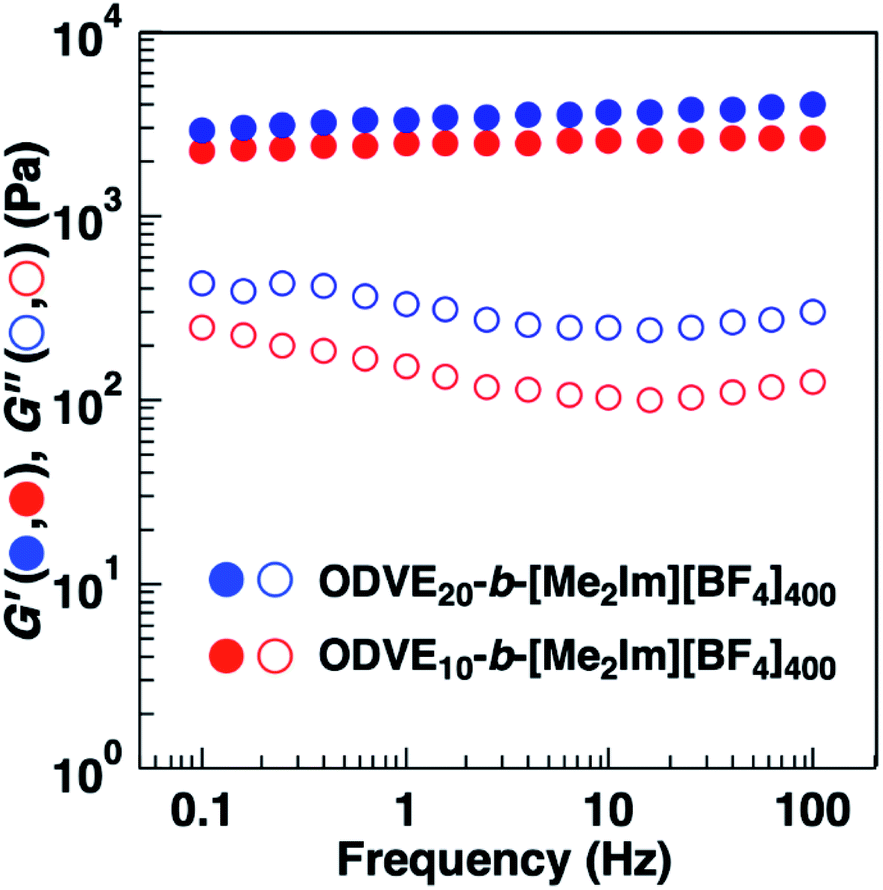 | ||
| Fig. 6 Frequency dependence of dynamic moduli, storage modulus G′ (filled symbols) and loss modulus G′′ (open symbols) of ODVEn-b-[Me2Im][BF4]400 (n = 10 and 20) in 10 wt% aqueous solutions at 55 °C. | ||
| Entry | Hydrophobic VE | Unit (n) | Cation | Anion | Unit (n) | Conc. (wt%) | |||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.4 | 0.2 | 0.1 | ||||||
| a Determined using the test-tube inversion method (gel: retention, sol: flow) at 50 °C. | |||||||||
| 1 | IBVE | 50 | Me2Im | BF4 | 400 | Gel | Sol | — | — |
| 2 | IBVE | 50 | Me2Im | Cl | 400 | Gel | Gel | Gel | Sol |
| 3 | IBVE | 50 | MeIm | BF4 | 400 | Gel | Gel | Gel | Sol |
| 4 | IBVE | 50 | MeIm | Cl | 400 | Gel | Gel | Gel | Sol |
| 5 | IBVE | 50 | Me2Im | BF4 | 800 | Gel | Gel | Gel | Sol |
| 6 | ODVE | 10 | Me2Im | BF4 | 800 | Gel | Gel | Sol | — |
| 7 | ODVE | 10 | Me2Im | Cl | 800 | Gel | Gel | Gel | Sol |
| 8 | ODVE | 10 | Me2Im | BF4 | 1200 | Gel | Gel | Sol | — |
| 9 | ODVE | 10 | Me2Im | Cl | 1200 | Gel | Gel | Sol | — |
| 10 | ODVE | 20 | Me2Im | BF4 | 1200 | Gel | Gel | Sol | — |
| 11 | ODVE | 20 | Me2Im | Cl | 1200 | Gel | Gel | Gel | Sol |
These results indicate that highly hydrophilic units, such as [MeIm] units, as cations or Cl− as counteranions, and long ionic liquid-type segments are effective for physical gelation at extremely low concentrations in water. In addition, hydrophobic segments with sufficient hydrophobicity, such as ODVE segments, and appropriate chain lengths are required. The balance between these factors is likely responsible for the formation of stable micelles composed of amphiphilic diblock copolymers and gelation due to the efficient packing or percolation of the micelles at low concentrations in water.
4 Conclusions
Amphiphilic diblock copolymers prepared by living cationic polymerization and subsequent chemical modification exhibited different physical gelation behaviors in water depending on the hydrophilicity of the ionic liquid-type segments and the substituents on the hydrophobic segments. In the well-controlled cationic block copolymerization of ODVE and CEVE, a suitable temperature and initiating system were important to avoid both gelation due to aggregation of poly(ODVE) chains and side reactions, such as dealcoholization. For example, the polymerization of ODVE at 0 °C ceased at a monomer conversion of approximately 40% due to aggregation of the poly(ODVE) chains. However, the well-controlled polymerization without the aggregations proceeded at 20 °C. Subsequently, the chlorine atoms in the precursor copolymers were quantitatively substituted by the introduction of various imidazole derivatives and the subsequent counteranion exchange. The physical gelation behavior of the amphiphilic copolymers in water varied depending on the structure and length of both the hydrophobic and hydrophilic segments. Amphiphilic diblock copolymers with highly hydrophilic units, such as [MeIm] units or Cl−, as an ionic liquid-type segment exhibited physical gelation behavior at 0.2 wt% in water. In addition, a sufficiently hydrophobic but short segment (i.e., ODVE units) was effective for the gelation of copolymers with long ionic liquid-type segments, such as 800 or 1200 units, at 0.2 wt% in water. These results indicate that the appropriate design of amphiphilic copolymers is important for physical gelation at extremely low concentrations in water. The results of this study provide a new platform for designing amphiphilic diblock copolymers that exhibit physical gelation in water.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the group of Prof. Tadashi Inoue (Osaka University) for dynamic viscoelasticity measurement. This work was partially supported by the JSPS KAKENHI Grant Number 17H03068.Notes and references
- H. Ringsdorf, B. Schlarb and J. Venzmer, Angew. Chem., Int. Ed. Engl., 1988, 27, 113 CrossRef.
- D. E. Descher and A. Eisenberg, Science, 2002, 297, 967 CrossRef.
- V. Breedveld, A. P. Nowak, J. Sato, T. J. Deming and D. J. Pine, Macromolecules, 2004, 37, 3943 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163 CrossRef CAS.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055 CrossRef CAS.
- S. Qin, Y. Geng, D. E. Discher and S. Yang, Adv. Mater., 2006, 18, 2905 CrossRef CAS.
- T. Yoshida, T. C. Lai, G. S. Kwon and K. Sako, Expert Opin. Drug Delivery, 2013, 10, 1497 CrossRef CAS.
- F. Chècot, S. Lecommandoux, H.-A. Klok and Y. Gnanou, Eur. Phys. J. E, 2003, 10, 25 CrossRef.
- X. Tong, G. Wang, A. Soldera and Y. J. Zhao, J. Phys. Chem. B, 2005, 109, 20281 CrossRef CAS.
- M. Galinski, A. Lewandowski and I. Stepniak, Electrochim. Acta, 2006, 51, 5567 CrossRef.
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef.
- B. Y. W. Man, J. M. Hook and J. B. Harper, Tetrahedron Lett., 2005, 46, 7641 CrossRef.
- D. A. Jaeger and C. E. Tucker, Tetrahedron Lett., 1989, 30, 1785 CrossRef.
- A. Zicmanis, S. Katkevica and P. Mekss, Catal. Commun., 2009, 10, 614 CrossRef.
- G. Zhao, T. Jiang, H. Gao, B. Han, J. Huang and D. Sun, Green Chem., 2004, 6, 75 RSC.
- K. Seno, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5724 CrossRef CAS.
- H. Yoshimitsu, A. Kanazawa, S. Kanaoka and S. Aoshima, Macrmolecules, 2012, 45, 9427 CrossRef CAS.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009 CrossRef CAS.
- J. Yuan and M. Antonietti, Polymer, 2011, 52, 1469 CrossRef CAS.
- S. Washiro, M. Yoshizawa, H. Kanajima and H. Ohno, Polymer, 2004, 45, 1577 CrossRef CAS.
- H. Nakajima and H. Ohno, Polymer, 2005, 46, 11499 CrossRef CAS.
- D. Yokota, A. Kanazawa and S. Aoshima, Polym. Chem., 2018, 9, 5080 RSC.
- P. Raffa, P. Brandenburg, D. A. Z. Wever, A. A. Breokhuis and F. Picchioni, Macromolecules, 2013, 46, 7106 CrossRef CAS.
- A. V. Korobko, W. N. Jesse, A. Lapp, S. U. Egelhaaf and J. R. C. Maarel, J. Chem. Phys., 2005, 122, 024902 CrossRef CAS.
- S. Misra, W. L. Mattice and D. H. Napper, Macromolecules, 1994, 27, 7090 CrossRef CAS.
- S. Sugihara, S. Matsuzono, H. Sakai, M. Abe and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3190 CrossRef CAS.
- J. V. Crivello and D. A. Conlon, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 1785 CrossRef CAS.
- S. Aoshima and T. Higashimura, Macromolecules, 1989, 22, 1009 CrossRef CAS.
- D. Valade, F. Boschet and B. Ameduri, Macromolecules, 2009, 42, 7689 CrossRef CAS.
- K. Vijayakrishna, S. K. Jewrajka, A. Ruiz, R. Marcilla, J. A. Pomposo, D. Mecerreyes, D. Taton and Y. Gnanou, Macromolecules, 2008, 41, 6299 CrossRef CAS.
- T. Yoshida, A. Kanazawa, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4288 CrossRef CAS.
- E. J. Goethals, W. Reyntjens, X. Zhang, B. Verdonck and T. Loontjens, Macromol. Symp., 2000, 157, 93 CrossRef CAS.
- X. Zhang, W. Reyntjens and E. J. Goethals, Polym. Int., 2000, 49, 277 CrossRef CAS.
- T. Yoshida, K. Seno, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1155 CrossRef CAS.
- K. Seno, A. Date, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4392 CrossRef CAS.
- L. Derici, S. Ledger, S. M. Mai, C. Booth, I. W. Hamley and J. S. Pedersen, Phys. Chem. Chem. Phys., 1999, 1, 2773 RSC.
- L. Crowhurst, P. R. Mawdsley, J. M. Perez-Arlandis, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2003, 5, 2790 RSC.
- S. Zhang, X. Qi, X. Ma, L. Lu and Y. J. Deng, J. Phys. Chem. B, 2010, 114, 3912 CrossRef.
- R. Macchieraldo, L. Esser, R. Elfgen, P. Voepel, S. Zahn, B. M. Smarsly and B. Kirchner, ACS Omega, 2018, 3, 8567 CrossRef.
- R. P. Matthews, C. Ashworth, T. Welton and P. A. Hunt, J. Phys.: Condens. Matter, 2014, 26, 284112 CrossRef.
- M. Galinski, A. Lewandowski and I. Stepniak, Electrochim. Acta, 2006, 51, 5567 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Polymerization results, 1H NMR spectra, and physical gelation behavior in water. See DOI: 10.1039/d0ra09163d |
| This journal is © The Royal Society of Chemistry 2020 |