
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt/nitrogen codoped carbon nanosheets derived from catkins as a high performance non-noble metal electrocatalyst for oxygen reduction reaction and hydrogen evolution reaction†
Luting Songab,
Jinquan Changa,
Yanhong Maa,
Xinghua Tanab,
Yuanqing Xua,
Limin Guoab,
Zhexue Chena,
Tingqiao Zhaoab,
Yueqi Lia,
Yanlin Liua,
Yong Zhang *a and
Weiguo Chu*ab
*a and
Weiguo Chu*ab
aNanofabrication Laboratory, CAS Key Laboratory for Nanosystems and Hierarchical Fabrication, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing, 100190, P. R. China
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, P. R. China. E-mail: wgchu@nanoctr.cn; zhangyong@nanoctr.cn
First published on 27th November 2020
Abstract
Novel energy devices which are capable of alleviating and/or solving the energy dilemma such as overall water splitting and fuel cells require the development of highly efficient catalysts, especially cheap high performance non-precious metal (NPM) catalysts. Here, we prepare highly efficient NPM catalysts of cobalt and nitrogen codoped carbon nanosheets (Co/N–CNSs) for oxygen reduction reaction (ORR) and hydrogen evolution reaction (HER) using harmful environment-polluting waste of biomass catkins as carbon precursors via a mild mechanical exfoliation and chemical process which is facile, low-cost, environmentally friendly and up-scalable. Compared with a commercial platinum-based catalyst (commercial 20% Pt/C), the Co/N–CNS electrocatalysts show outstanding ORR activity, acceptable HER activity and long term stability with an onset potential of 0.92 V versus the reversible hydrogen electrode (vs. RHE) and a half-wave potential of 0.83 V vs. the RHE in alkaline electrolytes. The excellent performance is closely related to the presence of abundant CoNx active sites. This work offers a novel and effective approach for preparing highly efficient ORR and HER NPM electrocatalysts from waste biomass materials.
Introduction
Developing novel energy conversion and storage systems such as overall water splitting, metal–air batteries and proton exchange membrane fuel cells (PEMFCs) may be taken as a relatively ideal and sustainable alternative for easing the energy and environmental crisis arising from traditional fossil fuels.1–4 Among the energy devices mentioned above, PEMFCs were commercialized in automobiles in 2014 and thus considered as one of the most efficient power sources for automotives.5 Such energy conversion systems mainly consist of four core electrocatalytic processes, which are the oxygen reduction reaction (ORR), oxygen evolution reaction (OER), hydrogen oxidation reaction (HOR) and hydrogen evolution reaction (HER).1,6 The kinetics of the ORR process is seriously sluggish because of slow reaction kinetics caused by the multistep proton-coupled electron transfer.7–9 Furthermore, even though the HER process is generally facial, it is still greatly limited owing to the inevitable large dynamic overpotential above the thermodynamic potential.3,10,11 Currently, expensive platinum-based materials are usually utilized as efficient electrocatalysts for the catalysis of the ORR and HER processes.12–14 Thus, as far as the scarcity, economics and service durability of materials, noble metal based electrocatalysts would not promote the extensive commercialization of energy conversion devices.13,15,16 It is therefore reasonable and urgent to explore novel less expensive non-precious metal (NPM) or metal-free electrocatalysts with high activity and long-term stability instead of noble metal based ones to realize bifunctional and highly efficient catalytic effects for ORR and HER.17–19Fifty years ago, Jasinski discovered that cobalt phthalocyanine could be utilized as electrocatalysts for the first time.20 Twenty years later, van Veen and Colijn discovered that metal–N4 macrocycles could efficiently improve the performance of ORR activity in both alkaline and acid electrolyte after high temperature treatment (600–900 °C) under inert atmosphere.21 Then, Yeager and the co-workers prepared ORR electrocatalysts by pyrolyzing separated metal, nitrogen and carbon precursors, which could significantly reduce the cost.22 Inspired by the pioneering work, some researchers have focused on non-precious metal (NPM) electrocatalysts for ORR and/or HER processes in fuel cells in the past decades such as Fe- and/or Co-based catalysts.12,17,23–25
It is well recognized that two key factors, concentration of accessible activity sites and their turnover frequency (TOF), would significantly affect the activity of state-of-the art electrocatalysts and further the electrocatalytic performance of NPM catalysts.26–31 The concentration of activity sites could be increased by enlarging the specific surface area of materials, suggesting the exposure of more activity centers.5,32 The TOF capable of characterizing the intrinsic properties of activity centers is normally determined by the coordination structure of metal ions and nonmetal atoms such as nitrogen and carbon. A good case in point is the structure of metal ions (Fe or Co) coordinated with four nitrogen atoms and integrated in disordered carbon structures (MeN4C10) or graphene matrix rings (MeN4C12), which can greatly enhance the performance of NPM electrocatalysts in fuel cells through defining and further deciphering precisely the activity sites in NPM electrocatalysts.11,33–35
Herein, we employ poplar catkins as carbon precursors, a kind of uniform and native microstructures of one dimensional (1D) hollow microtubes with abundant surface functional groups, which however may cause uncomfortable respiratory allergy and terrible air pollution in spring. Cobalt chloride (Co(Cl)2) and trimethylolaminomethane (Tris) as cobalt and nitrogen precursors, respectively, can be ball milled with catkins to achieve cobalt and nitrogen co-doped nanosheets in a large scale. The homogeneous distribution of cobalt and nitrogen precursors at atomic level in carbonaceous matrix enables to obtain Co/N co-doped carbon nanosheets (Co/N–CNSs) catalysts with abundant molecular CoNx active sites and porous structures by pyrolyzing and leaching the products from ball milling. The Co/N–CNSs catalysts are demonstrated to show the ORR activity comparable to that of commercial Pt/C catalysts in alkaline electrolyte and acceptable HER performance in acidic electrolyte and thus the superior comprehensive performance. Therefore, the current study not only can realize the superior performance of catalysts but also provide a facile, low-cost, environmentally friendly and up-scalable approach for preparing cheap catalysts by utilizing environmental waste-derived precursors.
Experimental methods
Chemicals
All the chemicals were of analytical grade and commercially available from Shanghai Chemical Reagent Co. Ltd and were used without further purification.Synthesis of cobalt and nitrogen co-doped carbon nanosheets (Co/N–CNSs)
In a typical fabrication process of Co/N–CNSs, 70 mg catkin, 10 mM cobalt chloride hexahydrate (Co(Cl)2·6H2O) and 40 mM trimethylolaminomethane (Tris) were mixed together and transferred into the agate jar for ball-milling (planetary ball mill, QM-3SP04, Nanjing LaiBu Technology Industry Co., Ltd, China) at 500 rpm for 12 h. Then, the as-prepared materials were washed with deionized water (DIW) and ethanol for several times to remove the impurities. The products were collected after drying overnight in the oven, followed by carbonization in argon (Ar) atmosphere at 700 °C, 800 °C and 900 °C for 2 h and 4 h, respectively, to produce CoNPs–Co/N–CNSs. To remove the cobalt-containing inorganic nanoparticles that were considered as the unstable and ORR and HER-unreactive phases, the CoNPs–Co/N–CNSs were then leached in 0.5 M H2SO4 at 80 °C for 8 h, followed by filtration collection, washing with deionized water and drying. Finally, the leached samples were pyrolyzed again in Ar atmosphere at the same temperature for 2 h.Characterization
Scanning electron microscopy (SEM) and energy filtered scanning electron microscopy (EFSEM) were performed on a Hitachi S-8200 microscope at an acceleration voltage of 5 kV. High-resolution transmission electron microscopy (HRTEM) test, energy filtered transmission electron microscopy (EFTEM) mapping and selected area electron diffraction (SAED) pattern were acquired on Tecnai G2 F20 U-TWIN electron microscopy operating at an accelerating voltage of 200 kV. X-ray photoelectron spectra (XPS) were recorded on an ESCALab250 MKII X-ray photoelectron spectrometer, using 220i-XL electron spectrometer from VG Scientific with 300 W Al Kα radiation. XRD data were recorded on a diffractometer (Rigaku D/max-2500) with Cu Kα radiation.Electrocatalytic measurements
The electrochemical measurements were carried out in a conventional three-electrode cell using electrochemical workstation (Autolab-302) at room temperature. A 4.0 mm diameter glassy carbon disk (disk geometric area 0.1256 cm2) and rotating ring-disk electrode (RRDE, disk diameter: 4.5 mm) served as the substrate for the working electrode, respectively, with the rotating rate varying from 100 to 1600 rpm. Ag/AgCl (3.5 M KCl) and Pt foil were used as reference and counter electrodes in the electrolyte, respectively. The Co-based catalysts or commercial Pt/C (20 wt%, Johnson Matthey) catalyst ink were prepared by blending the catalyst powder (2 mg) with 490 μL ethanol and 490 μL DIW containing 20 μL Nafion solution by sonication for at least 4 h. 20 μL of catalysts ink or 10 μL of commercial 20% Pt/C was then pipetted onto the glassy carbon surface.For the oxygen reduction reaction (ORR) test, the electrolyte was saturated with N2/O2 by bubbling N2/O2 prior to the start of each experiment for 30 min. The cyclic voltammetry (CV) tests were performed in Ar- and O2-saturated aqueous electrolyte solutions (0.1 M KOH) with a scan rate of 50 mV s−1. Rotating disk electrode (RDE) tests were performed in O2-saturated electrolyte with a sweep rate of 5 mV s−1. In order to estimate the double layer capacitance, the electrolyte was deaerated by bubbling with N2, and then the voltammogram was evaluated again in the deaerated electrolyte. The oxygen reduction current was taken as the difference between currents measured in the deaerated and O2-saturated electrolytes. Electron transfer numbers were calculated using the Koutecky–Levich equation:
 | (1) |
| B = 0.62nFC0(D0)2/3ν−1/6 | (2) |
| jK = nFkC0 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), C0 is the bulk concentration of O2 (1.2 × 10−6 mol cm−3 in 0.1 M KOH), D0 is the diffusion coefficient of O2 in electrolyte (1.9 × 10−5 cm2 s−1, in 0.1 M KOH), ν is the kinetic viscosity of the electrolyte (0.01 cm2 s−1, in 0.1 M KOH), ω is the angular velocity of the disk (ω = 2πN, N is the linear rotation speed) and k is the electron transfer rate constant.
485 C mol−1), C0 is the bulk concentration of O2 (1.2 × 10−6 mol cm−3 in 0.1 M KOH), D0 is the diffusion coefficient of O2 in electrolyte (1.9 × 10−5 cm2 s−1, in 0.1 M KOH), ν is the kinetic viscosity of the electrolyte (0.01 cm2 s−1, in 0.1 M KOH), ω is the angular velocity of the disk (ω = 2πN, N is the linear rotation speed) and k is the electron transfer rate constant.
To further detect the kinetic properties of the ORR, rotating ring-disk electrodes (RRDE) were employed to work in O2-saturated 0.1 M KOH with a scan rate of 5 mV s−1 and the ring-electron potential was controlled at 1.5 V vs. RHE. The electron transfer number (n) was calculated using the following equation:
 | (4) |
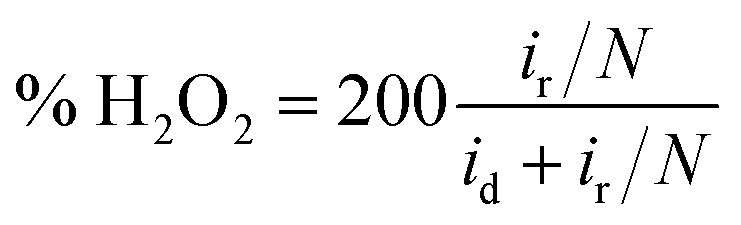 | (5) |
For the hydrogen evolution reaction (HER) test, the HER tests were performed with RDE in an Ar-saturated electrolyte (0.5 M H2SO4) at 1600 rpm with a sweep rate of 5 mV s−1. EIS spectra were recorded in an Ar-saturated electrolyte with a 5 mV AC potential from 10 kHz to 0.01 Hz at 1600 rpm.
Results and discussion
The typical synthetic procedure of Co/N–CNSs electrocatalysts is illustrated in Scheme 1. The catkins were directly collected from the field, and then washed with acetone, double-distilled water and absolute ethanol for several times to get rid of seeds and dusts involved. These pure carbonaceous microtubes with an inner diameter ca. 2 μm and an outer diameter of ca. 3 μm could be successfully obtained (Fig. S1†). The mixture of these catkin microtubes and Co and N precursors (Co(Cl)2 and Tris) was ball milled for 6 h to obtain Co/N–catkin nanosheets (step 1). The morphology of Co/N–catkin nanosheets thus obtained is shown by transmission electron microscopy (TEM) (Fig. 1a). These Co/N catkin nanosheets as-prepared above are pyrolyzed at 700–900 °C under the protection of inert atmosphere to form the composites in which Co nanoparticles are coated with Co/N–CNSs (CoNPs–Co/N–CNSs) (Scheme 1, step 2). To leach out these Co nanoparticles to obtain Co and N co-doped carbon nanosheets, the CoNPs–Co/N–CNSs were further treated with 0.5 M H2SO4 solution at 80 °C for 8 h, followed by the second pyrolysis. | ||
| Scheme 1 Schematic of preparation of Co/N–CNSs catalysts. | ||
 | ||
| Fig. 1 (a and b) TEM images of Co/N–catkin nanosheets and Co/N–CNSs, respectively. (c) HRTEM image of the Co/N–CNSs catalysts. The inset shows the SAED image. (d) EFTEM element mappings of the Co/N–CNSs catalysts. (e) XRD patterns of CoNPs–Co/N–CNSs-800 and Co/N–CNSs. | ||
The morphology of CoNPs–Co/N–CNSs is revealed by high angle annular dark-field scanning TEM (HAADF-STEM) images in Fig. S2a,† where some irregular nanoparticles dispersed at the nanosheets could be clearly observed.1 The TEM images could further confirm the structure with nanoparticles imbedded in CNSs (Fig. S2b†). The inset of Fig. S2b† displays the high resolution TEM (HRTEM) image to give the lattice spacings of nanoparticles of 0.205 nm and 0.180 nm, which match those of the (111) and (200) planes of metal cobalt phases, respectively. The selected area electron diffraction (SAED) pattern (in Fig. S2c†) further unambiguously identifies the nanoparticles as the cubic cobalt phase with the lattice spacings of 0.210 nm, 0.170 nm and 0.107 nm corresponding to the (111), (200) and (311) planes, respectively.7,9 After the acid leaching by 0.5 M H2SO4 at 80 °C for 8 hours and the second pyrolysis, the metal cobalt nanoparticles are hardly observed but the randomly orientated pores left can be easily seen by TEM instead (Fig. 1b). Additionally, the layer structures of pore walls were revealed by HRTEM (Fig. 1c) with an interlayer spacing of about 0.348 nm, corresponding to that of the (002) plane of graphitic carbon.2,10 One should bear in mind that only the diffracted rings are seen in the SADE pattern (inset of Fig. 1c), suggesting the removal of most cobalt nanoparticles by the acid leaching. Furthermore, elemental mappings by energy dispersive of X-ray (EDS) equipped energy filtered TEM (EFTEM) reveal Co and N to uniformly distribute across the whole carbon matrix of Co/N–CNSs after leaching out the cobalt nanoparticles, as shown in Fig. 1d. This is further evidenced by the severely weakening of the reflections of the cobalt phase from the X-ray diffraction (XRD) pattern (Fig. 1e and S3†). As shown in Fig. S3a,† no obvious crystal phase formed during the ball milling process. After the pyrolysis in Ar atmosphere for 700, 800 and 900 °C, we could only observe the crystal phase of metallic cobalt nanoparticles, which is in agreement with the HRTEM and SAED results (Fig. S3b†).2,16 The acid leaching followed by the second pyrolysis nearly leads to the absence of the crystalline cobalt phase, as demonstrated by the trace (111) reflections of the XRD pattern in Fig. S3c.†
Furthermore, we employed X-ray photoelectron spectroscopy (XPS) to identify the elements and their surface chemical states of the as-prepared electrocatalysts.1,2,15 Fig. 2a displays the XPS survey spectrum of the sample pyrolyzed at 800 °C, which confirms the existence of C, N, O, and Co, and the elemental contents of N and Co are also listed in Fig. 2b and Table S1.† In the first pyrolyzed sample of the CoNPs–Co/N–CNSs, the percentages of oxygen, cobalt and nitrogen are 4.607%, 1.023% and 1.785%, respectively. In contrast, for the acid-leached and then pyrolyzed sample of Co/N–CNSs, the percentages of oxygen and cobalt drop to 3.573% and 0.208%, respectively, accompanied by an increase of nitrogen content to 2.144%. This results reasonably from the disappearance of metal cobalt species and the decomposition of the oxygen-containing (–OH, –COOH, etc.) groups caused by the acid leaching and high temperature treatment process.32 The increase of nitrogen content arises from the successful doping of nitrogen into carbon.32 To further investigate the chemical states of the as-obtained electrocatalysts, the high-resolution N 1s and Co 2p spectra of CoNPs–Co/N–CNSs-800 and Co/N–CNSs are acquired in Fig. 2c–f. The N spectra of both CoNPs–Co/N–CNSs-800 and Co/N–CNSs could be deconvoluted into three components, which are assigned to the pyridinic-like N (398.4 eV), pyrrolic-like N (401.6 eV), and quaternary-like N (402.1 eV), respectively.12,17,36 The pyridinic N species are commonly accepted as the most important part of the catalytic active sites (CoNx), which could greatly affect the electrocatalytic performance of NPM electrocatalysts.11,37,38 The Co L-edge spectra display two separate peaks located around 772 and 787 eV, which correspond to L3 and L2 edges of core electrons excitation from the 2p3/2 and 2p1/2 hybrid orbits.1,39 The metallic Co0 (Co–Co bond), Co–C, and Co3+ peaks and the Co2+ peak for Co–O are centered at around 777.9, 778.8, 780.1 and 779.9 eV, respectively. Besides these peaks, an extra strong Co–N peak was observed at 781.1 eV.2,19,38 After the acid leaching and second pyrolysis, no metallic Co and Co2+ peaks were observed, suggesting that the remaining Co species are coordinated with N in Co–Nx moieties in the carbon matrix.1,32 Such Co–Nx coordination, which would generate a strong electron-withdrawing effect, could cause an electron density migration of adjacent C atoms.19,40 Consequently, the migration is expected to effectively adsorb intermediates, which would enhance the ORR/HER kinetics.4,19,40
 | ||
| Fig. 2 (a) XPS survey spectra of CoNPs–Co/N–CNSs-800 and Co/N–CNSs. (b) The contents of Co and N. (c and e) High-resolution XPS N 1s spectra of CoNPs–Co/N–CNSs-800 and Co/N–CNSs, respectively. (d and f) High-resolution XPS Co 2p spectra of CoNPs–Co/N–CNSs-800 and Co/N–CNSs, respectively. | ||
The Co/N–catkin nanosheets were first carbonized at 700, 800 and 900 °C, respectively, for optimization of the electrocatalytic performance of Co/N–CNSs for comparison with that of commercial 20% Pt/C tested under the same conditions. We achieved the best ORR activity of CoNPs–Co/N–CNSs at 800 °C (CoNPs–Co/N–CNSs-800), as demonstrated in Fig. S4a.† The linear sweep voltammetry (LSV) curves, which are taken by using rotating disk electrode (RDE) technique in basic electrolyte (0.1 M KOH), give the onset potential of 0.789 V (vs. RHE) and the diffusion-limited reaction current density of −4.58 mA cm−2, better than CoNPs–Co/N–CNSs-700 (0.77 V vs. RHE, −3.92 mA cm−2) and CoNPs–Co/N–CNSs-900 (0.68 V vs. RHE, −3.2 mA cm−2). Consequently, the CoNPs–Co/N–CNSs-800 is chosen for extra treatments for possible performance improvement, i.e. the acid leaching and second pyrolysis unless otherwise specified. The cyclic voltammograms (CVs) of both Co/N–CNSs and commercial 20% Pt/C were tested in N2- and O2-saturated 0.1 M KOH electrolyte. As shown in Fig. 3a, the CV curve of Co/N–CNSs displays a quasi-rectangular shape, and no obvious peaks for both Co/N–CNSs and commercial 20% Pt/C were observed in N2- saturated 0.1 M KOH. In contrast, a clear distinction occurs in O2-saturated electrolyte, where well-defined cathodic ORR peaks could be achieved for Co/N–CNSs at 0.706 V vs. RHE in and for commercial 20% Pt/C at 0.73 V vs. RHE in O2-saturated electrolyte. The presence of such a cathodic peak suggests a more pronounced ORR electrocatalytic activity of Co/N–CNSs compared to commercial 20% Pt/C. In Fig. 3b, significant enhancement of ORR activity could be observed for CoNPs–Co/N–CNSs-800 and Co/N–CNSs with the onset and half-wave potentials of 0.811 V and 0.789 V vs. RHE and 0.923 V and 0.835 V vs. RHE, respectively. Similarly, the limiting current density of the Co/N–CNSs is 5.21 mA cm−2, which is superior to that of CoNPs–Co/N–CNSs-800 (4.48 mA cm−2). The performance enhancement of Co/N–CNSs implies that the metallic Co0 phase doesn't favor the ORR performance. Compared with commercial 20% Pt/C with the onset, half-wave potentials and limiting current density of 0.976 V, 0.887 V vs. RHE and 5.51 mA cm−2, the Co/N–CNSs catalysts exhibit a great prospect of practical applications in fuel cells or metal–air batteries.
 | ||
| Fig. 3 (a) CV curves of commercial 20% Pt/C and Co/N–CNSs in N2-/O2-saturated 0.1 M KOH at the scan rate of 50 mV s−1. (b) LSV curves of commercial 20% Pt/C, CoNPs–Co/N–CNSs-800 and Co/N–CNSs in an O2-saturated 0.1 M KOH electrolyte at the scan rate of 5 mV s−1. (c) Time-dependent currents of commercial 20% Pt/C and Co/N–CNSs at 0.5 V (vs. RHE) in O2-saturated 0.1 M KOH with 1600 rpm. (d) Current–time (j–t) chronoamperometric responses of commercial 20% Pt/C and Co/N–CNSs in N2-/O2-saturated 0.1 M KOH under 1600 rpm with 1 M methanol addition. (e and f) Effects of the addition of 5 mM Br− and SCN− ions on the ORR activity of Co/N–CNSs catalysts in 0.1 M KOH. | ||
LSV curves (Fig. S4b†) are also shown to give insight into the ORR kinetics of Co/N–CNSs at various rotating speeds ranging from 100 to 1600 rpm in O2-saturated electrolyte. The limiting current density increased typically with the increase of rotation rate in that the oxygen molecules diffuse to the electrode surface efficiently, whereas the onset potential hardly changes.32,37,41 A good linearity was found between J−1/2 and ω−1/2 for the K–L plots in the voltage range from 0.1 to 0.7 V vs. RHE, as displayed by the K–L plots calculated using eqn (1)–(3) (Fig. S4c and inset†). The Co/N–CNSs displayed a slope of around 4, which implies a four-electron process for ORR. In Fig. S4d,† the rotating ring-disk electrode (RRDE) technique is further chosen to detect the selectivity of the four-electron reduction of oxygen for Co/N–CNSs catalysts (the potential of ring electrode is set 1.23 V to collect the formed peroxide species on the disk electrode).26 The electron-transfer number was calculated to be 3.98 according to eqn (4) and (5) (ESI†), in accord with the results derived from the K–L plots. The durability and the stability of methanol induction are also two key concerns for the performance evaluation of electrocatalysts in fuel cells.1,8,40 As shown in Fig. 3c, the chronoamperometric responses display only a slight activity loss of ca. 5.3% for Co/N–CNSs after 10000 s which is in sharp contrast with ca. 14.4% for the commercial 20% Pt/C. This clearly indicates the far better stability of Co/N–CNSs for ORR in alkaline electrolyte. Additionally, some issues may be encountered in practical applications in fuel cells in which both ORR and the fuel molecules (methanol) which pass through the membrane can be catalyzed to poison the commercial noble metal based catalysts.14,42 Therefore, both Co/N–CNSs and commercial 20% Pt/C were tested for further performance evaluation in practical direct methanol fuel cells (DMFCs).8,42 Fig. 3d displays the almost unchanged current of Co/N–CNSs whereas a drastic drop of current for 20% Pt/C occurred upon the introduction of 1 M methanol into an O2-saturated 0.1 M KOH electrolyte simultaneously. The undesirable activity drop could be ascribed to the methanol adsorption and de-hydrogenation CO which is strongly bonded to the surface of Pt sites.8,42 Consequently, the stability of Co/N–CNSs would not be influenced by the methanol, which makes it an ideal candidate as cathodic electrocatalysts for fuel cells. The active sites of the as-prepared Co/N–CNSs electrocatalysts could be further confirmed via a simple method, i.e. the ORR activity could be evaluated by introducing 5 × 10−3 mM Br− and SCN− into the electrolyte (Fig. 3e and f). The addition of 5 × 10−3 mM Br− caused the half-wave potential and the limited diffusion current density of Co/N–CNSs to decrease by about 3.2 mV and 1.2 mA cm−2, respectively. In sharp contrast, the introduction of 5 × 10−3 mM SCN− into the electrolyte caused the half-wave potential to decrease by about 16 mV, accompanied by an about 25% decrease in limited diffusion current density at 0.1 V vs. RHE. Obviously, Br− ions could only degrade the ORR activity slightly whereas SCN− ions could seriously poison the electrocatalytic sites. The difference of ORR performance also implies that the cobalt ionic species coordinated with N atoms to form highly active and stable CoNx sites are dispersed in the carbon matrix homogenously, as confirmed directly by the EDS elemental mappings and XPS results above.32
The HER electrocatalytic performance of catalysts was further examined with a typical three-electrode system in an Ar-saturated 0.5 M H2SO4 electrolyte, as shown in Fig. S5.†3,19 Similar to the ORR performance, CoNPs–Co/N–CNSs-800 showed the minimum overpotential of −0.430 V vs. RHE at the HER current density of −10 mA cm−2, far lower than −0.540 V vs. RHE for CoNPs–Co/N–CNSs-700 and -709 V vs. RHE for CoNPs–Co/N–CNSs-900. However, the acid leaching and second pyrolysis resulted in a significantly enhanced HER performance of Co/N–CNSs, −0.278 V vs. RHE at −10 mA cm−2, which is acceptable compared to the normal HER activity of the commercial 20% Pt/C with a near-zero overpotential of −0.093 V vs. RHE at −10 mA cm−2, as shown in Fig. 4a. The corresponding Tafel plots of CoNPs–Co/N–CNSs-800, Co/N–CNSs and commercial 20% Pt/C electrocatalysts are given in Fig. 4b in which the Tafel slop is normally taken as an important parameter for understanding the mechanisms or rate-determining steps of catalysts.1,28 The Tafel slope of Co/N–CNSs is 54.84 mV dec−1, very comparable to 52.7 mV dec−1 of commercial 20% Pt/C, and much lower than 150.62 mV dec−1 of CoNPs–Co/N–CNSs-800. To further understand the interfacial reactions and electrode kinetics of HER process, their electrochemical impedance spectra (EIS) were also acquired in Fig. 4c.1,19,41 From the Nyquist plots, the relatively lower solution resistance (Rs) of Co/N–CNSs suggests the rapid electron transfer and thus the superior HER kinetics toward the electrolyte interface.42 As shown in the inset of Fig. 4c, no semicircles are shown for commercial 20% Pt/C, CoNPs–Co/N–CNSs and Co/N–CNSs, which could be ascribed to the high electron mobility. Obvious semicircles are observed for all of the above electrodes in the high-frequency region.36 The CV curves under different scan rates are chosen to determine the intrinsic catalytic activity and electrochemical surface area which affect both the onset potential and current density of Co/N–CNSs.41,42 The electrochemical double-layer capacitance (Cdl) of Co/N–CNSs is calculated at 0.17 V vs. RHE from a series of CV curves with a scan rate from 10 to 140 mV s−1. No faradaic reactions occurred in the range of the mentioned-above scan rates40,41 and the electrochemical area of Co/N–CNSs is 5.67 mF cm−2, as shown in Fig. 4d. The HER kinetics of Co/N–CNSs is probably controlled predominantly by the Volmer–Heyrovsky reaction because of the direct correlation between the HER activity and the electrochemical area.1,19,42 Compared with other catalyst materials reported recently which are listed in Table S2,† the Co/N–CNSs displays the outstanding bi-functional ORR and HER electrocatalytic performances.
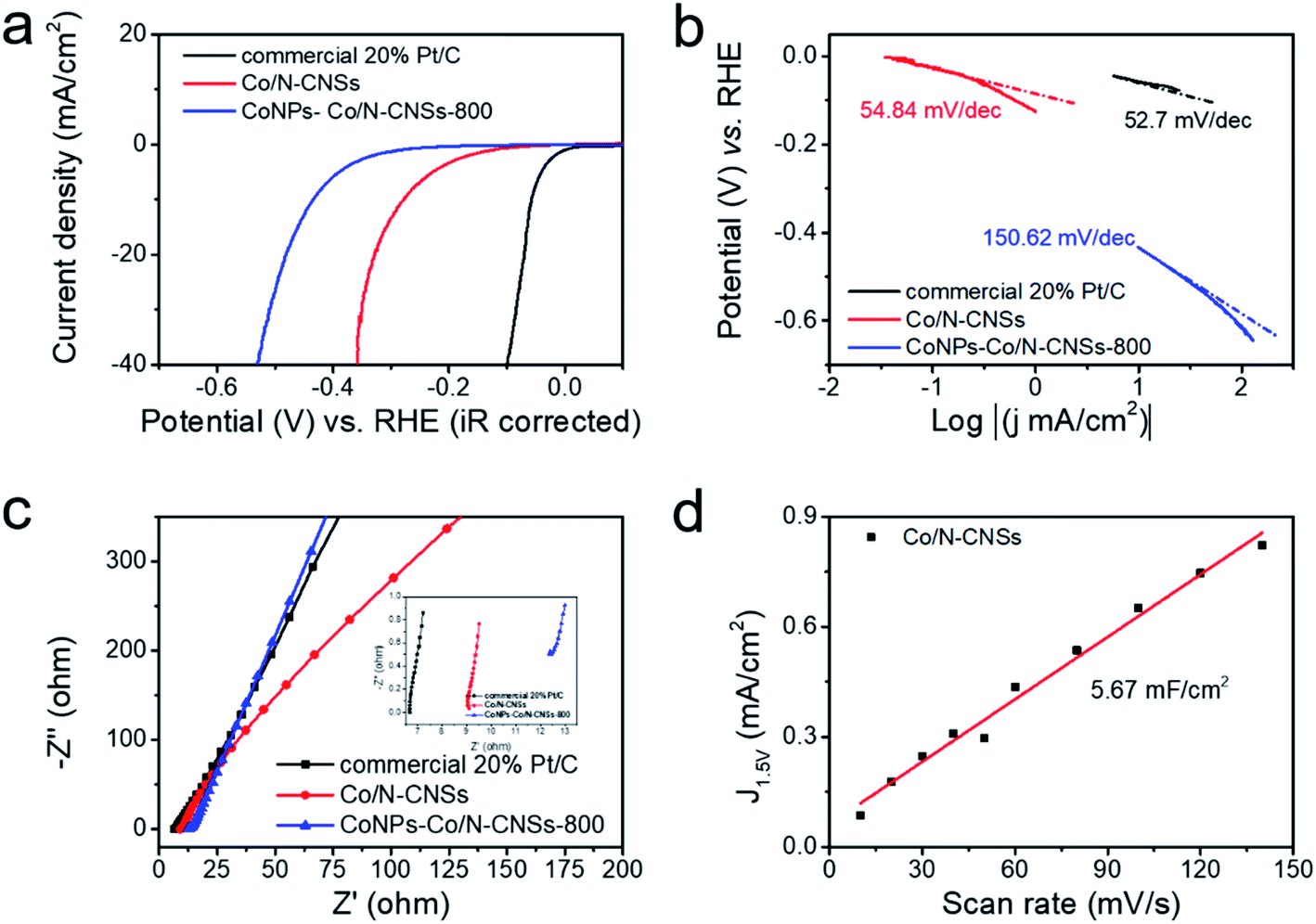 | ||
| Fig. 4 (a) HER LSV, (b) HER Tafel and (c) Nyquist plots (the inset: Nyquist plots in the high-frequency region) of commercial 20% Pt/C, CoNPs–Co/N–CNSs-800 and Co/N–CNSs. (d) The calculated Cdl for Co/N–CNSs. | ||
Conclusions
In conclusion, we prepared successfully heteroatoms Co and N codoped carbonaceous nanosheets nonprecious metal electrocatalysts with excellent electrocatalytic performance and long-term stability from environmentally hazardous biomass catkins using the mechanical exfoliation followed by acid leaching and pyrolysis. The Co/N–CNSs catalysts exhibit both the outstanding ORR and HER activity with the onset potential of 0.92 V vs. RHE and the half-wave potential of 0.83 V vs. RHE for ORR, and the potential of −0.278 V vs. RHE at −10 mA cm−2 for HER due to the presence of numerous active CoNx sites. The Co/N–CNSs display the far better long-term stability and tolerance to the methanol crossover compared to the commercial 20% Pt/C, which makes the Co/N–CNSs catalysts very promising in practical fuel cells. The present study provides a novel strategy for preparing the high performance NPM electrocatalysts from the biomass materials by combining the mechanical exfoliation and the chemical treatments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding support from the National Natural Science Foundation of China under grant no. 51802054, and Strategic Priority Research Program of the Chinese Academy of Sciences, grant no. Y9F3XD01ZX.References
- Z. Yang, C. Zhao, Y. Qu, H. Zhou, F. Zhou, J. Wang, Y. Wu and Y. Li, Adv. Mater., 2019, 31, e1808043 CrossRef.
- Q. Zhang and J. Q. Guan, Adv. Funct. Mater., 2020, 30, 2000768 CrossRef CAS.
- C. Wu, D. Liu, H. Li and J. Li, Small, 2018, 14, 1704227 CrossRef.
- X. D. Wen, Q. Zhang and J. Q. Guan, Coord. Chem. Rev., 2020, 409, 213214 CrossRef CAS.
- M. H. Shao, Q. W. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594 CrossRef CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060 RSC.
- H. Wang, S. Min, C. Ma, Z. Liu, W. Zhang, Q. Wang, D. Li, Y. Li, S. Turner, Y. Han, H. Zhu, E. Abou-Hamad, M. N. Hedhili, J. Pan, W. Yu, K. W. Huang, L. J. Li, J. Yuan, M. Antonietti and T. Wu, Nat. Commun., 2017, 8, 13592 CrossRef CAS.
- L. Hao, S. Zhang, R. Liu, J. Ning, G. Zhang and L. Zhi, Adv. Mater., 2015, 27, 3190 CrossRef CAS.
- X. Liu, L. Wang, P. Yu, C. Tian, F. Sun, J. Ma, W. Li and H. Fu, Angew. Chem., Int. Ed., 2018, 57, 16166 CrossRef CAS.
- M. Gorlin, P. Chernev, J. Ferreira de Araujo, T. Reier, S. Dresp, B. Paul, R. Krahnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603 CrossRef.
- A. Zitolo, N. Ranjbar-Sahraie, T. Mineva, J. Li, Q. Jia, S. Stamatin, G. F. Harrington, S. M. Lyth, P. Krtil, S. Mukerjee, E. Fonda and F. Jaouen, Nat. Commun., 2017, 8, 957 CrossRef.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443 CrossRef CAS.
- P. Jiang, J. Chen, C. Wang, K. Yang, S. Gong, S. Liu, Z. Lin, M. Li, G. Xia, Y. Yang, J. Su and Q. Chen, Adv. Mater., 2018, 30, 1705324 CrossRef.
- B. Y. Guan, Y. Lu, Y. Wang, M. Wu and X. W. Lou, Adv. Funct. Mater., 2018, 28, 1706738 CrossRef.
- H. Li, P. Wen, D. S. Itanze, M. W. Kim, S. Adhikari, C. Lu, L. Jiang, Y. Qiu and S. M. Geyer, Adv. Mater., 2019, e1900813 CrossRef.
- A. Aijaz, J. Masa, C. Roesler, W. Xia, P. Weide, A. J. R. Botz, R. A. Fischer, W. Schuhmann and M. Muhler, Angew. Chem., Int. Ed., 2016, 55, 4087 CrossRef CAS.
- H. Fei, J. Dong, M. J. Arellano-Jimenez, G. Ye, N. Dong Kim, E. L. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. J. Yacaman, P. M. Ajayan, D. Chen and J. M. Tour, Nat. Commun., 2015, 6, 8668 CrossRef CAS.
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688 CrossRef CAS.
- X. D. Wen, X. Y. Yang, M. Li, L. Bai and J. Q. Guan, Electrochim. Acta, 2019, 296, 830 CrossRef CAS.
- R. Jasinski, Nature, 1964, 201, 1212 CrossRef CAS.
- J. A. R. v. Veen and H. A. Colijn, Ber. Bunsenges. Phys. Chem., 1981, 85, 700 CrossRef.
- S. Gupta, D. Tryk, I. Bae, W. Aldred and E. Yeager, J. Appl. Electrochem., 1989, 19, 19 CrossRef CAS.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800 CrossRef CAS.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS.
- H.-W. Liang, W. Wei, Z.-S. Wu, X. Feng and K. Muellen, J. Am. Chem. Soc., 2013, 135, 16002 CrossRef CAS.
- H. W. Liang, S. Bruller, R. Dong, J. Zhang, X. Feng and K. Mullen, Nat. Commun., 2015, 6, 7992 CrossRef CAS.
- F. Jaouen, F. Charreteur and J. P. Dodelet, J. Electrochem. Soc., 2006, 153, A689 CrossRef CAS.
- F. Jaouen, E. Proietti, M. Lefevre, R. Chenitz, J. P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114 RSC.
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71 CrossRef CAS.
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian, J. Herranz and J.-P. Dodelet, Nat. Commun., 2011, 2, 416 CrossRef.
- L. T. Song, Z. Y. Wu, F. Zhou, H. W. Liang, Z. Y. Yu and S. H. Yu, Small, 2016, 12, 6398 CrossRef CAS.
- U. I. Kramm, M. Lefevre, P. Bogdanoff, D. Schmeisser and J. P. Dodelet, J. Phys. Chem. Lett., 2014, 5, 3750 CrossRef CAS.
- U. I. Kramm, I. Herrmann-Geppert, S. Fiechter, G. Zehl, I. Zizak, I. Dorbandt, D. Schmeißer and P. Bogdanoff, J. Mater. Chem. A, 2014, 2, 2663 RSC.
- U. I. Kramm, M. Lefevre, N. Larouche, D. Schmeisser and J. P. Dodelet, J. Am. Chem. Soc., 2014, 136, 978 CrossRef CAS.
- X. D. Wen, L. Bai, M. Li and J. Q. Guan, ACS Sustainable Chem. Eng., 2019, 7, 9249 CrossRef CAS.
- K. R. Yoon, J. Choi, S.-H. Cho, J.-W. Jung, C. Kim, J. Y. Cheong and I. D. Kim, J. Power Sources, 2018, 380, 174 CrossRef CAS.
- J. Xiao, C. Zhao, C. Hu, J. Xi and S. Wang, J. Power Sources, 2017, 348, 183 CrossRef CAS.
- Y. Lin, L. Yang, Y. Zhang, H. Jiang, Z. Xiao, C. Wu, G. Zhang, J. Jiang and L. Song, Adv. Energy Mater., 2018, 8, 1703623 CrossRef.
- J. Sanetuntikul, S. Hyun, P. Ganesan and S. Shanmugam, J. Mater. Chem. A, 2018, 6, 24078 RSC.
- X. Wang, F. Zhang, Y. Liu and Z. Wang, J. Electrochem. Soc., 2018, 165, J3052 CrossRef CAS.
- G. Li, J. Yu, J. Jia, L. Yang, L. Zhao, W. Zhou and H. Liu, Adv. Funct. Mater., 2018, 28, 1801332 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section for material synthesis, characterization and measurements. See DOI: 10.1039/d0ra08750e |
| This journal is © The Royal Society of Chemistry 2020 |