
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA photoredox catalysed Heck reaction via hole transfer from a Ru(II)-bis(terpyridine) complex to graphene oxide†
Marta Rosenthala,
Jörg K. N. Lindner b,
Uwe Gerstmannc,
Armin Meierd,
W. Gero Schmidtc and
René Wilhelm*d
b,
Uwe Gerstmannc,
Armin Meierd,
W. Gero Schmidtc and
René Wilhelm*d
aDepartment of Chemistry, University of Paderborn, Warburgerstr. 100, 33098 Paderborn, Germany
bDepartment Physics, Experimental Physics, University of Paderborn, Warburgerstr. 100, 33098 Paderborn, Germany
cDepartment of Physics, Theoretical Physics, University of Paderborn, Warburgerstr. 100, 33098 Paderborn, Germany
dInstitute of Organic Chemistry, Clausthal University of Technology, Leibnizstr. 6, 38678 Clausthal-Zellerfeld, Germany. E-mail: rene.wilhelm@tu-clausthal.de
First published on 25th November 2020
Abstract
The attachment of homoleptic Ru bis-terpy complexes on graphene oxide significantly improved the photocatalytic activity of the complexes. These straightforward complexes were applied as photocatalysts in a Heck reaction. Due to covalent functionalization on graphene oxide, which functions as an electron reservoir, excellent yields were obtained. DFT investigations of the charge redistribution revealed efficient hole transfer from the excited Ru unit towards the graphene oxide.
Introduction
Bis-terpyridine Ru complexes have a very short lifetime of their excited state1–4 which makes them less appealing for photocatalytic applications compared to other organic and metal organic dyes.5–9 Recently, Schubert and Dietzek were able to directly show a photoinduced charge-separation between a bis-terpyridine Ru complex covalently attached to a polyoxometalate unit.1 In case of an Ir complex with polyoxometalate Dietzek et al. showed that rather an increased yield instead of an increased charge separation is observed.10 Heteroleptic bis-terpyridine complexes with a donor and acceptor unit at each terpyridine moiety have been reported, where the bis-terpyridine metal center acts mainly as an electron relay.11–14 The latter complexes are considered promising for artificial photosynthetic applications.11,12,15–19 Nevertheless, a higher synthetic effort towards these complexes is necessary compared to homoleptic bis-terpyridine complexes. The latter are interesting due to their straightforward synthesis. Furthermore, they can be used to build linear structures without the danger to obtain geometric or optical isomers.2,11,12 Yet, just recently a homoleptic complex has been used to build a photocatalytic active MOF material for the reduction of carbon dioxide.20 Homoleptic bis-terpyridine iron complexes have been used to link nanoparticles.21 However, photocatalytic application with these homoleptic complexes are rare.20 Here, we demonstrate that the attachment of these complexes to graphene oxide results in a highly photocatalytic active material for a Heck reaction, via a hole transfer from the complex to graphene oxide.Graphene, i.e. single graphite sheets, is a carbon nanomaterial.22,23 Since its first synthesis and analysis in 2004,24 the number of publications concerning graphene, graphene oxide (GO) and reduced graphene oxide (reduced GO) has been exploding. GO can easily be prepared via the oxidation of graphite and graphene may be obtained via the treatment of graphite with appropriate solvents or via functionalization.22,23 Over the last few years an increasing number of reports have described the successful application of graphene/graphene oxide as promotors in photocatalysis,25 often doped with nitrogen26 or in combination with a semiconductor.27–31 In addition, 3D cross-linked graphene materials have been used in redox catalytic applications32 and functionalized graphene quantum dots have emerged for potential applications.33–36
Recently, photocatalytic selective organic transformations37–41 including homogenous photocatalyzed C–C cross-coupling reactions under visible-light have attracted much interest.42–53 Heterogeneous catalytic systems42,54–60 for C–C cross-coupling reactions have been reported in smaller numbers. Ligand-free palladium catalysed cross-couplings in general require higher temperatures.61–63 The photo-assisted reaction allows to perform the reaction at room temperature. The application of homoleptic terpyridine complexes would be of special interest but could not be realized so far.
Results and discussion
In order to create a new photocatalytic system with these complexes and in order to understand the interaction of this photoactive moiety and graphene oxide better, we first synthesized ligand 164 and the homoleptic Ru bis-terpyridine complex 2 according to literature procedures as depicted in Scheme 1.65 A UV/vis spectrum in acetonitrile showed a maximum absorption at 500 nm (see Fig. S2†). The in situ formation of a complex via metal salt addition to GO functionalized with terpyridine moieties was not considered in order to exclude the possibility of remnant metal salts in the material.66–68 The latter materials have already been reported for their potential in the generation of a photocurrent.67,68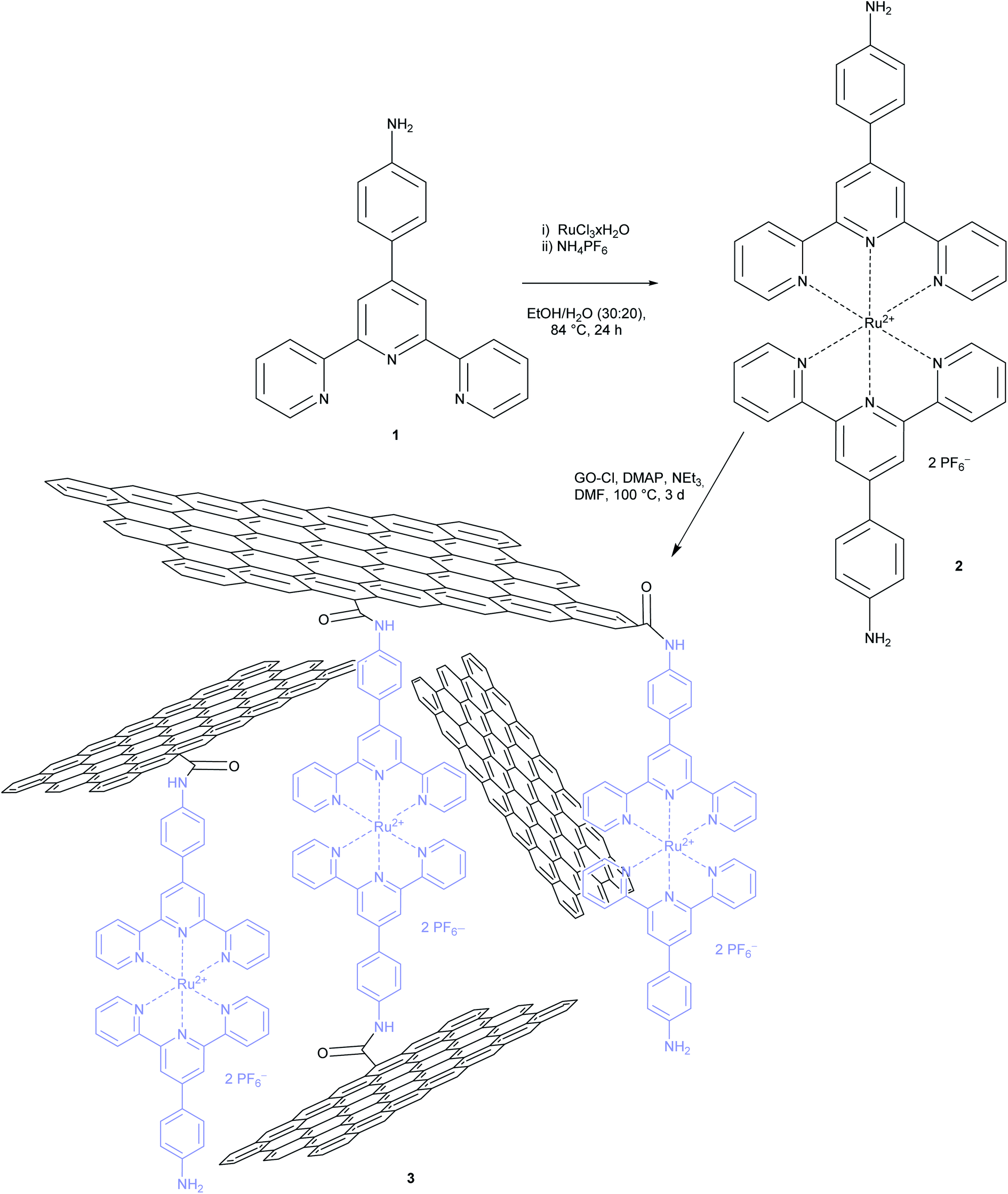 | ||
| Scheme 1 Synthesis of [Ru(bis-terpyridine)] complex 2 and functionalized GO material 3. | ||
Thereafter, in order to functionalize GO, its carboxylic acid functions were transformed to carboxylic acid chloride with oxalyl chloride.69 Treatment with complex 2 in the presence of DMAP and triethylamine resulted in the new material 3 as depicted in Scheme 1. For a high level of functionalization, GO and complex 2 were applied in the reaction with a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. After work-up and extensive washing the resulting material 3 was obtained and analysed. As can be seen in Scheme 1 it is assumed that an irregular network is formed. It consists of the complex and GO due to the presence of the two aniline groups in the complex. The material was insoluble in most solvents except DMSO, where a stable suspension was formed, and hence a good candidate for a heterogenous photocatalyst.
:1. After work-up and extensive washing the resulting material 3 was obtained and analysed. As can be seen in Scheme 1 it is assumed that an irregular network is formed. It consists of the complex and GO due to the presence of the two aniline groups in the complex. The material was insoluble in most solvents except DMSO, where a stable suspension was formed, and hence a good candidate for a heterogenous photocatalyst.
The material 3 was analysed via Raman, UV-vis, AFM, SEM, EDX and XRD, which are depicted in the ESI (Page S2–S8).† Fig. S1† shows the Raman spectrum of 3 in comparison to the commercially available GO.70 Graphitic materials exhibit a significant D-band for sp3-centers and a characteristic G-band for sp2-centers of carbon atoms. The ratio of the intensities of the D- and G-band allows to postulate the amount of graphene layers present in the material. While the GO consists of 16 layers according to the Raman spectrum the material 3 has a reduced number of 12 layers. AFM measurements of GO and material 3 are presented in Fig. S5.† A comparison of the two AFM images shows that the particle size was increased. This can be attributed to the fact, that the bifunctional complex could connect the GO sheets to larger particles. However, the sheets are not forming well defined layers. SEM images of material 3 showed no uniform structure (Fig. S6†). The edges of the particles are soft and small fiber structures. Hence, the morphology of 3 is different to GO, which can be attributed to the functionalization with complex 2. An EDX analysis (see Fig. S7 and S8†) showed that 3 contained ruthenium, nitrogen, fluorine and phosphor from the complex 2 next to carbon and oxygen. XRD spectra (Fig. S9 and S10†) showed that the typical peak of GO at 13° disappeared after the functionalization. Instead, a very broad peak between 23–27° appeared, which is specific for graphene material. This change indicated the disappearance of epoxide and carboxylic acid groups in 3. Cyclovoltammetry investigations failed with material 3.
For a structural characterization in the TEM material 3 was suspended in ethanol and the suspension dried on a carbon coated grid. An acceleration voltage of 80 kV was chosen to provide a good compromise between small beam damage at low voltages and sufficient lateral resolution. In fact, it turned out that material 3 was more stable than the supporting carbon foil of about 10 nm thickness. Graphene oxide is known to exhibit a crystalline lattice indistinguishable from the one of graphene.71 The high-resolution TEM analysis shows both agglomerates of small randomly oriented crystalline flakes with irregular shapes and sizes between about 1 and 10 nm (Fig. 1a) as well as staples of large ordered flakes few hundreds of nanometers in diameter (Fig. 1b). From an evaluation of contrasts (see ESI Fig. S11†) it can be concluded that one to three GO sheets are superimposed in Fig. 1b (regions I–III), where in the region (I) of one single GO layer the lattice fringes of 1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 00 type planes are visible. The Fourier transform (FFT) of the entire HRTEM image in Fig. 1b reveals the six-fold symmetry expected for a graphene lattice (inset in Fig. 1b) within the experimental errors of ±1° and lattice plane spacings 5.5% larger than reported for graphene in ref. 21.72 The FFTs of small agglomerates of flakes turn out to be difficult to interpret, most likely due to bending effects causing spot broadening and due to the 3D stacking of nearly 2D crystals resulting in higher order Laue zone effects.
00 type planes are visible. The Fourier transform (FFT) of the entire HRTEM image in Fig. 1b reveals the six-fold symmetry expected for a graphene lattice (inset in Fig. 1b) within the experimental errors of ±1° and lattice plane spacings 5.5% larger than reported for graphene in ref. 21.72 The FFTs of small agglomerates of flakes turn out to be difficult to interpret, most likely due to bending effects causing spot broadening and due to the 3D stacking of nearly 2D crystals resulting in higher order Laue zone effects.
 | ||
| Fig. 1 Transmission electron microscopy of material 3. (a) HRTEM image of an agglomerate of flakes, (b) lower magnification HRTEM image of a stack of larger GO sheets and Fourier transformed image in the inset. The numbers indicate the numbers of stacked GO sheets. (c) STEM-HAADF survey image, (d) magnified detail marked in red in (c) showing the distribution of high-Z elements, (e) magnified section of the region marked in green in (c) with the position of spectra (1–3), (f) STEM-EDX spectra taken at points (1–3) in (e). The red arrow in (e) marks contamination from a previous STEM-EDX line scan. | ||
In Fig. 1(c) and (d) STEM-HAADF images of a particularly large agglomerate are presented. In this mode, the local image intensity is proportional to ∫Z∼1.8dt, with Z being the atomic number and t the specimen thickness; hence a high brightness shows the presence of large amounts of heavy elements. This is obviously the case especially at the edges of flakes. A STEM-EDX analysis performed at the same sample gives notoriously small signals due to the limited beam interaction volume and due to large specimen contamination rates in the STEM mode, limiting the maximum signal integration time. Spectra (Fig. 1f) taken at three sample positions show the presence of ruthenium (Ru-Lα and Ru-Lβ at 2.558 and 2.683 keV, respectively) exclusively at particularly bright parts of the flake (Fig. 1e, blue spot) but not at the reference positions (red and green spots, Fig. 1e), confirming the preferential presence of Ru at the GO flake edges.
Finally, the photo-optical behaviour of material 3 was analysed via UV/vis spectroscopy in DMSO (Fig. S4†). The solvent was chosen because of the stable suspension of material 3 in this solvent. An absorption maximum was observed at 510 nm, which is close to the absorption maximum of the free complex at 500 nm in acetonitrile. The unfunctionalized GO showed a strong absorbance in the UV region, which continuously declined with longer wave length (Fig. S3†).
The new material 3 was investigated in the Heck reaction of 1-bromo-4-iodobenzene 4 with styrene 5 as shown in Scheme 2. First, different wave lengths for the reaction were evaluated in the presence of the heterogenous photocatalyst 3, Pd(OAc)2 and triethylamine. The highest yield of 60% was obtained with a cyan LED (495 nm), while a green LED (515 nm) resulted in a slightly lower yield of 50%. A blue LED (455 nm) gave a low yield of 34%. The highest yield was obtained with a wave length which corresponds close to the maximum absorption of the heterogenous complex 3 in DMSO (510 nm) and the free complex 2 in acetonitrile (500 nm). All the reactions resulted exclusively in the E-isomer.
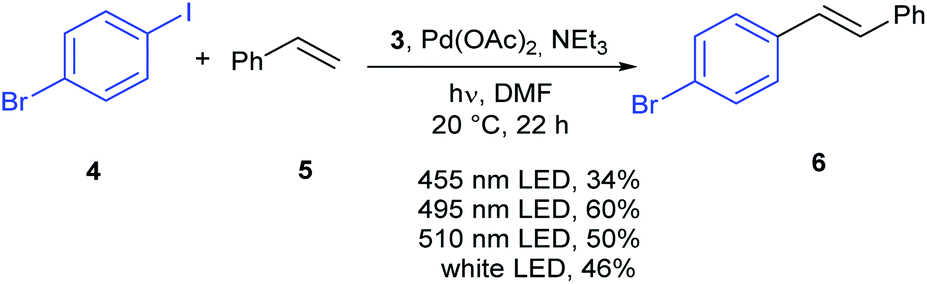 | ||
| Scheme 2 Heck reaction with 4 and 5. | ||
To optimize the reaction conditions the impact of the reaction time was investigated first as shown in Table 1. It was observed that conversion starts out slow but accelerates after an initial induction period, which could be explained by the time needed to generate a sufficient amount of Pd(0) species as indicated in Scheme 3. A reaction time of 24 h offered excellent results with a conversion of 96% (Table 1, entry 8). In addition, one reaction time was extended to 72 h in order to evaluate a possible E:Z conversion (Table 1, entry 9).73 This extension showed a slight increase in the E:Z ratio from 1:0.04 to 1:0.1.
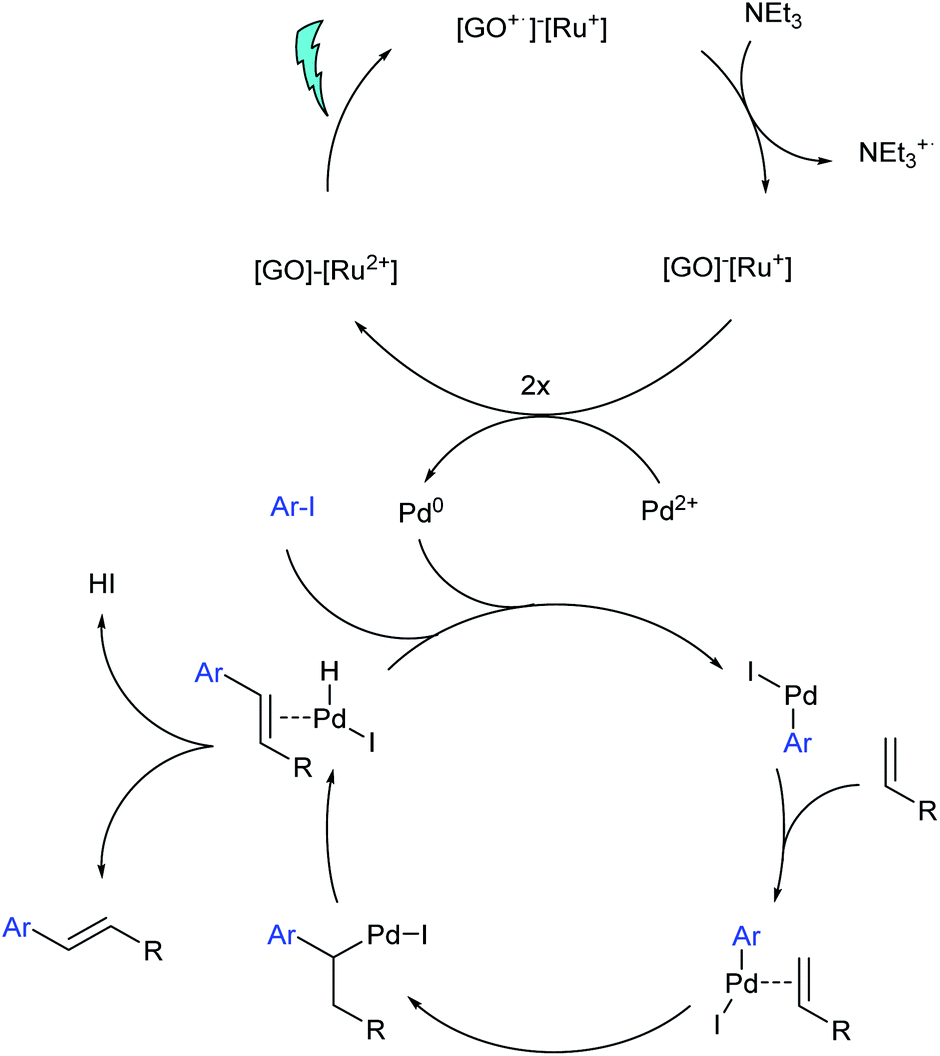 | ||
| Scheme 3 One possible mechanism for the photocatalytic cycle based on an SET. | ||
After the optimized reaction conditions were established, several control reactions were performed (Table 2). The reaction was first repeated in the absence of light, which resulted in only 7% yield (Table 2, entry 2). In order to evaluate the activity of material 3, a reaction with pure GO instead of 3 was carried out. A yield of 38% was obtained (Table 2, entry 3). The addition of GO and Ru complex 2 gave a yield of 53% (Table 2, entry 4), however, the expected 93% (Table 2, entry 1) were not reached. Obviously, the formation of a covalent bond between GO and Ru complex 2 in the material 3 is highly beneficial for its catalytic activity. The absence of material 3 and Pd(OAc)2 gave no product (Table 2, entry 5).
When Pd(OAc)2 was omitted from the reaction, only 18% yield were observed (Table 2, entry 6) while a yield of 30% was isolated without 3, alongside various side products (Table 2, entry 7). Clearly, both catalytic components are needed in order to achieve a high yield. Reducing the load of 3 from 5 wt% to 0.5 wt% resulted in a decrease of the yield to 33%.
In order to evaluate the catalytic activity of the material 3 further, the heterogenous catalyst 3 was recovered after the reaction and analysed. An SEM measurement revealed that the material did not change its morphology. However, small amounts of Pd were detectible via EDX. Nevertheless, the recovered material 3 without the addition of Pd(OAc)2 resulted in only 18% yield when applied in the reaction. When the reaction was performed with the recycled material and the normal addition of Pd(OAc)2 the same yield as in the beginning could be achieved. Hence, the photocatalytic activity of material 3 was not compromised after the recycling.
A possible mechanism is shown in Scheme 3. As supported by our DFT calculations (see below) it is based on hole transfer towards the GO. An alternative, first considered mechanism would have been that the excited complex 2 transfers its electron to the GO, and that the resulting Ru(III) species is reduced with triethylamine. As proposed in a homogenous photo-assisted Heck reaction,43 the electron enriched GO could finally transfer its electron to Pd(II) and reduce it to the active species Pd(0). In Scheme 3 the electron is transferred from the Ru+ species. Taking into account that the so far proposed mechanism would need only the light for the formation of Pd(0) which could then catalyse the reaction in the dark, a control reaction with material 3 was carried out. The material was first irradiated for 2 h with 495 nm light and stirred under exclusion of light for 24 h which provided a yield of only 6%. This implies that the catalyst 3 would also be needed to support the oxidative addition step or the elimination step in the palladium catalysed cycle. Hence, next to the proposed SET mechanism in Scheme 3, an additional subsequent transfer of a triplet state from the Ru unit to GO cannot be ruled out. The excited GO would then act as a sensitizer and would transfer its energy to the palladium–olefin intermediate, which has not been explicitly depicted in Scheme 3.
In order to determine the actual mechanism behind the photo-induced energy transfer, density functional theory (DFT) simulations of the excitation process have been performed using the Quantum ESPRESSO package.74 First, the excitation process of the pure Ru complex is investigated. An efficient excitation is possible by a HOMO–LUMO transition. Due to the strong hybridization of the involved Ru-4d states with carbon (and nitrogen) orbitals, the charge redistribution is not limited to the Ru ion, but extends over the whole molecular unit (see Fig. 2a). Upon this excitation an electron from the axial part of the complex is transferred to the NC cage surrounding the Ru ion enabling an efficient intra-molecular charge separation. As suggested by the observed UV/vis-spectra we expect this intra Ru-4d transition to be also responsible for photoexcitation of material 3, where the Ru unit is used to functionalize GO (see Fig. 2b). Here, the hole is further transferred from the axial part of the Ru complex towards the GO. The LUMO (i.e. the electron) in contrast remains almost unaffected. It is important to note that this charge transfer is almost independent of the O-content of the attached graphene flake. In Fig. 2b it is exemplarily shown for an O/C ratio of about 30%: the hole is transferred in large amount to the GO where it is mainly (de)localized over the O-free, graphene-like parts of the material. In other words, the Ru unit serves as an efficient hole pump towards the attached GO. This mechanism also appears to be robust with respect to the size of the attached GO flake (see ESI†). Only for very large GO flakes we expect the excited electron to be transferred into GO related empty states below the molecular LUMO, resulting in an effective induction of a triplet exciton into the GO. In any event, the calculations show that the initial excitation step consists of a hole transfer from the Ru complex to the GO.
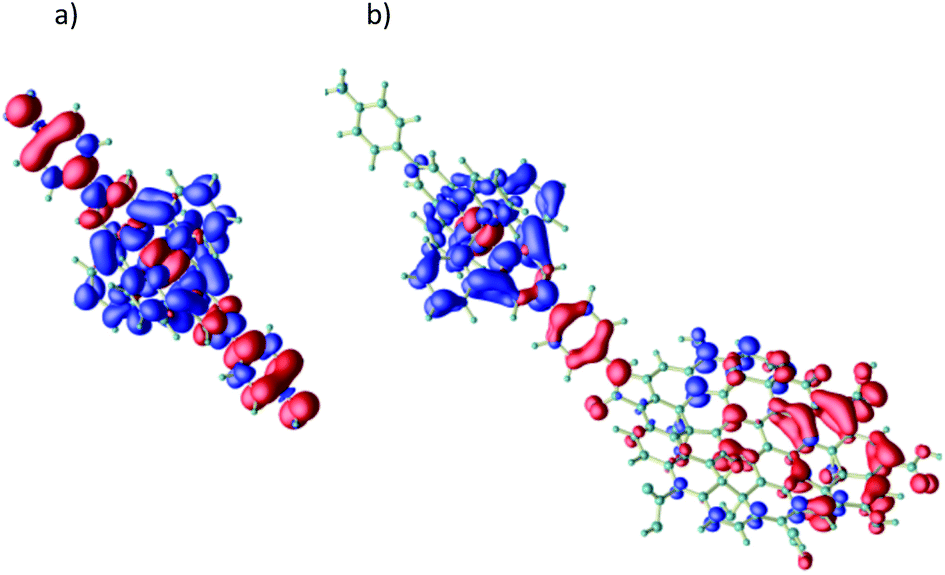 | ||
| Fig. 2 Frontier orbitals involved in a molecular HOMO–LUMO transition (intra Ru-4d excitation) for the pure Ru complex (a) and for a Ru unit functionalized with a GO-flake ((b), C/O ratio of 0.30); for further examples see ESI.† The electron is transferred from the HOMO (red) into the LUMO (blue). | ||
This is strong evidence that the photocatalytic cycle is probably based on a hole transfer and/or an energy transfer of a triplet state.75,76 The formation of an aryl radical from the iodoarene is not favourable under the reaction conditions.43 A control reaction in the presence of TEMPO showed no significant decrease in the yield, supporting the proposed absence of radical intermediates. The optimized conditions were then applied to further aryl iodides and olefins. The results are summarized in Table 3. Good to excellent yields could be obtained for each derivative.
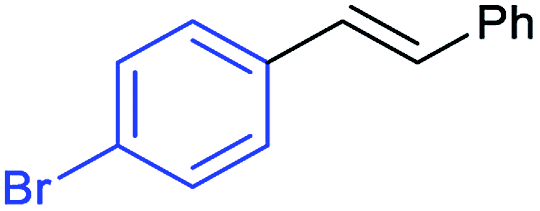
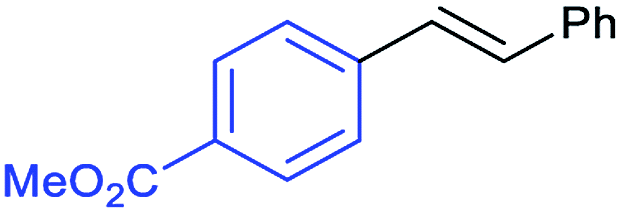
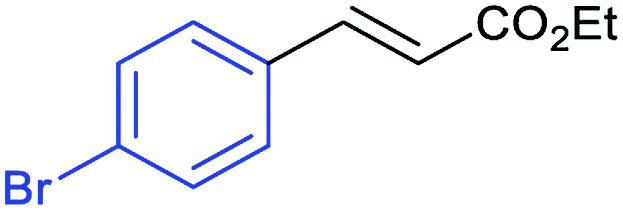
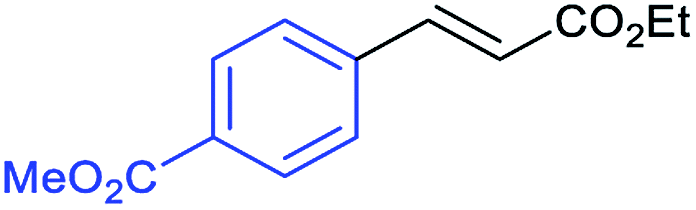
Conclusions
In summary, we have shown that the attachment of a Ru bis-terpy complex 2 on graphene oxide significantly improved the photocatalytic activity of these complexes in a Heck reaction resulting in excellent yields. HAADF-STEM and STEM-EDX measurements show the presence of Ru mainly at the edges of GO flakes. The improvement of photocatalytic activity is attributed to a covalent functionalization to graphene oxide. In principle, the latter can function as an electron sink and reservoir. Although a photoinduced dark catalytic cycle could be possible, it was observed that light is needed in order to maintain the product. DFT simulations of the photoexcited state identify hole transfer from the metal complex moiety to the attached GO as the initial step within the photocatalytic cycle. Hence, we presented a deeper understanding of the energy transfer between the metal complex 2 and GO.Experimental
Materials and reagents
All reagents such as NEt3 (triethylamine) and DMAP (4-(dimethylamino)-pyridine) and solvents such as DMF (dimethylformamide) were purchased from commercial sources like Fluka AG, Merck AG, Lancaster, Alfa Aesar, Riedel de Haën and Sigma Aldrich. Unless specified otherwise the reagents were further purified by standard procedures. GO (powder, 0.5–20 μm, elemental analysis: C: 55–65 at%, O: 35–45 at%, S: 0–2 at%, N: 0–1 at%) was purchased from Graphitene Ltd. SEM (Scanning Electron Microscope) measurements were performed on a NEON 40 from Zeiss, TEM (Transmission Electron Microscope) measurements were performed on a JEM-ARM200F NEORAM from JOEL. AFM (Atomic Force Microscope) measurements were performed on a Veeco Dimension Icon from Bruker. 1H-NMR spectra were measured at room temperature by Avance 500 and Ascent 700 from Bruker using methanol as internal standard. Fourier transform infrared (FTIR) and attenuated total reflection (ATR) spectra were measured with VERTEX 700 by Bruker using monocrystalline NaCl or KBr plates. Ligand 1 was prepared according to the literature.64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :6′,2′′-terpyridin]-4′-yl)aniline)Ru(II)](PF6)2 (2). 4-([2,2′:6′,2′′-Terpyridin]-4′-yl)aniline 1 (0.3 g, 0.93 mmol, 1 eq.) was added to a solution of Ru(III)Cl3·3H2O (0.61 mmol, 0.6 eq.) in a mixture of 50 mL ethanol/water (30:20) and refluxed overnight. Excess of NH4PF6 (0.1 g, 0.81 mmol, 0.8 eq.) was added to the reaction mixture and stirred for 5 h at room temperature. After evaporation of ethanol, the resulting salt was washed with water (2 × 20 mL), ethanol (2 × 20 mL) and diethyl ether (2 × 20 mL). Yield: 410 mg (0.39 mmol, 85%) as black crystalline solid. 1H-NMR (700 MHz, CD3CN-d3, TMS): δ [ppm] = 4.77 (sbr, 4H, NH2), 6.95–6.97 (m, 4H, H3′′′, H5′′′), 7.14–7.16 (m, 4H, H5, H5′′), 7.41–7.42 (m, 4H, H6, H6′′), 7.90–7.93 (m, 4H, H4, H4′′), 8.00–8.02 (m, 4H, H2′′′, H6′′′), 8.61 (d, J = 8.0 Hz, 4H, H3, H3′′), 9.91 (s, 4H, H3′, H5′). 13C-NMR (176 MHz, CD3CN-d3, TMS): δ [ppm] = 115.73 (C-3′′′, C-5′′′), 120.76 (C-3′, C-5′), 122.74 (C-1′′′), 125.16 (C-3, C-3′′), 128.17 (C-5, C-5′′), 129.77 (C-2′′′, C-6′′′), 138.76 (C-4, C-4′′), 149.38 (C-4′′′), 151.78 (C-6, C-6′′′), 153.31 (C-4′), 156.15 (C-2′, C-6′), 159.47 (C-2, C-2′′). HRMS(ESI): found m/z 375.0907 ([M]+); calc. for [C42H32N8Ru]2+: 375.0907. Raman ν = 1991, 1500–3400 cm−1. FTIR ν = 742, 785, 826, 1186, 1244, 1412, 1466, 1524, 1599, 3369 cm−1. The spectral data were consistent with literature values.65
:6′,2′′-terpyridin]-4′-yl)aniline)Ru(II)](PF6)2 (2). 4-([2,2′:6′,2′′-Terpyridin]-4′-yl)aniline 1 (0.3 g, 0.93 mmol, 1 eq.) was added to a solution of Ru(III)Cl3·3H2O (0.61 mmol, 0.6 eq.) in a mixture of 50 mL ethanol/water (30:20) and refluxed overnight. Excess of NH4PF6 (0.1 g, 0.81 mmol, 0.8 eq.) was added to the reaction mixture and stirred for 5 h at room temperature. After evaporation of ethanol, the resulting salt was washed with water (2 × 20 mL), ethanol (2 × 20 mL) and diethyl ether (2 × 20 mL). Yield: 410 mg (0.39 mmol, 85%) as black crystalline solid. 1H-NMR (700 MHz, CD3CN-d3, TMS): δ [ppm] = 4.77 (sbr, 4H, NH2), 6.95–6.97 (m, 4H, H3′′′, H5′′′), 7.14–7.16 (m, 4H, H5, H5′′), 7.41–7.42 (m, 4H, H6, H6′′), 7.90–7.93 (m, 4H, H4, H4′′), 8.00–8.02 (m, 4H, H2′′′, H6′′′), 8.61 (d, J = 8.0 Hz, 4H, H3, H3′′), 9.91 (s, 4H, H3′, H5′). 13C-NMR (176 MHz, CD3CN-d3, TMS): δ [ppm] = 115.73 (C-3′′′, C-5′′′), 120.76 (C-3′, C-5′), 122.74 (C-1′′′), 125.16 (C-3, C-3′′), 128.17 (C-5, C-5′′), 129.77 (C-2′′′, C-6′′′), 138.76 (C-4, C-4′′), 149.38 (C-4′′′), 151.78 (C-6, C-6′′′), 153.31 (C-4′), 156.15 (C-2′, C-6′), 159.47 (C-2, C-2′′). HRMS(ESI): found m/z 375.0907 ([M]+); calc. for [C42H32N8Ru]2+: 375.0907. Raman ν = 1991, 1500–3400 cm−1. FTIR ν = 742, 785, 826, 1186, 1244, 1412, 1466, 1524, 1599, 3369 cm−1. The spectral data were consistent with literature values.65Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the DFG (413541925) for financial support. The numerical DFT calculations have been performed at the Paderborn Center for Parallel Computing (PC2).Notes and references
- Y. Luo, M. Wächtler, K. Barthelms, A. Winter, U. S. Schubert and B. Dietzek, Chem. Commun., 2018, 54, 2970–2973 RSC.
- K. Barthelmes, M. Sittig, A. Winter and U. S. Schubert, Eur. J. Inorg. Chem., 2017, 3698–3706 CrossRef CAS.
- J.-P. Collin, S. Guillerez, J.-P. Sauvage, F. Barigelletti, L. DeCola, L. Flamigni and V. Balzani, Inorg. Chem., 1991, 30, 4230–4238 CrossRef CAS.
- S. Chakraborty, T. J. Wadas, H. Hester, R. Schmehl and R. Eisenberg, Inorg. Chem., 2005, 44, 6865–6878 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS.
- R. A. Angnes, Z. Li, C. R. D. Correia and G. B. Hammond, Org. Biomol. Chem., 2015, 13, 9152–9167 RSC.
- I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS.
- Chemical Photocatalysis, ed. B. König, De Gruyter, Berlin, Boston, 2013 Search PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS.
- Y. Luo, S. Maloul, S. Schçnweiz, M. Wächtler, C. Streb and B. Dietzek, Chem.–Eur. J., 2020, 26, 8045–8052 CrossRef CAS.
- E. Baranoff, J.-P. Collin, L. Flamigni and J.-P. Sauvage, Chem. Soc. Rev., 2004, 33, 147–155 RSC.
- V. Duprez, M. Biancardo, H. Spanggaard and F. C. Krebs, Macromolecules, 2005, 38, 10436–10448 CrossRef CAS.
- Y. Luo, M. Wächtler, K. Barthelmes, A. Winter, U. S. Schubert and B. Dietzek, Chem. Commun., 2019, 55, 5251–5254 RSC.
- Y. Luo, J. H. Tran, M. Wächtler, M. Schulz, K. Barthelms, A. Winter, S. Rau, U. S. Schubert and B. Dietzek, Chem. Commun., 2019, 55, 2273–2276 RSC.
- S. Sinn, B. Schulze, C. Friebe, D. G. Brown, M. Jäger, J. Kübel, B. Dietzek, C. P. Berlinguette and U. S. Schubert, Inorg. Chem., 2014, 53, 1637–1645 CrossRef CAS.
- M. Rupp, T. Auvray, E. Rousset, G. M. Mercier, V. Marvaud, D. G. Kurth and G. S. Hanan, Inorg. Chem., 2019, 58, 9127–9134 CrossRef CAS.
- T. Auvray, R. Sahoo, D. Deschênes and G. S. Hanan, Dalton Trans., 2019, 48, 15136–15143 RSC.
- A. Winter and U. S. Schubert, ChemCatChem, 2020, 12, 2890–2941 CrossRef CAS.
- P. Labra-Vázquez, M. Bocé, M. Tassé, S. Mallet-Ladeira, P. G. Lacroix, N. Farfán and I. Malfant, Dalton Trans., 2020, 49, 3138–3154 RSC.
- M. E. Mahmoud, H. Audi, A. Assoud, T. H. Ghaddar and M. Hmadeh, J. Am. Chem. Soc., 2019, 141, 7115–7121 CrossRef.
- M. R. Dewi, T. A. Gschneidtner, S. Elmas, M. Ranford, K. Moth-Poulsen and T. Nann, ACS Nano, 2015, 9, 1434–1439 CrossRef CAS.
- H.-X. Wang, Q. Wang, K.-G. Zhou and H.-L. Zhang, Small, 2013, 9, 1266–1283 CrossRef CAS.
- S. Eigler and A. Hirsch, Angew. Chem., Int. Ed., 2014, 53, 7720–7738 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS.
- Y. Tong, H. Pan, W. Huang, W. Qiu, Z. Ding, C. Xu and R. Yuan, New J. Chem., 2019, 43, 8741–8745 RSC.
- A. Savateev and M. Antonietti, ACS Catal., 2018, 8, 9790–9808 CrossRef CAS.
- S. Z. Liu, H. Q. Sun, S. M. Liu and S. B. Wang, Chem. Eng. J., 2013, 214, 298–303 CrossRef CAS.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS.
- N. Zhang, M.-Q. Yang, S. Liu, Y. Sun and Y.-J. Xu, Chem. Rev., 2015, 115, 10307–10377 CrossRef CAS.
- M.-Q. Yang, N. Zhang, M. Pagliaro and Y.-J. Xu, Chem. Soc. Rev., 2014, 43, 8240–8254 RSC.
- M. E. Malefane, B. Ntsendwana, P. J. Mafa, N. Mabuba, U. Feleni and A. T. Kuvarega, ChemistrySelect, 2019, 4, 8379–8389 CrossRef CAS.
- Y. Lu, Y. Ma, T. Zhang, Y. Yang, L. Wei and Y. Chen, J. Am. Chem. Soc., 2018, 140, 11538–11550 CrossRef CAS.
- T. Scharl, A. Ferrer-Ruiz, A. Saura-Sanmartin, L. Rodriguez-Perez, M. A. Herranz, N. Martin and D. M. Guldi, Chem. Commun., 2019, 55, 3223–3226 RSC.
- A. Cadranel, J. T. Margraf, V. Strauss, T. Clark and D. M. Guldi, Acc. Chem. Res., 2019, 52, 955–963 CrossRef CAS.
- S. J. Phang and L.-L. Tan, Catal. Sci. Technol., 2019, 9, 5882–5905 RSC.
- C. Kütahya, P. Wang, S. Li, S. Liu, J. Li, Z. Chen and B. Strehmel, Angew. Chem., Int. Ed., 2020, 59, 3166–3171 CrossRef.
- J.-Y. Li, Y.-H. Li, M.-Y. Qi, Q. Lin, Z.-R. Tang and Y.-J. Xu, ACS Catal., 2020, 10, 6262–6280 CrossRef CAS.
- N. Zhang, Y. Zhang, M.-Q. Yang and Y.-J. Xu, Curr. Org. Chem., 2013, 17, 2503–2515 CrossRef CAS.
- M.-Q. Yangab and Y.-J. Xu, Phys. Chem. Chem. Phys., 2013, 15, 19102–19118 RSC.
- C. Han, Y.-H. Li, M.-Y. Qi, F. Zhang, Z.-R. Tang and Y.-J. Xu, Sol. RRL, 2020, 4, 1900577 CrossRef CAS.
- C. Han, Z.-R. Tang, J. Liu, S. Jin and Y.-J. Xu, Chem. Sci., 2019, 10, 3514–3522 RSC.
- Q. Gu, Q. Jia, J. Long and Z. Gao, ChemCatChem, 2019, 11, 669–683 CrossRef CAS.
- H. Zhang and X. Huang, Adv. Synth. Catal., 2016, 358, 3736–3742 CrossRef CAS.
- I. Abdiaj, L. Huck, J. M. Mateo, A. d. l. Hoz, M. V. Gomez, A. Diaz-Ortiz and J. Alcázar, Angew. Chem., Int. Ed., 2018, 57, 13231–13236 CrossRef CAS.
- M. O. Konev, T. A. McTeague and J. W. Johannes, ACS Catal., 2018, 8, 9120–9124 CrossRef CAS.
- A. Sagadevan and K. C. Hwang, Adv. Synth. Catal., 2012, 354, 3421–3427 CrossRef CAS.
- M. Majek and A. J. v. Wangelin, Angew. Chem., Int. Ed., 2013, 52, 5919–5921 CrossRef CAS.
- A. Sagadevan, V. P. Charpe and K. C. Hwang, Catal. Sci. Technol., 2016, 6, 7688–7692 RSC.
- P. Chuentragool, D. Kurandina and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 11586–11598 CrossRef CAS.
- X.-J. Wei, I. Abdiaj, C. Sambiagio, C. Li, E. Zysman-Colman, J. Alczar and T. Noel, Angew. Chem., Int. Ed., 2019, 58, 13030–13034 CrossRef CAS.
- T. E. Schirmer, A. Wimmer, F. Wolfgang, C. Weinzierl and B. König, Chem. Commun., 2019, 55, 10796–10799 RSC.
- Z.-J. Wang, S. Zheng, E. n. Romero, J. K. Matsui and G. A. Molander, Org. Lett., 2019, 21, 6543–6547 CrossRef CAS.
- G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, J. Am. Chem. Soc., 2017, 139, 18307–18312 CrossRef CAS.
- N. Marina, A. E. Lanterna and J. C. Scaiano, ACS Catal., 2018, 8, 7593–7597 CrossRef CAS.
- B. W. Crabbe, O. P. Kuehm, J. C. Bennett and G. L. Hallett-Tapley, Catal. Sci. Technol., 2018, 8, 4907–4915 RSC.
- S. Sarina, H.-Y. Zhu, Q. Xiao, E. Jaatinen, J. Jia, Y. Huang, Z. Zheng and H. Wu, Angew. Chem., Int. Ed., 2014, 53, 2935–2940 CrossRef CAS.
- A. Eskandari, M. Jafarpour and A. Rezaeifard, Appl. Organomet. Chem., 2019, 33, e5093 CrossRef.
- Z. Zhang, C. R. Rogers and E. A. Weiss, J. Am. Chem. Soc., 2020, 142, 495–501 CrossRef CAS.
- S. Rohani, A. Ziarati, G. M. Ziarani, A. Badiei and T. Burgi, Catal. Sci. Technol., 2019, 9, 3820–3827 RSC.
- X.-W. Guo, C.-H. Hao, C.-Y. Wang, S. Sarina, X.-N. Guo and X.-Y. Guo, Catal. Sci. Technol., 2016, 6, 7738–7743 RSC.
- X.-Y. Zhou, X. Chen and L.-G. Wang, Synthesis, 2017, 49, 5364–5370 CrossRef CAS.
- M. Blanco, D. Mosconi, C. Tubaro, A. Biffis, D. Badocco, P. Pastore, M. Otyepka, A. Bakandritsos, Z. Liu, W. Ren, S. Agnoli and G. Granozzi, Green Chem., 2019, 21, 5238–5247 RSC.
- M. Mirza-Aghayan, M. Mohammadi, A. Addad and R. Boukherroub, Appl. Organomet. Chem., 2020, 34, e5524 CAS.
- T. Mutai, J.-D. Cheon, S. Arita and K. Araki, J. Chem. Soc., Perkin Trans. 2, 2001, 1045–1050 RSC.
- G. D. Storrier, S. B. Colbran and D. C. Craig, J. Chem. Soc., Dalton Trans., 1997, 3011–3028 RSC.
- D. Zhou, Q.-Y. Cheng, Y. Cui, T. Wang, X. Li and B.-H. Han, Carbon, 2014, 66, 592–598 CrossRef CAS.
- T. Matsui, M. R. Karim, H. Takehira, M. Nakamura, R. Ohtani, M. Koinuma and S. Hayami, Chem. Lett., 2016, 45, 365–367 CrossRef CAS.
- S. Song, Y. Xue, L. Feng, H. Elbatal, P. Wang, C. N. Moorefield, G. R. Newkome and L. Dai, Angew. Chem., Int. Ed., 2014, 53, 1415–1419 CrossRef CAS.
- R. Sekiya, Y. Uemura, H. Murakami and T. Haino, Angew. Chem., Int. Ed., 2014, 53, 5619–5623 CrossRef CAS.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- N. R. Wilson, P. A. Pandey, R. Beanland, R. J. Young, I. A. Kinloch, L. Gong, Z. Liu, K. Suenaga, J. P. Rourke, S. J. York and J. Sloan, ACS Nano, 2009, 3, 2547–2556 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. B. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS.
- J. Xu, N. Liu, H. Lv, C. He, Z. Liu, X. Shen, F. Cheng and B. Fan, Green Chem., 2020, 22, 2739–2743 RSC.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- M. Kudisch, C.-H. Lim, P. Thordarson and G. M. Miyake, J. Am. Chem. Soc., 2019, 141, 19479–19486 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and analytical data. See DOI: 10.1039/d0ra08749a |
| This journal is © The Royal Society of Chemistry 2020 |