
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-long cycle life and binder-free manganese-cobalt oxide supercapacitor electrodes through photonic nanostructuring†
Madhu Gaire *,
Binod Subedi,
Shiva Adireddy and
Douglas Chrisey
*,
Binod Subedi,
Shiva Adireddy and
Douglas Chrisey
Tulane University, Physics and Engineering Physics, School of Science and Engineering 6400, Freret St, 2001 Percival Stern Hall, New Orleans, Louisiana 70118, USA. E-mail: mgaire@tulane.edu
First published on 4th November 2020
Abstract
We report a novel photonic processing technique as a next-generation cost-effective technique to instantaneously synthesize nanostructured manganese-cobalt mixed oxide reduced graphitic oxide (Mn-Co-rGO) for supercapacitor electrodes in energy storage applications. The active material was prepared directly on highly conductive Pt–Si substrate, eliminating the need for a binder. Surface morphological analysis showed that the as-prepared electrodes have a highly porous and resilient nanostructure that facilitates the ion/electron movement during faradaic redox reactions and buffers the volume changes during charge–discharge processes, leading to the improved structural integrity of the electrode. The presence of distinct redox peaks, due to faradaic redox reactions, at all scan rates in the cyclic voltammetry (CV) curves and non-linear nature of the charge–discharge curves suggest the pseudocapacitive charge storage mechanism of the electrode. The electrochemical stability and the life cycle were examined by carrying out galvanostatic charge–discharge (GCD) measurements at 0.40 mA cm−2 constant areal current density for 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, and the electrode showed 95% specific capacitance retention, exhibiting excellent electrochemical stability and an ultra-long cycle life. Such remarkable electrochemical performance could be attributed to the enhanced conductivity of the electrode, the synergistic effect of metal ions with rGO, and the highly porous morphology, which provides large specific surface area for electrode/electrolyte interaction and facilitates the ion transfer.
000 cycles, and the electrode showed 95% specific capacitance retention, exhibiting excellent electrochemical stability and an ultra-long cycle life. Such remarkable electrochemical performance could be attributed to the enhanced conductivity of the electrode, the synergistic effect of metal ions with rGO, and the highly porous morphology, which provides large specific surface area for electrode/electrolyte interaction and facilitates the ion transfer.
1. Introduction
Metal oxide based supercapacitors (SCs), also known as pseudocapacitors, have received increasing attention as energy storage devices because of their high specific capacitance. Due to the fast and reversible redox reactions at the electrode/electrolyte interface, pseudocapacitors have higher power density, faster charge/discharge characteristics, and higher energy density than conventional carbon-based electric double-layer capacitors (EDLCs).1,2 These attributes make their application wide-ranging from consumer electronics to electric vehicles. Among metal oxides, RuO2 has high conductivity, long cycle life, and excellent electrochemical performance; however, toxicity, high cost, and low abundance limit its practical applications.3,4 Other metal oxides, such as MnO2, Fe2O3, NiO, Co3O4, etc. have been studied as alternative electrode materials.5,6The preparation of mixed transition metal oxides for supercapacitor electrodes, by incorporating two or more individual metal ions, has received an upsurge of interest as an effective method to improve the performance of supercapacitors.7,8 The mixed oxide approach enhances the electrochemical performance more than using single oxides because they offer advantageous structural defects, improved chemical stability and a wide working potential window.9 Mixed oxides also offer synergistic effects from the pure oxides leading to superior conductivity and richer redox reactions.9–11 Moreover, the coupling of metal oxides offers various combinations of cations and tunable stoichiometric/non-stoichiometric compositions, making it easier to study the electrochemical performance by manipulating the physical and chemical properties.12,13 Preparation of binary and ternary metal oxide mixtures has been reported by this approach.1,4,7–10,12–17
Among mixed transition metal oxides, Mn–Co mixed oxide is an excellent candidate for supercapacitor electrodes owing to its high electrochemical activity, high capacitance benefitting from multiple redox reactions than the individual Mn or Co oxides, and long cycle life.18,19 Furthermore, the addition of Co impedes the dissolution of Mn into the electrolyte and improves the redox reversibility and stability of the electrode.20,21 Despite their many advantages, drawbacks including their low conductivity, which also hinders ion diffusion and electron transfer,22 poor structural stability, small specific surface area, and volume changes during the redox reactions, still, need to be addressed. These issues could be resolved by: (i) combining highly conductive material with the metal oxides, (ii) designing and fabricating novel electrode structures, and (iii) growing the active material directly on a highly conductive substrate.23,24
Firstly, incorporating carbonaceous materials with metal oxide is an effective method to improve the overall performance of the electrode as carbon offers low cost, high electronic conductivity, high power density, high surface area, controlled pore size distribution and high chemical stability.25–30 Secondly, since the performance of the electrode is highly dependent on its structure and morphology, designing and preparing electrode with novel nanostructure is crucial for high performance of the supercapacitor. Therefore, reducing the particle size to the nanoscale is an important advantage as nanostructures offer a high surface to volume ratio, which ensures better electrode/electrolyte interaction and increased utilization of the active material during electrochemical processes. Thirdly, by directly growing the active material on a highly conductive substrate, the need for a binder is eliminated and better mechanical adhesion and electrical contact between active materials and the substrate is ensured, facilitating the electrode/electrolyte interfacial charge transfer.
In this work, we have focused on preparing active materials directly on conductive substrates for the fabrication of highly porous 3D nanostructured manganese-cobalt oxide reduced graphitic oxide (Mn-Co-rGO) electrodes. The presence of rGO improves the electronic conductivity, makes the electrode structure more robust, offers synergistic effect with metal oxides, enhancing the electrochemical activity and results in improved overall electrochemical performance of the electrode by exploiting the double-layer capacitance from rGO as well as the pseudocapacitance from both Mn and Co oxides. Furthermore, such porous structures offer: (i) increased effective utilization of active materials,31 (ii) shortened ion diffusion lengths and increased mass transport, and (iii) reduced phase transformation during faradaic redox reactions.32,33
While the idea of preparing mixed metal oxides electrodes is not novel, and different groups have already reported such work following various techniques,12–17 what we are presenting here is novel and unique in that our photonic processing technique yields unique nanostructure and, in turn, improved properties:
(1) uses high energy pulsed light to photonically synthesize the electrodes almost instantaneously as opposed to the already reported conventional techniques which take several hours,15,18,34–36
(2) allows for the use of inexpensive precursor and substrates,
(3) can be accomplished at room temperature and is amenable to roll to roll processing, and
(4) due to the extremely short irradiation time of the high energy light-pulse, the material can quickly attain high temperatures, reaching the metastable phases without damaging the substrate, which is not possible otherwise.
With the aforementioned advantages, the as-processed electrode has unique and robust highly porous 3D nanostructures that offers high specific surface area and shortened ion diffusion length, advantageous for electrochemical performance and long cycle life, both of which are essential requirements for supercapacitor applications. Since the decrease in capacitance over the time seen elsewhere20,22,23 is mainly due to structural damage caused by lattice expansion during the charge–discharge processes, as prepared porous nanostructures help alleviate the volume changes, leading to the robust structural integrity of the electrode, which in turn results in improved capacitance retention even after a high number of charge discharge cycles. To our knowledge, such a quick processing technique has not been published before for the preparation of mixed metal oxides electrodes. This work provides a promising approach for an easy and fast synthesis of mixed transition metal oxide electrodes, which can potentially be applied in consumer electronics, energy storage devices, electric vehicles, etc. It is well understood that the lifetime of the entire device based on an electrochemical capacitor would be limited by cycle life. Moreover, the device would have to be engineered around any changes in capacitance. Therefore, it is crucial that electrodes have high electrochemical stability and long cycle life. In this regard, by following the aforementioned techniques, we fabricated the nanostructured Mn-Co-rGO electrodes with excellent electrochemical stability and an ultra-long lifetime.
The as-prepared electrodes were evaluated as potential supercapacitor electrodes and showed an initial specific capacitance of 11 mF cm−2 at a constant areal current density of 0.4 mA cm−2. The electrochemical measurements were performed in 1 M KOH as the electrolyte because of its safety, high ionic conductivity, and high mobility K+ ions. We performed charge discharge cycling test for 80000 cycles, and the electrode showed 95% capacitance retention, exhibiting excellent electrochemical stability, and ultra-long cycle life.
2. Experimental section
2.1 Electrode preparation by spray-coating
Organometallic precursors Mn(III) and Co(III) acetylacetonate (acac) powder (Sigma Aldrich) was mixed in two different compositions (Sample A = 80% Mn-acac and 20% Co-acac and Sample B = 50% Mn-acac and 50% Co-acac) and dissolved in 20 mL acetone (Sigma Aldrich) to prepare solution with concentration of 5 wt% and the solution was put in ultrasonication system for 30 minutes. Then the precursor solution was spray-coated on highly conductive Pt-coated silicon substrate (University Wafer, USA) using an air-spray gun (Paasche Airbrush, Chicago) with nitrogen gas at room temperature. After the solvent vaporized, the precursor remained as powder thin film, ready for processing. Fig. 1(a) shows the schematic representation of spray-coating technique.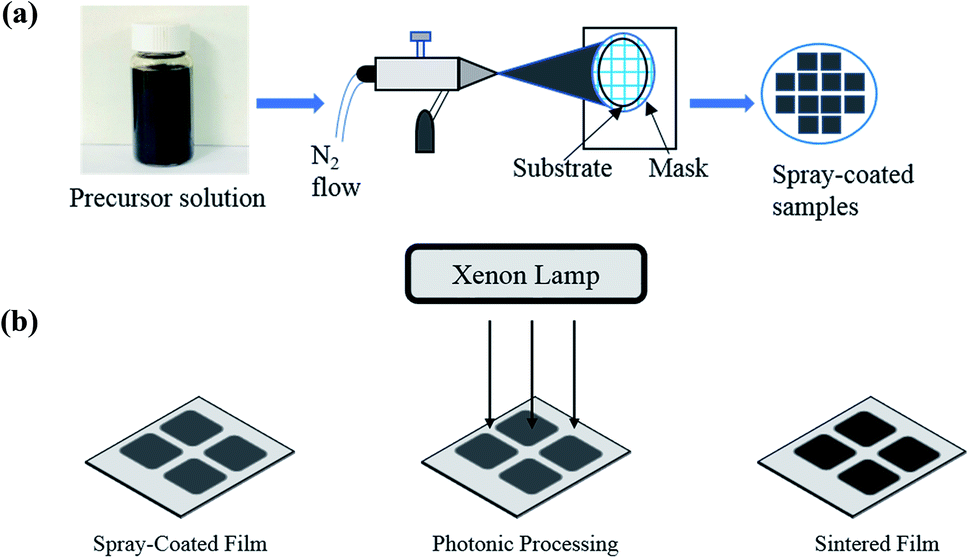 | ||
| Fig. 1 (a) Schematic representation of thin film deposition using an air spray and (b) schematic of the film processing using a photonic curing system. | ||
2.2 Processing of as-sprayed electrodes
Processing of the as-spray-coated powder films was carried out via photonic curing technique using a PulseForge 1300 (NovaCentrix Corp., Austin, Texas) which uses the flashlight from xenon lamp with an output spectrum of 220–1000 nm. The parameters of pulsed light irradiation were controlled with the help of a computer and are shown in Table S1 (see the ESI†). Before the irradiation, the energy density of the light pulse was measured by an in situ integrated bolometer. The film processing was done in ambient conditions and was accomplished in several seconds. Once the irradiation was complete, the powdered and non-conductive precursor films transformed into solid and conductive thin film electrodes. After the photonic processing, the mass loading of the electrodes was calculated to be 0.5 mg. Fig. 1(b) shows the schematic representation of photonic processing technique.2.3 Photonic processing mechanism
Traditional thermal processing techniques rely on exposure to high temperatures for minutes to hours for rapid thermal processing and ovens/furnaces, respectively. We have used a novel photonic processing approach as a next-generation and low-cost production technique to instantaneously synthesize metal oxides electrodes and the mechanism for mixed oxide electrode nucleation and growth is, in a concomitant manner, also novel. It is well known that transition metal acetylacetonate (acac) absorbs visible light making the photonic curing technique a viable approach to process the electrodes as it is mainly based on the absorption of mostly visible photonic energy from the xenon flash lamp by the precursor materials. Because of the extremely short duration of the irradiation and the high intensity (i.e., 0.01 s total and 5.6 J cm−2, respectively), the precursor materials absorb sufficient energy for the curing process to start without damaging the substrate. While the initial irradiation of the pulse train will heat and evaporate the remaining solvent, the subsequent irradiation of the precursor material will absorb sufficient thermal energy to initiate the organic decomposition and mixed oxide local crystallization, reaching a transient condition. With further increase in time/energy, fusing and growth of the mixed oxide atoms occurs to create nanotexture formation. In our past work,37 we have reported a detailed sintering mechanism for pure metal oxides, and we believe that similar process applies for the mixed oxides electrodes as well.2.4 Characterization
The morphology was analyzed by using a field-emission scanning electron microscope (Hitachi S-4800, Hitachi Corp., Tokyo, Japan). The crystal structure was analyzed by using a DXR Raman Microscope (Thermo Scientific) with a 532 nm laser as the excitation source, and X-ray diffraction (XRD) measurement by using a powder X-ray diffractometer (Rigaku Smart Lab, USA) with a Cu Kα radiation at 40 kV voltage and 40 mA current. The composition of the electrode was verified by energy dispersive X-ray spectroscopy (EDX).2.5 Electrochemical measurements
Electrochemical measurements were performed in a three-electrode workstation (Model M204, Autolab, Metroohm, The Netherlands) with the as-prepared Mn-Co-rGO electrode, Hg/HgO and Pt wire as working, reference and counter electrodes, respectively. Cyclic voltammetry (CV), galvanostatic charge–discharge cycling (GCD) and electrochemical impedance spectroscopy (EIS) were performed in 1 M KOH aqueous electrolyte at room temperature (23 °C). The CV curves were recorded in the working potential range of 0.0–0.6 V (vs. Hg/HgO) at various scan rates of 5–100 mV s−1. The GCD measurements were taken at various current densities in the same potential range. The EIS measurements were performed with an AC amplitude of 10 mV over the frequency range from 1 MHz to 10 mHz.3. Results and discussion
3.1 Structural and morphological properties
The surface morphology of the supercapacitor electrodes is important as all the electrochemical reactions take place on or near the surface, and different morphologies have different electrode/electrolyte interface properties as well as ion transfer rates. Therefore, the morphologies of the Mn-Co-rGO (Fig. 2) govern its electrochemical properties. From the SEM images of the electrodes before and after processing, we noticed a significant change in morphology as the dense precursor films (before processing) transformed into a highly conductive and porous nanostructured thin films (after processing) with an average particle size of 50 nm. Such porous morphology is ideal for supercapacitor as it: (a) shortens the ion diffusion length, (b) allows for ions diffusion into the inner pores as well as the bulk of the active material, (c) offers increased mass transport and intercalation and de-intercalation of electrolyte due to the capillary force mechanism, resulting in a lower series resistance, which is crucial for better electrochemical behavior,38,39 and (d) provides a large specific surface area for electrode/electrolyte interaction and prevents aggregation of active material, ensuring enhanced utilization of electrode material. Additionally, highly porous morphology also helps alleviate the volume changes during charge–discharge processes, enhancing the structural integrity of the electrode. From SEM images after the cycling tests (Fig. 2(c and f)) for both the samples, it was observed that the Sample A still retains porous morphology whereas Sample B has relatively dense morphology in comparison to the SEM images before cycling test. The EDX measurements of the electrodes, as shown in Fig. 2(g) and (h), show the elemental composition and their atomic percentages for both the electrodes. Expectedly, the EDX images clearly show that Sample A and B are composed of Mn and Co with ratios of around 4:1 and 1:1, respectively. The peaks around 2 keV are present due to Pt and Si from the substrate.
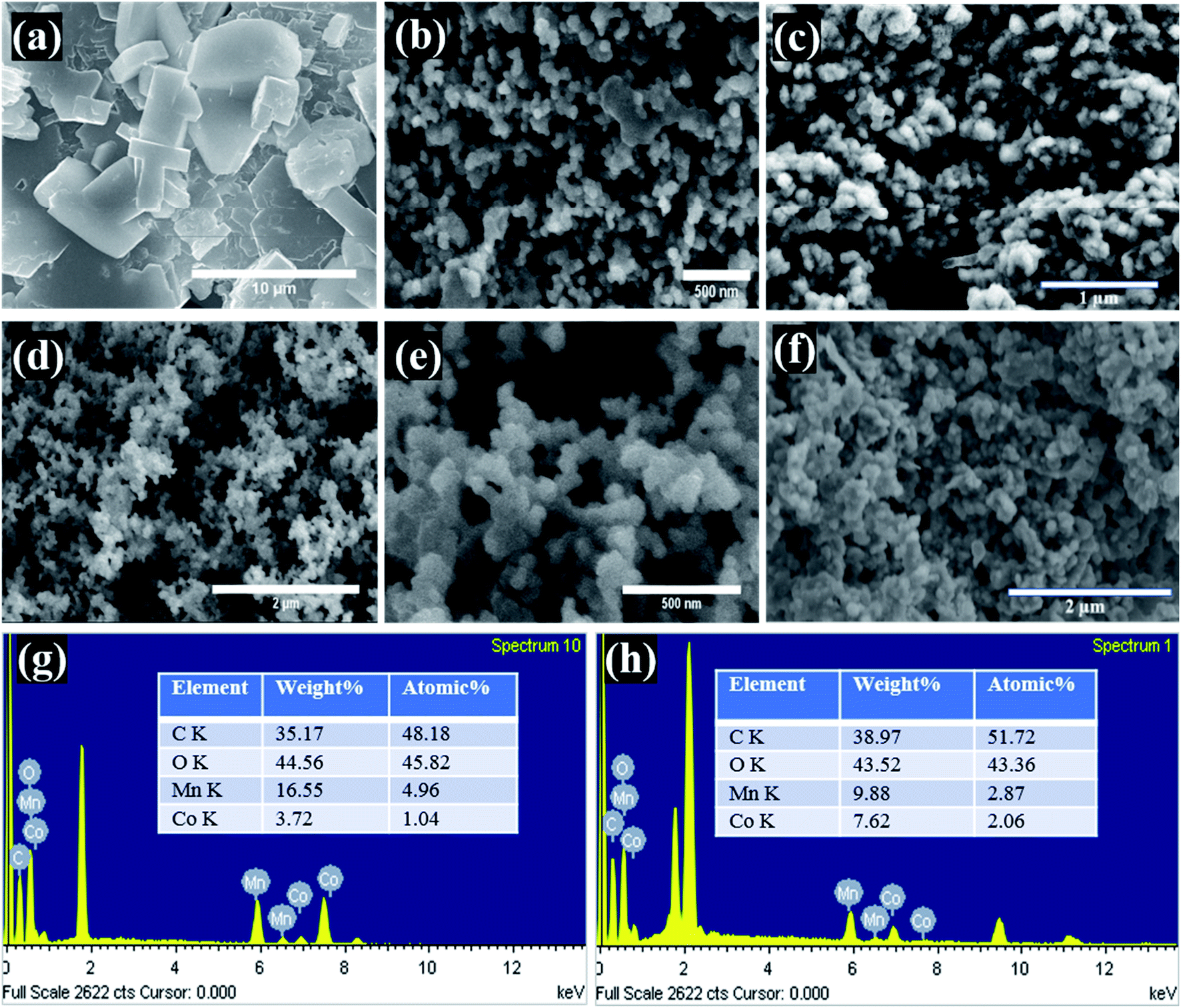 | ||
| Fig. 2 SEM images of (a) precursor film before curing, (b) and (c) Sample A before and after cycling test, respectively, (d and e) and (f) Sample B before and after cycling test, respectively. EDX spectrum of (g) Sample A and (h) Sample B. | ||
The crystalline structure and the chemical composition of the Mn-Co-rGO electrodes was studied by performing Raman spectroscopy and X-ray diffraction spectroscopy, which are shown in Fig. 3. From Fig. 3(a), the peaks at 311 cm−1 and 351 cm−1 are attributed to the out of plane bending modes of Mn–O,40–42 and the peaks at 484 and 653 cm−1 correspond to stretching vibration modes of Mn–O and Co–O, which indicates the existence of MnCo2O4.43 In addition to these four peaks, the spectrum shows distinct D and G peaks centered at 1340 and 1590 cm−1, respectively, which confirms the presence of graphene oxide. While D peak corresponds to the structural defect of the carbon, G peak corresponds to in-plane vibration of doubly degenerate E2g mode phonon sp2 hybridized graphitic carbon.44 The presence of carbon components in the film, as confirmed by Raman and EDX analysis, improves the conductivity and structural stability of the as-prepared electrode, which results in improved electrochemical stability and long cycle life.
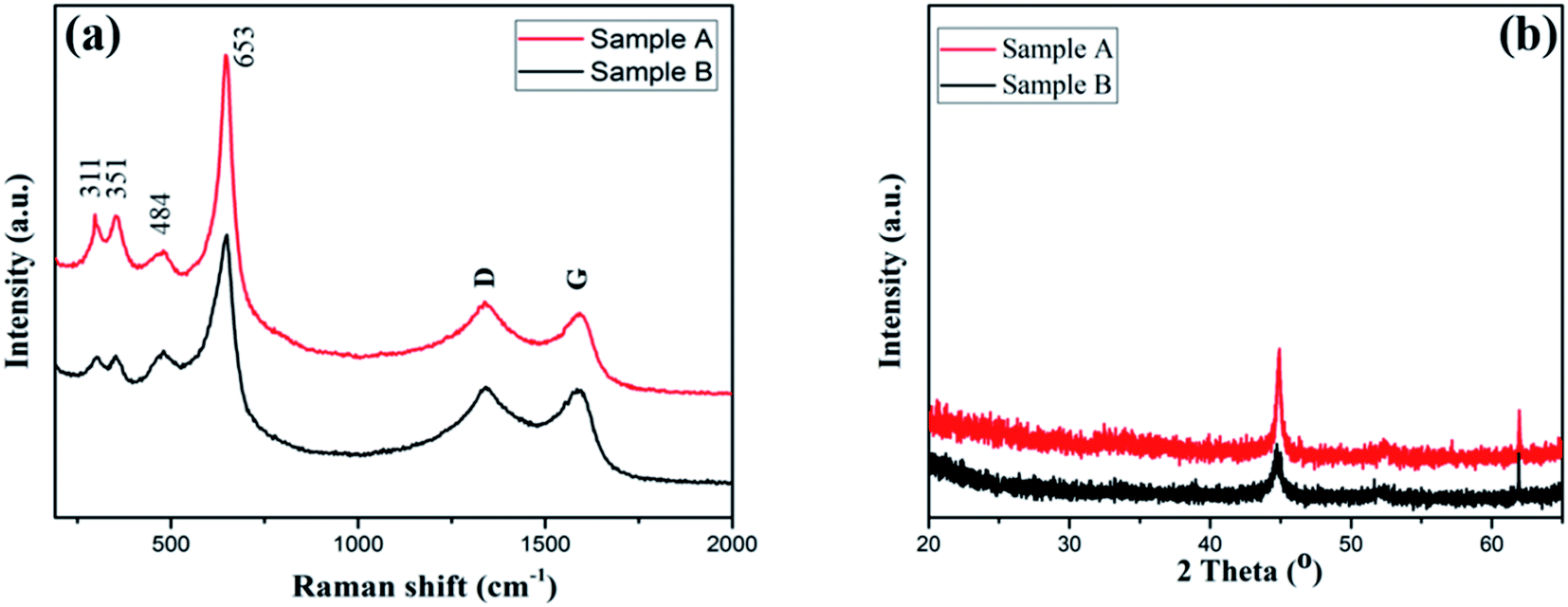 | ||
| Fig. 3 Raman spectra (a) and XRD patterns (b) for the as-prepared Mn-Co-rGO electrodes. | ||
Fig. 3(b) shows the XRD patterns of the as-prepared electrodes. Two characteristics peaks at 45 and 62° were observed for both samples. While the height at 45° is well-indexed with randomly oriented grains of MnCo2O4,45,46 the peak at 62° corresponds to the presence of MnCoOx phase with rock salt like structure.47 Although Sample A with less Mn content showed better electrochemical behavior (discussed later), we did not observe any significant difference in XRD patterns.
3.2 Electrochemical properties
To study the electrochemical performance of the Mn-Co-rGO electrodes, cyclic voltammetry (CV) measurements were performed in the potential window of 0–0.6 V in a three-electrode configuration at room temperature. The CV measurements were recorded at various scan rates, ranging from 5 to 100 mV s−1 in 1 M KOH aqueous electrolyte. To investigate if there was any capacitance contribution from the substrate, the CV curves of the substrate were recorded at various scan rates and after various number of CV cycles, as shown in Fig. S1 (see the ESI†). Since the current density response of the substrate is negligible at all scan rates, it has no contribution to the overall capacitance of the electrode. Fig. 4(a) and (b) represent the CV curves at various scan rates, for Sample A and B, respectively. For both the samples, CV curves show non-rectangular shapes with well-defined characteristic oxidation and reduction peaks within 0.35–0.5 V range indicating pseudocapacitive charge storage mechanism of the electrodes via fast faradaic redox reactions. Additionally, even at high scan rates, the CV curves maintained similar shape without any obvious distortions, exhibiting excellent capacitive behavior and rate capability of the electrodes. The presence of the redox peaks could be attributed to the following chemical reactions between the active material and the electrolyte:17,48| (Co,Mn)3O4 + H2O + OH−1 = CoOOH + MnOOH + e−1 | (1) |
| MnOOH + CoOOH + 2OH−1 = CoO2 + MnO2 + 2H2O + 2e−1 | (2) |
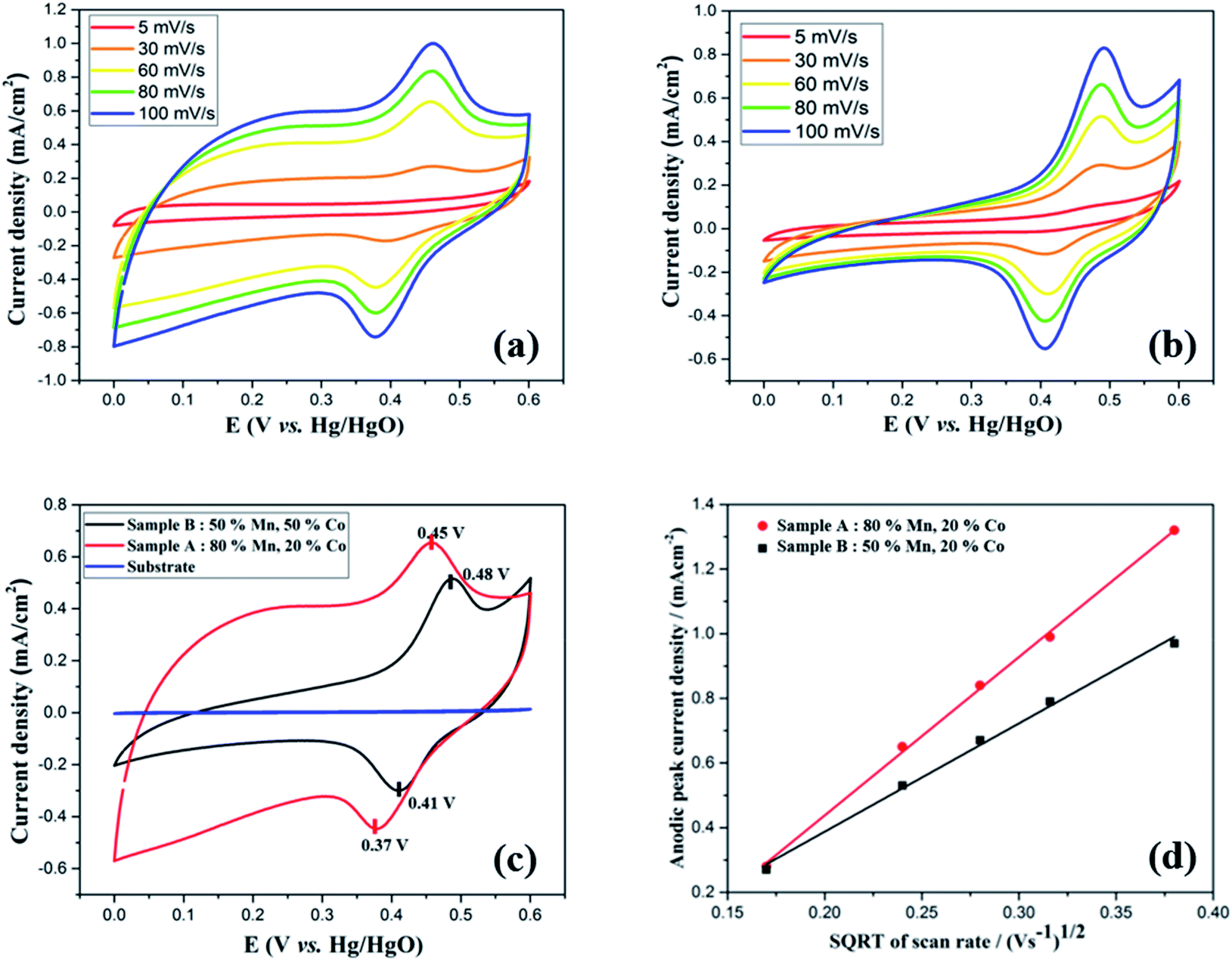 | ||
| Fig. 4 (a and b) CV curves at various scan rates for Sample A and B respectively, (c) CV curves at 60 mV s−1 for both the samples and the substrate, and (d) Randles–Sevcik analysis for both the samples. | ||
Upon increasing the scan rate, redox current and thereby the area of the CV curves increased, suggesting relatively low resistance and good reversibility of the electrode. It is to be noted that oxidation and reduction peaks showed slight shifts towards positive and negative sides, respectively, with increasing scan rates, showing the asymmetric nature of the CV curves. These peak shifts could be attributed to the kinetic irreversibility of the redox processes caused by polarization and an increase in internal resistance at higher scan rates.13,49,50 As obtained CV curves are consistent with the literature16,19,21,22,51 for Mn–Co mixed oxides with a slight difference in the redox peak potentials, which could be attributed to composition and morphology of the electrode. Fig. 4(c) compares the CV curves of both samples and the substrate at 60 mV s−1 scan rate. For both samples, CV curves show similar shapes with a slight difference in redox peak potentials. It is notable that the CV curve of the bare substrate shows a flat line, indicating its negligible electrochemical performance. Interestingly, at the same scan rate, the CV for Sample A shows a larger integrated area than Sample B, signifying the former's higher capacity.
To understand the relation between ion diffusivity and the electron mobility, we performed Randles–Sevcik analysis by plotting anodic peak current densities (ip,a) vs. square root of the scan rate (√v) as shown in Fig. 4(d). Both the samples showed linear relationship between anodic peak current density and the square root of the scan rate, which is in agreement with the Cottrell equation, suggesting the capacitance mainly originates from the diffusion-controlled mechanism taking place throughout the bulk of the electrode material.52,53 The diffusion coefficient (D) of OH− ions for both the samples could be calculated by using the following Randles–Sevcik equation:54–56
| ip = 2.69 × 105 × n3/2 × A × D1/2 × C × v1/2 | (3) |
| D(Sample A)/D(Sample B) = [(Ip/√v)(Sample A)/(Ip/√v)(Sample B)]2 = (4.95/3.34)2 = 2.2 |
From the above calculation, the diffusion coefficient of Sample A was calculated to be 2.2 times that of Sample B, revealing higher ion mobility and better electrolyte penetration of the former.22,55
To further examine the electrochemical properties of the electrodes, galvanostatic charge/discharge (GCD) measurement was performed in the potential range of 0–0.6 V (vs. Hg/HgO) at different areal current densities. To investigate any capacitive contribution from the bare substrate, GCD measurement of the bare substrate was performed. Fig. 5(a) compares the GCD curves of the substrate and Sample A at a constant areal current density of 0.3 mA cm−2, which, in agreement with CV analysis, further confirms that the substrate has no contribution to the overall capacitance of the electrode. Fig. 5(b) and (c) show GCD plots at various current densities for Sample A and B, respectively. At all current densities, the discharge curves show nonlinearity with the presence of a voltage plateau, confirming the pseudocapacitive characteristics of the electrodes, which is consistent with the CV curves. Such nonlinear nature of the discharge curves is attributed to the faradaic redox processes based on voltage-dependent charge transfer reactions.57 At the start of the discharge process, a sudden potential drop, also known as internal resistance (IR) drop, was noticed, which represents the energy losses arising from internal or equivalent series resistance.58 Overall, a discharge curve can be divided into three regions: (i) sudden potential drop region (due to internal resistance), (ii) slow potential decay region (due to pseudocapacitive behavior), and (iii) fast potential drop region (due to EDLC behavior).
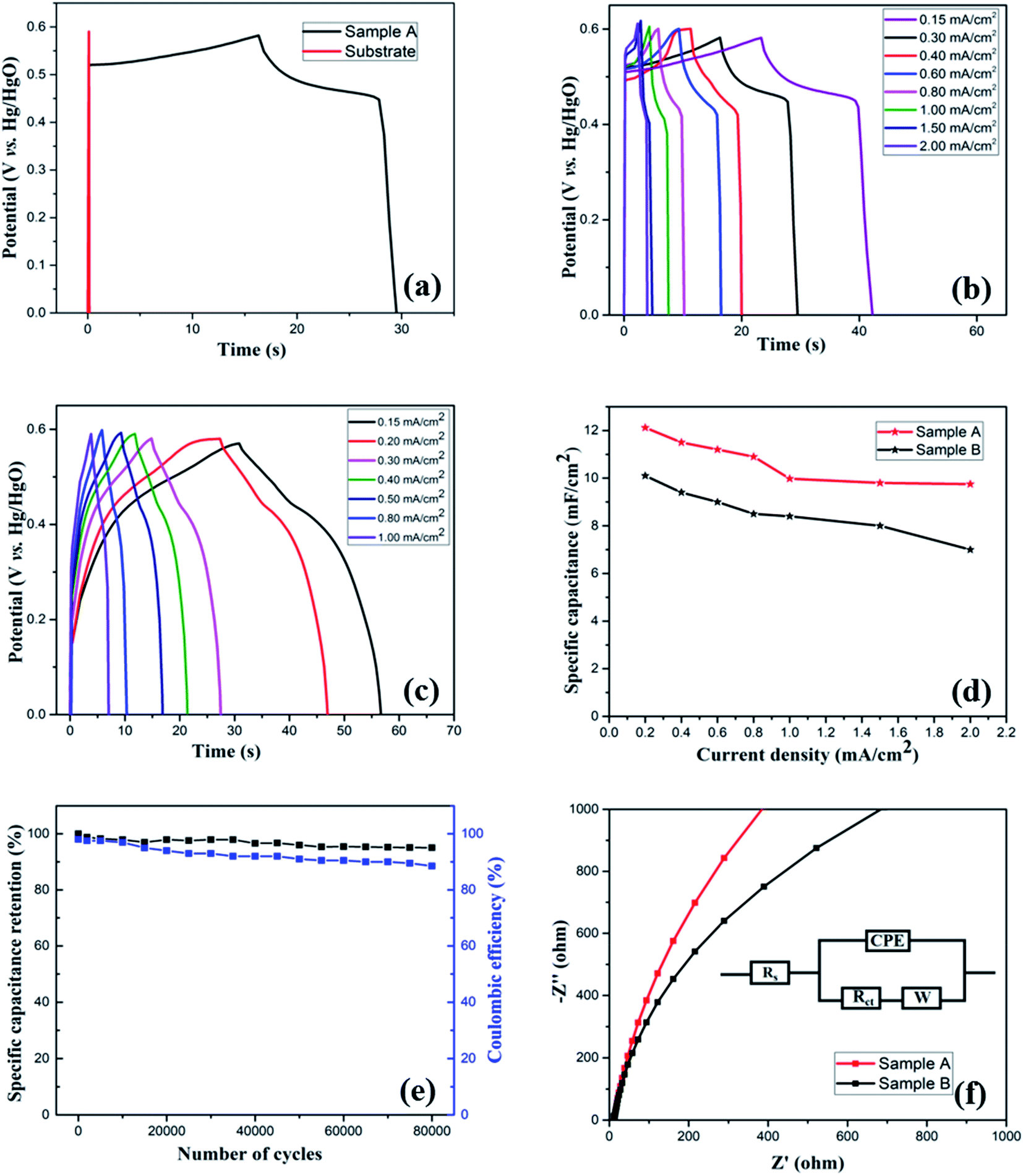 | ||
| Fig. 5 (a) GCD curves for (a) substrate and Sample A at 0.3 mA cm−2 current density, (b and c) Samples A and B, respectively, at various current densities, and (d) rate capability test for both the samples, (e) specific capacitance retention vs. number of cycles for Sample A and (f) EIS analysis for both the samples. | ||
With increasing discharge currents, the IR drop was enhanced, which could be due to high ionic resistance. The slopes of the plots IR drop vs. discharge currents can be used to estimate the resistance of the electrodes: the higher the slope, the greater the total resistance of the electrode.54,59 From Fig. S2 (see the ESI†), we observed that Sample B has a higher overall resistance than Sample A.
It is well known that electrode materials with pseudocapacitive charge storage mechanisms, as represented peak-shaped CV curves and non-linear GCD curves, have capacitances dependent on certain working potential windows.60,61 Therefore, the correct method to calculate the specific capacitance (C) is from the GCD curves by using the following equation:34
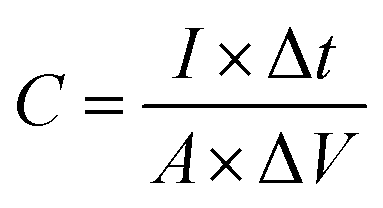 | (4) |
As a crucial parameter for a supercapacitor electrode, rate capability was analyzed by performing the charge–discharge measurement at different areal current densities, as shown in Fig. 5(d). Even after increasing the applied current from 0.4 to 2 mA cm−2, the electrode (Sample A) retained 80% of initial capacitance, indicating its good rate capability. In general, at higher current densities, the electrolyte ions suffer from low diffusion and cannot access the bulk of the active material; therefore, only the surface of the electrode can take part in redox reactions, resulting in decreased effective interaction. At lower current densities, electrolyte ions can interact with the bulk of the electrode material, which leads to an increased rate of intercalation/deintercalation process, resulting in high specific capacitance.
Cyclic stability is a crucial parameter to analyze the performance of the supercapacitor electrodes. It is critical that the electrodes have a long cycle life as lifetime of the entire device based on an electrochemical capacitor would be limited by cycle life. Moreover, the device would have to be engineered around any changes in capacitance. To study the long-term cycling stability of the as-prepared electrodes, we performed continuous GCD measurements at 0.4 mA cm−2 areal current density for 80000 and 30000 cycles, respectively, for Samples A and B. Fig. S3(a) (see the ESI†) shows the trend of areal specific capacitance vs. the number of cycles for both the samples, while Fig. 5(e) shows specific capacitance retention percentage vs. number of cycles for Sample A. Because of the improved conductivity and enhanced stability due to the presence of carbon component and a desirable porous nanostructure, the electrode (Sample A) retains 95% capacitance even after 80000 cycles, indicating excellent electrochemical stability and an ultra-long lifetime. On the other hand, Sample B shows 55% capacitance retention (see the ESI, Fig. S3(b)†) after 30000 cycles.
After a high number of cycles, it is natural for the electrodes to show a decrease in performance. For the as-prepared Sample A, the possible reasons for a slight decrease in the specific capacitance are: (i) poor ion diffusion in the electrolyte, (ii) irreversible faradaic reactions, (iii) degradation of the electrolyte, and (iii) dissolution of active materials into the electrolyte due to long time immersion.
Fig. 5(e) also shows the coulombic efficiency of the electrode (Sample A) after various number of GCD cycles, which was evaluated from the GCD curves by using the following equation:34
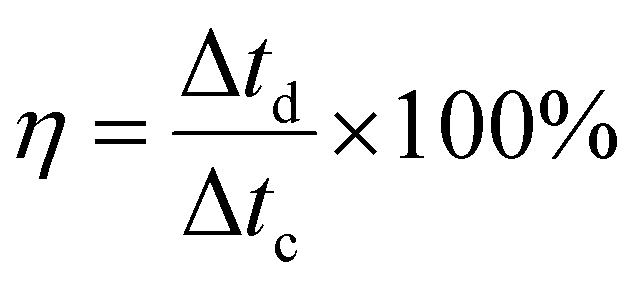 | (5) |
The electrode retained the coulombic efficiency of 90% even after 80000 GCD cycles, which further signifies its excellent kinetic reversibility and cycling stability, contributing to the improvement of the energy utilization.62 We have carried out a significantly higher number of GCD cycles than what is reported in the literature for most of the Mn–Co mixed oxide electrodes.13,17,19,20,48,52,63 The excellent performance of the as-prepared electrode in terms of long-term cycling stability is superior to the Mn–Co oxide supercapacitor electrodes previously reported. Overall, the excellent capacitive performance of the electrodes could be attributed to the following things: (i) synergistic effect of manganese and cobalt ions with rGO, (ii) enhanced conductivity of the electrode, (iii) enhanced specific surface area and shortened ions diffusion due to a highly porous morphology which allows for easy intercalation and de-intercalation of high-mobility K+, which is beneficial for high pseudocapacitance due to its smaller bare ionic size,64 (iv) improved structural integrity as porous structures buffer the volume changes during the redox processes, and (v) enhanced mechanical adhesion and interfacial charge transfer due to the direct growth of the active material on the substrate. These results demonstrate that the as-prepared electrode has the potential for applications in long-term electronics.
To further understand the electrochemical behavior of the Mn-Co-rGO electrodes, electrochemical impedance spectroscopy (EIS) measurements were performed over the frequency range from 1 MHz to 10 mHz by applying 10 mV input signal. Fig. 5(f) shows the EIS curves in the form of Nyquist plots with an equivalent circuit (inset). The presence of a constant phase element (CPE) in the equivalent circuit is due to the redox processes and diffusion of ions through active material. A typical Nyquist plot contains a semicircle in the high-frequency region and a straight-line region in the low-frequency region. The real-axis intercept of the semicircle gives total internal resistance (Rs) which is a combination of the ionic resistance of the electrolyte, intrinsic resistance of the electrode and contact resistance at the electrode/electrolyte interface, while its diameter gives faradaic charge transfer resistance (Rct), resulting from the faradaic reactions and the double layer capacitance on the electrode surface.25,57,62,65 The linear section in low-frequency region is due to diffusion of OH− ions in the active material and is represented by Warburg impedance (W) and the phase angle of the line is indicative of the capacitive behavior as phase angles less than 90° suggest pseudocapacitive behavior.54,66,67 The steeper the slope, the better the capacitive behavior of the electrode, indicating that the diffusion takes place through the bulk of the active material. From this measurement, the series resistance of Sample A and Sample B was calculated to be 10 and 12.85 Ω, respectively. The absence of a semicircle at the high-frequency region in the Nyquist plots is suggestive of a very low or negligible charge transfer resistance of the electrodes, indicating excellent ion transport. The steeper slope for Sample A than for Sample B in the low-frequency region signifies former's excellent capacitance behavior due to the fast diffusion of OH− ions. This could also be another factor contributing to the superior electrochemical performance of Sample A.
From the above analysis and characterization, we observed that Sample A showed significantly better performance than Sample B, in terms of both the life cycle and specific capacitance retention. This could be due to: (a) optimum composition of manganese and cobalt, resulting in better electrochemical activity than for Sample B, which can be seen in Fig. 4(c) as CV curve for Sample A shows significantly higher integrated area than that of Sample B, (b) higher diffusion coefficient D, which means higher OH− ions mobility and better electrolyte penetration than Sample B, (c) lower overall resistance of the electrode as shown in the Fig. S2 (see ESI†), and (d) better capacitive behavior, as signified by the steeper slope of the EIS curve at the low-frequency region than Sample B.
4. Conclusions
In summary, we have presented an easy and highly scalable technique to spray coat the precursor thin films and process them almost instantaneously to prepare binder-free and highly porous 3D Mn-Co-rGO composite nanostructure. By using a novel photonic curing technique, we have significantly reduced the processing time from hours (in case of conventional methods) to just seconds. The initial specific capacitance of the as-prepared binder-free Mn-Co-rGO electrode was calculated to be 11 mF cm−2 at a constant current density of 0.4 mA cm−2. After 80000 GCD cycles, which is significantly higher than what is reported for the same material, the electrode retains 95% of the initial capacitance, exhibiting excellent electrochemical stability and ultra-long cycle life. Such excellent performance of the electrode could be attributed to the optimized processing parameters, synergistic effect of manganese and cobalt ions with the carbon component, unique and highly porous nanostructure, enhanced electronic conductivity and structural integrity. The easy and quick thin film electrode fabrication method discussed here can be followed to fabricate and optimize other promising nanostructured-binary and ternary metal oxide electrodes for their potential application as supercapacitor electrodes
Conflicts of interest
There are no conflicts to declare.References
- G. Zhang and X. W. D. Lou, Adv. Mater., 2013, 25(7), 976–979, DOI:10.1002/adma.201370044.
- L. Liu, J. Lang, P. Zhang, B. Hu and X. Yan, ACS Appl. Mater. Interfaces, 2016, 8(14), 9335–9344, DOI:10.1021/acsami.6b00225.
- P. Yang and W. Mai, Nano Energy, 2014, 8, 274–290 CrossRef CAS.
- L. Zhu, Z. Chang, Y. Wang, B. Chen, Y. Zhu, W. Tang and Y. Wu, J. Mater. Chem. A, 2015, 3(44), 22066–22072, 10.1039/c5ta05556c.
- X. C. Dong, H. Xu, X. W. Wang, Y. X. Huang, M. B. Chan-Park, H. Zhang, L. H. Wang, W. Huang and P. Chen, ACS Nano, 2012, 6(4), 3206–3213, DOI:10.1021/nn300097q.
- Z. Yu, B. Duong, D. Abbitt and J. Thomas, Adv. Mater., 2013, 25(24), 3302–3306, DOI:10.1002/adma.201300572.
- C. Yuan, H. Bin Wu, Y. Xie and X. W. Lou, Angew. Chem. Int. Ed., 2014, 53(6), 1488–1504 CrossRef CAS.
- D. Guo, P. Zhang, H. Zhang, X. Yu, J. Zhu, Q. Li and T. Wang, J. Mater. Chem. A, 2013, 1(32), 9024–9027, 10.1039/c3ta11487b.
- Y. Zhang, L. Li, H. Su, W. Huang and X. Dong, J. Mater. Chem. A, 2015, 3(1), 43–59 RSC.
- J. Zhang, F. Liu, J. P. Cheng and X. B. Zhang, ACS Appl. Mater. Interfaces, 2015, 7(32), 17630–17640, DOI:10.1021/acsami.5b04463.
- R. Wang and X. Yan, Sci. Rep., 2014, 4, 3712, DOI:10.1038/srep03712.
- H. Bin Wu, H. Pang and X. W. Lou, Energy Environ. Sci., 2013, 6(12), 3619–3626, 10.1039/c3ee42101e.
- S. Jiang, T. Shi, H. Long, Y. Sun, W. Zhou and Z. Tang, Nanoscale Res. Lett., 2014, 9(1), 492, DOI:10.1186/1556-276X-9-492.
- X. Wang, C. Yan, A. Sumboja and P. S. Lee, Nano Energy, 2014, 3, 119–126, DOI:10.1016/j.nanoen.2013.11.001.
- J. Du, G. Zhou, H. Zhang, C. Cheng, J. Ma, W. Wei, L. Chen and T. Wang, ACS Appl. Mater. Interfaces, 2013, 5(15), 7405–7409, DOI:10.1021/am4017335.
- J. Liu, J. Jiang, C. Cheng, H. Li, J. Zhang, H. Gong and H. J. Fan, Adv. Mater., 2011, 23(18), 2076–2081, DOI:10.1002/adma.201100058.
- S. H. Kim, Y. Il Kim, J. H. Park and J. M. Ko, Int. J. Electrochem. Sci, 2009, 4, 1489–1496 CAS.
- J. Chen, Y. Cui, X. Wang, M. Zhi, M. Lavorgna, A. P. Baker and J. Wu, Electrochim. Acta, 2016, 188, 704–709, DOI:10.1016/j.electacta.2015.12.052.
- F. Yunyun, L. Xu, Z. Wankun, Z. Yuxuan, Y. Yunhan, Q. Honglin, X. Xuetang and W. Fan, Appl. Surf. Sci., 2015, 357, 2013–2021, DOI:10.1016/j.apsusc.2015.09.176.
- Q. Tian, X. Wang, G. Huang and X. Guo, Nanoscale Res. Lett., 2017, 12(1), 214, DOI:10.1186/s11671-017-1977-0.
- D. Yang, J. Power Sources, 2012, 198, 416–422, DOI:10.1016/j.jpowsour.2011.10.008.
- S. Liu and S. C. Jun, J. Power Sources, 2017, 342, 629–637, DOI:10.1016/j.jpowsour.2016.12.057.
- M. Huang, Y. Zhang, F. Li, L. Zhang, Z. Wen and Q. Liu, J. Power Sources, 2014, 252, 98–106, DOI:10.1016/j.jpowsour.2013.12.030.
- C. Guan, X. Qian, X. Wang, Y. Cao, Q. Zhang, A. Li and J. Wang, Nanotechnology, 2015, 26(9), 094001, DOI:10.1088/0957-4484/26/9/094001.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48(13), 3825–3833, DOI:10.1016/j.carbon.2010.06.047.
- L. Bao, J. Zang and X. Li, Nano Lett., 2011, 11(3), 1215–1220, DOI:10.1021/nl104205s.
- Y. He, W. Chen, X. Li, Z. Zhang, J. Fu, C. Zhao and E. Xie, ACS Nano, 2013, 7(1), 174–182, DOI:10.1021/nn304833s.
- G. Yu, L. Hu, N. Liu, H. Wang, M. Vosgueritchian, Y. Yang, Y. Cui and Z. Bao, Nano Lett., 2011, 11(10), 4438–4442, DOI:10.1021/nl2026635.
- M. Zhi, C. Xiang, J. Li, M. Li and N. Wu, Nanoscale, 2013, 5(1), 72–88, 10.1039/c2nr32040a.
- S. Bose, T. Kuila, A. K. Mishra, R. Rajasekar, N. H. Kim and J. H. Lee, J. Mater. Chem., 2012, 22(3), 767–784 RSC.
- L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38(9), 2520–2531, 10.1039/b813846j.
- L. Shen, Q. Che, H. Li and X. Zhang, Adv. Funct. Mater., 2014, 24(18), 2630–2637, DOI:10.1002/adfm.201303138.
- G. Q. Zhang, H. Bin Wu, H. E. Hoster, M. B. Chan-Park and X. W. Lou, Energy Environ. Sci., 2012, 5(11), 9453–9456, 10.1039/c2ee22572g.
- W. W. Wang, Mater. Chem. Phys., 2008, 108(2–3), 227–231, DOI:10.1016/j.matchemphys.2007.09.022.
- J. Fan, S. W. Boettcher and G. D. Stucky, Chem. Mater., 2006, 18(26), 6391–6396, DOI:10.1021/cm062359d.
- C. Yuan, J. Li, L. Hou, J. Lin, X. Zhang and S. Xiong, J. Mater. Chem. A, 2013, 1(37), 11145–11151, 10.1039/c3ta11949a.
- M. Gaire, K. Liang, S. Luo, B. Subedi, S. Adireddy, K. Schroder, S. Farnsworth and D. B. Chrisey, RSC Adv., 2020, 10(28), 16817–16825, 10.1039/d0ra01081b.
- P. Iamprasertkun, A. Krittayavathananon, A. Seubsai, N. Chanlek, P. Kidkhunthod, W. Sangthong, S. Maensiri, R. Yimnirun, S. Nilmoung, P. Pannopard, S. Ittisanronnachai, K. Kongpatpanich, J. Limtrakul and M. Sawangphruk, Sci. Rep., 2016, 6, 1–12, DOI:10.1038/srep37560.
- X. Lu, D. Zheng, T. Zhai, Z. Liu, Y. Huang, S. Xie and Y. Tong, Energy Environ. Sci., 2011, 4(8), 2915–2921, 10.1039/c1ee01338f.
- F. Xiao and Y. Xu, Int. J. Electrochem. Sci, 2012, 7, 7440–7450 CAS.
- C. M. Julien, M. Massot and C. Poinsignon, Spectrochim. Acta Mol. Biomol. Spectrosc., 2004, 60(3), 689–700, DOI:10.1016/S1386-1425(03)00279-8.
- J. Jiang and A. Kucernak, Electrochim. Acta, 2002, 47(15), 2381–2386, DOI:10.1016/S0013-4686(02)00031-2.
- C. Sun, J. Yang, Z. Dai, X. Wang, Y. Zhang, L. Li, P. Chen, W. Huang and X. Dong, Nano Res., 2016, 9(5), 1300–1309, DOI:10.1007/s12274-016-1025-x.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97(18), 187401, DOI:10.1103/PhysRevLett.97.187401.
- D. Wang, Q. Zhang, K. Zhou, W. Yang, Y. Hu and X. Gong, J. Hazard. Mater., 2014, 278, 391–400, DOI:10.1016/j.jhazmat.2014.05.072.
- Y. Liang, H. Wang, J. Zhou, Y. Li, J. Wang, T. Regier and H. Dai, J. Am. Chem. Soc., 2012, 134(7), 3517–3523, DOI:10.1021/ja210924t.
- M. Gliech, A. Bergmann, C. Spöri and P. Strasser, J. Energy Chem., 2016, 25(2), 278–281, DOI:10.1016/j.jechem.2016.01.002.
- P. Xu, K. Ye, D. Cao, J. Huang, T. Liu, K. Cheng, J. Yin and G. Wang, J. Power Sources, 2014, 268, 204–211, DOI:10.1016/j.jpowsour.2014.06.014.
- W. Li, B. Zhang, R. Lin, S. M. Ho-Kimura, G. He, X. Zhou, J. Hu and I. P. Parkin, Adv. Funct. Mater., 2018, 28(23), 1705937, DOI:10.1002/adfm.201705937.
- X. Wang, X. Liu, H. Rong, Y. Song, H. Wen and Q. Liu, RSC Adv., 2017, 7(47), 29611–29617 RSC.
- Y. Guo, C. Wu, N. W. Li, S. Yuan and L. Yu, J. Mater. Chem. A, 2019, 7(44), 25247–25253, 10.1039/c9ta05790k.
- A. N. Naveen, P. Manimaran and S. Selladurai, J. Mater. Sci. Mater. Electron., 2015, 26(11), 8988–9000, DOI:10.1007/s10854-015-3582-2.
- G. Zeng, Y. Chen, L. Chen, P. Xiong and M. Wei, Electrochim. Acta, 2016, 222, 773–780, DOI:10.1016/j.electacta.2016.11.035.
- S. G. Krishnan, M. V. Reddy, M. Harilal, B. Vidyadharan, I. I. Misnon, M. H. A. Rahim, J. Ismail and R. Jose, Electrochim. Acta, 2015, 161, 312–321, DOI:10.1016/j.electacta.2015.02.081.
- K. Wang, J. Yang, J. Zhu, L. Li, Y. Liu, C. Zhang and T. Liu, J. Mater. Chem. A, 2017, 5(22), 11236–11245, 10.1039/c7ta01457k.
- J. Shi, X. Li, G. He, L. Zhang and M. Li, J. Mater. Chem. A, 2015, 3(41), 20619–20626, 10.1039/c5ta04464b.
- G. S. Gund, D. P. Dubal, N. R. Chodankar, J. Y. Cho, P. Gomez-Romero, C. Park and C. D. Lokhande, Sci. Rep., 2015, 5, 12454, DOI:10.1038/srep12454.
- B. Bhujun, M. T. T. Tan and A. S. Shanmugam, Results Phys., 2017, 7, 345–353, DOI:10.1016/j.rinp.2016.04.010.
- A. Daraghmeh, S. Hussain, L. Servera, E. Xuriguera, A. Cornet and A. Cirera, Electrochim. Acta, 2017, 245, 531–538, DOI:10.1016/j.electacta.2017.05.186.
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45(21), 5925–5950 RSC.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3(9), 1294–1301 RSC.
- W. Yang, Z. Gao, J. Ma, X. Zhang, J. Wang and J. Liu, J. Mater. Chem. A, 2014, 2(5), 1448–1457, 10.1039/c3ta14488g.
- F. Li, G. Li, H. Chen, J. Q. Jia, F. Dong, Y. B. Hu, Z. G. Shang and Y. X. Zhang, J. Power Sources, 2015, 296, 86–91, DOI:10.1016/j.jpowsour.2015.07.029.
- E. J. Lee, L. Lee, M. A. Abbas and J. H. Bang, Phys. Chem. Chem. Phys., 2017, 19(31), 21140–21151, 10.1039/c7cp03546b.
- M. S. Wu and H. H. Hsieh, Electrochim. Acta, 2008, 53(8), 3427–3435, DOI:10.1016/j.electacta.2007.12.005.
- Y. Lei, J. Li, Y. Wang, L. Gu, Y. Chang, H. Yuan and D. Xiao, ACS Appl. Mater. Interfaces, 2014, 6(3), 1773–1780, DOI:10.1021/am404765y.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7(5), 1597–1614 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08510c |
| This journal is © The Royal Society of Chemistry 2020 |