
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of SrTiO3 submicron cubes with simultaneous and competitive photocatalytic activity for H2O splitting and CO2 reduction†
Haoshan Weiab,
Jingyi Caiab,
Yong Zhang *ab,
Xueru Zhangc,
Elena A. Baranovade,
Jiewu Cuiab,
Yan Wangab,
Xia Shuab,
Yongqiang Qinab,
Jiaqin Liubf and
Yucheng Wu*ab
*ab,
Xueru Zhangc,
Elena A. Baranovade,
Jiewu Cuiab,
Yan Wangab,
Xia Shuab,
Yongqiang Qinab,
Jiaqin Liubf and
Yucheng Wu*ab
aSchool of Materials Science and Engineering, Hefei University of Technology, Hefei 230009, Anhui, China. E-mail: zhangyong.mse@hfut.edu.cn; ycwu@hfut.edu.cn
bKey Laboratory of Advanced Functional Materials and Devices of Anhui Province, Hefei 230009, Anhui, China
cInstrumental Analysis Center, Hefei University of Technology, Hefei 230009, China
dChina International S&T Cooperation Base for Advanced Energy and Environmental Materials, Hefei 230009, Anhui, China
eDepartment of Chemical and Biological Engineering, Centre for Catalysis Research and Innovation (CCRI), University of Ottawa, 161 Louis-Pasteur, Ottawa, ON K1N 6N5, Canada
fInstitute of Industry & Equipment Technology, Hefei University of Technology, Hefei 230009, Anhui, China
First published on 24th November 2020
Abstract
Single crystalline strontium titanate (SrTiO3) submicron cubes have been synthesized based on a molten salt method. The submicron cubes showed superior photocatalytic activity towards both water splitting and carbon dioxide reduction, in which methane (CH4) and hydrogen (H2) were simultaneously produced. The average production rate of methane up to 8 h is 4.39 μmol g−1 h−1 but drops to 0.46 μmol g−1 h−1. However, the average production rate of hydrogen is 14.52 before 8 h but then increases to 120.23 μmol g−1 h−1 after 8 h. The rate change of the two processes confirms the competition between the H2O splitting and CO2 reduction reactions. Band structure and surface characteristics of the SrTiO3 submicron cubes were characterized by diffuse reflective UV-Vis spectroscopy, Mott–Schottky analysis, X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared spectroscopy (FTIR). The results reveal that the simultaneous and competitive production of methane and hydrogen is due to a thermodynamics factor, as well as the competition between the adsorption of carbon dioxide and water molecules on the surface of the faceted SrTiO3. This work demonstrates that SrTiO3 photocatalysts are efficient in producing sustainable fuels via water splitting and carbon dioxide reduction reactions.
Introduction
Fossil fuel depletion and increased anthropogenic CO2 emission have been identified as two future challenges triggering global economic and environmental problems.1,2 Therefore, the development of sustainable energy resources is both essential and urgent. By utilizing the abundant solar energy, it is possible to split H2O or/and reduce CO2 using photocatalysts.3,4 This process produces hydrogen or/and other value-added chemicals from CO2, as well as simultaneously addressing environmental issues.5–8Perovskite-type oxides are the materials with the general formula ABO3, in which the larger A cation is coordinated to 12 anions and the B cation occupies 6 coordinated sites forming a network of corner-sharing BO6 octahedra.9 The site of A can be larger cations of rare-earth elements – alkaline earth or alkali, whereas the smaller B cations include transition metals.10 The distortions present in the crystal structure are determined by the ionic radii and valence of cations, which are responsible for the chemical and physical properties of the materials.10 Due to their broad adaptability, perovskite-type materials have been extensively studied and used for a wide range of applications as catalytic, oxygen transport, ferroelectric, piezoelectric, and dielectric materials.11–14 Recently, the photocatalytic water splitting and CO2 reduction properties have been explored in some typical perovskite-type materials.15–20 For example, BaTiO3 nanoparticles synthesized at room temperature and ambient pressure showed 7 and 1.9 times higher photocatalytic hydrogen evolution than the commercial BaTiO3 in the presence and absence of TEOA as a sacrificial agent, respectively.21 Beata's group reported that silver deposited on the surface of LiNbO3 in the form of Ag2O was responsible for the enhanced photocatalytic activity in the studied reaction.22 Balasubramanian et al. reported photocatalytic reduction of CO2 by water under UV-visible light over La modified NaTaO3, which showed a stable performance for up to 20 hours.23
SrTiO3 has a theoretical band gap width of 3.24 eV (ref. 24 and 25) in which both the conduction and the valence band potential satisfy the thermodynamic conditions of photocatalytic water splitting and photocatalytic reduction of carbon dioxide.26 This makes SrTiO3 a good candidate for various photocatalytic processes.27,28 However, the pure SrTiO3 suffers from a low utilization of visible light, because it only responds in the ultraviolet region, difficulty in recovery, complex preparation methodology and low photocatalytic activity due to the easy recombination of photogenerated electrons and holes.29
Many efforts have been done to improve chemical and physical properties of SrTiO3 by altering its composition through additional oxygen vacancies and element doping.30–32 For example, Ye et al. prepared a Ti3+ self-doped SrTiO3−δ photocatalyst with enhanced activity for artificial photosynthesis under visible light by oxygen vacancy accommodated in perovskite.33 Another work investigated the influence of N, S and Fe doping on photocatalytic CO2 reduction performance of strontium titanates.34 Up to date, there are very few reports that investigate the simultaneous water splitting and CO2 reduction and their competitive nature over SrTiO3 nano- or submicron cubes. It is known that pure CH4 has characteristics of low flame propagation speed, small flammable limit range, and low thermal efficiency, resulting in low combustion efficiency and large fuel consumption. However, the mixture gas of H2 to CH4 has the advantages of increasing the flammability limit, speeding up the spread of flames, and reducing the emission of pollutants.35 The simultaneous CO2 conversion and water splitting through photocatalysts can provide a simple and energy-saving method for obtaining the mixed gas of CH4 and H2. Moreover, the selectivity of photocatalysts to produce mixed gas with different mixing ratios deserves more attention which is of great significance for unveiling the origin of catalytic selectivity as well as finding great potential applications. The unveiling the origin of the crystal facets of the nano- or submicron on the catalyst performance is a key question for the efficient synthesis of useful chemicals.
In this work, we report the preparation of perovskite strontium titanate submicron cubes photocatalysts by the molten salt synthesis method, which is one of the attractive techniques with merits of one-step, facile, time-saving, and versatile.36 Photocatalysis measurements suggest that methane and hydrogen are simultaneously and competitively produced on the SrTiO3 submicron cubes surface during photocatalytic reduction of carbon dioxide with water. The competitive relationship between the two reactions is investigated and the reaction mechanism is proposed.
Experimental
Chemicals and materials
SrCO3 (AR, 99.8%), H2PtCl6·6H2O (AR, Pt 37.5%), triethanolamine and ethanol (AR, 99.5%) were purchased from Sinopharm Chemical Reagent Company. P25 TiO2 (AR, 99.8%), NaCl (AR, 99.5%) and KCl (AR, 99.5%) were purchased from Aladdin Reagent Company. All chemicals were used as received without any further purification.Synthesis of SrTiO3 submicron cubes
SrTiO3 submicron cubes were prepared by the molten salt synthesis method as reported previously.37 In the typical synthesis of SrTiO3 submicron cubes, SrCO3, TiO2, NaCl and KCl were mixed in an overall stoichiometric ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1: 50:50. Then the mixture was put into the mortar and grinded thoroughly for 30 min. Next, the mixture was put into an alumina crucible and then heated to 700 °C at a rate of 5 °C min−1 in a muffle furnace and maintained at this temperature for 5 h. After cooling down to room temperature, the sample was washed with deionized water (18.25 MΩ cm) several times to remove NaCl and KCl. Then washed with diluted hydrochloric acid (0.05 M) to remove unreacted impurity of SrCO3. Finally, dried in an oven at 60 °C for 12 h.
:1: 50:50. Then the mixture was put into the mortar and grinded thoroughly for 30 min. Next, the mixture was put into an alumina crucible and then heated to 700 °C at a rate of 5 °C min−1 in a muffle furnace and maintained at this temperature for 5 h. After cooling down to room temperature, the sample was washed with deionized water (18.25 MΩ cm) several times to remove NaCl and KCl. Then washed with diluted hydrochloric acid (0.05 M) to remove unreacted impurity of SrCO3. Finally, dried in an oven at 60 °C for 12 h.
Preparation of Pt nanoparticles loaded SrTiO3 submicron cubes
Pt nanoparticles loaded SrTiO3 submicron cubes were prepared in a traditional method of photodeposition.38 In a typical experiment, 100 mg as-prepared SrTiO3 submicron cubes sample was dispersed in 100 mL of deionized water containing 20 mL of triethanolamine (TEOA) by sonication for 10 min. The prepared solution H2PtCl6 (20 mmol L−1) were added into the above dispersion solution with the mass ratio of Pt/SrTiO3 3%. The solution was then sealed and evacuated with a vacuum pump to remove oxygen from the solution and stirred in dark for SrTiO3 submicron cubes fully adsorbing metal ions. After 30 min, the solution was irradiated from the top by a 300 W Xe lamp (PLS-SXE 300, Beijing Trusttech Co. Ltd, China) with simulated sunlight for 4 h. The sample was collected by centrifugation and washed with deionized water and ethanol. Then, dried in a vacuum drying oven oven at 60 °C for 12 h. The as-obtained samples were marked as STO-Pt.Physicochemical characterization
The X-ray diffraction (XRD, 2θ = 3° min−1, 2 h = 10–90°, D/MAX2500V, Rigaku Corporation, Japan) analyses were performed to characterize the crystal structure. The microstructure of the SrTiO3 nanocrystal was investigated by scanning electron microscopy (SEM, SU8020, HITACHI, Ltd., Japan). The valence states of the elements in the SrTiO3 nanocrystals were analyzed using an ESCALAB250 spectrometer equipped with an Al Ka (1486.6 eV) radiation source. Transmission electron microscopy (TEM) analysis with selected-area diffraction was carried out on a JEOL 2100F field-emission transmission electron microscope operated at 200 kV. The UV-Vis absorption spectra were recorded on a UV-3600 UV-Vis-NIR scanning spectrophotometer (Shimadzu).Photocatalytic activity measurements
50 mg of photocatalyst STO-Pt was placed into an aqueous triethanolamine (TEOA) solution (100 mL, 10 vol%) in a closed gas circulation system (Perfect Light Company Labsolar-III (AG)). The CO2 (99.99%) gas flow rate into the reactor was controlled at 10 mL min−1 for 45 min using a mass flow controller (D08-3F, Sevenstar, China). UV-Vis light and visible light irradiation were obtained from a 300 W Xe lamp (PLS-SXE 300, Beijing Trusttech Co. Ltd, China) with Simulated sunlight. The evolved gases were detected in situ using an online gas chromatograph (GC-7890B, Agilent) equipped with a thermal conductivity detector (TCD) and flame ionization detector (FID). In addition, a control experiment of water splitting was carried out under the same conditions but in the absence of carbon dioxide.Mott–Schottky measurements
SrTiO3 thin film photoelectrodes were prepared by electrophoretic deposition on the FTO substrate as reported previously.39 In a typical process, FTO glasses (3 cm × 1 cm × 0.22 cm) were ultrasonically cleaned sequentially with acetone, ethanol and water for 30 minutes each. Electrophoretic deposition was carried out in 70 mL acetone solution containing 30 mg iodine. The FTO electrode, with 1 cm2 deposition area immersed into the solution, was parallel to the Pt electrode. The distance between two electrodes was 80 mm. A 40 V bias was applied for 3 min between two electrodes using a potentiostat (ITECH IT6834). The deposited SrTiO3 electrodes were dried in the air at room temperature and subjected to calcination at 350 °C for 30 min. Mott–Schottky experiments were carried out in a three-electrode electrochemical setup in 0.5 mol L−1 Na2SO4 electrolyte. The deposited SrTiO3 electrode, the platinum electrode and silver/silver chloride electrode (Ag/AgCl) were used as a working, counter and reference electrode, respectively.Results and discussion
The crystallite structure of the as-prepared sample was characterized by X-ray diffraction. Fig. 1 shows strong peaks are very similar to purchased SrTiO3 commercial powders, which correspond to cubic SrTiO3 phase (JCPDS no. 86-0179) with space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (221). No additional diffraction peaks are observed, suggesting the high purity of the obtained SrTiO3 crystals. Fig. S1† presents the XRD Rietveld refinement patterns of the SrTiO3 crystal. The refinement of the sample yields χ2, wRp and Rp values of 1.817, 6.54% and 5.8%, respectively, indicating close agreements with the experimental data. The crystal cell parameter of the prepared SrTiO3 is 3.10931 (15) Å, and the unit cell volume is 59.379 (7) Å3. Based on the experimental and calculated results, it was confirmed that the synthesized sample has a perovskite structure.
m (221). No additional diffraction peaks are observed, suggesting the high purity of the obtained SrTiO3 crystals. Fig. S1† presents the XRD Rietveld refinement patterns of the SrTiO3 crystal. The refinement of the sample yields χ2, wRp and Rp values of 1.817, 6.54% and 5.8%, respectively, indicating close agreements with the experimental data. The crystal cell parameter of the prepared SrTiO3 is 3.10931 (15) Å, and the unit cell volume is 59.379 (7) Å3. Based on the experimental and calculated results, it was confirmed that the synthesized sample has a perovskite structure.
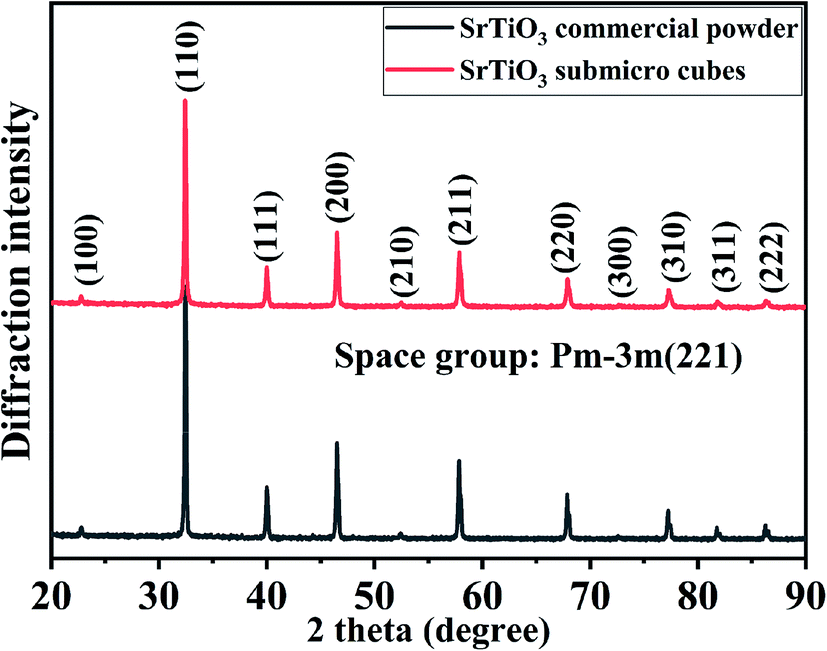 | ||
| Fig. 1 XRD pattern of the as-prepared SrTiO3 synthesized by molten salt method. | ||
The morphology of the SrTiO3 sample was observed by the scanning electron microscopy (SEM). Fig. 2(a) and (b) show that as-synthesized SrTiO3 sample has a cubic shape. Fig. 2(c) and (d) shows the particle size distribution and high resolution TEM images of SrTiO3, respectively. TEM images reveal that the side average length of the cubes is about 79 nm, which have been shown in Fig. S2.† The grain size calculated by the Scherrer's formula, D = Kγ/Bcosθ, is 83 nm. These two values are very similar. So, it is proved that the prepared cubes have single crystal nature with a clear lattice fringe spacing of 0.28 nm in the direction of (110) and (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) and a lattice fringe spacing of 0.19 nm for (200) and (020). In addition, HRTEM characterization shown in Fig. 2(d) suggests the faceted shape of the cubic particle, which is enclosed with (200) planes at the edge and planes at the corner. The single crystal nature of SrTiO3 cubes can be confirmed by the selected area electron diffraction (SAED) pattern as shown in Fig. 2(e). Furthermore, Fig. 2(f)–(h) show TEM and HRTEM images of the SrTiO3 submicron cubes loaded with Pt nanoparticles of 2–4 nm in size.
0) and a lattice fringe spacing of 0.19 nm for (200) and (020). In addition, HRTEM characterization shown in Fig. 2(d) suggests the faceted shape of the cubic particle, which is enclosed with (200) planes at the edge and planes at the corner. The single crystal nature of SrTiO3 cubes can be confirmed by the selected area electron diffraction (SAED) pattern as shown in Fig. 2(e). Furthermore, Fig. 2(f)–(h) show TEM and HRTEM images of the SrTiO3 submicron cubes loaded with Pt nanoparticles of 2–4 nm in size.
 | ||
| Fig. 2 (a and b) SEM of SrTiO3, (c) low resolution TEM of SrTiO3, (d) high resolution TEM of SrTiO3 lattice fringes, (e) selected area electron diffraction pattern of SrTiO3, (f, g) TEM of STO-Pt, (h) high resolution TEM of STO-Pt lattice fringes. | ||
X-ray photoelectron spectroscopy (XPS) was used to characterize the elemental surface composition of the photocatalyst. All XPS spectra were fitted using a Shirley-type background subtraction method. The background functions were fitted using 80% Gaussian and 20% Lorentzian functions.40 Fig. 3(a) shows that titanium is present in the Ti4+ state in the SrTiO3 sample, which is fitted out using two peaks at 458.2 and 463.9 eV, corresponding to Ti4+ 2p3/2 and Ti4+ 2p1/2, respectively. However, the XPS peaks of titanium in the Pt loaded SrTiO3 sample are shifted positively to 458.51 and 464.26 eV (Fig. 3(a)). This shows that part of the electrons on the Ti element are transferred onto the surface of the Pt nanoparticles in the Pt loaded photocatalyst. This peak shift also occurs for Sr and O elements. The Sr2+ 3d5/2 and 3d3/2 peaks, shown in Fig. 3(b) are observed at 132.75 and 134.51 eV, respectively and are shifted to 132.92 and 134.63 eV in the presence of Pt. High resolution XPS spectrum of O 1s in Fig. 3(c) illustrates two peaks at 529.4 and 531.5 eV. The peak at 529.4 eV is associated with the lattice O2− ions in the crystal structure. And the peak at 531.4 eV is attributed to O2− in the oxygen defects –OH at cubic SrTiO3 surface, which may be beneficial for photocatalytic water splitting and photocatalytic reduction of carbon dioxide.41,42 The two peaks are also shifted positively to 529.66 and 531.83 eV, respectively for Pt loaded sample. Notably, the significant increase of the peak attributing to O2− in the oxygen defects –OH indicate the more absorption site for H2O, which is conducive to the photocatalytic reaction. The peak at 530.48 eV corresponds to Pt–O. Fig. S3† shows the higher resolution XPS spectra of Pt. It can be seen that platinum has three valence states, which is fitted using six peaks at 70.8 and 74.42 eV corresponding to Pt0, 71.34 and 75.98 eV corresponding to Pt2+ and 72.27 and 78.08 eV corresponding to Pt4+, respectively. The appearance of high-valence Pt may be due to oxidation of part of Pt during photodeposition by the high activity holes. It confirmed that the Schottky junction is formed at the interface between SrTiO3 and Pt nanoparticles.43 The Tauc plot of SrTiO3 samples is shown in Fig. S4.† The optical bandgap of SrTiO3 is about 3.23 eV.
 | ||
| Fig. 3 High resolution XPS spectra of (a) Sr 3d, (b) Ti 2p, (c) O 1s in SrTiO3 sample. | ||
Photocatalytic activity of the SrTiO3 submicron cubes was investigated for CO2 reduction and water splitting. Our experimental results indicate that the submicron cubes show superior activity for a simultaneous production of CH4 and H2. Fig. 4 shows the production rate of CH4 and H2 over SrTiO3 loaded with 3 wt% Pt under UV-Vis light (300 W Xe lamp) using 20 vol% triethanolamine (TEOA) as sacrificial agent. From the plots, it can be seen that the SrTiO3 submicron cubes exhibit superior activity towards both CO2 reduction and water splitting. As known, crystal facets play a crucial role in photocatalysis due to their different surface energy. As discussed in the HRTEM and SAED characterization section (Fig. 2), the obtained SrTiO3 submicron cubes are enclosed with (100) plane at the edge and (110) plane at the corner of the SrTiO3 cubes. The SrTiO3 (100) surface is the most thermodynamically stable facet, which can be terminated by a SrO or TiO2 plane with a similar surface energy of 6.85 eV nm−2.44–46 For the (110) facet, there are two alternating layers of SrTiO4+ and O24−, forming an unbalanced dipole with a high surface energy of 20.02 eV nm−2.46,47 The terminal plane of the two facets is shown in Fig. S5.† Directional separation of photogenerated electron–hole pairs is due to differences in surface energy, which is conducive to the separation of photogenerated carriers48–50 thereby enhancing the photocatalytic activity of the submicron cubes for the CO2 reduction and water splitting.
 | ||
| Fig. 4 (a) Photocatalytic reduction of CO2 over STO-Pt with under simulated sunlight with 20% volume ratio of triethanolamine (TEOA) as sacrificial agent, (b) production rate change before and after eighth hour. | ||
Quantitively analysis on the photocatalytic activity of the submicron cubes indicated that for the CO2 reduction, the production of CH4 increases rapidly with an average production rate of 4.39 μmol g−1 h−1 at the first 8 h as shown in Fig. 4(b), then the CH4 amount tends to reach a plateau with the average CH4 production rate down to 0.46 μmol g−1 h−1 when 8 ≤ t ≤ 28 h. On the other hand, the rate of H2 production by photocatalytic water splitting increases gradually at the first 8 h with an average rate of 14.52 μmol g−1 h−1, then the H2 production rate increases rapidly to 120.23 μmol g−1 h−1 from the 8th to the 28th hrs. Interestingly, a negative correlation between the production of CH4 and H2 can be observed in Fig. 4(a), demonstrating a clear competitive relationship between the two reactions. In addition, the competitive correlation is altered with the reaction time. From the results of the third repetitive test of photocatalytic reduction of CO2 on the same sample (Fig. S6†), it can be obviously seen that the law of the production of CH4 and H2 is almost the same as the results in the first round test. Therefore, it can be inferred that the surface chemistry evolution during the photocatalytic reaction is recoverable. Nevertheless, there is a certain attenuation of the production rates of CH4 and H2, which may be caused by possible property fading of the catalysts after the long-time reaction. Some references have been listed for comparison with our experiments in the ESI† (Table S1†). Based on the information in the table, it can be seen that the material we prepared has a good performance of photocatalytic reduction of carbon dioxide with simultaneous photocatalytic water splitting, demonstrating that our method is a rational strategy to design Ti-based photocatalysts for photocatalytic reduction of CO2.
To determine whether the generated CH4 and H2 come from the photocatalytic decomposition of the scarifying reagent of triethanolamine in water, two comparative experiments have been implemented. First one is to control for all other conditions the same except for the absence of carbon dioxide. The results are shown in the Fig. S7.† The H2 production rate is 426.41 μmol g−1 h−1, but no methane is produced. The other one was performed in pure phase triethanolamine with all other conditions the same, but no gaseous products were detected. Based on the above results, it suggests that the generated CH4 and H2 are not supposed to come from the photocatalytic decomposition of the scarifying reagent of triethanolamine in water.
To get insight into the correlation between the CH4 and H2 production, the band structure characteristics of the obtained SrTiO3 submicron cubes were investigated by the Mott–Schottky measurement. This approach is as a powerful and widely used tool to determine capacitance, carrier density and flat band potential.51 The presented 1/C2 obtained from impedance spectroscopy at three frequencies, 1000, 2000 and 5000 Hz, shown in Fig. 5. The flat-band potential of the semiconductor could be extracted from the x-axis intersection. As illustrated in Fig. 5(a), the flat band potential of the prepared SrTiO3 sample measured at three different frequencies is almost identical at approximately −0.14 vs. RHE, which is in agreement with the reported values.52–54 Combined with the measured band gap value (Fig. S4†), the band structure of prepared SrTiO3 sample has been obtained in Fig. 5(b). A proper equivalent circuit (Fig. S8†) has been used to fit the EIS plots for further understanding the conductivity of the photocatalysts. Rct can be assigned to the charge-transfer resistance in the photocatalyst. The charge-transfer resistances for SrTiO3 submicro cubes is 104590 Ω, which is smaller than that of SrTiO3 commercial powder, 118170 Ω. This proves the more effective separation and transportation of photo-generated carriers in SrTiO3 submicro cubes. As we all know, the thermodynamic condition of the photocatalytic reduction reaction is that the conduction band edge of the semiconductor is higher than the standard reduction potential of the reactant, and the photocatalytic oxidation reaction occurs only when the valence band edge of the semiconductor is lower than the standard oxidation potential of the reactant. As shown in Fig. 5(b), the potential of H+/H2 (0 V vs. NHE at pH 7) for the redox reaction is more negative than that of CO2/CH4 (0.21 V vs. NHE at pH 7), resulting in easier production of methane than hydrogen.7 Therefore, from the perspective of thermodynamics, the photo-reduction of CO2 to CH4 is more likely to occur than H2O to H2.
 | ||
| Fig. 5 Band structure of SrTiO3 nanocrystals. (a) The Mott–Schottky curves of SrTiO3 electrodes measured in Na2SO4 solution. (b) The scheme of the band structure of SrTiO3 nanocrystals. | ||
To confirm the reason for the selectivity changes, SrTiO3 with different Pt loadings were used to test the performance of photocatalytic reduction of CO2. When no Pt NPs are loaded, the signal of CH4 and H2 cannot be detected in the gas chromatography. This is due to the characteristics of SrTiO3 that can only respond in the ultraviolet region and the high recombination efficiency of photogenerated carriers. So, the samples with 1 wt% and 5 wt% Pt loaded were used for the photocatalytic CO2 reduction test (Fig. 6). No obvious inflection point was found for the amount of hydrogen generated in the sample loaded 1 wt%. However, the pattern similar to the sample with 3 wt% Pt loaded has been found for the sample 5 wt% Pt loaded. Therefore, this selectivity change may be closely related to Pt. The production rate of CH4 and H2 have an obvious dependence on the load amount of Pt. The production rate of CH4 is arranged in order as 3 wt% Pt > 1 wt% Pt > 5 wt% Pt; for H2 is 3 wt% Pt > 5 wt% Pt > 1 wt% Pt. During the reactions, the appropriate Pt NPs loading can improve the separation efficiency of photogenerated electron–hole pairs and act as a catalytic site for water splitting. When the loading amount of Pt NPs is low, the water splitting efficiency and carrier separation are relatively low. However, when the loading amount is too large, Pt NPs will increase the recombination efficiency of carriers, which is not conducive to the progress of the photocatalytic reaction. And also reduce the utilization efficiency of the Pt. Additionally, the photocatalytic reduction of CO2 performance over STO-Pt under simulated sunlight with 10% and 100% volume ratio of TEOA as sacrificial agent have been tested. The results tested in 10% volume ratio of TEOA has been obtained in Fig. S9,† which are similar to the performance tested in the reaction solution containing 20% volume ratio of TEOA. No gas is generated in the system of 100% volume ratio of TEOA. These results demonstrate that the selectivity changes are not necessarily related to TEOA.
 | ||
| Fig. 6 (a) Photocatalytic reduction of CO2 over SrTiO3 loaded with 1 wt% Pt under simulated sunlight with 20% volume ratio of triethanolamine (TEOA) as sacrificial agent, (b) photocatalytic reduction of CO2 over SrTiO3 loaded with 5 wt% Pt under simulated sunlight with 20% volume ratio of triethanolamine (TEOA) as sacrificial agent. | ||
As is known, when the Pt NPs are loaded onto the SrTiO3, the surface electron density of SrTiO3 increases because of the lower Fermi energy of Pt NPs and the surface nanojunctions of Pt-SrTiO3 are formed. After Pt NPs were loaded, the photocatalytic performance was significantly improved, as shown in Fig. 4 and 6. The formed nanojunctions at the interface of Pt NPs and SrTiO3 maybe a crucial factor in performance improvement. For the water splitting reaction, Pt itself is a good hydrogenation and dehydrogenation catalyst, and it has good adsorption and desorption of hydrogen. It is reasonable to deduce that the reaction sites of water splitting are mainly on the surface of Pt NPs. However, the sites of CO2 reduction on Pt loaded materials are not clear because of the divergent research conclusions that have been reported.55–58 It should be noted that both hydrogen generation and methane production occurred simultaneously in our experiments, however, it looks that the evolution of methane suppressed the production of hydrogen. While the evolution of methane declined, production of hydrogen was obviously enhanced. The reaction process may be as follows: in the early stage of the reaction, the concentration of CO2 molecules adsorbed on the surface of SrTiO3 was sufficiently high, which is the main reason for the high rate of CH4 generation during this period; however, as the reaction proceeded, surface chemistry or microstructure of the adsorption site of CO2 might change, making it more inclined to promote the hydrogen production. This can be confirmed by the repetitive experiments results (Fig. S6†) and subsequent X-ray photoelectron spectroscopy tests (Fig. 7). That is the reason why the competitive mechanism between the two reactions was proposed.
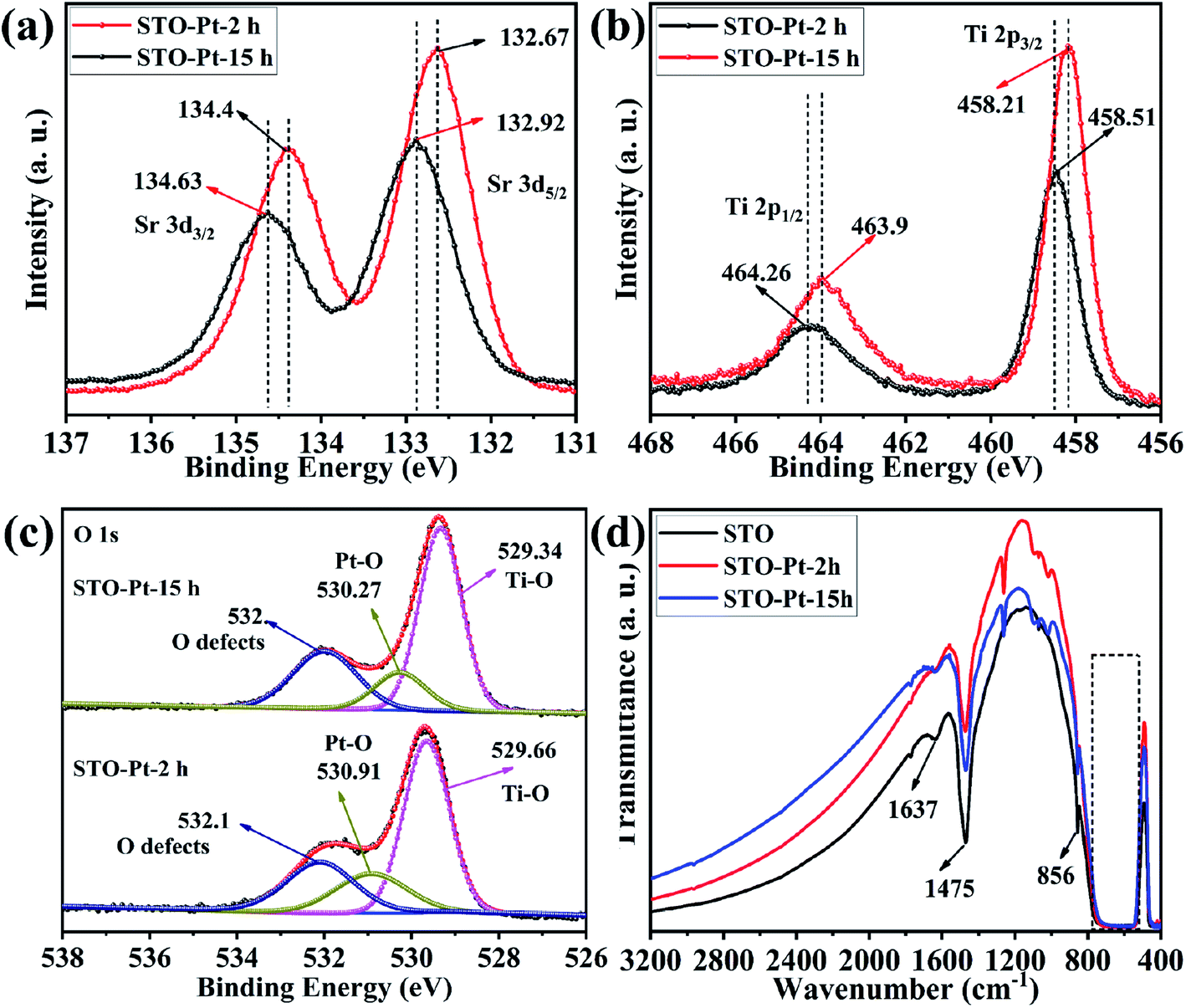 | ||
| Fig. 7 High resolution XPS spectra of (a) Sr 3d, (b) Ti 2p, (c) O 1s in STO-Pt-2h and STO-Pt-15h samples. (d) FTIR absorption spectra of SrTiO3, STO-Pt-2h and STO-Pt-15h. | ||
To confirm our assumption, changes of the surface chemistry and the electronic structure of the samples vs. reaction time were determined by XPS as shown in Fig. 7. In this figure, STO-Pt-2h and STO-Pt-15h correspond to the samples of STO-Pt after 2 h and 15 h reaction, respectively. The XPS peaks of Ti in STO-Pt-15h are negatively shifted from 458.51 and 464.26 eV, to 458.21 and 463.9 eV, respectively (Fig. 7(a)). This peak shift also occurs for the Sr and O. The Sr2+ 3d5/2 and 3d3/2 peaks, shown in Fig. 7(b) are shifted to 134.4 and 132.67 eV, respectively. High resolution XPS spectrum of O 1s, illustrated in Fig. 7(c), indicates that the lattice O2− ions in the crystal structure shifted to 529.34 eV from 529.66 eV, which implies breakdown of the Schottky junction. And the peak attributed to O2− in the oxygen defects –OH on the cubic SrTiO3 surface becomes stronger with the reaction time, indicating that the adsorption of H2O for the sample of STO-Pt-15h is easier than the sample of STO-Pt-2h.
FTIR was carried out to measure the vibration mode of the bonds in the as-prepared STO, STO-Pt-2h and STO-Pt-15h. As shown in Fig. 7(d), the absorption peak at 1637 cm−1 originates from hydroxyl vibration. It is speculated that the hydroxyl group (H–O–H bending) was derived from the adsorption water on the surface of the sample. The transmission peaks at 1475 cm−1 probably corresponds to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (mode in Sr-OOC). The broad absorption band at 760 to 557 cm−1 and the absorption peak at 856 cm−1 belong to the characteristic stretching vibration of the Ti–O bond (stretching vibration) and Sr–O/Ti–O (octahedron bending), respectively. The absorption peak of Ti–O bond is widened probably due to the presence of several Sr–Ti–O absorption peak.59 Notably, the absorption peak derived from the adsorption of water on the surface is apparently stronger for the sample STO-Pt-15h than STO-Pt-2h, which also confirms the assumption that adsorption of water onto the surface of SrTiO3 submicron cubes is favored over CO2 adsorption as the reaction proceed at the surface chemical modification of the photocatalyst take place with time.
O stretching (mode in Sr-OOC). The broad absorption band at 760 to 557 cm−1 and the absorption peak at 856 cm−1 belong to the characteristic stretching vibration of the Ti–O bond (stretching vibration) and Sr–O/Ti–O (octahedron bending), respectively. The absorption peak of Ti–O bond is widened probably due to the presence of several Sr–Ti–O absorption peak.59 Notably, the absorption peak derived from the adsorption of water on the surface is apparently stronger for the sample STO-Pt-15h than STO-Pt-2h, which also confirms the assumption that adsorption of water onto the surface of SrTiO3 submicron cubes is favored over CO2 adsorption as the reaction proceed at the surface chemical modification of the photocatalyst take place with time.
Conclusion
In this work, SrTiO3 submicron cubes were successfully prepared by a molten salt synthesis method. A simultaneous and competitive photocatalytic generation of CH4 and H2 were observed on the submicron cubes originated from the reduction of CO2 and water splitting. The generation of CH4 is higher in the first 8 h with the average production rate of 4.39 μmol g−1 h−1, then gradually reaches a plateau area between 8 and 28 hours with the average production rate of 0.46 μmol g−1 h−1. On the contrary, evolution of H2 is evidently suppressed in the first 8 h and increases significantly between 8 to 28 hours, with the average production rate increase from 14.52 μmol g−1 h to 120.23 μmol g−1 h−1. The investigated surface chemistry and electronic structure of SrTiO3 submicron cubes, photo-reduction reaction thermodynamic conditions and competing relationship of reactant molecular adsorption on the surface of SrTiO3 are proposed as the main causes of product rate changes. The current work expands our understanding of the application of photocatalyst for photocatalytic reduction of carbon dioxide and water.Conflicts of interest
There are no conflicts to declare.Statement of contributions
Haoshan Wei: Writing-Original Draft, Methodology, Investigation. Jingyi Cai: Methodology, Investigation. Yong Zhang: Resources, Investigation, Supervision, Writing-Reviewing and Editing. Xueru Zhang: Methodology, Investigation. Elena A. Baranova: Writing-Reviewing, Software. Jiewu Cui: Validation, Investigation. Yan Wang: Validation, Software. Xia Shu: Methodology, Investigation Yongqiang Qin: Investigation. Jiaqin Liu: Investigation, Supervision. Yucheng Wu: Resources, Project administration, Editing.Acknowledgements
This project is supported by Key Project of Changfeng-HFUT Industrial Innovation Guidance Funds (JZ2020YDZJ0122), the National Natural Science Foundation of China (Grant No. 51772072, 51672065, U1810204), Fundamental Research Funds for the Central Universities (PA2019GDQT002, PA2019GDZC0096). We also would like to thank the financial support from the 111 Project “New Materials and Technology for Clean Energy” (B18018).References
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- A. Goeppert, M. Czaun, J. P. Jones, G. K. S. Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC.
- M. Marszewski, S. Cao, J. Yu and M. Jaroniec, Mater. Horiz., 2015, 2, 261–278 RSC.
- C. Dong, M. Xing and J. Zhang, Mater. Horiz., 2016, 3, 608–612 RSC.
- S. Xie, Q. Zhang, G. Liu and Y. Wang, Chem. Commun., 2015, 52, 35–59 RSC.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- N. U. Stefan, M. A. Juan Antonio and G. Hermenegildo, Int. J. Mol. Sci., 2014, 15, 5246–5262 CrossRef.
- K. Karuppiah and A. M. Ashok, Nanomater. Nanotechnol., 2019, 8, 51–58 Search PubMed.
- A. R. West, Chem. Rec., 2006, 6, 206–216 CrossRef CAS.
- C. D. Chandler, C. Roger and M. J. Hampden-Smith, Chem. Rev., 1993, 93, 1205–1241 CrossRef CAS.
- A. Bhalla, R. Guo and R. Roy, Mater. Res. Innovations, 2000, 4, 3–26 CrossRef CAS.
- M. Pena and J. Fierro, Chem. Rev., 2001, 101, 1981–2018 CrossRef CAS.
- F. Kyoichi, K. Kiyofumi, K. Jumpei, H. Tadashi and K. Kunihito, Nanoscale, 2010, 2, 2080–2083 RSC.
- M. Zhou, J. Chen, Y. Zhang, M. Jiang, S. Xu, Q. Liang and Z. Li, J. Alloys Compd., 2020, 817, 152796 CrossRef CAS.
- E. Can and R. Yildirim, Appl. Catal., B, 2019, 242, 267–283 CrossRef CAS.
- R. Shi, G. I. N. Waterhouse and T. Zhang, Sol. RRL, 2017, 1, 1700126 CrossRef.
- K. M. Macounová, R. Nebel, M. Klusáčková, M. Klementová and P. Krtil, ACS Appl. Mater. Interfaces, 2019, 11, 16506–16516 CrossRef.
- A. Alzahrani, D. Barbash and A. Samokhvalov, J. Phys. Chem. C, 2016, 120, 19970–19979 CrossRef CAS.
- F. Yang, Q. Zhang, L. Zhang, M. Cao, Q. Liu and W.-L. Dai, Appl. Catal., B, 2019, 117901 CrossRef CAS.
- T. Chen, J. Meng, S. Wu, J. Pei, Q. Lin, X. Wei, J. Li and Z. Zhang, J. Alloys Compd., 2018, 754, 184–189 CrossRef CAS.
- B. Zielińska, Bull. Mater. Sci., 2014, 37, 911–916 CrossRef.
- V. Jeyalakshmi, S. Tamilmani, R. Mahalakshmy, P. Bhyrappa, K. R. Krishnamurthy and B. Viswanathan, J. Mol. Catal. A: Chem., 2016, 420, 200–207 CrossRef CAS.
- C. E. Ekuma, M. Jarrell, J. Moreno and D. Bagayoko, AIP Adv., 2012, 2, 129 Search PubMed.
- S. Piskunov, E. Heifets, R. Eglitis and G. Borstel, Comput. Mater. Sci., 2004, 29, 165–178 CrossRef CAS.
- S. Zeng, P. Kar, U. K. Thakur and K. Shankar, Nanotechnology, 2018, 29, 052001 CrossRef.
- J. Zhang, J. H. Bang, C. Tang and P. V. Kamat, ACS Nano, 2009, 4, 387–395 CrossRef.
- M. Miyauchi, M. Takashio and H. Tobimatsu, Langmuir, 2004, 20, 232–236 CrossRef CAS.
- J. Jiang, Y. Jia, Y. Wang, R. Chong, L. Xu and X. Liu, Appl. Surf. Sci., 2019, 486, 93–101 CrossRef CAS.
- M. Ahmadi, M. S. Dorraji, M. Rasoulifard and A. Amani-Ghadim, Sep. Purif. Technol., 2019, 228, 115771 CrossRef CAS.
- A. Kudo, R. Niishiro, A. Iwase and H. Kato, Chem. Phys., 2007, 339, 104–110 CrossRef CAS.
- A. B. Djurišić, Y. H. Leung and A. M. Ching Ng, Mater. Horiz., 2014, 1, 400–410 RSC.
- K. Xie, N. Umezawa, Z. Ning, P. Reunchan, Y. Zhang and J. Ye, Energy Environ. Sci., 2011, 4, 4211–4219 RSC.
- V. Jeyalakshmi, R. Mahalakshmy, K. R. Krishnamurthy and B. Viswanathan, Catal. Today, 2017, 300, 152–159 CrossRef.
- C. Tang, Y. Zhang and Z. Huang, Renewable Sustainable Energy Rev., 2014, 30, 195–216 CrossRef CAS.
- Y.-J. Gu, W. Wen, S. Zheng and J.-M. Wu, Mater. Chem. Front., 2020, 4, 2744–2753 RSC.
- G. Zhang, W. Jiang, S. Hua, H. Zhao, L. Zhang and Z. Sun, Nanoscale, 2016, 8, 16963–16968 RSC.
- S. Bhardwaj and B. Pal, Adv. Powder Technol., 2018, 29, 2119–2128 CrossRef CAS.
- L. Mu, Y. Zhao, A. Li, S. Wang, Z. Wang, J. Yang, Y. Wang, T. Liu, R. Chen and J. Zhu, Energy Environ. Sci., 2016, 9, 2463–2469 RSC.
- Y. Li, Y. Wang, W. Doherty, K. Xie and Y. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 8553–8562 CrossRef CAS.
- H. Tan, Z. Zhao, W.-b. Zhu, E. N. Coker, B. Li, M. Zheng, W. Yu, H. Fan and Z. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 19184–19190 CrossRef CAS.
- T. Kawabe, K. Tabata, E. Suzuki, Y. Yamaguchi and Y. Nagasawa, J. Phys. Chem. B, 2001, 105, 4239–4244 CrossRef CAS.
- D. J. Seong, M. Jo, D. Lee and H. Hwang, Electrochem. Solid-State Lett., 2007, 10, H168–H170 CrossRef CAS.
- R. I. Eglitis and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 155439 CrossRef.
- R. Eglitis and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 155420 CrossRef.
- S. Woo, H. Jeong, S. A. Lee, H. Seo, M. Lacotte, A. David, H. Y. Kim, W. Prellier, Y. Kim and W. S. Choi, Sci. Rep., 2015, 5, 8822 CrossRef.
- G. S. Foo, Z. D. Hood and Z. Wu, ACS Catal., 2017, 8, 555–565 CrossRef.
- R. Li, H. Han, F. Zhang, D. Wang and C. Li, Energy Environ. Sci., 2014, 7, 1369–1376 RSC.
- J. L. Giocondi and G. S. Rohrer, J. Am. Ceram. Soc., 2003, 86, 1182–1189 CrossRef CAS.
- T. Tachikawa and T. Majima, Chem. Commun., 2012, 48, 3300–3302 RSC.
- T. W. Kim, Y. Ping, G. A. Galli and K.-S. Choi, Nat. Commun., 2015, 6, 8769 CrossRef CAS.
- H.-C. Chen, C.-W. Huang, J. C. Wu and S.-T. Lin, J. Phys. Chem. C, 2012, 116, 7897–7903 CrossRef CAS.
- W. Wei, Y. Dai, M. Guo and B. Huang, Appl. Surf. Sci., 2011, 257, 6607–6611 CrossRef CAS.
- W. Wei, Y. Dai, K. Lai, M. Guo and B. Huang, Chem. Phys. Lett., 2011, 510, 104–108 CrossRef CAS.
- X. Wu, C. Wang, Y. Wei, J. Xiong, Y. Zhao, Z. Zhao, J. Liu and J. Li, J. Catal., 2019, 377, 309–321 CrossRef CAS.
- S. Xie, Y. Wang, Q. Zhang, W. Deng and Y. Wang, ACS Catal., 2014, 4, 3644–3653 CrossRef CAS.
- Y. Wei, X. Wu, Y. Zhao, L. Wang, Z. Zhao, X. Huang, J. Liu and J. Li, Appl. Catal., B, 2018, 236, 445–457 CrossRef CAS.
- M. Tasbihi, F. Fresno, U. Simon, I. J. Villar-García, V. Pérez-Dieste, C. Escudero and V. A. de la Peña O'Shea, Appl. Catal., B, 2018, 239, 68–76 CrossRef CAS.
- T. P. Xie, Y. Wang, C. L. Liu and L. J. Xu, Materials, 2018, 11, 17 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08246e |
| This journal is © The Royal Society of Chemistry 2020 |