DOI:
10.1039/D0RA08244A
(Paper)
RSC Adv., 2020,
10, 41336-41352
Adhesive functionalized ascorbic acid on CoFe2O4: a core–shell nanomagnetic heterostructure for the synthesis of aldoximes and amines†
Received
26th September 2020
, Accepted 6th November 2020
First published on 11th November 2020
Abstract
This paper reports on the simple synthesis of novel green magnetic nanoparticles (MNPs) with effective catalytic properties and reusability. These heterogeneous nanocatalysts were prepared by the anchoring of Co and V on the surface of CoFe2O4 nanoparticles coated with ascorbic acid (AA) as a green linker. The prepared nanocatalysts have been identified by scanning electron microscopy, transmission electron microscopy, energy-dispersive X-ray spectroscopy, X-ray atomic mapping, thermogravimetric analysis, X-ray powder diffraction, vibrating sample magnetometer analysis, coupled plasma optical emission spectrometry and Fourier transform infrared spectroscopy. The impact of CoFe2O4@AA-M (Co, V) was carefully examined for NH2OH·HCl oximation of aldehyde derivatives first and then for the reduction of diverse nitro compounds with sodium borohydride (NaBH4) to the corresponding amines under green conditions. The catalytic efficiency of magnetic CoFe2O4@AA-M (Co, V) nanocatalysts was investigated in production of different aldoximes and amines with high turnover numbers (TON) and turnover frequencies (TOF) through oximation and reduction reactions respectively. Furthermore, the developed environment-friendly method offers a number of advantages such as high turnover frequency, mild reaction conditions, high activity, simple procedure, low cost and easy isolation of the products from the reaction mixture by an external magnetic field and the catalyst can be reused for several consecutive runs without any remarkable decrease in catalytic efficiency.
1. Introduction
One of the important goals in the field of catalysis in terms of green chemistry is to make progress with environmentally benign, practical, clean, economical and efficient processes for catalyst separation and recycling.1 From this aspect, nanocatalysts have exhibited good catalytic activity because of their large surface area, small size, selectivity, reusability and recovery from reaction mixtures.2,3 In addition, MNPs have been used as alternatives to conventional heterogeneous supports. For this purpose, magnetic ferrite nanoparticles have been utilized as separable magnetic catalysts as they have large magnetic anisotropies, high thermal stability, high surface areas, moderate saturation magnetization and chemical stability.4,5 A literature review shows that MNPs have been used in biology,6 biomedicine,7 material sciences,8,9 biochips and biosensors.10 Bimetal oxide MNPs or metal ferrite nanoparticles, MFe2O4 (M: Mn, Co, Fe, Ni and Zn), are a group of MNPs which have numerous applications in various fields. They show a more tunable particle size and better morphology than Fe3O4 MNPs. Among metal ferrite MNPs, cobalt ferrite (CoFe2O4) MNPs have been recognized interesting due to their mechanical hardness high magnetization and high chemical and thermal stability.11 It is worth noting the organic coat owing catalytic sites along with structural approach, improve the catalytic activity of such nanocomposite.12 Vitamin C (ascorbic acid, AA) as a bio-organic coater is well known for its anti-oxidant property and is water soluble. Therefore, it is of much interest to synthesize stable water-soluble magnetic nanocomposite using ascorbic acid.13 The AA is a sugar acid with a cis-enediol group on the sugar ring and adjacent alcoholic hydroxyl groups on the side chain available for binding to iron oxide nanoparticles.14
On the other hand, aldoximes and ketoximes have been widely used in industry, medicine and analytical chemistry; and they are very effective and versatile intermediates in synthetic organic chemistry. The typical synthetic method for oximes is oximation of aldehydes and ketones with hydroxylamine.15 This method is also used for the industrial production of ε-caprolactam from cyclohexanone.16 However, hydroxylamine is easily explosive and unstable during heating. To minimize the risks, hydroxylamine is often replaced by diverse salts such as NH2OH·HCl or NH2OH·H2SO4.17 Nevertheless, these methodologies show several shortcomings, such as drastic reaction conditions and complicated operation, which mostly yield many side products and lead to drastic environmental contamination.18,19 Hence, it is important to prepare efficient and highly selective catalysts that can enhance the yield of the corresponding oximes and suppress the formation of side products as much as possible. In recent years, several efficient catalysts were implemented for oximation reaction such as: [Co(L)2]2Na2[β-Mo8O26]·9H2O and [Fe(L)2]2Na2[β-Mo8O26]·9H2O,20 [K(H2O)6]2[Co(H2O)5]4[WZn3(H2O)2(ZnW9O34)2]·19H2O,21 pyridine-chloroform,22 ethanol-pyridine,23 NaOH with or without solvent24–27 and hyamine (10 mol%).28 For further reviews on catalytic oximation systems see ref. 29–45. Also, one important class of starting and intermediate materials are aromatic amines which are used for producing a great diversity of chemicals such as agrochemicals and rubber materials, pharmaceuticals, biological active molecules, synthetic resins, dyes, plastics and paints.46 Among some of the reducing agents such as NaBH4, ammonium formate, acetic acid, ethylene glycol, hydrazine hydrate, and formic acid as hydrogen sources, NaBH4 showed higher activity compared to the others.47 It is noteworthy that the alone NaBH4 does not reduce nitro groups under mild reaction conditions but reducing ability of this reagent increases dramatically through reduction of nitro compounds when metal complexes or another promoter are added. Up to now, some catalytic systems have been reported such as magnetically nano core–shell Fe3O4@Cu(OH)x/NaBH4,46 bis-thiourea complexes of bivalent cobalt, nickel, copper and zinc chlorides48 Ni2B/NaBH4,49 Cu NPs/NaBH4,50 NaBH4/Fe3O4@PAMAM/Ni(0)-b-PEG,51 and TiO2@Au/NH2NH2.52 However, to study more reported reduction protocols see ref. 53–68. In spite of their merits, the most commonly reported methods in both oximation and reduction process, suffer from environmental pollution, the use of hazardous reagents, unsatisfactory yields, large amounts, poor recovery of expensive catalysts and long reaction times, which ultimately lead to the production of large amount of toxic waste.
In continuation of synthesis of task-specific catalysts with nanomagnetic properties, herein, we wish to introduce CoFe2O4@AA-M (Co, V) as a novel and active MNPs and concerning the application of prepared nano catalysts, we have explored their efficacy in the oximation of aldehyde with NH2OH·HCl and reduction of various nitro compounds to the corresponding amines with NaBH4 under eco-friendly conditions. It is notable that, the aim of current work is to introduce magnetic nano-ascorbic acid in the trapping and stabilizing of the Co and V nanoparticles as a proficient, harmless to the environment, recyclable and magnetic powerful solid catalysts with good stability which does not have any of the aforementioned drawbacks for the synthesis of mentioned organic reactions.
2. Results and discussion
2.1 Catalyst preparation
Development of reusable nanocatalysts and their application in the progress of organic reaction are our main goal. CoFe2O4@AA-M (Co, V) was synthesized as illustrated in Scheme 1. First, we prepared CoFe2O4 by co-precipitation method. Then to prepare functionalized magnetic nanocatalysts, the surface of MNPs was coated using ascorbic acid as green linker. Lastly, CoFe2O4@AA-M (Co, V) nanocomposites were generated by stable interaction of Co and V with functional group of ascorbic acid. To confirm the successful synthesis of heterogeneous catalysts, they were identified by several characterization equipment such as FT-IR, XRD, FE-SEM, TEM, EDX, TGA, ICP-OES, X-ray atomic mapping and VSM.
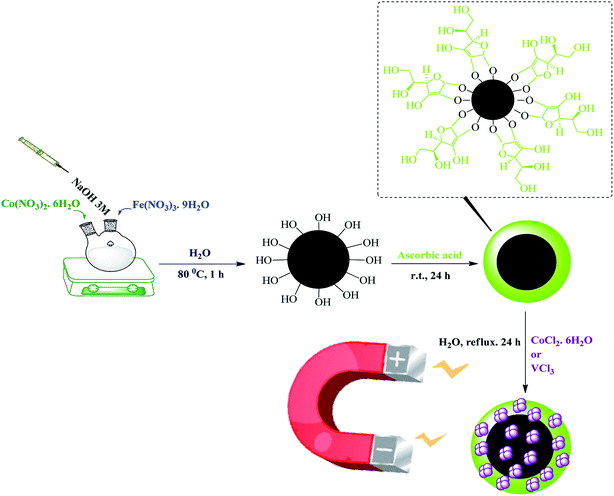 |
| | Scheme 1 Preparation of CoFe2O4@AA-M (Co, V). | |
2.2 Catalyst characterization
2.2.1 FE-SEM and TEM studies. The morphology of the CoFe2O4@AA-M (Co, V) catalysts were achieved using scanning electron microscopy technique. FE-SEM images of these nanostructures are shown in Fig. 1a and b. As illustrated in this figure, the prepared compounds were formed with uniform nanometers-sized particles. It should be noted that the synthesized samples were obtained with approximately spherical shape. Also, the transmission electron microscopy (TEM) confirm the core–shell structure and as shown in Fig. 1c the size of the nanocatalysts is around 50 nm.
 |
| | Fig. 1 FE-SEM images of CoFe2O4@AA-Co at (a) 500 nm, (b) 200 nm, FE-SEM images of CoFe2O4@AA-V at (c) 500 nm, (d) 200 nm and TEM image of CoFe2O4@AA-Co nanocatalyst. | |
2.2.2 EDX and X-ray atomic map studies. The atomic percentage of different elements of the synthesized nanoparticles was investigated by energy-dispersive X-ray spectroscopy (EDX). The EDX analysis of CoFe2O4@AA-Co nanocatalyst confirmed the presence of O, C, Fe, Co, and analysis of CoFe2O4@AA-V nanocatalyst confirmed the presence of O, C, Fe, Co, V species in the obtained catalysts (Fig. 2a and b). Also, the X-ray atomic mapping of CoFe2O4@AA-Co and CoFe2O4@AA-V were examined in order to measure the dispersion of Co and V active sites anchored over ascorbic acid along with the other core elements in the catalyst. It was illustrated that the elemental map images were showed the good dispersion of Co and V on surface of the catalyst.
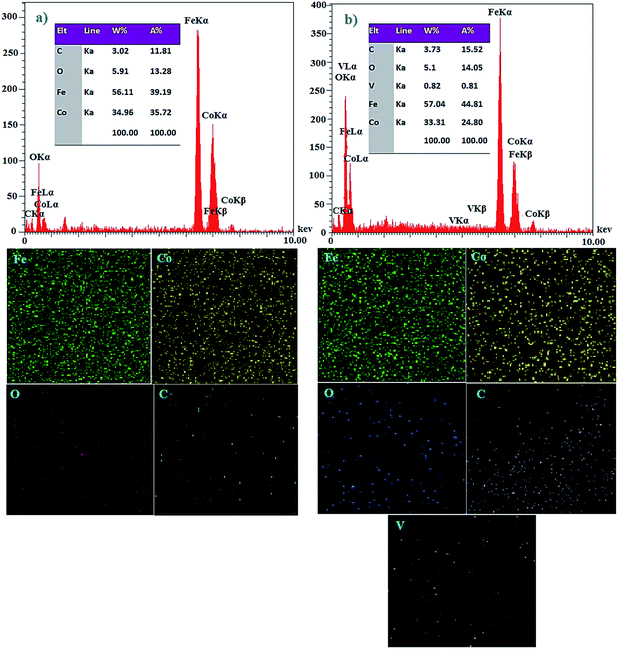 |
| | Fig. 2 EDX spectra and X-ray atomic map analysis of (a) CoFe2O4@AA-Co (b) CoFe2O4@AA-V nanocatalyst. | |
2.2.3 TGA-DTA studies. The thermal stability of CoFe2O4@AA-M (Co, V) were determined using thermo-gravimetric analysis (TGA) in the temperature range of 30–700 °C (Fig. 3). The TGA pattern shows an initial small weight loss below 200 °C. In the all samples, the weight loss below 200 °C is related to the elimination of the chemically and physically adsorbed solvents or surface hydroxyl groups, and the other weight loss in the range of 200–500 °C is belonging to the disintegration of the organic layers on the surface of CoFe2O4 magnetic nanocomposite.69 The reduction of weight in the next step (500–720 °C) was associating to the further decomposition of ascorbic acid residues. The TGA profile demonstrate that the nanocatalysts have a sensible stableness up to 200 °C. According to the results of these analyses, the good grafting of AA-M (Co, V) onto the surface of CoFe2O4 nanoparticles is verified.
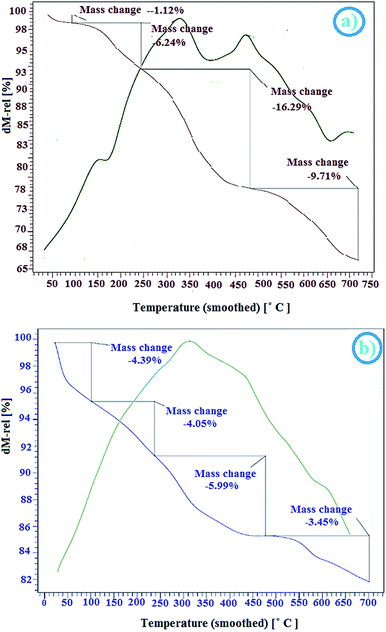 |
| | Fig. 3 TGA-DTA profile of (a) CoFe2O4@AA-Co, (b) CoFe2O4@AA-V nanocatalyst. | |
2.2.4 XRD studies. XRD profile of CoFe2O4 and CoFe2O4@AA-M are shown below. XRD analyses were carried out to characterize the phase formation and crystalline structure of cobalt ferrite nanoparticles. The pattern confirms formation of pure cubic cobalt ferrite by the peak positions of 2θ values at 22.12°, 3039°, 42.18°, 49.85°, 61.89°, 66.79° and 75.20° with JCPDS 22 1086 (Fig. 4a). As illustrated in XRD pattern of CoFe2O4@AA-Co presence of peaks at 2θ = 43.6° and 50.7° indicated successful modification of Co and presence of peaks at 2θ = 44.2° and 51.3° indicated successful V at the surface of CoFe2O4 nanoparticles (Fig. 4b and c).
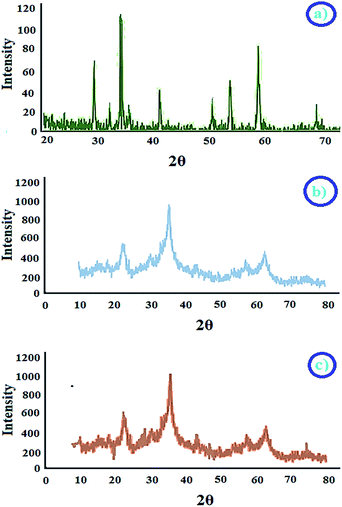 |
| | Fig. 4 XRD patterns of (a) CoFe2O4, (b) CoFe2O4@AA-Co, (c) CoFe2O4@AA-V nanocatalyst. | |
2.2.5 VSM studies. To compare magnetic property of bare CoFe2O4 with CoFe2O4@AA-M (Co, V) nanoparticles, vibrating sample magnetometer (VSM) technique has been used (Fig. 5). As shown, the saturation magnetization value (Ms) of the bare CoFe2O4 is 74.4 emu g−1 while after NPs coating, Ms amount of CoFe2O4@AA-Co and CoFe2O4@AA-V nanoparticles are 33.7 and 22.8 emu g−1 respectively. It is clear that the magnetic saturation for CoFe2O4 nanoparticles is higher than that of prepared nanocatalysts which is due to the loading of organic layers and metal complexes on CoFe2O4 nanoparticles. Despite the decline in magnetic character of CoFe2O4@AA-M (Co, V), it is sensible enough to be easily separated from the reaction mixture by inducing an external magnetic field.
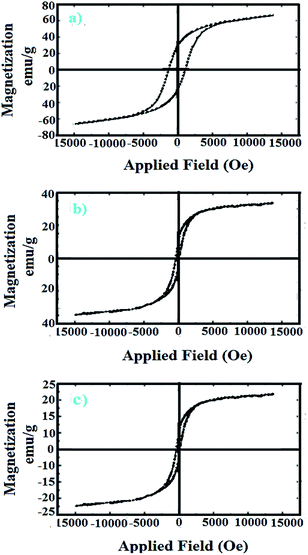 |
| | Fig. 5 Magnetization curves of (a) CoFe2O4, (b) CoFe2O4@AA-Co, (c) CoFe2O4@AA-V nanocatalyst. | |
2.2.6 ICP-OES studies. The exact amount of Co and V anchored on the surface of modified CoFe2O4 are measured to be 0.52 mmol g−1 and 0.50 mmol g−1 using the ICP atomic emission spectroscopy method.
2.2.7 FT-IR studies. Fig. 6 presents FT-IR spectrum of bare CoFe2O4 nanoparticles (a), CoFe2O4@AA (b), CoFe2O4@AA-Co (c) and CoFe2O4@AA-V (d). Spectrum of the CoFe2O4 clearly shows absorption bands around 586 and 3464 cm−1, which are characteristic of the presence of metal–oxygen bond and hydroxyl functional group (O–H), respectively. As shown in Fig. 6b, absorption peaks at approximately 2870–2980 cm−1 (C–H stretching vibration) and absorption peak at approximately 1650 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration) in FT-IR spectrum is due to the loading of ascorbic acid on the surface of the prepared CoFe2O4 nanoparticles. As can be seen from FT-IR spectra of CoFe2O4@AA-Co (c) and CoFe2O4@AA-V (d), the shift on spectrum to lower wavenumbers attributed to asymmetrical and symmetrical modes of the metal–oxygen bonds is happened; that is because of a robust interaction between the O group of Co and V complexes on the MNPs.
O stretching vibration) in FT-IR spectrum is due to the loading of ascorbic acid on the surface of the prepared CoFe2O4 nanoparticles. As can be seen from FT-IR spectra of CoFe2O4@AA-Co (c) and CoFe2O4@AA-V (d), the shift on spectrum to lower wavenumbers attributed to asymmetrical and symmetrical modes of the metal–oxygen bonds is happened; that is because of a robust interaction between the O group of Co and V complexes on the MNPs.
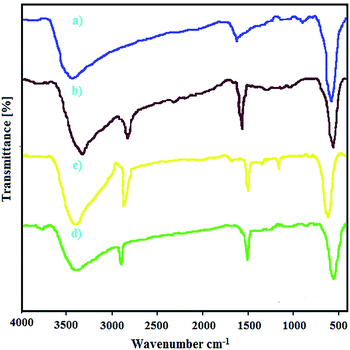 |
| | Fig. 6 FT-IR spectra of (a) CoFe2O4, (b) CoFe2O4@AA, (c) CoFe2O4@AA-Co, (d) CoFe2O4@AA-V nanocatalyst. | |
2.3 The catalytic activity
To investigate the catalytic activity of CoFe2O4@AA-Co and CoFe2O4@AA-V, first these nanostructures were examined by NH2OH·HCl oximation of aldehyde derivatives. To optimize the reaction condition, oximation of benzaldehyde with NH2OH·HCl as model substrate was tested. The influence of reaction-solvent, temperature and the amount of nanocatalyst.
On the course of the reaction has been illustrated in Table 1. In spite of the great capabilities of NH2OH·HCl, oximation of aldehydes with the alone reagent did not proceed at all (entry 1). Also, CoFe2O4 was not efficient catalyst to progress the reaction perfectly (entry 2). n-Hexane as aprotic solvent was not able to progress the reaction well (entries 3 and 13). Entries 4–7 and 13–17 show that oximation of benzaldehyde with NH2OH·HCl in the presence of CoFe2O4@AA-Co and CoFe2O4@AA-V respectively, in the protic solvents such as THF, EtOAc, CH3CN and EtOH did not take place completely even under drastic conditions. Interestingly, using H2O as a sole solvent dramatically accelerated the rate of the oximation process (entries 8–12, 18 and 22). These observations revealed that H2O was the best solvent of choice and the best results were achieved in the presence of 2 mmol hydroxylamine hydrochloride with 20 mg of nano CoFe2O4@AA-M (Co, V) in H2O and at room temperature condition and it showed a full efficiency for complete conversion of 1 mmol benzaldehyde to benzaldoxime immediately (entries 11 and 21).
Table 1 Optimization experiments for oximation of benzaldehyde to benzaldoxime with NH2OH·HCl/CoFe2O4@AA-M (Co, V)
| Entry |
NH2OH·HCl (mmol) |
Catalyst (mg) |
Conditiona |
Time (min) |
Conversion (%) |
| All reactions were performed with 1 mmol of benzaldehyde, 1.5 mL of solvent. |
| 1 |
1.5 |
— |
H2O/50 °C |
24 h |
— |
| 2 |
1.5 |
CoFe2O4 (20) |
H2O/50 °C |
120 |
30 |
| 3 |
1.2 |
CoFe2O4@AA-Co (20) |
n-Hexane/50 °C |
120 |
30 |
| 4 |
1.2 |
CoFe2O4@AA-Co (20) |
THF/50 °C |
25 |
80 |
| 5 |
1.2 |
CoFe2O4@AA-Co (20) |
EtOAc/50 °C |
20 |
88 |
| 6 |
1.2 |
CoFe2O4@AA-Co (20) |
CH3CN/50 °C |
25 |
88 |
| 7 |
1.2 |
CoFe2O4@AA-Co (20) |
EtOH/50 °C |
20 |
85 |
| 8 |
1 |
CoFe2O4@AA-Co (20) |
H2O/50 °C |
5 |
93 |
| 9 |
1.2 |
CoFe2O4@AA-Co (10) |
H2O/r.t. |
25 |
85 |
| 10 |
1.2 |
CoFe2O4@AA-Co (15) |
H2O/r.t. |
10 |
93 |
| 11 |
1.2 |
CoFe2O4@AA-Co (20) |
H2O/r.t. |
20 s |
98 |
| 12 |
1.2 |
CoFe2O4@AA-Co (25) |
H2O/r.t. |
10 s |
99 |
| 13 |
1.2 |
CoFe2O4@AA-V (20) |
n-Hexane/50 °C |
125 |
25 |
| 14 |
1.2 |
CoFe2O4@AA-V (20) |
THF/50 °C |
28 |
80 |
| 15 |
1.2 |
CoFe2O4@AA-V (20) |
EtOAc/50 °C |
22 |
87 |
| 16 |
1.2 |
CoFe2O4@AA-V (20) |
CH3CN/50 °C |
30 |
88 |
| 17 |
1.2 |
CoFe2O4@AA-V (20) |
EtOH/50 °C |
23 |
86 |
| 18 |
1 |
CoFe2O4@AA-V (20) |
H2O/50 °C |
5 |
90 |
| 19 |
1.2 |
CoFe2O4@AA-V (10) |
H2O/r.t. |
26 |
84 |
| 20 |
1.2 |
CoFe2O4@AA-V (15) |
H2O/r.t. |
10 |
91 |
| 21 |
1.2 |
CoFe2O4@AA-V (20) |
H2O/r.t. |
20 s |
96 |
| 22 |
1.2 |
CoFe2O4@AA-V (25) |
H2O/r.t. |
15 s |
97 |
Furthermore, the progress of the oximation of benzaldehyde to benzaldoxime with NH2OH·HCl in the presence of CoFe2O4@AA-V MNPs was also investigated and confirmed by UV-Vis analysis using the optimized reaction conditions (mentioned in Table 1, entry 11). As shown in Fig. 7, during the oximation reaction, benzaldehyde which is shown by the blue curve (λmax = 250 nm) was fully converted to benzaldoxime (λmax = 270 nm) which is intimated by the red curve.
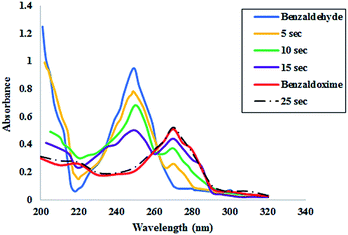 |
| | Fig. 7 UV-Vis analysis of benzaldehyde oximation under optimized reaction conditions. | |
It should also be mention that the oximation of benzaldehyde to corresponding oxime could be considered as pseudo-first-order kinetics because the concentration of NH2OH·HCl was in excess. Notably, the pseudo-first-order kinetics equation can be defined as ln(Ct/C0) = −kappt, where C0 and Ct are the initial and instantaneous concentration of benzaldehyde, respectively. As well as the t and kapp stand for the apparent reaction time and rate constant in turn. Thus, the reaction apparent rate constant (kapp) can be simply calculated with the slope of ln(Ct/C0) versus reaction time. In this study, the kapp for oximation of benzaldehyde was 0.153 s−1 (Fig. 8).
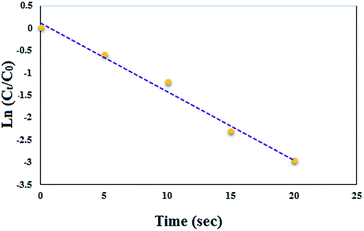 |
| | Fig. 8 Relationship between ln(Ct/C0) and reaction time for the oximation of benzaldehyde. | |
In order to investigate the effects of the NH2OH·HCl amount on the reaction rate, the concentration of benzaldehyde and CoFe2O4@AA-Co was kept constant and various amounts of NH2OH·HCl (1.2, 1.5 and 1.7 mmol) were added to the reaction vessel. According to the UV-visible spectra (Fig. 9), the reaction was completed at 20, 15 and 10 s, respectively.
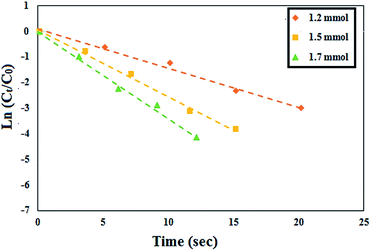 |
| | Fig. 9 Effect of the concentration of NH2OH·HCl on the reaction rate. | |
Also, Table 2 shows kapp for these reactions. Based on the obtained results, we observed that with increasing the concentration of NH2OH·HCl, the amount of the kapp increased. Notably, although the increase in the amount of NH2OH·HCl increases, the speed of the oximation reaction, based on the green chemistry protocols, especially economic efficiency, we selected 1.2 mmol of the NH2OH·HCl as an optimal amount for the mentioned reaction.
Table 2 Effect of the concentration of NH2OH·HCl on the reaction rate
| Entry |
NH2OH·HCl (mmol) |
Time (s) |
kapp (s) |
| 1 |
1.2 |
20 |
0.153 |
| 2 |
1.5 |
15 |
0.262 |
| 3 |
1.7 |
12 |
0.338 |
The effect of various nanocatalyst amounts on the reaction rate was also studied (Fig. 10). All the other reaction parameters during the mentioned reaction were kept constant. Based on the results obtained from Table 3, it can be seen that the kapp increased from 0.153 to 0.516 s−1 as increasing the concentration of CoFe2O4@AA-Co from 20 to 30 mg. Notably, the results clearly showed that the dependence of the reaction rate on various CoFe2O4@AA-Co amounts is much higher than NH2OH·HCl various value.
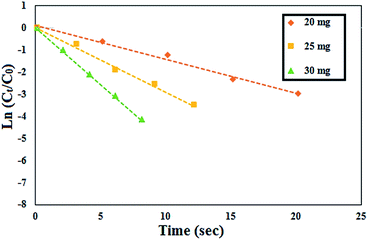 |
| | Fig. 10 Effect of the concentration of CoFe2O4@AA-Co on the reaction rate. | |
Table 3 Effect of the concentration of CoFe2O4@AA-Co on the reaction rate
| Entry |
CoFe2O4@AA-Co (mg) |
Time (s) |
kapp (s) |
| 1 |
20 |
20 |
0.153 |
| 2 |
25 |
12 |
0.292 |
| 3 |
30 |
8 |
0.516 |
Encouraged by the achieved outcomes, the oximation ability of NH2OH·HCl/CoFe2O4@AA-M (Co, V) system were examined for oximation of diverse aldehyde derivatives to the corresponding aldoximes at the optimized reaction conditions. The outcomes of this examination are shown in Tables 4 and 5.
Table 4 Oximation of aldehydes with NH2OH·HCl in the presence of CoFe2O4@AA-Coa,b,c,d,e
Table 5 Oximation of aldehydes with NH2OH·HCl in the presence of CoFe2O4@AA-Va,b,c,d,e
As illustrated, all reactions were done successfully using the molar equivalents of aldehyde![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NH2OH·HCl (1:1.2) in the presence of 20–40 mg of the nanocatalysts producing the products in excellent yields within immediate to 7 min. The result shows that benzaldehyde can be transformed to benzaldoxime in 96–99% yield (Tables 4 and 3, entry 1). In the case of electron-releasing substitutions on aromatic rings the corresponding aldoximes can be also obtained in excellent yields. As well, aromatic aldehydes with electron-withdrawing functionalities were also successfully changed to the corresponding aldoximes in high yields using CoFe2O4@AA-M (Co, V) system. Entry 11 represents that this method is also effective for the oximation of aliphatic aldehyde to corresponding oxime. The catalyst turnover frequency (TOF) and turnover number (TON) are two important factors that are used for measuring the efficiency of the heterogeneous and homogenous catalysts. In heterogeneous catalytic reactions, the TON and TOF can be determined on the basis of the amount of product which was formed.
:NH2OH·HCl (1:1.2) in the presence of 20–40 mg of the nanocatalysts producing the products in excellent yields within immediate to 7 min. The result shows that benzaldehyde can be transformed to benzaldoxime in 96–99% yield (Tables 4 and 3, entry 1). In the case of electron-releasing substitutions on aromatic rings the corresponding aldoximes can be also obtained in excellent yields. As well, aromatic aldehydes with electron-withdrawing functionalities were also successfully changed to the corresponding aldoximes in high yields using CoFe2O4@AA-M (Co, V) system. Entry 11 represents that this method is also effective for the oximation of aliphatic aldehyde to corresponding oxime. The catalyst turnover frequency (TOF) and turnover number (TON) are two important factors that are used for measuring the efficiency of the heterogeneous and homogenous catalysts. In heterogeneous catalytic reactions, the TON and TOF can be determined on the basis of the amount of product which was formed.
A conceivable mechanism for the nano CoFe2O4@AA-M (Co, V) catalyzed was shown in Scheme 2. The nanocatalysts simplify the oximation process via coordination of its M (Co, V) with O of carbonyl. The condensation made by linkage of R1R2CO and O-coordination of hydroxylamine and subsequent intramolecular nucleophilic attack. As described, CoFe2O4 was not capable to progress the reaction perfectly and anchoring Co and V lead to synthesis efficient nanocatalysts which accelerated the rate of the oximation process. According to these illustrations, Co and V are the main active sites of the nanocatalysts.
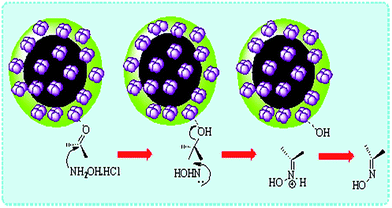 |
| | Scheme 2 A conceivable mechanism for the nano CoFe2O4@AA-M (Co, V) towards oximation of aldehydes. | |
Thereafter, catalytic activity of CoFe2O4@AA-Co and CoFe2O4@AA-V system was distinguished by comparison of the achieved result for benzaldehyde with other reported oximation systems. The oximation of benzaldehyde with hydroxylamine hydrochloride in the presence of diverse catalysts is selected as reaction model in all cases. The results in Table 6 demonstrate that in terms of short reaction times, easy work-up procedure, mild reaction conditions, suppression of any side product, excellent reusability, handle and inexpensiveness of the nanocatalysts, the present system illustrates comparable or better efficiency than the previous reported systems.
Table 6 Comparison of the promoter activity of CoFe2O@AA-M/NH2OH·HCl system for oximation of benzaldehyde with other reported protocols
| Entry |
Catalyst |
Condition |
Yield (%) |
Time (min) |
References |
| 1 |
SiO2@FeSO4 |
NH2OH·HCl/s.f/oil bath/70–80 °C |
96 |
12 |
34 |
| 2 |
Bi2O3 |
NH2OH·HCl/s.f |
95 |
1.5 |
40 |
| 3 |
Cu–SiO2 |
NH2OH·HCl/aq. EtOH |
90 |
2–3 h |
41 |
| 4 |
Nano Fe3O4 |
NH2OH·HCl/s.f/oil bath/70–80 °C |
100 |
20 |
33 |
| 5 |
H2C2O4 |
NH2OH·HCl/CH3CN/reflux |
95 |
60 |
35 |
| 6 |
CoFe2O4@AA-Co |
NH2OH·HCl/H2O/r.t. |
99 |
Immediate |
This work |
| 7 |
CoFe2O4@AA-V |
NH2OH·HCl/H2O/r.t. |
96 |
Immediate |
This work |
Then the catalytic activity of CoFe2O4@AA-M (Co, V) nanoparticles was examined in NaBH4 reduction of diverse nitro compounds. Reduction of nitrobenzene as a model compound with NaBH4 was performed under different reaction conditions, in order to optimize reaction conditions. The results have been indicated in Table 7. The illustrated results in Table 7 show that reduction of nitrobenzene with the alone NaBH4 in the absence of catalyst did not proceed at all (entry 1). Also, CoFe2O4 was not efficient catalyst to progress the reaction perfectly (entry 2). The illustrated results show that reduction of nitrobenzene with NaBH4 in the presence of CoFe2O4@AA-M (Co, V) in aprotic solvent was not efficient (entries 4 and 13). Analysis of the results indicates that H2O was the best solvent of choice and the reaction take place with high speed and the corresponding product was separated in high yield with 30 mg of the CoFe2O4/AA-M (Co, V) at room temperature (entries 10 and 19).
Table 7 Optimization experiments for reduction of nitrobenzene using CoFe2O4@AA-M (Co, V) nanocomposite
| Entry |
NaBH4 (mmol) |
Catalyst (mg) |
Conditiona |
Time (min) |
Conversion (%) |
| All reactions were proceeded with 1 mmol of nitrobenzene, 1.5 mL of solvent. |
| 1 |
2.5 |
— |
H2O/50 °C |
24 h |
— |
| 2 |
2.5 |
CoFe2O4 (40) |
H2O/50 °C |
120 |
40 |
| 3 |
2.5 |
CoFe2O4@AA-Co (40) |
THF/50 °C |
25 |
75 |
| 4 |
2.5 |
CoFe2O4@AA-Co (40) |
n-Hexane/50 °C |
120 |
10 |
| 5 |
2.5 |
CoFe2O4@AA-Co (40) |
EtOAc/50 °C |
20 |
78 |
| 6 |
2.5 |
CoFe2O4@AA-Co (40) |
CH3CN/50 °C |
25 |
80 |
| 7 |
2.5 |
CoFe2O4@AA-Co (40) |
EtOH/50 °C |
20 |
75 |
| 8 |
1.5 |
CoFe2O4@AA-Co (40) |
H2O/50 °C |
10 |
91 |
| 9 |
2 |
CoFe2O4@AA-Co (35) |
H2O/r.t. |
4 |
96 |
| 10 |
2 |
CoFe2O4@AA-Co (30) |
H2O/r.t. |
5 |
98 |
| 11 |
2.5 |
CoFe2O4@AA-Co (30) |
H2O/r.t. |
5 |
98 |
| 12 |
2.5 |
CoFe2O4@AA-V (40) |
THF/50 °C |
28 |
75 |
| 13 |
2.5 |
CoFe2O4@AA-V (40) |
n-Hexane/50 °C |
125 |
10 |
| 14 |
2.5 |
CoFe2O4@AA-V (40) |
EtOAc/50 °C |
22 |
76 |
| 15 |
2.5 |
CoFe2O4@AA-V (40) |
CH3CN/50 °C |
30 |
80 |
| 16 |
2.5 |
CoFe2O4@AA-V (40) |
EtOH/50 °C |
23 |
75 |
| 17 |
1.5 |
CoFe2O4@AA-V (40) |
H2O/50 °C |
11 |
90 |
| 18 |
2 |
CoFe2O4@AA-V (35) |
H2O/r.t. |
5 |
95 |
| 19 |
2 |
CoFe2O4@AA-V (30) |
H2O/r.t. |
6 |
98 |
| 20 |
2.5 |
CoFe2O4@AA-V (30) |
H2O/r.t. |
6 |
98 |
In continue, this protocol was used in reduction of diverse nitro compounds derivatives. The summarized results in Tables 8 and 9 indicate that reduction of nitro compounds having electron-releasing or withdrawing functionalities was accomplished successfully using 2–3 molar equivalents of NaBH4 and 30–40 mg of CoFe2O4@AA-M (Co, V) within 3–15 min in H2O at room temperature. Also, the case of nitroarenes having carbonyl and nitro groups show did not any selectivity and the ketones and aldehydes can both be readily reduced to alcohols (entries 8–10). It is considerable that the successful reduction of nitroaldehydes and nitroketones needed higher molar equivalents of NaBH4 and CoFe2O4@AA-M (Co, V) complexes. A more investigation showed that the present system was also effective for reduction of aliphatic nitro compounds (entry 11). The corresponding product were obtained exclusively with high turnover numbers (TON) and turnover frequencies (TOF).
Table 8 Reduction of nitro compounds with NaBH4 in the presence of CoFe2O4@AA-Co nanocompositea,b,c,d,e
Table 9 Reduction of nitro compounds with NaBH4 in the presence of CoFe2O4@AA-V nanocompositea,b,c,d,e
A conceivable mechanism for the nano CoFe2O4@AA-M (Co, V) catalyzed was shown in Scheme 3. The nanocatalysts facilitate the reduction of nitro group. It should be mentioned that there are four steps in the nitro reduction process.
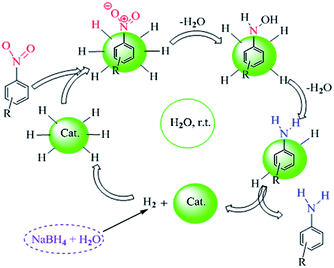 |
| | Scheme 3 A conceivable mechanism for the nano CoFe2O4@AA-M (Co, V) towards reduction of nitro compounds. | |
First hydrogen absorption occurred, then adsorbed on the metal surfaces. In the third stage, electron transfer through metal surfaces from BH4− to aromatic nitro compounds. Eventually, aromatic amino compounds desorbed from catalyst surface. Here the B–H bond cleavage occurs on the surface of CoFe2O4@AA-M (Co, V) nanocatalysts to give the [M]–H species. Such high reactive intermediates are responsible for the fast reduction of nitro compounds into the corresponding phenylhydroxylamine with very rapid reaction kinetics and possibly skips the compounds intermediate.
Thereafter, catalytic activity of CoFe2O4@AA-Co and CoFe2O4@AA-V system was distinguished by comparison of the achieved result for nitrobenzene with other reported reduction systems. The reduction of nitrobenzene in the presence of different catalysts is the selected reaction model in all cases. The results in Table 10 indicate that in terms of mild reaction conditions, easy work-up procedure, short reaction times, high recyclability, suppression of any side product, handle and inexpensively of the nanocatalysts, the present system illustrates better or comparable efficiency than the previous reported systems.
Table 10 Comparison the reduction of nitrobenzene using CoFe2O@AA-M (Co, V) nanocomposite with other reported protocols
| Entry |
Catalyst |
Condition |
Yield (%) |
Time (min) |
References |
| 1 |
Fe3O4@Cu(OH)x |
NaBH4/H2O/55–60 °C |
95 |
3 |
46 |
| 2 |
Fe3O4@PAMAM/Ni(0)-b-PEG |
NaBH4/H2O/40 °C |
91 |
120 |
51 |
| 3 |
SS-Pd |
NaBH4/MeOH·H2O/50 °C |
98 |
60 |
54 |
| 4 |
Ag/α-Fe2O3–rGO |
N2H4·H2O/H2O/r.t. |
98 |
30 |
57 |
| 5 |
Ag/Fe2O3 |
NaBH4/H2O/100 °C |
99 |
30 |
58 |
| 6 |
Fe–Cu@MCC |
NaBH4/H2O/r.t. |
93 |
8 |
59 |
| 7 |
Pd-NPs@oak gum |
NaBH4/EtOH·H2O/50 °C |
96 |
60 |
60 |
| 8 |
Pd/Fe3O4@C |
NaBH4/EtOH/25 °C |
100 |
60 |
61 |
| 9 |
UiO-66-D-PANI–AgPd |
Formic acid/H2O/90 °C |
99 |
6 h |
62 |
| 10 |
CoPd@CNT |
NaBH4/MeOH:H2O/r.t. |
99 |
3 |
63 |
| 11 |
Graphene–ZnO–Au |
H2O/r.t./(UV-Vis lamp) |
97 |
140 |
64 |
| 12 |
Pd–NHC–γ-Fe2O3–n-butyl SO3H |
NaBH4/EtOH·H2O/r.t. |
93 |
3 h |
65 |
| 13 |
Fe3O4@APTMS@ZrCp2 |
Glycerol/H2O/r.t. |
96 |
40 |
67 |
| 14 |
Fe3O4@Ni |
NaBH4/H2O/r.t. |
95 |
25 |
68 |
| 15 |
CoFe2O4@AA-Co |
NaBH4/H2O/r.t. |
96 |
3 |
This work |
| 16 |
CoFe2O4@AA-V |
NaBH4/H2O/r.t. |
95 |
3 |
This work |
2.4 Recycling CoFe2O4@AA-M (Co, V)
As one of the interesting advantages of nonmagnetic catalysts is their easy isolation from the reaction mixture by applying an external magnet, the possibility of the magnetic recycling of catalysts were also tested. Thus, initially the recovery and reusability of CoFe2O4@AA-Co and CoFe2O4@AA-V were investigated towards NH2OH·HCl oximation of benzaldehyde and then for reduction of nitrobenzene with NaBH4. For this reason, after completion of the reactions, the catalysts were easily separated from the reaction mixture using an external magnet and washed carefully with ethanol then dried under air atmosphere for additional using at the subsequent runs. It is clear that the efficiency of the investigated catalysts was restored after several successive runs with a low decreasing of its catalytic activity (Fig. 11 and 12).
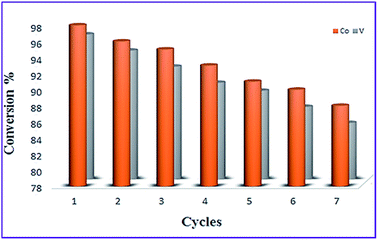 |
| | Fig. 11 Reusability of CoFe2O4@AA-M (Co, V) towards NH2OH·HCl oximation of benzaldehyde under optimize conditions. | |
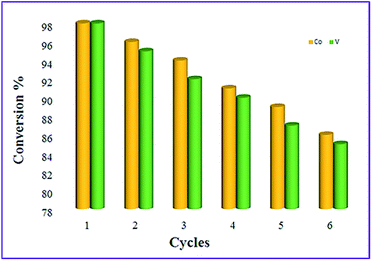 |
| | Fig. 12 Reusability of CoFe2O4@AA-M (Co, V) for the reduction of nitrobenzene with NaBH4 under optimized conditions. | |
In order to identify the changes in the chemical structure of the catalysts towards NH2OH·HCl oximation of benzaldehyde during the six cycles, FE-SEM and FT-IR analyses were performed and the results are demonstrated below (Fig. 13a and b). These investigations clarified that CoFe2O4@AA-M (Co, V) catalysts retained its chemical structure after the longevity of experiments. It is noteworthy that investigation of catalyst reusability after reduction of nitro compounds also display the same results and confirmed the catalyst efficiency after several runs.
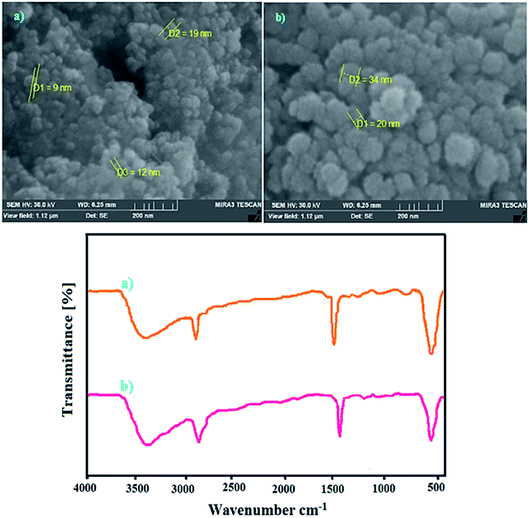 |
| | Fig. 13 FE-SEM and FT-IR analyses of reused catalysts for reduction of nitrobenzene with NaBH4 under optimized conditions after 6 runs (a) CoFe2O4@AA-Co and (b) CoFe2O4@AA-V. | |
2.5 Catalyst leaching study
The value of Co and V leaching in reduction of nitro compounds was investigated by comparing the metal anchoring value before and after recovering of the catalyst by ICP-OES method. Result showed that the value of Co in fresh catalyst and the recycled one after 6 times is 0.52 and 0.49 mmol g−1 respectively and the value of V in fresh catalyst and the recycled one after 6 times is 0.50 and 0.47 mmol g−1 respectively which illustrated the minimal Co and V leaching in the catalytic process demonstrated the efficiency and stability of the catalyst.
2.6 Hot filtration test
In continuation of current protocol, efficiency of introduced catalysts were examined with hot filtration test. The oximation of benzaldehyde in the presence of reported catalysts was selected to carried out hot filtration test. Under optimized conditions, oximation of benzaldehyde was proceeded in the presence of CoFe2O4@AA-Co and CoFe2O4@AA-V in which the yields of products in the half time of the reaction were 70 and 68%, respectively. Then the reaction was repeated and the nanocatalyst was isolated in half time of the reaction. Thereupon, continuous reaction was carried out without CoFe2O4@AA-Co and CoFe2O4@AA-V in which the yields of reaction were 73 and 70%, respectively. These observations corroborated that the leaching of metal has not been happened.
3. Experimental
3.1 Instruments and materials
All chemicals and solvents were purchased from Sigma-Aldrich and Merck chemical companies and were used without extra purification. The surface morphology and diameter of the CoFe2O4/AA-M (Co, V) nanocatalysts were studied by scanning electron microscopy (FE-SEM) analysis data was recorded SEM-TESCAN MIRA3. Transmission electron microscopy (TEM) was carry out using a FEI CM200 field emission at accelerating voltage of 80 kV. The XRD was recorded on JEOL JSM-6100 microscope with (Cu Kα radiation, λ = 1.54 Å). The thermal gravimetric analysis (TGA) of nanocomposite was carried out on a Shimadzu analyzer DTG-60. Magnetic properties of the samples were determined using a vibrating sample magnetometer (VSM) at room temperature (MDKFD, University of Kashan, Iran). The amounts of Co and V in synthesized the nanocatalysts were evaluated by plasma-optical emission spectrometry (ICP-OES). Infrared (FT-IR) spectra of all samples were recorded on PerkinElmer Spectrum one instruments, using KBr pellets in the range of 400–4000 cm−1.
3.2 Synthesis of CoFe2O4@AA nanocomposite
To synthesize superparamagnetic CoFe2O4 as catalyst support, a solution of 50 mL of Fe(NO3)3·9H2O (16 mmol, 6.46 g) was added to a solution of 25 mL of Co(NO3)2·6H2O (8 mmol, 2.32 g) and the mixture was vigorously stirred. Then, sodium hydroxide 3 M was added dropwise by stirring until the reaction mixture reached pH = 11–12 then stirring was continued at 80 °C for one hour. Subsequently, the final product was isolated using an external magnet and washed three times with double distilled hot water and ethanol. At the end, the magnetic nanocomposites (CoFe2O4) dried in oven at 60 °C.
Similarly, to produce CoFe2O4@AA, a solution of ascorbic acid (1 g in 25 mL double distilled water) was added dropwise to suspensions of 2 g CoFe2O4 in 30 mL double distilled water. Then, the reaction mixture was stirred for 24 hours at room temperature. Finally, the prepared nanocomposite was isolated using an external magnet washed with double distilled water/ethanol several times and dried in vacuum oven at 60 °C.
3.3 Synthesis of CoFe2O4@AA-Co nanostructure
To expand the scope of the processes, we produced the CoFe2O4@AA-Co by blending sonicated CoFe2O4@AA (1 g) in 50 mL of double distilled water and a solution of 5 mL CoCl2·6H2O (0.476 g, 2 mmol) was added to mentioned mixture and stirred under reflux for 24 hours. Then, the achieved product was isolated using an external magnet and washed with double distilled water/ethanol several times then dried in a vacuum oven at 60 °C.
3.4 Synthesis of CoFe2O4@AA-V nanostructure
In this step, CoFe2O4@AA (1 g) was dispersed in 50 mL of double distilled water with ultrasonication for 10 min. Subsequently, a solution of 5 mL VCl3 (0.314 g, 2 mmol) was added to mentioned mixture and stirred under reflux for 24 hours. The resultant product separated using an external magnet and washed with double distilled water/ethanol several times and, finally, dried in a vacuum oven at 70 °C.
3.5 A general procedure for oximation of aldehydes with NH2OH·HCl in the presence of CoFe2O4@AA-M (Co, V) system
In a round-bottom flask (10 mL) equipped with a magnetic stirrer, a solution of aldehyde (1 mmol) in H2O (1.5 mL) was provided. After 1 min, hydroxylamine hydrochloride (1 mmol, 0.069 g) was added and the solution was stirred at room temperature for 30 s. To the prepared solution CoFe2O4@AA-Co (30 mg) was added and stirring of the reaction mixture was carried on at room temperature. Progress of the reaction was monitored using TLC (eluent: n-hexane/EtOAc: 5/2). After completion of the reaction, H2O (3 mL) was added and the mixture was stirred for 5 min. The aldoxime product was extracted with EtOAc (2 × 4 mL) then the organic layer was dried over anhydrous Na2SO4. Evaporation of the solvent caused the pure aldoxime in excellent yield (Table 4, entry 1). The pure product was obtained upon column chromatography.
3.5.1 Benzaldoxime. Yellow oil; 1H NMR (500 MHz, CDCl3) δ 9.26 (br, 1H), 8.28 (s, 1H), 7.64–7.60 (m, 2H), 7.46–7.43 (m, 3H). 13C NMR (125 MHz, CDCl3) δ 153.35, 132.79, 131.05, 129.02, 126.78.
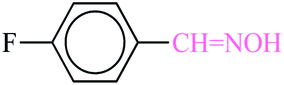
3.5.2 4-Fluorobenzaldoxime. White solid; 1H NMR (500 MHz, CDCl3) δ 8.22 (s, 1H), 7.64–7.59 (m, 2H), 7.15–7.1 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 164, 162.3, 149.45, 129, 127.89, 115.96.
3.5.3 4-Nitrobenzaldoxime. White solid; 1H NMR (500 MHz, DMSO-d6) δ 11.8 (br, 1H), 8.27 (s, 1H), 8.32 (d, J 1/4 8.5 Hz, 2H), 7.92 (d, J 1/4 8.5 Hz, 2H). 13C NMR (125 MHz, DMSO-d6) δ 146.75, 147, 140, 127.14, 124.04.
3.5.4 2-Chlorobenzaldoxime. White solid; 1H NMR (500 MHz, CDCl3) δ 8.75 (br, 1H), 7.92 (dd, J 1/4 7.5, 1.5 Hz, 1H), 7.43 (dd, J 1/4 7.5, 1.2 Hz, 1H), 7.42 (td, J 1/4 7.5, 1.2 Hz, 1H), 7.36–7.31 (m, 1H). 13C NMR (125 MHz, CDCl3) δ 148.1, 134.06, 130.81, 129.74, 129.8, 126.87, 127.19.
3.6 A general procedure for reduction of nitro compounds with NaBH4 in the presence of CoFe2O4@AA-M (Co, V) system
In a round-bottom flask (10 mL) equipped with a magnetic stirrer, a solution of nitrobenzene (1 mmol) in H2O (1.5 mL) was provided. After 1 min, sodium borohydride (2 mmol, 0.076 g) was added and the solution was stirred at room temperature for 30 s. To the prepared solution CoFe2O4@AA-Co (30 mg) was added and stirring of the reaction mixture was carried on at room temperature. Progress of the reaction was monitored using TLC (eluent: n-hexane/EtOAc: 5/2). After completion of the reaction, H2O (3 mL) was added and the mixture was stirred for 5 min. The aniline product was extracted with EtOAc (2 × 4 mL) then the organic layer was dried over anhydrous Na2SO4. Evaporation of the solvent caused aniline in excellent yield (Table 8, entry 1) and the column chromatography afforded the pure aniline.
3.6.1 Aniline. Yellow oil; 1H NMR (400 MHz, CDCl3) δ = 3.48 (s, 2H), 6.62–6.79 (m, 3H), 7.14 ppm (t, J = 8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ = 115.1, 117.88, 129.8, 146.5 ppm.
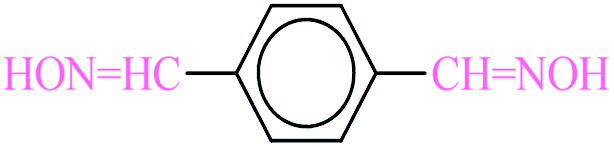
3.6.2 Benzene-1,4-diamine. White solid; 1H NMR (400 MHz, CDCl3) δ = 3.38 (s, 4H), 6.61 (s, 4H); 13C NMR (100 MHz, CDCl3) δ = 117.2, 138.7 ppm.
3.6.3 4-Hydroxymethylaniline. Colorless solid; 1H NMR (CDCl3, 500 MHz): δ 7.21–7.18 (m, 2H), 6.72–6.66 (m, 2H), 4.78 (s, 2H) 3.67 (br, 2H); 13C NMR (CDCl3, 125 MHz): δ 146.1, 128.9, 128.6, 115.2, 65.1.
3.6.4 2-Amino-phenol. White solid; 1H NMR (500 MHz, DMSO-d6) δ: 8.96 (s, 1H), 6.63–6.61 (m, 1H), 6.54–6.59 (m, 1H), 6.52–6.55 (m, 1H), 6.31–6.35 (m, 1H), 4.40 ppm (s, 2H); 13C NMR (125 MHz, DMSO-d6) δ = 145, 137, 119.8, 116.7, 115, 114.7 ppm.
4. Conclusions
In present work we successfully exhibited an effectual practice for the synthesis of two efficient and green reusable heterogeneous catalytic system, CoFe2O4@AA-Co and CoFe2O4@AA-V obtained by anchoring Co or V onto the face of CoFe2O4. Next, these nanomaterials were identified by FT-IR, FE-SEM, TEM, EDX, TGA, XRD, VSM, X-ray atomic mapping and ICP-OES techniques. The catalytic status of these nanostructures was obtained as a recyclable system for the oximation of multiple aldehyde combinations by hydroxylamine hydrochloride in H2O/r.t. conditions. Aldoximes were produced in great yields within immediate to 7 min. In addition, the catalytic efficiency of introduced nanocatalysts were tested toward NaBH4 reduction of different nitro compounds. All reduction reactions were performed in H2O within 3–15 min to obtain amines in high yields. CoFe2O4@AA-M (Co, V) were easily separated from the reaction combination using an external magnet and reused in both oximation and reduction reactions several times without considerable loss of its catalytic activity. Finally, these catalysts are preferable over the other catalysts because of their supreme properties, such as easy preparation, short reaction times, clean reaction conditions, suppression of any side product and simple work-up method. So, we believe that nano CoFe2O4@AA-Co and CoFe2O4@AA-V systems could be considered as useful and new addition to the present methodologies in these scopes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the support of this work by University of Kurdistan.
References
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford university, Oxford, 1998, vol. 1 Search PubMed.
-
(a) R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481 RSC;
(b) A. Schatz, T. R. Long, R. N. Grass, W. J. Stark, P. R. Hanson and O. Reiser, Adv. Funct. Mater., 2010, 20, 4323 CrossRef.
-
(a) M. B. Gawande, A. K. Rathi, P. S. Branco, T. M. Potewar, A. Velhinho, I. D. Nogueira, A. Tolstogouzov, C. Amjad, A. Ghumman and O. M. N. D. Teodoro, RSC Adv., 2013, 3, 3611 RSC;
(b) M. B. Gawande, S. N. Shelke, A. Rathi, P. S. Branco and R. K. Pandey, Appl. Organomet. Chem., 2012, 26, 395 CrossRef CAS;
(c) M. B. Gawande, V. D. B. Bonifácio, R. S. Varma, I. D. Nogueira, N. Bundaleski, C. Amjad, A. Ghumman, O. M. N. D. Teodoro and P. S. Branco, Green Chem., 2013, 15, 1226 RSC.
- R. Sato Turtelli, G. V. Duong, W. Nunes, R. Grossinger and M. Knobel, J. Magn. Magn. Mater., 2008, 320, 339 CrossRef.
- E. S. Murdock, R. F. Simmons and R. Davidson, IEEE Trans. Magn., 1992, 28, 3078 CAS.
- P. Majewski and B. Thierry, Crit. Rev. Solid State Mater. Sci., 2007, 32, 203 CrossRef CAS.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995 CrossRef CAS.
- S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428 CrossRef CAS.
- V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743 RSC.
- D. L. Graham, H. A. Ferreira and P. P. Freitas, Trends Biotechnol., 2004, 22, 455 CrossRef CAS.
- C. Pereira, A. M. Pereira, C. Fernandes, M. Rocha, R. Mendes, M. P. Fernández-García, A. Guedes, P. B. Tavares, J. M. Grenèche and J. o. P. Araújo, Chem. Mater., 2012, 24, 1496 CrossRef CAS.
- L. Wu, A. Mendoza-Garcia, Q. Li and S. Sun, Chem. Rev., 2016, 116, 10473 CrossRef CAS.
- H. Pardoe, W. Chua-anusorn, T. G. S. Pierre and J. Dobson, J. Magn. Magn. Mater., 2001, 225, 41 CrossRef CAS.
- B. Zumreoglu-Karan, Coord. Chem. Rev., 2006, 250, 2295 CrossRef.
- Sh. Zhao, L. Huang and Y. F. Song, Eur. J. Inorg. Chem., 2013, 1659 CrossRef CAS.
- R. Raja, G. Sankar and J. M. Thomas, J. Am. Chem. Soc., 2001, 123, 8153 CrossRef CAS.
- S. Zhao, L. Liu and Y. F. Song, Dalton Trans., 2012, 41, 9855 RSC.
- R. García-Álvarez, A. E. Díaz-Álvarez, J. Borge, P. Crochet and V. Cadierno, Organometallics, 2012, 31, 6482 CrossRef.
- D. Tyagi, R. K. Rai, A. D. Dwivedi, S. M. Mobina and S. K. Singh, Inorg. Chem. Front., 2015, 2, 116 RSC.
- P. Han, J. Li, C. Li Xing, M. Zhao, Q. Xia Han and M. Xue Li, Inorg. Chem. Commun., 2019, 110, 107592 CrossRef CAS.
- S. Z. Xing, Q. X. Han, Z. L. Shi, S. G. Wang, P. P. Yang, Q. Wu and M. X. Li, Dalton Trans., 2017, 46, 11537 RSC.
- G. Sosnovsky, J. A. Krogh and S. G. Umhoefer, Synthesis, 1979, 9, 722 CrossRef.
- P. Miller and D. H. Kaufman, Synlett, 2000, 8, 1169 Search PubMed.
- R. S. Ramon, J. Bosson, S. Diez-Gonzalez, N. Marion and S. P. Nolan, J. Org. Chem., 2010, 75, 1197 CrossRef CAS.
- E. Abele, R. Abele, O. Dzenitis and E. Lukevics, Chem. Heterocycl. Compd., 2003, 39, 3 CrossRef CAS.
- I. Damljanovic, M. Vukicevic and R. D. Vukicevic, Monatsh. Chem., 2006, 137, 301 CrossRef CAS.
- I. M. Osadchenko and A. P. Tomilov, Russ. J. Appl. Chem., 2002, 75, 511 CrossRef CAS.
- U. P. Lad, M. A. Kulkarni and R. S. Patil, Rasayan J. Chem., 2010, 3, 425 Search PubMed.
- D. Setamdideh, B. Khezri and S. Esmaeilzadeh, J. Chin. Chem. Soc., 2012, 59, 1119 CrossRef CAS.
- J. J. Xia and G. W. Wang, Molecules, 2007, 12, 231 CrossRef CAS.
- J. J. Guo, T.-S. Jin, S.-L. Zhang and T. S. Li, Green Chem., 2001, 3, 193 RSC.
- H. Sharghi and M. Hosseini, Synthesis, 2002, 8, 1057 CrossRef.
- B. Zeynizadeh and M. Karimkoshteh, J. Nanostruct. Chem., 2013, 3, 57 CrossRef.
- M. Karimkoshteh, M. Bagheri and B. Zeynizadeh, Nanochem. Res., 2016, 1, 57 CAS.
- P. N. Ghozlojeh and D. Setamdideh, Orient. J. Chem., 2015, 31, 1823 CrossRef.
- G. L. Kad, M. Bhandari, J. Kaur, R. Rathee and J. Singh, Green Chem., 2001, 3, 275 RSC.
- B. Zeynizadeh and E. Amjadi, Asian J. Chem., 2009, 21, 3611 CAS.
- B. R. Kim, G. H. Sung, J. J. Kim and Y. J. Yoon, J. Korean Chem. Soc., 2013, 57, 295 CrossRef CAS.
- J. T. Li, X. L. Li and T. S. Li, Ultrason. Sonochem., 2006, 13, 200 CrossRef CAS.
- L. Saikia, J. M. Baruah and A. J. Thakur, Org. Med. Chem. Lett., 2011, 1, 1 CrossRef.
- K. Ramanjaneyulu, P. Seshagiri Rao, T. Rambabu, K. Jayarao, C. B. T. Sundari Devi and B. Venkateswara Rao, Der Pharma Chem., 2012, 4, 473 Search PubMed.
- H. Sharghi and M. H. Sarvari, J. Chem. Res., 2000, 1, 24 CrossRef.
- A. Elmakssoudi, K. Abdelouahdi, M. Zahouily, J. Clark and A. Solhy, Catal. Commun., 2012, 29, 53 CrossRef CAS.
- M. Ait Taleb, R. Mamouni, N. Saffaj, A. Mouna, M. L. Taha, A. Benlhachemi, B. Bakiz, M. Ezahri and S. Villain, J. Mater. Environ. Sci., 2016, 7, 4580 Search PubMed.
- B. Zeynizadeh and S. Sorkhabi, Curr. Chem. Lett., 2020, 9, 121 CrossRef.
- Z. Shokri, B. Zeynizadeh, S. A. Hosseini and B. Azizi, J. Iran. Chem. Soc., 2017, 14, 101 CrossRef CAS.
- M. Niakan and Z. Asadi, Catal. Lett., 2019, 149, 2234 CrossRef CAS.
- B. Zeynizadeh and S. Sorkhabi, J. Chem. Soc. Pak., 2016, 38, 679 CAS.
- B. Zeynizadeh and M. Zabihzadeh, J. Iran. Chem. Soc., 2015, 12, 1221 CrossRef CAS.
- B. Zeynizadeh, M. Zabihzadeh and Z. Shokri, J. Iran. Chem. Soc., 2016, 13, 1487 CrossRef CAS.
- S. J. Tabatabaei Rezaei, A. Mashhadi Malekzadeh, S. Poulaei, A. Ramazani and H. Khorramabadi, Appl. Organomet. Chem., 2018, 32, e3975 CrossRef.
- P. L. Gkizis, M. Stratakis and I. N. Lykakis, Catal. Commun., 2013, 36, 48 CrossRef CAS.
- Y. Motoyama, Y. Lee, K. Tsuji, S. H. Yoon, I. Mochida and H. Nagashima, ChemCatChem, 2011, 3, 1578 CrossRef CAS.
- A. K. Shil, D. Sharma, N. R. Guha and P. Das, Tetrahedron Lett., 2012, 53, 4858 CrossRef CAS.
- R. Nie, J. Wang, L. Wang, Y. Qin, P. Chen and Z. Hou, Carbon, 2012, 50, 586 CrossRef CAS.
- E. Kim, H. S. Jeong and B. M. Kim, Catal. Commun., 2014, 45, 25 CrossRef CAS.
- B. Paul, D. D. Purkayastha, S. S. Dhar, S. Das and S. Haldar, J. Alloys Compd., 2016, 681, 316 CrossRef CAS.
- A. K. Patra, N. T. Vo and D. Kim, Appl. Catal., A, 2017, 538, 148 CrossRef CAS.
- S. Karami, B. Zeynizadeh and Z. Shokri, Cellulose, 2018, 25, 3295 CrossRef CAS.
- H. Veisi, N. H. Nasrabadi and P. Mohammadi, Appl. Organomet. Chem., 2016, 30, 890 CrossRef CAS.
- N. Mei and B. Liu, Int. J. Hydrogen Energy, 2016, 41, 17960 CrossRef CAS.
- V. Babel and B. L. Hiran, Catal. Lett., 2020, 150, 1865 CrossRef CAS.
- H. Goksu, B. Celik, Y. Yildiz, F. Sen and B. Kilbas, ChemistrySelect, 2016, 1, 2366 CrossRef.
- P. Roy, A. P. Periasamy, C. T. Liang and H. T. Chang, Environ. Sci. Technol., 2013, 47, 6688 CrossRef CAS.
- S. Sobhani, F. O. Chahkamalia and J. M. Sansano, RSC Adv., 2019, 9, 1362 RSC.
- M. R. Nabid, Y. Bide and M. Niknezhad, ChemCatChem, 2014, 6, 538 CrossRef CAS.
- B. Zeynizadeh and F. Sepehraddin, J. Iran. Chem. Soc., 2017, 14, 2649 CrossRef CAS.
- P. S. Rathore, R. Patidar, T. Shripathi and S. Thakore, Catal. Sci. Technol., 2015, 5, 286 RSC.
- M. Hajjami, A. Ghorbani-Choghamarani, R. Ghafouri-Nejad and B. Tahmasbi, New J. Chem., 2016, 40, 3066 RSC.
- Z. Rappoport, CRC Handbook of Tables for Organic Compound Identification, Boca Raton, 3rd edn, 1966 Search PubMed.
- L. Brehm and J. Watson, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 3646 CrossRef CAS.
- O. Mazaheri and R. J. Kalbasi, RSC Adv., 2015, 5, 34398 RSC.
- http://www.sigmaaldrich.com, accessed 24 Sep 2020.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08244a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Mohammad Ghadermazi
,
Mohammad Ghadermazi