
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceObservation of glucose-6-phosphate anomeric exchange in real-time using dDNP hyperpolarised NMR†
Sivaranjan Uppala,
Ayelet Gamliel,
Gal Sapir ,
Jacob Sosna,
J. Moshe Gomori and
Rachel Katz-Brull*
,
Jacob Sosna,
J. Moshe Gomori and
Rachel Katz-Brull*
Department of Radiology, Hadassah Medical Center, Hebrew University of Jerusalem, The Faculty of Medicine, Jerusalem, Israel. E-mail: rkb@hadassah.org.il
First published on 11th November 2020
Abstract
A hyperpolarised-NMR acquisition approach that is sensitive to the process of glucose-6-phosphate anomerization is presented. Using selective depolarisation of one of the anomer's signals, it is possible to observe the replenishing of this signal due to the fast anomeric exchange of this compound. The forward to reverse reaction rate constants ratio was ca. 1.6.
The anomerization rate of glucose (Glc) intrigued researchers back at the turn of the 20th century.1 Numerous studies into both Glc and glucose-6-phosphate (G6P) anomerization have been performed since,2–15 due to their importance for the activities of anomer specific enzymes such as G6P dehydrogenase (which converts D-G6P to 6-phospho-D-glucono-1,5-lactone and is specific to β-G6P2,16), phosphoglucomutase (which converts G6P to G1P, and vice versa, and is specific to α-G6P2), and G6P isomerase (which interconverts G6P and fructose-6-phosphate (F6P) and is specific to α-G6P2 and α-F6P17). G6P is the product of Glc phosphorylation which is carried out by hexokinases including glucokinase. From basic principles, the G6P anomer that is formed in this reaction is determined by the Glc anomer that serves as a precursor for this reaction.2,14 Nevertheless, anomeric equilibrium of G6P will lead to the formation of the other G6P anomer, even if starting from a single Glc anomer (ESI, Scheme S1†). The Glc anomeric selectivity of hexokinase from different organisms and organs have been extensively studied and was found to depend on multiple variables such as temperature and the hexose concentration.18–20 Recently, we have shown that yeast hexokinase shows selectivity for the β anomer of 2-deoxyglucose (2DG), a Glc derivative.21 Additionally, we have shown that following the production of β-2DG-6-phosphate (β-2DG6P), anomeric equilibrium produces α-2DG6P.21 Since G6P is a branch point for Glc metabolism and because G6P dehydrogenase, phosphoglucomutase, and G6P isomerase focus on different anomers of G6P, the anomerization of G6P is mechanistically important for the understanding of metabolic pathways routing.
However, the methodology for monitoring the anomerization process of the phosphorylated sugar is not trivial, specifically due to the speed of this process, which is at least two orders of magnitude faster than that of Glc.2,3,22 In the 1960s, the methodologies for determining the anomeric conversion rates for Glc and G6P were indirect, for example necessitating a conversion to trimethylsilyl ethers for gas chromatography.6,7 Other methods included calorimetry2 and spectrophotometric measurements.2,4,9,15 Later studies observed the two anomers of Glc and its derivatives using 1H-,6 31P-5 and 13C-nuclear magnetic resonance (NMR) in solution.
Simultaneous detection and differentiation between the anomers of Glc and G6P is done by 13C NMR using the signals of the C1 position. The C1 signals of both anomers of are ca. 4 ppm apart and therefore can be resolved. Due to the low sensitivity of 13C NMR, observing the anomerization process of sugars is challenging at thermal equilibrium, even when using 13C-labeled compounds. Nevertheless, the two anomers have been observed in vivo in animal models23 and in the human skeletal muscle24 and brain.25,26 However, using the C1 signals it is typically not possible to discern between 13C-labeled Glc and G6P in vivo due to the small chemical shift difference.
Mechanistic studies of Glc metabolism anomer specificity have typically used a specific anomer as a substrate. However, anomerization readily takes place as soon as the solid Glc or G6P are dissolved in water. At equilibrium, at 37 °C, a Glc solution contains 64% of the β and 36% of the α anomers. At 25 °C, the half-life for the anomeric exchange is 30–60 min (α to β and β to α, respectively),3 and reports on the half-life in other temperatures vary.27,28 Nevertheless, it is likely and generally assumed that at lower temperatures this half-life is longer. Therefore, experiments aiming at assessing the anomeric specificity of D-Glc metabolism were reported using low temperatures.18 However, studies at lower temperatures affect enzyme activity. Another approach is to carry out the reaction in D2O which was found to prolong the life time of the Glc anomers in solution.7,12 However, D2O may affect enzyme activities14 due to H–D exchange of protein bound protons. In addition, the binding of Glc anomers was studied by saturation transfer difference NMR (STD NMR).29
We note that NMR methods that are based on 1H detection per se, in the absence of isotopic label, whether direct or via chemical exchange saturation transfer (CEST)30 cannot differentiate between Glc anomers. The transition states for the ring-opening and ring closing in the mutarotation reactions of Glc were studied by saturation transfer 13C NMR,31 and mutarotase activity on Glc32 and on its sulfonated derivatives33 were studied by 1H and 13C NMR exchange spectroscopy (EXSY) in one and two dimensions. Rate constants for anomerization of furanose sugars in D2O were obtained by 1H and 13C saturation-transfer NMR spectroscopy34 but the use of D2O has been shown to slow down the anomerization of G6P.4
The dissolution-dynamic nuclear polarisation (dDNP) approach35 has revolutionised the ability of NMR to monitor chemical and biochemical processes much faster and using physiologically relevant substrate concentrations. Furthermore, kinetic analysis through selective dDNP enhanced signal saturation or inversion has been reported.36 Using the dDNP approach, the distribution of stable isotope-labelled Glc and its metabolites can be investigated at 3–4 orders of magnitude higher signal-to-noise ratio (SNR) compared to thermal equilibrium NMR spectroscopy studies.21,37 [13C6,D7]Glc has been utilised in several dDNP studies both as a metabolic agent14,21,38–41 and as an imaging agent.19,37 The labelling with 13C is required for increasing the 13C-NMR SNR. The deuteration of the 13C sites (deuterium substitution of directly bound protons) is necessary to prolong the T1 of these directly bound 13C nuclei,37,42 to enable observation of the hyperpolarised state and thus allow detection of chemical and enzymatic processes. Still, the lifetimes of the hyperpolarised sites of this stable-isotope labelled Glc analogue are relatively short, with T1 relaxation times of about 10 to 20 s.21 Therefore, this technique is suitable for investigation of fast processes, of the order of seconds. Although the C1 positions of [13C6,D7]Glc and [13C6,D7]G6P anomers have been previously monitored in dDNP studies of hexokinase reactions,13,21 their anomeric exchange rates were not characterised. Here we have devised a selective RF pulse excitation strategy and an experimental dDNP approach for monitoring the anomeric exchange of [13C6,D7]G6P in real-time.
13C-NMR of [13C6,D7]Glc and [13C6,D7]G6P offers a unique capability to observe both the α and β anomers of these compounds in solution. This is because for both these compounds the chemical shift difference between the C1α and the C1β signals is large enough to be observed, even in the presence of the complex split signals due to the J-coupling with the adjacent 13C and D nuclei.21 Hyperpolarization of [13C6,D7]Glc yields NMR signals that can be monitored with a temporal resolution of the order of seconds which is important when attempting to observe fast anomeric exchange processes. In addition, we have capitalised on the properties of a uniformly labelled compound in a hyperpolarised state, where there is a potential to quench one of the signals specifically. This is because the quenched signal cannot return to its starting level as the signal will relax to thermal equilibrium and the hyperpolarised signal is 3–4 orders of magnitude larger than that.
Our aim in this study was to null the signal of one of the anomers in a reaction mixture that contained both anomers in equilibrium and then observe and quantify the build-up of the nullified anomer signal as the anomeric equilibrium took place. It is important to note that nulling the signal of one of the anomers does not modify the reaction composition or the equilibrium process, but only the presentation in the NMR spectrum: the remaining hyperpolarised state in one of the anomers operates as a label, to highlight the formation of the other. To the best of our knowledge, this is the first investigation of anomeric exchange using hyperpolarised NMR.
First, we investigated this approach on hyperpolarised [13C6,D7]Glc which was introduced into a hexokinase reaction medium, but without the enzyme. The following acquisition strategy was taken: first, a series of ten frequency selective RF pulses was applied to fully excite only the C1 signal of the β anomer. The lower panel in ESI Fig. S1A† (time 1–10 s), demonstrates that indeed only this signal was excited and decayed quickly due to this massive depolarisation, it also demonstrates the need for the application of several selective pulses. Immediately after this selective depolarisation, a series of hard (non-selective) RF pulses was applied to excite the entire 13C bandwidth with a low flip angle (10°). The resulting spectra displayed all of the expected hyperpolarised [13C6,D7]Glc signals except for that of the C1β signal (which was depolarised by the previous selective pulse series). This result indicated that once depolarised, the C1β signal of hyperpolarised [13C6,D7]Glc cannot regain its intensity, even though all of the other 13C sites retain their hyperpolarised state. We note that we have specifically chosen to use formulations in which the starting material, [13C6,D7]Glc, was already in anomeric equilibrium in order to monitor the solution composition at equilibrium (ESI Note S3,† Materials and methods, dDNP).
To validate this experimental approach and make sure that the application of the frequency selective RF pulses does not affect the decay of other hyperpolarised sites, we acquired hyperpolarised signals from a similar hyperpolarised solution of [13C6,D7]Glc using only the series of non-selective, low flip-angle pulses. Indeed, all of the signals of hyperpolarised 13C were observed (ESI Fig. S1B†) and appeared to decay at a similar decay rate (ESI Fig. S1C and Table S1†). This acquisition approach was further validated as described in the ESI S2.†
Next, we applied the same acquisition strategy (selective pulses on C1β followed by non-selective acquisition) on a hyperpolarised [13C6,D7]Glc solution which was combined with a solution containing the enzyme hexokinase. This reaction led to a quick conversion of [13C6,D7]Glc to [13C6,D7]G6P, as confirmed by the immediate appearance of the C6 signal of [13C6,D7]G6P (Fig. 1A). As opposed to the experiment without the enzyme, a signal at the C1β chemical shift was observed as well, despite the preceding depolarisation of this signal by the selective RF pulses. However, both C1β and C1α signals appeared at a relatively lower intensity compared to those of hyperpolarised [13C6,D7]Glc which did not undergo an enzymatic reaction and did not undergo selective anomer depolarisation (Fig. S1B†). In the non-selective acquisition (Fig. 1A, upper panel), the signal of C6 position showed a higher level of [13C6,D7]G6P than [13C6,D7]Glc already in the first spectrum. This suggested that a significant proportion of the reaction occurred during the application of the selective pulses. Because the anomeric exchange rate of G6P is higher than that of Glc3 and because in the experiments without the enzyme the C1β signal was fully depolarised, it appeared likely that the C1β observed in this experiment is of [13C6,D7]G6P and not of [13C6,D7]Glc.
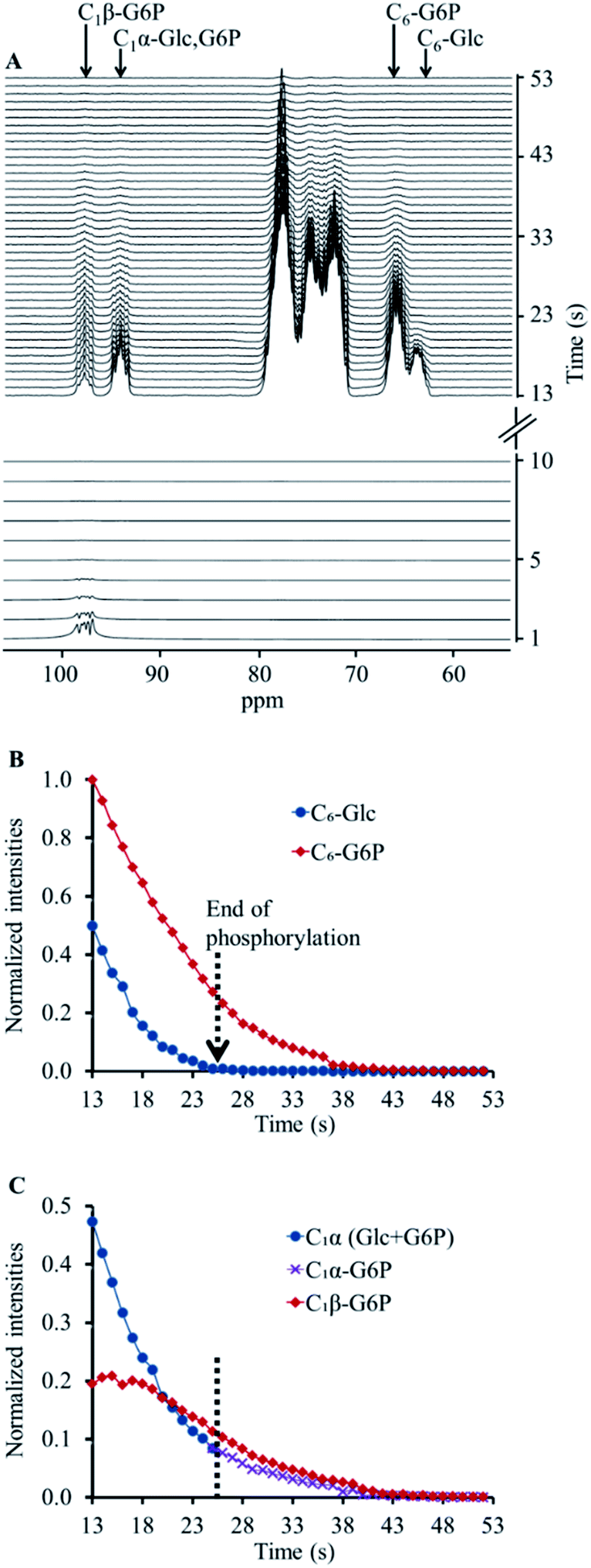 | ||
| Fig. 1 Hyperpolarised [13C6,D7]Glc in a reaction mixture with hexokinase (5.43 mg). (A) Lower panel, selective excitation and depolarisation of the C1β position for 10 s with a TR of 1 s. Upper panel, subsequent non-selective (hard pulse) excitations (from the 13th second) with the same TR. (B) A time course showing the signal decay for the chemical shifts of C6-[13C6,D7]Glc and C6-[13C6,D7]G6P determined using the non-selective excitations (upper panel in A). The dotted arrow indicates the time at which the phosphorylation reaction ended, as determined by the disappearance of the C6-[13C6,D7]Glc signal from the spectra. (C) A time course showing the signal decay for the chemical shifts of C1α and C1β positions determined using the non-selective excitations (upper panel in A). The dotted arrow indicates the time at which the phosphorylation reaction ended (according to B). From this time onwards, the C1α and C1β signals can be considered those of [13C6,D7]G6P and were used in the analysis of [13C6,D7]G6P anomeric exchange. | ||
The appearance of hyperpolarised C1β signal in this experiment and the overall lower intensity of the C1 position signals can be explained as follows. Hyperpolarised [13C6,D7]Glc underwent phosphorylation during the application of the selective depolarizing pulses on C1β. The C1β of [13C6,D7]Glc and of the newly formed [13C6,D7]G6P were continuously depolarised. Due to the anomeric equilibrium of [13C6,D7]G6P, the signal of the C1α position of [13C6,D7]G6P was also affected as it exchanged with a depolarised position. In agreement, the signal of the C1β position of [13C6,D7]G6P exchanged with a hyperpolarised position and therefore appeared in the spectra. Thus, this acquisition strategy appeared to be sensitive to the anomeric exchange of [13C6,D7]G6P in real-time. Further testing and validation of this explanation is provided in ESI S3.†
The decay of the C1α and C1β signals of [13C6,D7]G6P in two enzymatic experiments (Fig. 1 and ESI S3†) is shown in ESI Fig. S4A and B,† respectively. These data served to determine the anomeric exchange rate of [13C6,D7]G6P using a kinetic model previously developed to determine reaction rates of hyperpolarised substrates and products, whereas the respective NMR signals decay during the reaction according to each site's specific T1.43 Here, the C1α was taken as the substrate and the anomeric exchange was taken as the reaction leading to C1β with a reaction rate constant kforward. The reverse reaction, where C1β is converted back to C1α was accounted for in the kinetic model with a reaction rate constant termed kreverse. The T1 of both compounds was fitted simultaneously with the forward and reverse reaction (anomeric exchange) rates. The intensities of C1α and C1β signals of [13C6,D7]G6P were taken from the time point at which entire [13C6,D7]Glc pool was converted to [13C6,D7]G6P. The results of the fit to the kinetic model are summarised in the ESI Table S2.†
In these experiments we have shown the ability to detect the anomeric exchange of [13C6,D7]G6P in real-time. The dDNP hyperpolarisation technology provided the required orders of magnitude increase in SNR in order to be able to observe the process quickly and in concentrations that are physiologically relevant. In addition, the unique ability to quench the signal of one of the anomers enabled the direct observation of this process. For Glc, the time required to achieve complete equilibrium between the α and β anomers is a few hours.7 Therefore, we could not observe this anomeric exchange process, as the hyperpolarised measurements can only take place while the hyperpolarised state of the site of interest exists. For [13C6,D7]Glc 13C sites, this time window was typically 30–50 s. However, as the G6P anomeric exchange rate is much faster,2,3 observing its anomeric exchange in real-time was possible using the current approach. For such a fast anomeric equilibrium the approach taken here was uniquely useful because it is much more challenging for observation with conventional assays.
As the anomeric equilibrium of both Glc and G6P has a preference for the β anomeric form,3 we targeted here the exchange of the α to the β anomer (arbitrarily termed here as the forward reaction). To this end, the selective pulses were applied in the C1β frequency (98.6 ppm) and its signal was completely depolarised. Further consecutive acquisition with hard pulses enabled visualisation of the exchange of the C1α position into C1β, but only in the experiments where [13C6,D7]G6P was formed and not on [13C6,D7]Glc solutions without enzymatic activity.
On hyperpolarised 13C spectra, the signals of [13C6,D7]Glc and [13C6,D7]G6P typically overlap, specifically because we could not use deuterium decoupling. Nevertheless, in a previous study, these C1 signals were discerned using a careful deconvolution procedure.21 Nevertheless, it is not trivial to discern the newly formed 13C signals of [13C6,D7]G6P in this type of spectra. To clearly and uniquely observe the anomerization and quantify the C1-[13C6,D7]G6P peak integrals we combined the selective depolarisation approach with a time window selection that was determined by monitoring the evolution of the C6 position- for which the substrate ([13C6,D7]Glc) and the product ([13C6,D7]G6P) signals were resolved. The data used for determining the anomeric exchange process were taken only from those spectra that did not show the substrate signal, which suggested that the C1 signals were due to G6P only.
As regards to the forward and reverse anomeric exchange rates (ESI Table S2†), it is interesting to note that the ratio of these rates was 1.64 ± 0.01 (n = 2). This ratio is in a good agreement with previous studies performed at thermal equilibrium on non-deuterated compounds.3,5 However, the actual rates are not directly comparable due to differences in experimental conditions.3,5 Because deuterium substitution may lead to an isotopic effect – i.e. an effect on metabolic and chemical exchange rates,44 this agreement suggests that the results obtained here are relevant to non-labelled G6P as well. We believe that using the technique described here the anomerization of [13C6,D7]G6P in other biological systems and models can be determined.
The T1 of the C1 position for the α and β anomers upon interaction with ca. 5 mg of the hexokinase enzyme was found to be 10 s (Table S2†). This determination is in agreement with the values determined previously on a similar reaction with a similar amount of enzyme, at the same magnetic field, but sampled with hard pulses only and analysed with a kinetic model that included the phosphorylation reaction rate and not the anomerization rate.21 This strong similarity further validates the results of the kinetic model analysis done here. In addition, upon interaction with a higher enzyme content, the T1 values decreased to ca. 7 s. This trend towards a decrease in T1 upon interaction with the hexokinase enzyme was also observed previously for this reaction.21 The use of a kinetic model that encompasses freedom for T1 of the specific sites and simultaneous fit of the substrate and the product decay data enabled this determination of the anomerization exchange rate from the hyperpolarised data.
In conclusion, a methodology for observing the anomerization reaction of a stable-isotope labelled G6P in real-time and at physiological concentrations is presented. To the best of our knowledge, this is the first report in which the dDNP technology has been harnessed for researching anomerization reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Talia Harris for her assistance in the course of this study. This project has received funding from the European Union's Horizon 2020 Research and Innovation Program under grant agreement no. 667192, and from the Israel Innovation Authority, KAMIN Incentive program, grant agreement no. 63361.References
- T. M. Lowry, J. Chem. Soc., Trans., 1903, 83, 1314–1323 RSC.
- M. Salas, E. Vinuela and A. Sols, J. Biol. Chem., 1965, 240, 561–568 CAS.
- K. J. Schray and S. J. Benkovic, Acc. Chem. Res., 1978, 11, 136–141 CrossRef CAS.
- K. J. Schray and E. E. Howell, Arch. Biochem. Biophys., 1978, 189, 102–105 CrossRef CAS.
- R. S. Balaban and J. A. Ferretti, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 1241–1245 CrossRef CAS.
- C. Y. Lee, T. E. Acree and R. S. Shallenberger, Carbohydr. Res., 1969, 9, 356–360 CrossRef CAS.
- H. Jacin, J. M. Slanski and R. J. Moshy, J. Chromatogr., 1968, 37, 103–107 CrossRef CAS.
- B. Wurster and B. Hess, FEBS Lett., 1974, 40, S105–S111 CrossRef.
- B. Wurster and B. Hess, Eur. J. Biochem., 1973, 36, 68–71 CrossRef CAS.
- C. Simmerling, T. Fox and P. A. Kollman, J. Am. Chem. Soc., 1998, 120, 5771–5782 CrossRef CAS.
- J. N. Brönsted and E. A. Guggenheim, J. Am. Chem. Soc., 1927, 49, 2554–2584 CrossRef.
- W. J. Malaisse, I. Verbruggen, M. Biesemans and R. Willem, Int. J. Mol. Med., 2004, 13, 855–857 CAS.
- E. Miclet, D. Abergel, A. Bornet, J. Milani, S. Jannin and G. Bodenhausen, J. Phys. Chem. Lett., 2014, 5, 3290–3295 CrossRef CAS.
- A. Sadet, M. M. Weber Emmanuelle, A. Jhajharia, D. Kurzbach, G. Bodenhausen, E. Miclet and D. Abergel, Chem.–Eur. J., 2018, 24, 5456–5461 CrossRef CAS.
- J. M. Bailey, P. H. Fishman and P. G. Pentchev, Biochemistry, 1970, 9, 1189–1194 CrossRef CAS.
- J. E. Smith and E. Beutler, Proc. Soc. Exp. Biol. Med., 1966, 122, 671–673 CrossRef CAS.
- K. J. Schray, S. J. Benkovic, P. A. Benkovic and I. A. Rose, J. Biol. Chem., 1973, 248, 2219–2224 CAS.
- W. J. Malaisse, Adv. Biol. Chem., 2012, 2, 1–9 CrossRef CAS.
- F. Fichaux, J. Marchand, B. Yaylali, V. Leclercq-Meyer, J. Catala and W. J. Malaisse, Int. J. Pancreatol., 1991, 8, 151–167 CAS.
- V. Leclercq-Meyer, J. Marchand and W. J. Malaisse, Horm. Metab. Res., 1991, 23, 257–261 CrossRef CAS.
- G. Sapir, T. Harris, S. Uppala, A. Nardi-Schreiber, J. Sosna, J. M. Gomori and R. Katz-Brull, Sci. Rep., 2019, 9, 19683–19696 CrossRef CAS.
- J. M. Bailey, P. H. Fishman and P. G. Pentchev, Biochemistry, 1970, 9, 1189–1194 CrossRef CAS.
- B. Künnecke, E. Küstermann and J. Seelig, Magn. Reson. Med., 2000, 44, 556–562 CrossRef.
- R. Roussel, P. G. Carlier, C. Wary, G. Velho and G. Bloch, Magn. Reson. Med., 1997, 37, 821–824 CrossRef CAS.
- R. Gruetter, E. J. Novotny, S. D. Boulware, D. L. Rothman, G. F. Mason, G. I. Shulman, R. G. Shulman and W. V. Tamborlane, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 1109–1112 CrossRef CAS.
- B. Ross, A. Lin, K. Harris, P. Bhattacharya and B. Schweinsburg, NMR Biomed., 2003, 16, 358–369 CrossRef CAS.
- J. Okuda and I. Miwa, J. Clin. Biochem. Nutr., 1986, 1, 189–199 CrossRef CAS.
- W. J. Malaisse, M. H. Giroix, S. P. Dufrane, F. Malaisse-Lagae and A. Sener, Biochim. Biophys. Acta, 1985, 847, 48–52 CrossRef CAS.
- A. Blume, M. Fitzen, A. J. Benie and T. Peters, Carbohydr. Res., 2009, 344, 1567–1574 CrossRef CAS.
- S. Walker-Samuel, R. Ramasawmy, F. Torrealdea, M. Rega, V. Rajkumar, S. P. Johnson, S. Richardson, M. Gonçalves, H. G. Parkes, E. Årstad, D. L. Thomas, R. B. Pedley, M. F. Lythgoe and X. Golay, Nat. Med., 2013, 19, 1067–1072 CrossRef CAS.
- B. E. Lewis, N. Choytun, V. L. Schramm and A. J. Bennet, J. Am. Chem. Soc., 2006, 128, 5049–5058 CrossRef CAS.
- P. W. Kuchel, B. T. Bulliman and B. E. Chapman, Biophys. Chem., 1988, 32, 89–95 CrossRef CAS.
- P. Abayakoon, J. P. Lingford, Y. Jin, C. Bengt, G. J. Davies, S. G. Yao, E. D. Goddard-Borger and S. J. Williams, Biochem. J., 2018, 475, 1371–1383 CrossRef CAS.
- A. S. Serianni, J. Pierce, S. G. Huang and R. Barker, J. Am. Chem. Soc., 1982, 104, 4037–4044 CrossRef CAS.
- J. H. Ardenkjaer-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158–10163 CrossRef CAS.
- H. F. Zeng, Y. Lee and C. Hilty, Anal. Chem., 2010, 82, 8897–8902 CrossRef CAS.
- H. Allouche-Arnon, T. Wade, L. F. Waldner, V. N. Miller, J. M. Gomori, R. Katz-Brull and C. A. McKenzie, Contrast Media Mol. Imaging, 2013, 8, 72–82 CrossRef CAS.
- T. Harris, H. Degani and L. Frydman, NMR Biomed., 2013, 26, 1831–1843 CrossRef CAS.
- S. Meier, M. Karlsson, P. R. Jensen, M. H. Lerche and J. O. Duus, Mol. BioSyst., 2011, 7, 2834–2836 RSC.
- K. Kumagai, M. Akakabe, M. Tsuda, M. Tsuda, E. Fukushi, J. Kawabata, T. Abe and K. Ichikawa, Biol. Pharm. Bull., 2014, 37, 1416–1421 CrossRef CAS.
- K. N. Timm, J. Hartl, M. A. Keller, D. E. Hu, M. I. Kettunen, T. B. Rodrigues, M. Ralser and K. M. Brindle, Magn. Reson. Med., 2015, 74, 1543–1547 CrossRef CAS.
- H. Allouche-Arnon, M. H. Lerche, M. Karlsson, R. E. Lenkinski and R. Katz-Brull, Contrast Media Mol. Imaging, 2011, 6, 499–506 CrossRef CAS.
- H. Allouche-Arnon, Y. Hovav, L. Friesen-Waldner, J. Sosna, J. Moshe Gomori, S. Vega and R. Katz-Brull, NMR Biomed., 2014, 27, 656–662 CrossRef CAS.
- C. E. Grimshaw and W. W. Cleland, Biochemistry, 1980, 19, 3153–3157 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08022e |
| This journal is © The Royal Society of Chemistry 2020 |