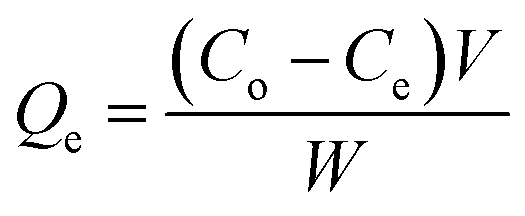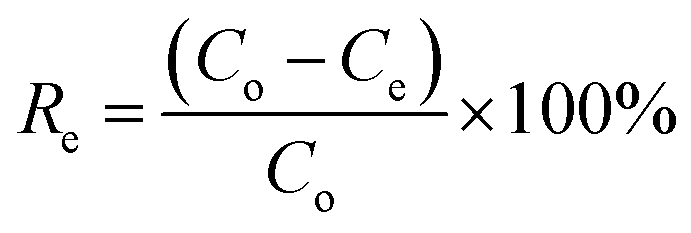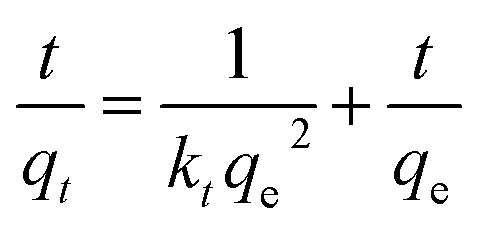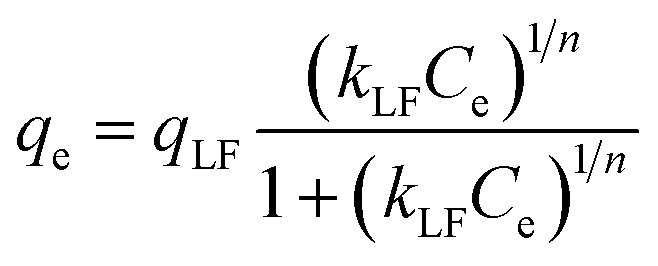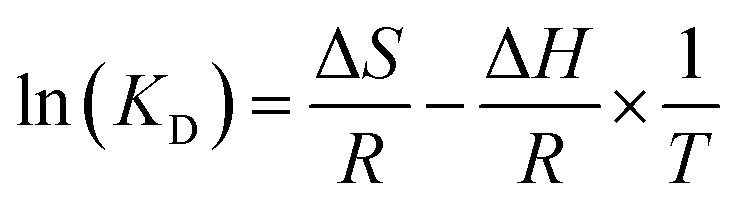DOI:
10.1039/D0RA07862J
(Paper)
RSC Adv., 2020,
10, 40329-40340
Evaluation of adsorptive desulfurization performance and economic applicability comparison of activated carbons prepared from various carbon sources†
Received
14th September 2020
, Accepted 26th October 2020
First published on 5th November 2020
Abstract
Adsorptive desulfurization (ADS) using activated carbon (AC) as adsorbent presents competitive potential in separating thiophenic sulfur from liquid fuels with high selectivity under mild operation conditions. It is also a highly economic remedy in ultra-low sulfur content situations. Most importantly, a suitable feedstock for macroscopic quantity preparation of AC adsorbents with good adsorptive desulfurization performance and low-cost is required to satisfy the requirements of this field. In this work, four representative substances (i.e., coal, coconut shell, polyurethane plastic waste, and petroleum coke) were selected as the carbon source for the preparation of various AC adsorbents. The physicochemical properties of the prepared AC adsorbents were characterized using BET, SEM, XRD, XPS, elemental analysis and Boehm's method. The corresponding adsorptive desulfurization performance was investigated. The corresponding desulfurization capacity obtained was in the order: CS-ACA > PUPW-ACA > PC-ACA > AT-ACA. Under the optimal conditions of 30 °C and 30 min contact time, the desulfurization rate of 0.5 g PUPW-ACA can reach about 98%. The HHV of non-condensable gas generated during the experiment was calculated, and the HHV of the pyrolysis oil was measured. The results showed that the by-products produced by PC had the highest HHV. The economics of the desulfurization of the four kinds of activated carbon were analyzed and evaluated. From a comprehensive analysis, PUPW-ACA has the highest economic production value and has the potential for industrial production. This plays a dual role in environmental protection.
1 Introduction
Considering the context of the worsening global environment, environmental protection has become one of the world's most recognized issues with an increasing emphasis on the atmospheric environment.1–4 Although immense attention has been devoted to research on alternative energy from new sources such as wind, ocean tides, and solar power, to ease the stress of environmental protection, traditional fossil energy (coal and petroleum) is still the dominant energy source of modern society.5,6 Petroleum and its derived fuels contain sulfur, which results in severe environmental pollution.7–9 Consequently, the combustion of these fuels with sulfur-containing compounds would generate SOx, which is responsible for acid rain and sequential irreversible damage to the surface environment.10–12 To eliminate this problem, the desulfurization of fossil fuel has long been one of the most important practices and remedies in petroleum refineries.
As a rousingly competitive desulfurization technology, adsorptive desulfurization (ADS) has attracted significant attention from academics and enterprises.13–20 Various desulfurization adsorbents, for instance, metal–organic frameworks (MOFs),21 zeolites,22 and activated carbons (ACs),23 have been constantly developed to fulfill the requirements of desulfurization. AC adsorbents (ACA) have the qualities of simple preparation processes, low-cost, relatively high adsorption capability, and earth-abundant sources for manufacture.24–26 Saleh et al. used polymer waste to prepare ACA,27 while Yang et al. used biomass-based ACA for desulfurization characterization.12 Besides, coal,28 petroleum coke,29 and plastic waste30 have been used to prepare ACAs. The usage of plastic waste as a carbon source reduces and recycles solid waste,31 which further favors environmental protection from other perspectives.
The carbon source, activation method, surface modification method, and experimental conditions of activated carbons for adsorption desulfurization have been extensively studied. However, these studies only evaluated the desulfurization performances and feasibility of activated carbons but did not evaluate the economic and industrial applications in detail. Therefore, in this study, the desulfurization performance of adsorbents prepared from different carbon sources and the economic applications of their by-products (non-condensable gas and pyrolysis oil) were evaluated in detail. The prices of corresponding adsorbents in practical applications, which is an important indicator for assessing whether a carbon source can be used in industry, were accurately calculated. The economic applicability information has immense potential to drive the commercialization of ACAs. Representatively, anthracite, coconut shell, petroleum coke, and a new rising plastic waste (i.e., polyurethane plastic waste, PUPW) were selected to prepare ACAs in this study.
The main objective was to systematically study the impact of carbon source on the structure and corresponding desulfurization performance of resultant ACAs via comparison characterizations, including SEM, BET, XRD, elemental analysis and XPS. Finally, a promising feedstock for ACA manufacture was recommended based on the comprehensive analysis of the yield, adsorptive desulfurization capacity, and regeneration of ACAs, higher heat value (HHV) of by-product, and economical factors. This study provides a more systematic and environmentally friendly method for preparing desulfurization adsorbents, which has more industrial production potential.
2 Experimental
2.1 Materials
Dibenzo[b,d]thiophene (99%, DBT), n-hexane (99%), n-heptane (99%), NaOH (4 mol), nitric acid (4 mol) and toluene (99%) were purchased from Shanghai Macklin Reagent Co. Ltd. CO2 gas was purchased from Petrotech Chemical Technology Co. Ltd. (Qingdao, China). Polyurethane plastic waste (PUPW) was provided by China Everbright Greentech Limited. Petroleum coke, coconut shell, and anthracite were provided by Rainy Sunshine Economics & Trade Company of Qingdao.
2.2 Preparation of ACA
Firstly, PUPW, petroleum coke, anthracite, and coconut shell were crushed and then sieved to acquire powdered feedstocks of 100 mesh. The powdered feedstocks were pyrolyzed at 800 °C under N2 atmosphere for 4 h to acquire temperature equilibrium. The obtained carbonaceous residuum was activated at a CO2 flow of 100 ml min−1 at 800 °C for 6 h. 1 g of AC and 10 ml of 4 mol nitric acid solution were accurately weighed and mixed. The mixture was stirred and then refluxed at 60 °C for 3 h, as suggested by Saleh.7 After that, the modified AC was washed with deionized water repeatedly to be neutralized. Finally, the AC sample was vacuum dried at 50 °C for 5 h to obtain the resultant ACA (PUPW-ACA) for further desulfurization performance evaluation. Similarly, petroleum coke-based ACA, anthracite-based ACA, and coconut shell-based ACA were prepared and labelled as PC-ACA, AT-ACA, and CS-ACA, respectively. The pyrolysis oil and the non-condensable gas generated in the preparation process were collected and characterized.
2.3 The characterizations of products
The N2 adsorption–desorption isotherms were obtained by an ASAP 2020 Plus HD88 instrument. The specific surface area was obtained by the Brunauer–Emmett–Teller (BET) method, while the pore size distribution was found by the NL-DFT method. The surface structures of the AC adsorbents were obtained using SEM observation (Sirion 200, FEI Electron Optics Co., U. S.). The surficial information was supplementarily analyzed via X-ray photoelectron spectroscopy (XPS). Analysis of the crystalline phase of the adsorbent by X-ray diffraction (XRD). The number of acidic-oxygenated groups on activated carbon surface was determined according to the Boehm's method (Base titration).
The higher heating value (HHV) of pyrolysis oil was measured using an adiabatic oxygen bomb calorimeter (IKA C6000, Germany).
The composition of non-condensable gas was analyzed using a gas chromatography analyzer (GC-TCD Agilent 6090, American). The detailed procedure has been presented in previous literature.32 The higher heating value (HHV) of the non-condensable gas was calculated by the following equation:
where HHV
gas is the overall HHV of non-condensable gas,
Vi is the volumetric fraction of gas species (i) detected in gas chromatography analyzer, and HHV
i is the HHV of each gas species (i).
Vapor adsorption experiments were used to characterize the hydrophilicity of samples. Approximately 500 mg of ACAs were put into a 5 ml glass bottle and dried under 70 °C for 24 h. The samples were cooled at room temperature in a desiccator before determining their accurate weights (500 mg). The ACAs were placed in an Erlenmeyer flask with a frosted glass joint, which was filled with saturated solvent vapor. The amount of solvent used was 60 ml. The ACAs powder was placed carefully in a way that it did not touch the wall of the Erlenmeyer flask. After 24 hours, the ACAs were taken out of the Erlenmeyer flask, and then thoroughly dried using laboratory tissues. Finally, the weight was obtained, and the weight gain of ACAs during storage was the maximal adsorption of vapor.33,34 The values of pH at the point of zero charge (pHpzc) of the materials were analyzed using ZetaPlus Zeta Potential Analyzer.35
2.4 Adsorption performance test
Adsorption performance tests were performed using the static adsorption method. A model oil was formulated from n-hexane and DBT, with sulfur content between 50 μg g−1 and 250 μg g−1. Prior to the test, ACA was vacuum dried at 100 °C for 1 h. In the adsorption test, 20 mg of the adsorbent was mixed with 50 ml of the model oil, and then continuously stirred at 10–50 °C for a designated time. Each adsorption test was performed three times to acquire the average adsorption performance. Gas Chromatography (GC) equipped with a flame photometric detector was used to determine the concentration of sulfur compounds. The adsorption capacity of AC adsorbent (Qe, mg g−1) and the sulfur removal rate (Re, %) at equilibrium were calculated using eqn (2) and (3):| |
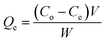 | (2) |
| |
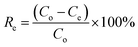 | (3) |
where Co and Ce (mg L−1) are the initial concentration and equilibrium concentration of sulfur compounds, respectively, V is the volume of the model oil, and W is the weight of the AC adsorbent. Besides, the impact of aromatic compounds on the desulfurization adsorption was studied. 2–10% toluene was introduced into the model oil and adsorption tests were implemented following the aforementioned procedure.
2.5 Regeneration experiments
The regeneration and reuse performance of ACAs were studied by performing regeneration experiments with five runs was performed for each adsorbent. Toluene was used to regenerate the used adsorbent, and 1 g of adsorbent and 5 ml of toluene were stirred at room temperature for 30 minutes. Then the adsorbent was separated from toluene, and the separated adsorbent was dried at 100 °C.
3 Results and discussion
3.1 Product distribution of ACA preparation
The feedstocks were disintegrated into solid (i.e., ACA), liquid (pyrolysis oil or pyrolysis water), and gas (non-condensable gas) products during the preparation process. The property of each product (gas, pyrolysis oil, and ACA) was greatly influenced by the preparation process,36 meanwhile, the preparation scenario used in the study was kept constant for the sake of comparison. Fig. 1 indicates the product distributions obtained in four cases. Higher yields of ACA were observed in both cases of anthracite and petroleum coke, compared to that of PUPW and coconut shell. Specifically, the yield of four ACAs followed the order: AT-ACA (70.5 wt%) > PC-ACA (66.7 wt%) > CS-ACA (23.7 wt%) > PUPW-ACA (13.5 wt%). This could be rationalized by the carbon species contained in the feedstocks. Although anthracite and petroleum coke belonged to natural substances and man-made substances, respectively, they were both characterized with high carbonization proved by their carbon contents (anthracite, 79.70 wt%; petroleum coke 76.09 wt%). Furthermore, it has been shown in previous researches that anthracite and petroleum coke are aromatic carbon dominated. This usually does not favor pyrolysis performance, which results in the fairly higher pyrolysis carbonaceous residue. Furthermore, the microcrystalline consisted most of aromatic carbon, and were against CO2 activation. In the cases of PUPW and coconut shell, the reverse applied. From the perspective of liquid yield, the pyrolysis oil yields achieved in the cases of PUPW and coconut shell were significantly higher than those achieved in the cases of anthracite and petroleum coke. It further supported the argument via the negative correlation of aromatic carbon ratio with pyrolysis oil yield. Notably, considerable pyrolysis water was formed in the case of CS, mainly due to the abundant hydroxyl groups. Meanwhile, the high heat value (HHV) of CS-based pyrolysis oil was wakened due to the high oxygen content of the parent substance (i.e., CS, 47.5 wt%), compared to the HHVs of PC-based and AT-based pyrolysis oils, as shown in Fig. 1. Complementary oil-water separation and deoxygenation would be necessary if the by-product pyrolysis liquid generated in the preparation of CS-ACA pursued comprehensive utilization. Extra investigation cost was expected. Although the HHVs of pyrolysis oils from PC and AT were qualified for liquid fuel, their yields remained depressing. The yield of pyrolysis oil from PUPW was rather high (48.2 wt%), while the HHV of pyrolysis oil ranked three (32.9 MJ kg−1), making it a candidate for energy resource. This was because the compositional characteristics of PUPW-based pyrolysis oil enabled upgrading under mild conditions. Although PUPW-based pyrolysis oil inherited a fair number of oxygens from PUPW, it, for the most part, existed in the form of aliphatic oxygen,37 which could be easily removed, compared to the phenoxy oxygen contained in biomass-based pyrolysis oil.
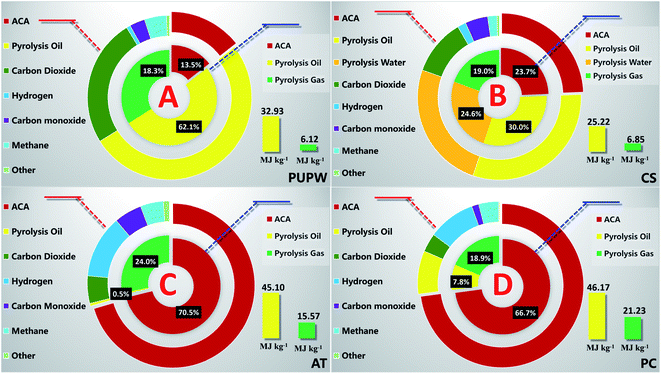 |
| | Fig. 1 Product distribution of ACA preparation. | |
Another so-called by-product, pyrolysis gas without condensable components, was gathered and sent for analysis. As shown in Fig. 1, the pyrolysis gas from various origins was mainly composed of CO2, H2, CO, and CH4. The yields and HHVs of four pyrolysis gases were calculated, as suggested by Chen et al.32 and presented in Fig. 1 as well. The yield of PUPW-based pyrolysis gas was the highest, while the HHV was the lowest (6.14 MJ kg−1) due to the dominant CO2. The finding in this study was similar to that of Zhang et al.37 The ratio of CO2 in the CS-based pyrolysis gas decreased by 14.5 wt%, making the HHV higher. Furthermore, the HHV improved as H2 dominated in the cases of AT and PC. The HHV of AT-based pyrolysis gas was lower than that of the PC-based pyrolysis gas mainly due to the relatively higher ratio of CO. Although the dominant component in the pyrolysis gas varied, the pyrolysis gas could still serve as a complementary energy source, for instance, via combustion.
3.2 Characterization of ACAs
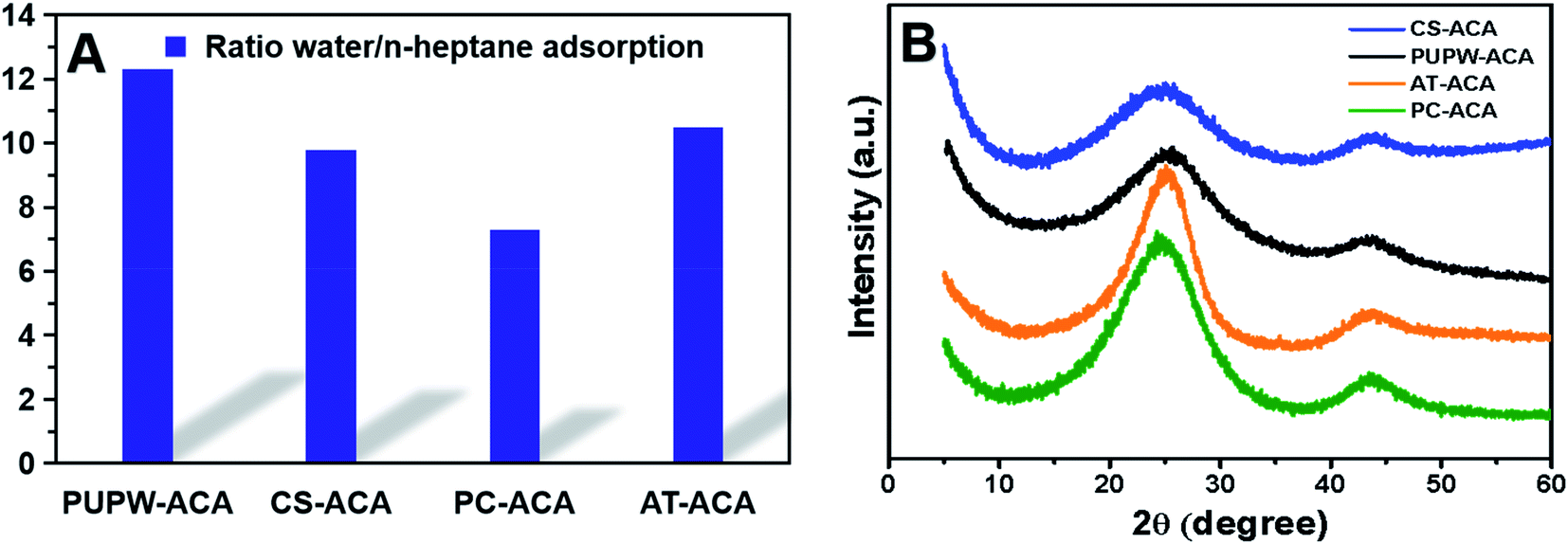
The N2 adsorption–desorption isotherms and pore size distributions of four ACAs are displayed in Fig. 2, in which the sequence of SBET is: CS-ACA > PUPW-ACA > PC-ACA > AT-ACA. According to the IUPAC classification, the adsorption isotherms of PUPW-ACA and CS-ACA are Type I, while the adsorption isotherms of PC-ACA and AT-AC are Type IV.27,38–41 It was shown that micropore dominated in the PUPW-ACA and CS-ACA, while mesopore dominated in the PC-ACA and AT-AC, as supported by the microporosity in Table 1. AT and PC have high crystallinity, high degree of ordering, compact structure, and partial graphitization. As a result, the activation of CO2 mainly occurs on the surface of carbon, and CO2 has poor diffusion ability on its surface. In the process of CO2 activation, amorphous carbon has high reaction activity with CO2, while microcrystalline carbon has low reaction activity. Therefore, only a few macroporous structures can be formed on the surface. For PUPW and CS, the degree of graphitization is lower after carbonization, and less microcrystalline carbon is formed. Therefore, CO2 can react with more amorphous carbon. Due to the porous structure of PUPW and CS, more pore structures can be formed on the surface and inside. Therefore, the specific surface area of PUPW-ACA and CS-AC is higher than that of AT-ACA and PC-ACA. Scanning Electron Microscopy (SEM) was used to characterize and analyze the microstructures of the four materials. As shown in Fig. 3, both CS-ACA and PUPW-ACA have similar morphologies with more pore structures. AT-ACA and PC-ACA have similar morphology, with the same layered structure. This is confirmed by the results of BET, CS-ACA and PUPW-ACA have a larger specific surface area. This also shows that PC-ACA and AT-ACA have less desulfurization capacity.
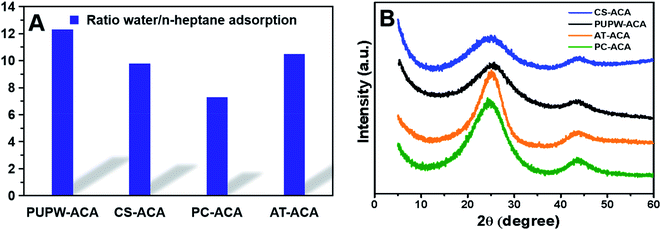 |
| | Fig. 2 Ratio of water and n-heptane adsorption values (A), XRD spectra of PUPW-ACA, CS-ACA, PC-ACA, and AT-ACA (B). | |
Table 1 Pore information and specific surface area of various ACAs
| ACA |
SBETa (m2 g−1) |
Smic (m2 g−1) |
Vtotalb (cm3 g−1) |
V<1 nm (cm3 g−1) |
Savec (nm) |
| SBET: specific surface area using BET. Vtotal: total pore volume (at P/Po ≅ 0.99). Save: average pore size. |
| PUPW-ACA |
1082 |
1006 |
0.499 |
0.413 |
2.2 |
| CS-ACA |
1150 |
1097 |
0.625 |
0.525 |
1.9 |
| PC-ACA |
136 |
40 |
0.118 |
0.008 |
8.5 |
| AT-ACA |
52 |
0.5 |
0.080 |
0.002 |
9.1 |
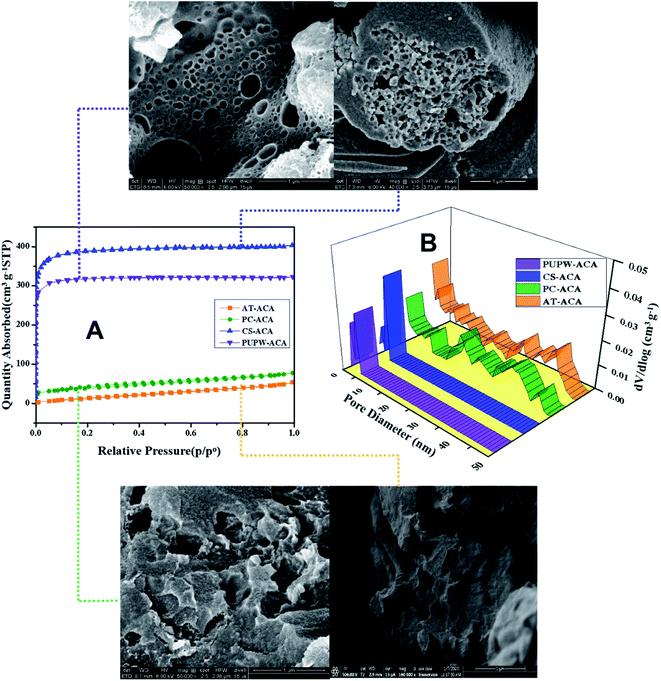 |
| | Fig. 3 The N2 adsorption–desorption isotherms (A), pore size distribution diagram (B) and SEM of PUPW-ACA, CS-ACA, PC-ACA, and AT-ACA, respectively. | |
The type and content of the oxygen-containing group on the surface of ACAs were qualitatively analyzed based on a reaction of alkali and acid with the surface oxide, termed as the Boehm's method.42–44 NaHCO3 only neutralizes the special carboxyl groups on the surface of carbon, while Na2CO3 can neutralize the carboxyl group and the lactone group. NaOH could neutralize the carboxyl groups, lactone groups, and phenolic hydroxyl groups on the surface. According to the difference in alkali consumption, the content of the corresponding functional group can be calculated. The total acidity can be calculated from the sum of carboxyl, hydroxyl and phenol. The analysis results of four ACAs are presented in Table 2. Obviously, the contents of the phenol and lactone groups were relatively lower in ACAs, while the content of the carboxyl group on the surface of all ACAs occupied a high proportion. It was probably due to the oxidation modification process, in which the carboxyl group was productively formed. Both the total content of oxygen-containing groups and the content of the carboxyl group on the surface of PUPW-ACA were higher than that of the other three ACAs. Rich surface groups, especially the carboxyl group, might serve as adsorption active sites to provide the prerequisite for high desulfurization possibility, as suggested by Yang et al.,45 and Ania and Bandosz.46 The highest carboxyl acidity acquired in this study was 1.25 mmol g−1. It was equivalent to 11.55 mmol m−2 × 104 if based on the specific surface area of corresponding ACA, which was higher than that of desulfurization ACA (9.77 mmol m−2 × 104) with similar SBET from Bu et al.44
Table 2 Surface acidity of PUPW-ACA, CS-ACA, PC-ACA, and AT-ACA
| ACAs |
Surface aciditya (mmol g−1) |
pHpzc |
| Phenol |
Lactonic |
Carboxyl |
Total acidity |
| Determined by Boehm's method. |
| PUPW-ACA |
0.29 |
0.36 |
1.25 |
1.90 |
5.05 |
| CS-ACA |
0.22 |
0.25 |
1.12 |
1.59 |
5.73 |
| PC-ACA |
0.15 |
0.21 |
0.90 |
1.26 |
6.30 |
| AT-ACA |
0.24 |
0.20 |
1.18 |
1.62 |
5.77 |
The hydrophilicity of the ACAs surface was characterized through the adsorption experiment of water and n-heptane vapor. The higher the affinity of the ACAs to water, the higher the polarity of the surface and the stronger the hydrophilicity of the surface.33,34 The Fig. 2 shows the ratio of the absorption mass of water to the absorption mass of n-heptane. It could be seen that the adsorption capacities of all ACAs for water were much stronger than those of n-heptane. This showed that ACAs had strong hydrophilicity. Thus, they had strong polarity. The pHpzc results of ACAs in Table 2 were also consistent with the surface acid group content and hydrophilicity results. It strongly implied that PUPW-based ACA qualified for desulfurization.
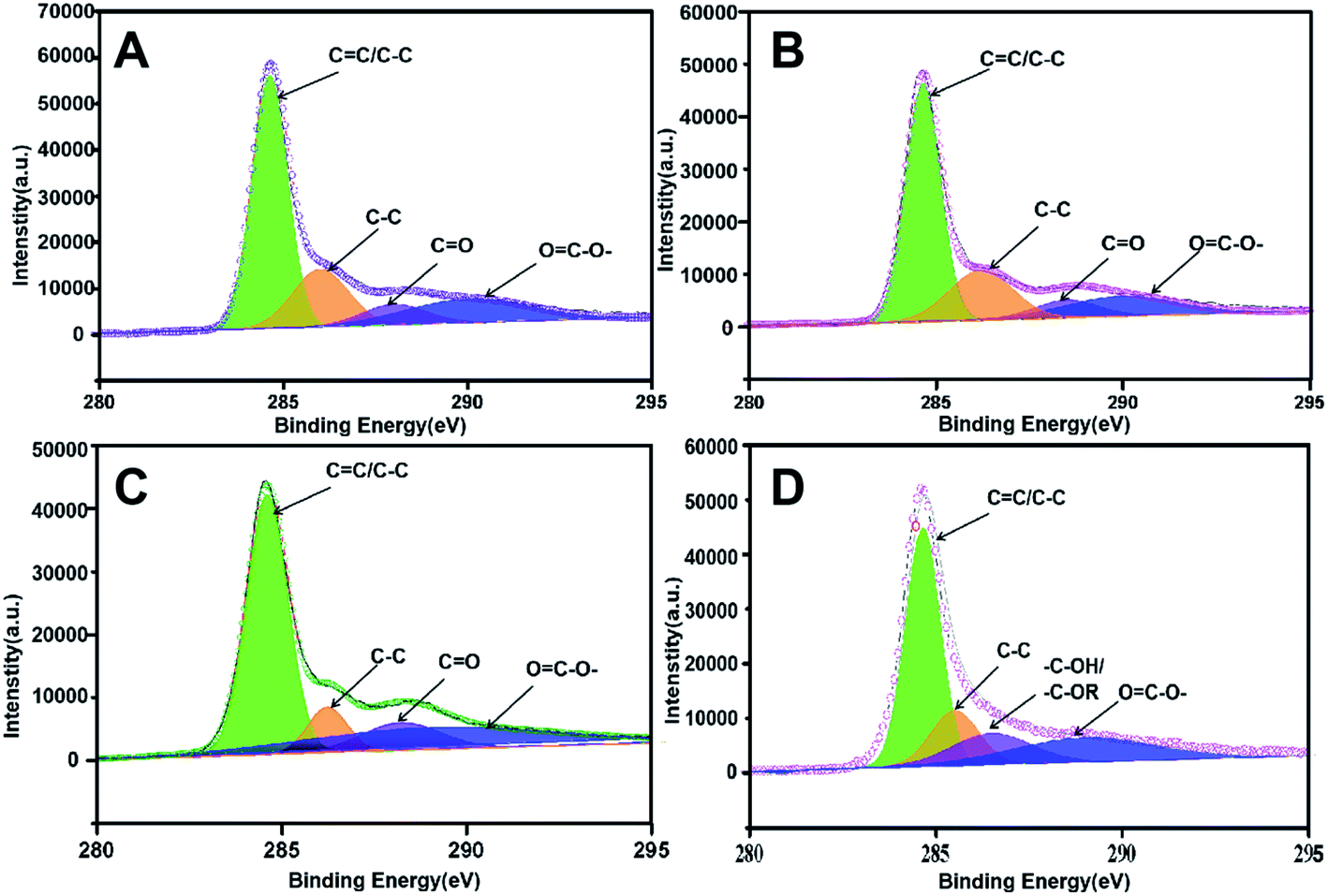
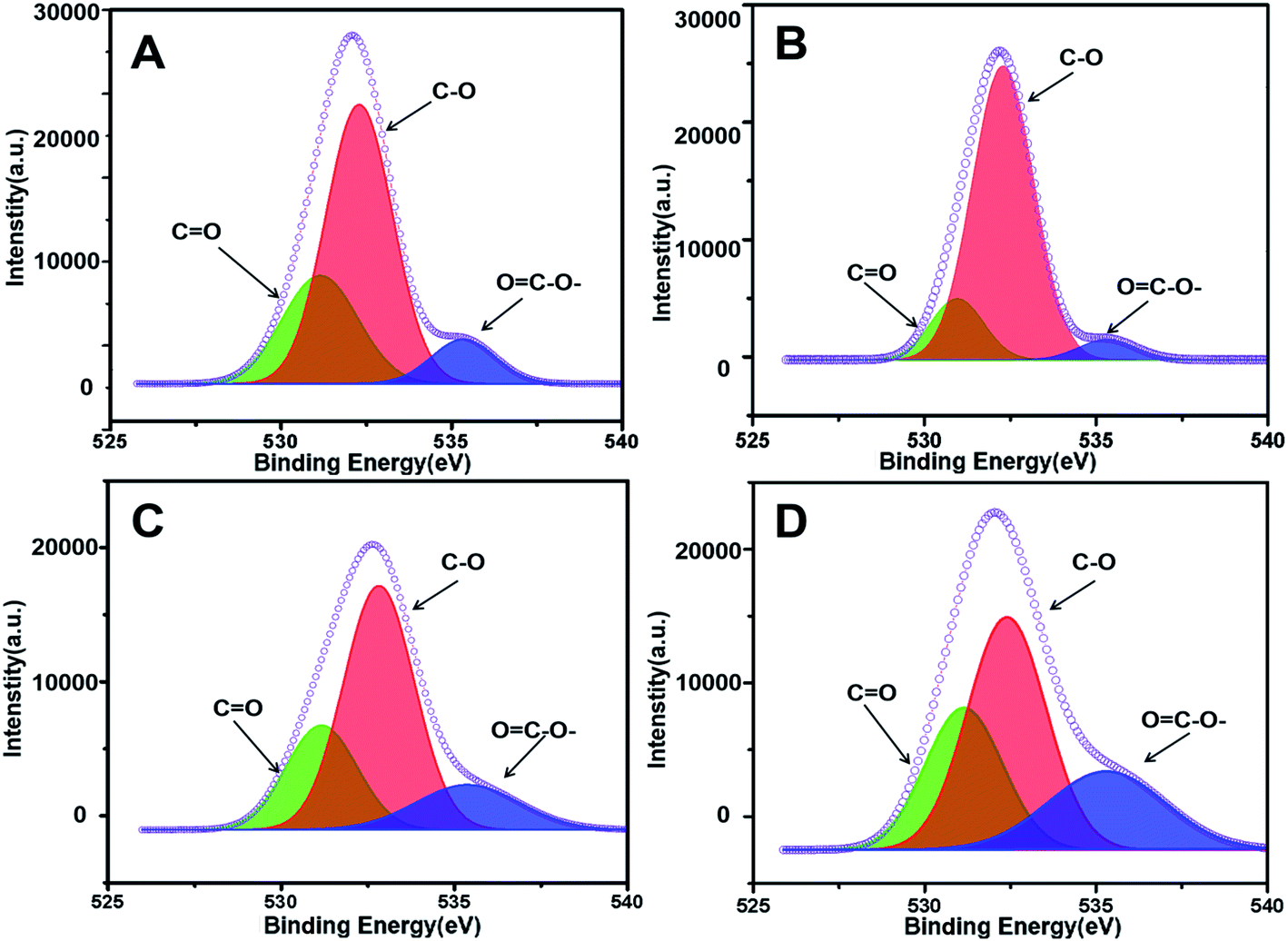
X-ray photoelectron spectroscopy (XPS) was further implemented to characterize the chemical valence of C in the four prepared adsorbents as support. As shown in Fig. 4, the C 1s peak of PUPW-ACA (A), CS-ACA (B), and PC-ACA (C) could be fitted to four peaks centered at 284.6, 285.6, 287.6, and 288.9 eV, corresponding to another serial carbon chemical environments, i.e., sp2 C, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and OC–O-.38,47,48 And the C 1s peak of AT-ACA (Fig. 4D) could be fitted to four peaks centered at 284.6, 285.6, 286.5, and 288.9 eV, corresponding to various carbon chemical environments, i.e., sp2 C, C–O, –C–OH/–C–OR, and OC–O–, respectively. It was suggested from Fig. 4 that although the sp2 hybridized type of carbon (284.6 eV, graphitic/aromatic carbon) was dominant due to the elementary characteristics of their origins, abundant oxygen-containing groups decorated the carbonaceous surfaces. As shown in Fig. 5, the O 1s peak of PUPW-ACA (A), CS-ACA (B), PC-ACA (C) and AT-ACA (D) could be fitted to three peaks centered at 531.1, 532.2, and 535.3 eV, corresponding to another serial oxygen chemical environments, i.e., CO, C–O, and OC–O–. Their corresponding oxygen contents are 13.2, 8.7, 8.5, and 7.6% respectively. The results of the XPS spectra further supported the findings from Boehm titration.
O, and OC–O-.38,47,48 And the C 1s peak of AT-ACA (Fig. 4D) could be fitted to four peaks centered at 284.6, 285.6, 286.5, and 288.9 eV, corresponding to various carbon chemical environments, i.e., sp2 C, C–O, –C–OH/–C–OR, and OC–O–, respectively. It was suggested from Fig. 4 that although the sp2 hybridized type of carbon (284.6 eV, graphitic/aromatic carbon) was dominant due to the elementary characteristics of their origins, abundant oxygen-containing groups decorated the carbonaceous surfaces. As shown in Fig. 5, the O 1s peak of PUPW-ACA (A), CS-ACA (B), PC-ACA (C) and AT-ACA (D) could be fitted to three peaks centered at 531.1, 532.2, and 535.3 eV, corresponding to another serial oxygen chemical environments, i.e., CO, C–O, and OC–O–. Their corresponding oxygen contents are 13.2, 8.7, 8.5, and 7.6% respectively. The results of the XPS spectra further supported the findings from Boehm titration.
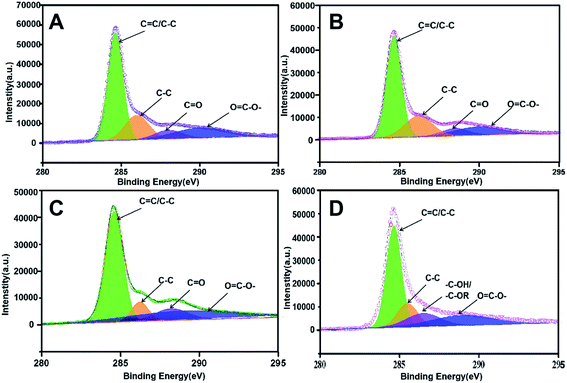 |
| | Fig. 4 C 1s XPS spectra of PUPW-ACA (A), CS-ACA (B), PC-ACA (C), and AT-ACA (D). | |
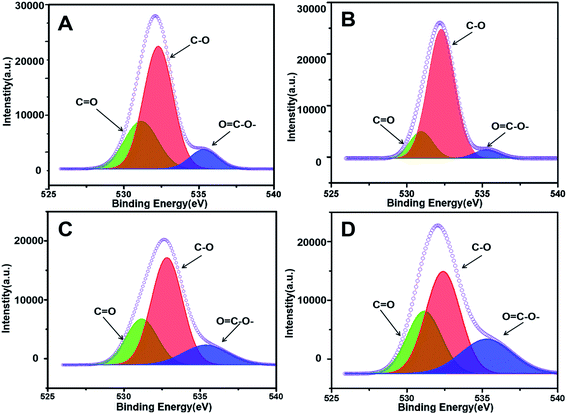 |
| | Fig. 5 O 1s XPS spectra of PUPW-ACA (A), CS-ACA (B), PC-ACA (C), and AT-ACA (D). | |
The XRD spectra of the four samples shown in Fig. 2 showed two broad peaks at 25.5 and 43.5°. These two peaks correspond to the (002) plane, (100) plane and (101) plane of the hexagonal graphite lattice, respectively. PUPW-ACA and CS-ACA have similar peak shapes, with lower peak intensities and crystallinities. The XRD spectra of PC-ACA and AT-ACA have higher peak intensities than those of PUPW-ACA and CS-ACA, and their crystallinities are also higher. This indicates that PUPW-ACA and CS-ACA have more amorphous carbon.
3.3 Desulfurization performance of ACAs
Static adsorption method was used for the desulfurization performance evaluation of the four desulfurization-ACAs prepared in this study.
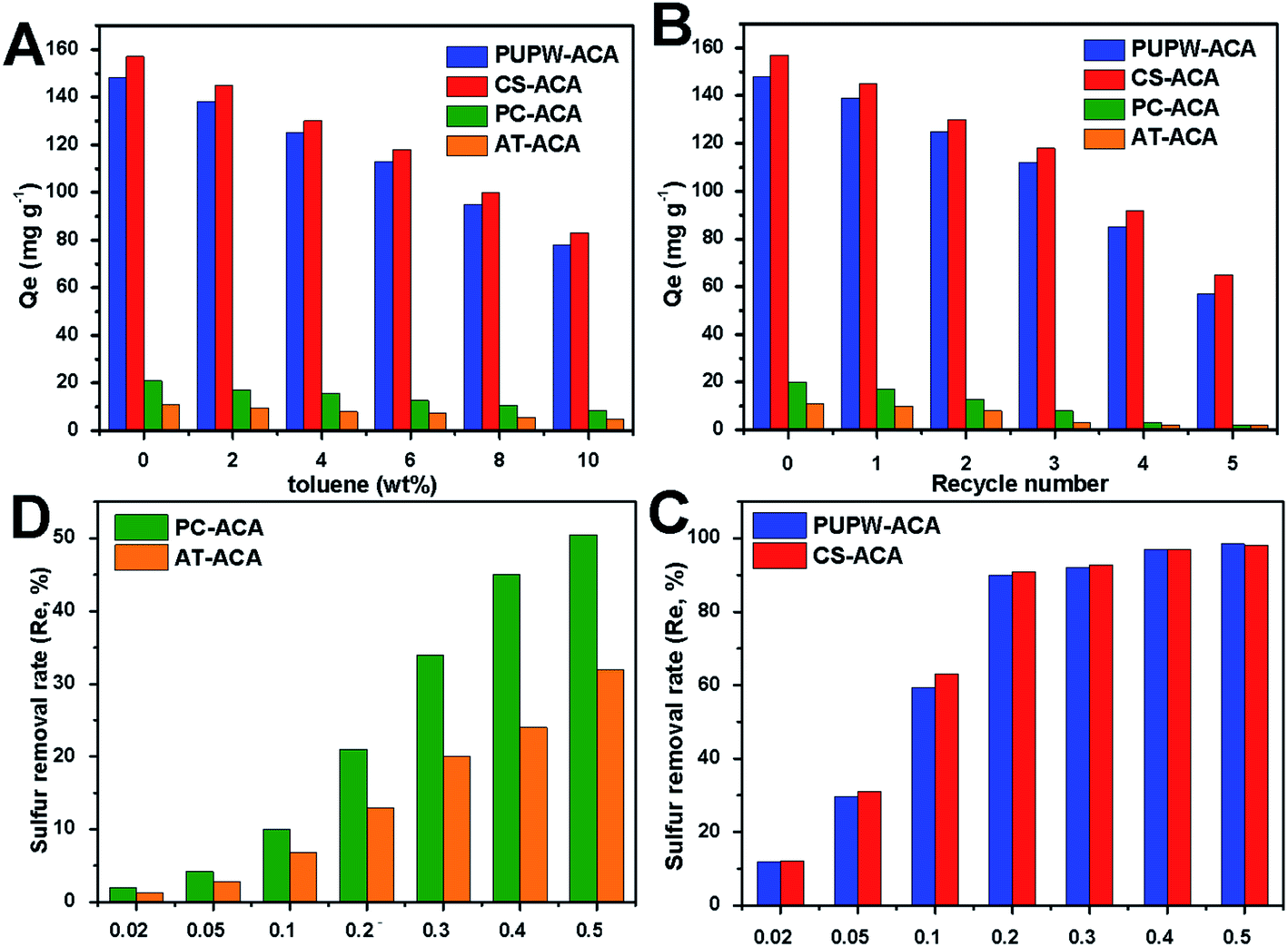
It could be intuitively observed from Fig. 6 that the adoption capacity of ACAs followed the order: CS-ACA > PUPW-ACA > PC-ACA > AT-ACA. Both CS-ACA and PUPW-ACA had fairly higher adsorption capacities (157 mg DBT g−1 and 146 mg DBT g−1) than those of PC-ACA and AT-ACA (21 mg DBT g−1 and 11 mg DBT g−1). However, the time for CS-ACA and PUPW-ACA to reach adsorption equilibrium was higher than that of PC-ACA and AT-ACA. It could be observed in this study that the specific surface area occupied was the main influencing factor. The reason why PC-ACA and AT-ACA reached the equilibrium of adsorption earlier might be related to the more mesopore and macropore structures; it was easier to reach the adsorption saturation state.
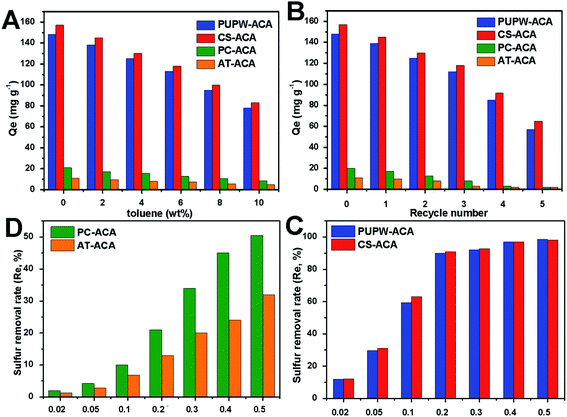 |
| | Fig. 6 Effect of toluene content (A) and recycle number (B) on the desulfurization performance of PUPW-ACA, CS-ACA, PC-ACA, and AT-ACA; effect of ACA dosage on the sulfur removal rate (C and D). | |
Different weights of adsorbent were added to 100 g 250 μg g−1 of model oil solution to perform adsorption experiments to characterize the relationship between the amount of adsorbent and the desulfurization rate. It can be seen from Fig. 6 that as the amount of adsorbent increased from 0.02 g to 0.5 g, the desulfurization rate increased significantly. However, if the amounts of CS-ACA and PUPW-ACA continuously increase, the desulfurization rate would not increase significantly. But the desulfurization rate of AT-ACA and PC-ACA continued to increase. These results are reasonable because as the amount of adsorbent increases, the number of active sites increases, and the adsorption performance becomes better. For these four adsorbents, the optimum dosage is when the desulfurization rate is about 90%. To further verify the desulfurization capabilities of these four adsorbents in actual fuel oil, a competitive adsorption experiment was implemented. The composition in fuel oil is very complicated; among them, aromatics are the main compounds that compete with DBT for adsorption. As the toluene content increased from 0 to 10%, the adsorption capacity of the four adsorbents for DBT decreased significantly. When the toluene content is 10%, the adsorption capacity of each adsorbent for DBT will be about 50%. This is probably because the π-electrons provided by toluene can be adsorbed on the adsorbents through π–π-interactions or π-complexation.
Finally, the regeneration performance of the four desulfurization adsorbents prepared was measured. The desulfurization effect after five regenerations is shown in Fig. 6. It can be seen from Fig. 6 that after five regeneration experiments of the four adsorbents, the desulfurization capacity of CS-ACA and PUPW-ACA decreased more obviously; both decreased by about 45%. This may be related to their mechanical strength, pore-clogging and loss of surface functional groups. The SEM images of the four adsorbents after five regenerations are shown in Fig. 1S.† It can be seen that there are some blockage and collapse of the aperture. In addition, the results of the elemental analysis showed that a certain amount of S element was detected in the regenerated adsorbents. The S content in PC-ACA, PUPW-ACA, CS-ACA, and AT-ACA were 0.56, 0.90, 1.01, 0.41%, respectively. This may be due to the failure to remove the DBT completely during the regeneration process, resulting in the blockage of a part of the aperture. Compared with the result of the O content in XPS, the oxygen content of the adsorbent after regeneration has a certain amount of decrease. The O content in PC-ACA, PUPW-ACA, CS-ACA, and AT-ACA were 6.4, 11.2, 7.7, 6.9%, respectively. This may be caused by the loss of some oxygen-containing functional groups on the surface, which in turn led to a decrease in the adsorption desulfurization capacity. Table 3 shows the results of desulfurization research available in the literature and current work. In this study, the desulfurization abilities of PU and CS are higher, however, the preparation process is more environmentally friendly and has higher economic benefits.
Table 3 Comparison of the results of desulfurization studies available in the literature and the present work
| Adsorbent material |
Specific area |
Adsorbates |
Adsorption capacities |
Ref. |
| Iron modified activated carbon with Mn as additive |
825 m2 g−1 |
DBT DMBT |
16.24 mg S g−1 |
19 |
| MIL-101(Cr) |
3711 m2 g−1 |
DBT |
28.9 mg S g−1 |
49 |
| Activated Al2O3 |
143.6 m2 g−1 |
DBT |
21.0 mg S g−1 |
50 |
| Aluminum impregnated activated carbon |
1146 m2 g−1 |
DBT |
34.48 mg S g−1 |
51 |
| Composites of activated carbon and zinc and nickel oxides |
62 m2 g−1 |
Thiophene, BT, DBT |
4.98 mg S g−1 |
52 |
| CS-ACA |
1150 m2 g−1 |
DBT |
27.27 mg S g−1 |
In this work |
| PUPW-ACA |
1082 m2 g−1 |
DBT |
25.36 mg S g−1 |
In this work |
3.4 Adsorption kinetics
Kinetic study is essential in adsorption experiments and industrial adsorption production; it can provide important relevant data for reactor design. In the initial stage of adsorption, DBT is first adsorbed by the macropores and mesopores of the adsorbent, which shows that the initial adsorption rate is fast. When the large and mesopores of the adsorbent are adsorbed and saturated, DBT will be adsorbed by the micropores. Increased adsorption resistance is manifested by a slower adsorption rate. The pseudo-first-order model and pseudo-second-order model were used to fit and analyse the adsorption data of the four adsorbents to study the adsorption kinetics and adsorption pathways. The pseudo-first-order model assumes that DBT does not desorb on the surface of the adsorbent and that no DBT exists on the surface of the adsorbent.53
Pseudo-first-order dynamics is expressed by the following equation:
| |
ln(qe − qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) qe − k1t qe − k1t
| (4) |
Pseudo-second-order was used to describe the entire adsorption process and the total amount of adsorption. The pseudo-second-order is shown by eqn (5):
| |
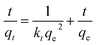 | (5) |
where
qe (mg g
−1) and
qt (mg g
−1) represent the equilibrium quantity and quantity of sulfur adsorbed at contact time
t (min) respectively.
k1 (min
−1) and
kt (g mg
−1 min
−1) are the rate constants for the pseudo-first-order and pseudo-second-order, respectively.
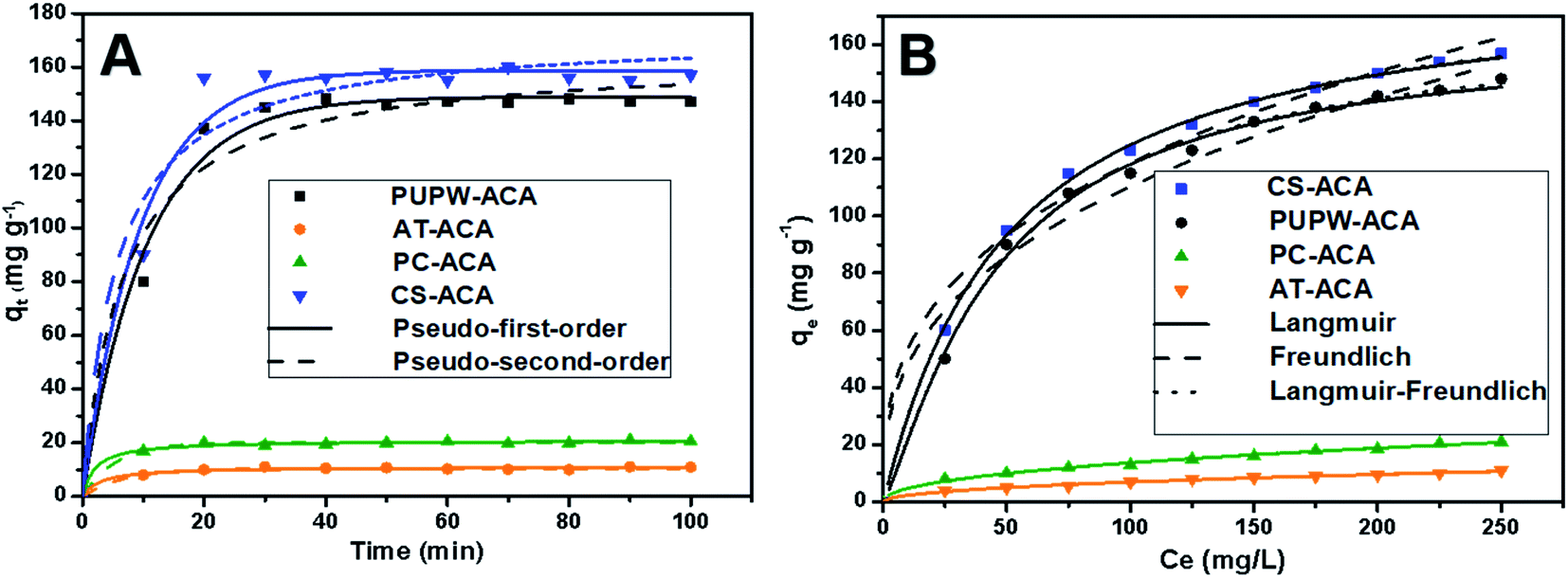
All kinetic parameters of the above four adsorbents have been given in Table 4. From the data in Table 4, it can be observed that the R2 of the pseudo-first-order is significantly larger than that of the pseudo-second-order, and the theoretical value of the pseudo-first-order is closer to the experimental value. The results show that the adsorption of DBT by the four adsorbents prepared in this experiment conforms to a pseudo-first-order kinetic model. The migration of the adsorbed substance from the solution to the surface of the adsorbent may be controlled by a single mechanism or multiple mechanisms. For example, there may be membrane diffusion, external diffusion, surface diffusion, pore diffusion, or any combination of the two. This can explain the existence of a chemisorption mechanism at the active site of the adsorbent.
Table 4 Kinetic parameters for the adsorption process with PUPW-ACA, PC-ACA, AT-ACA, and CS-ACA for DBT
| Compound |
qe (exp.) (mg g−1) |
Pseudo-first order |
Pseudo-second order |
| k1 (min−1) |
qe (cal.) (mg g−1) |
R2 |
K2 (g mg−1 min−1) |
qe (cal.) (mg g−1) |
R2 |
| PUPW-ACA |
148 |
0.0943 |
148.88 |
0.9856 |
0.0009 |
163.99 |
0.9584 |
| AT-ACA |
11 |
0.1455 |
10.56 |
0.9856 |
0.0298 |
11.10 |
0.9748 |
| PC-ACA |
21 |
0.1888 |
20.08 |
0.9913 |
0.0235 |
20.85 |
0.9902 |
| CS-ACA |
157 |
0.1051 |
158.77 |
0.9765 |
0.0010 |
172.50 |
0.9469 |
3.5 Adsorption models and thermodynamics
The adsorption isotherms were plotted to follow Langmuir, Freundlich and L–F equation35,54–56 and the fitting parameters were summarized in Fig. 7(B). The equilibrium expression of the Langmuir model is:| | |
qe = qmax × KL × Ce/(1 + KL × Ce)
| (7) |
where KL (kg mg−1) represents Langmuir constant that relates to the affinity of the binding sites which describes the intensity of the adsorption process, and qmax is maximum adsorption capacity.
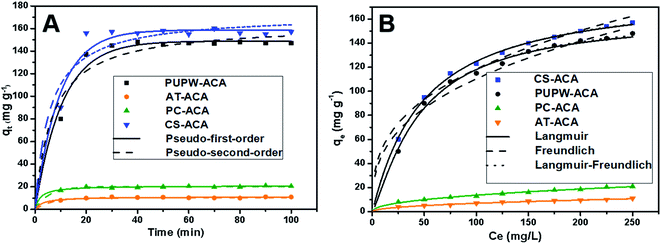 |
| | Fig. 7 (A) Kinetic curves of adsorption of DBT on PC-ACA, PUPW-ACA, CS-ACA, and AT-ACA. (B) Isotherms of adsorption of DBT on PC-ACA, PUPW-ACA, CS-ACA, and AT-ACA. (adsorbent dosage 0.6 g L−1; time of contact fixed at 30 min; temperature, 30 °C). | |
Freundlich equation is given as follows:
where
KF and
n are Freundlich constants indicative of adsorption capacity and adsorption intensity, respectively.
The Langmuir–Freundlich (L–F) model is developed from the Langmuir and Freundlich models. This model, due to considerable heterogeneity of the surface of the adsorbent, is more appropriate. The L–F equation is:
| |
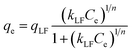 | (9) |
where
qLF is maximum adsorption capacity and
KLF is L–F constant.
Fig. 7(B) shows the equilibrium adsorption isotherms of four adsorbents. Table 5 shows the detailed data of equilibrium adsorption isotherms. It can be seen that the regression coefficients of L–F isotherm of the four adsorbents are all higher than Langmuir and Freundlich, suggesting that the L–F model can better fit the adsorption data. And the qLF value is closer to the experimental value. Based on the L–F isotherm, the maximum adsorption capacity of DBT (mg g−1) onto adsorbents followed this order: CS-ACA > PUPW-ACA > PC-ACA > AT-ACA. This is because the Langmuir model is only suitable for describing the single-molecule adsorption process, and the Freundlich model is suitable for describing the multi-molecular layer adsorption process. However, the L–F model has wider applicability.
Table 5 Adsorption isotherm parameters for the adsorption process with PUPW-ACA, PC-ACA, AT-ACA, and CS-ACA for DBT. (temperature, 30 °C)
| Compound |
Langmuir isotherm |
Freundlich isotherm |
L–F isotherm |
| KL (L g−1 min−1) |
qmax (mg g−1) |
R2 |
KF (g mg−1) |
n |
R2 |
KLF |
n |
qLF (mg g−1) |
R2 |
| PUPW-ACA |
0.0178 |
139.75 |
0.9861 |
14.20 |
2.899 |
0.9837 |
0.0203 |
1.9709 |
147.89 |
0.9988 |
| AT-ACA |
0.0090 |
12.56 |
0.9889 |
11.00 |
2.776 |
0.9787 |
0.0212 |
1.8293 |
10.96 |
0.9966 |
| PC-ACA |
0.0008 |
18.72 |
0.9856 |
1.657 |
2.180 |
0.9876 |
0.0086 |
2.154 |
20.53 |
0.9936 |
| CS-ACA |
0.0112 |
148.37 |
0.9811 |
0.7987 |
2.127 |
0.9836 |
0.0092 |
2.092 |
156.88 |
0.9914 |
Determine the thermodynamic parameters through the following equations.35
| | |
ΔG = RTln(KD)
| (11) |
| |
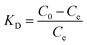 | (12) |
| |
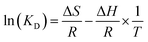 | (13) |
where
R is the universal gas constant (8.314 J K
−1 mol
−1);
T is the absolute temperature (Kelvin);
C0 is the initial concentration,
Ce is the equilibrium concentration; and
KD is the distribution equilibrium constant (dimensionless). By plotting a graph of ln(
KD)
versus 1/
T, a straight line is obtained. From the intercept and slope of the plot, it is possible to calculate the changes in entropy Δ
S and changes in enthalpy (Δ
H), respectively. According to E.C. Lima
et al.,
57 we calculated the thermodynamic parameters of the adsorbent in detail.
Table 6 shows the detailed thermodynamic data of PC-ACA, PUPW-ACA, CS-ACA, and AT-ACA. The values of Δ
G, Δ
H, and Δ
S are all negative numbers, which indicates that the adsorption process of the adsorbent to DBT is spontaneous, exothermic, and a decrease in the degree of freedom.
Table 6 Thermodynamic parameters for the adsorption process with PUPW-ACA, PC-ACA, AT-ACA, and CS-ACA for DBT
| Temperature (K) |
283 |
293 |
303 |
313 |
323 |
| PUPW-ACA |
| ΔG (kJ mol −1) |
−8.56 |
−8.17 |
−7.78 |
−7.39 |
−7.00 |
| ΔH (kJ mol −1) |
— |
— |
−19.57 |
— |
— |
| ΔS (J K −1 mol −1) |
— |
— |
−38.91 |
— |
— |
|
| AT-ACA |
| ΔG (kJ mol −1) |
−1.62 |
−1.48 |
−1.34 |
−1.20 |
−1.07 |
| ΔH (kJ mol −1) |
— |
— |
−5.54 |
— |
— |
| ΔS (J K −1 mol −1) |
— |
— |
−13.85 |
— |
— |
|
| PC-ACA |
| ΔG (kJ mol −1) |
−2.91 |
−2.75 |
−2.60 |
−2.44 |
−2.28 |
| ΔH (kJ mol −1) |
— |
— |
−7.35 |
— |
— |
| ΔS (J K −1 mol −1) |
— |
— |
−15.69 |
— |
— |
|
| CS-ACA |
| ΔG (kJ mol −1) |
−13.10 |
−12.74 |
−12.38 |
−12.03 |
−11.67 |
| ΔH (kJ mol −1) |
— |
— |
−23.21 |
— |
— |
| ΔS (J K −1 mol −1) |
— |
— |
−35.74 |
— |
— |
3.5 Adsorption mechanism
The adsorption of DBT in model oil by activated carbon is mainly affected by pore structure and surface chemical groups. It is known that the dynamic diameter of DBT is 0.55 nm, and the smaller pore structure may be one of the most important driving forces for DBT adsorption. Shi et al. reported that the pore structure smaller than 1 nm has a strong correlation with the maximum adsorption desulfurization capacity.56 This also confirms that the small pore size is beneficial to the adsorption of DBT. According to the Lewis acid–base theory, most of the thiophene sulfur compounds in fuel are Lewis bases.58 DBT is a kind of Lewis base, which is easy to adsorb at the center of Lewis acid. The higher the content of acidic compounds in the oxygen-containing functional groups on the surface of activated carbon, the stronger the acidity. Ania et al. have proposed that the increase of oxygen-containing functional groups can promote specific oxygen–sulfur interactions, and the increase of acidic sites will also disturb the charge distribution on the surface of activated carbon so that active sites for adsorption of sulfur-containing compounds may be formed.46 Zhou et al. also suggested that the increase of oxygen-containing functional groups on the surface of activated carbon also plays an important role in improving its desulfurization ability.59 Starting from the correlation between the structure of carbon materials and the adsorption capacity of organic sulfides, Sano et al. proposed that specific oxygen functional groups are key to adsorb sulfides.60 The above-mentioned studies have also confirmed this theory.
3.6 Economic analysis
In this work, four different desulfurization sorbents were prepared using four different raw materials under the same experimental conditions, so the cost of heat consumption and instrument loss during the experiment were ignored. The desulfurization performance and yield of the adsorbents prepared by the four raw materials were evaluated in detail, and the HHV values of the by-products (non-condensable gas and pyrolysis oil) produced by the four raw materials during the experiment were calculated in detail. Based on the aforementioned two analysis results, the kind of raw material which is most suitable for the preparation of the adsorption desulfurization agent was finally evaluated.
The prices of raw materials and chemicals used in this study were obtained from the Chemical Materials Network. The current market prices of PUPW, AT, PC and dried CS are 7 $ t−1, 79 $ t−1, 145 $ t−1 and 216 $ t−1, respectively. The price of PUPW is that of PUPW compressed into blocks, to increase its density and facilitate transportation. As a common plastic waste, PUPW's price is extremely low, so its cost as a waste is a huge advantage. In this experiment, the yields of the adsorbents prepared by PUPW, AT, PC, and CS were 13.5%, 70.7%, 66.7%, and 23.7%, respectively. So, the prices of PUPW-ACA, AT-ACA, PC-ACA, and CS-ACA are 51.9 $ t−1, 112 $ t−1, 217 $ t−1, and 911 $ t−1, respectively. If 1 t fuel with 10000 μg g−1 sulfur content is reduced to 10 μg g−1, the mass used for PUPW-ACA, AT-ACA, PC-ACA, and CS-ACA will be 0.068 t, 0.908 t, 0.476 t, and 0.064 t, respectively. If these four adsorbents are reused 3 times, then the service quality of PUPW-ACA, AT-ACA, PC-ACA, and CS-ACA will be reduced to 0.0191 t, 0.252 t, 0.127 t, and 0.0182 t, respectively. This decreases the prices to 0.99 $ t−1, 28.22 $ t−1, 27.56 $ t−1 and 16.58 $ t−1, respectively. Therefore, it can be seen that PUPW-AC has more obvious economic benefits. Besides, non-condensable gases may be burnt in turbines and boilers. They can also supply heat to pyrolysis equipment to achieve energy self-sufficiency. The pyrolysis oil contains a variety of components, which can be directly used as fuel oil or refined into high-quality liquid fuel. In addition, the stability of the four adsorbents was evaluated in acid and alkali environments. Table S1† shows the change of the pore structure of the adsorbent after acid, base treatment, indicating that PUPW-ACA has high stability. In summary, in terms of economics and the wide range of raw material sources, the desulfurizer prepared with PUPW has higher economic benefits and practical significance.
4 Conclusions
The order of the specific surface area of the four adsorbents is CS-ACA > PUPW-ACA > PC-ACA > AT-ACA. The results of XPS indicated that each adsorbent contains the same kind of surface functional groups. The Base titration results revealed that the order of the number of functional groups on the surface is CS-ACA > AT-ACA > PUPW-ACA > PC-ACA. The results of adsorption desulfurization experiments show that the desulfurization capabilities of the four desulfurization agents are in the order: AT-ACA > CS-ACA > PC-ACA > PUPW-ACA. This shows that using activated carbon as a desulfurization agent is very feasible; its desulfurization ability is controlled by many factors, including specific surface area, pore structure, and surface functional groups. Based on the above conditions, the economic analysis of these four desulfurizers was carried out. If PUPW-ACA is used to reduce 1 t fuel oil with a sulfur content of 10000 μg g−1 to 10 μg g−1, the cost is only 0.99 $. And the by-products produced by PUPW have the highest HHV value and higher economic benefits. The overall analysis proves that PUPW has the possibility of realizing large-scale production of fuel oil desulfurization agent. This method not only solves the problem of difficult treatment of the waste PUPW, and makes high-quality resource utilization, but also provides a practical method for reducing the sulfur content of fuel oil. This is also in line with the concept of the development of a contemporary environment-friendly society.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (20CX02206A), the Development Fund of State Key Laboratory of Heavy Oil Processing and Graduate student innovation projects of China University of Petroleum (YCX2020049).
References
- Y. X. Li, J. X. Shen, S. S. Peng, J. K. Zhang, J. Wu, X. Q. Liu and L. B. Sun, Nat. Chem., 2020, 11(1), 3206 CAS.
- L.-X. Cai, S.-C. Li, D.-N. Yan, L.-P. Zhou, F. Guo and Q.-F. Sun, J. Am. Chem. Soc., 2018, 140(14), 4869–4876 CrossRef CAS.
- H. Li, J. Liu, J. Li, Y. Hu, W. Wang, D. Yuan, Y. Wang, T. Yang, L. Li, H. Sun, S. Ren, X. Zhu, Q. Guo, X. Wen, Y. Li and B. Shen, ACS Catal., 2017, 7(7), 4805–4816 CrossRef CAS.
- M. Behl, J. Yeom, Q. Lineberry, P. K. Jain and M. A. Shannon, Nat. Nanotechnol., 2012, 7(12), 810–815 CrossRef CAS.
- G. F. S. Barbarelli, M. Amelio and N. M. Scornaienchi, Appl. Energy, 2018, 224, 717–730 CrossRef.
- Y. L. Qi Li, S. Guo and H. Zhou, Nano Today, 2017, 16, 46–60 CrossRef.
- T. A. Saleh, J. Cleaner Prod., 2018, 172, 2123–2132 CrossRef CAS.
- G. I. Danmaliki and T. A. Saleh, Chem. Eng. J., 2017, 307, 914–927 CrossRef CAS.
- G. I. Danmaliki, T. A. Saleh and A. A. Shamsuddeen, Chem. Eng. J., 2017, 313, 993–1003 CrossRef CAS.
- D. M. Xue, S. C. Qi, Q. Z. Zeng, R. J. Lu, J. H. Long, C. Luo, X. Q. Liu and L. B. Sun, Chem. Eng. J., 2019, 374, 1005–1012 CrossRef CAS.
- A. Mansouri, A. A. Khodadadi and Y. Mortazavi, J. Hazard. Mater., 2014, 271, 120–130 CrossRef CAS.
- Y. Yang, G. Lv, L. Deng, B. Lu, J. Li, J. Zhang, J. Shi and S. Du, J. Cleaner Prod., 2017, 161, 422–430 CrossRef CAS.
- B. K. Jung and S. H. Jhung, Fuel, 2015, 145, 249–255 CrossRef CAS.
- T. A. Saleh and G. I. Danmaliki, Process Saf. Environ. Prot., 2016, 102, 9–19 CrossRef CAS.
- F. Subhan, S. Aslam, Z. Yan, L. Zhen, M. Ikram, R. Ullah, U. J. Etim and A. Ahmad, Chem. Eng. J., 2018, 339, 557–565 CrossRef CAS.
- X. Jiang, W. Xu, W. Liu, M. Yue, Y. Zhu and M. Yang, Fuel, 2019, 241, 777–785 CrossRef CAS.
- L. Fan, J. Chen, J. Guo, X. Jiang and W. Jiang, J. Anal. Appl. Pyrolysis, 2013, 104, 353–360 CrossRef CAS.
- Y. Zhao, J. Dou, X. Duan, H. Chai, J. Oliveira and J. Yu, Adverse Environ. Sci. Technol., 2020, 54(3), 1973–1981 CrossRef CAS.
- J. A. Arcibar-Orozco, A. A. Acosta-Herrera and J. R. Rangel-Mendez, J. Cleaner Prod., 2019, 218, 69–82 CrossRef CAS.
- M. Moradi, R. Karimzadeh and E. S. Moosavi, Fuel, 2018, 217, 467–477 CrossRef CAS.
- J. K. Hye-Young Cho, Se-Na Kim and W.-S. Ahn, Microporous
Mesoporous Mater., 2013, 169, 180–184 CrossRef.
- K. X. Lee and J. A. Valla, Appl. Catal., B, 2017, 201, 359–369 CrossRef CAS.
- T. A. Saleh, S. A. Al-Hammadi, A. Tanimu and K. Alhooshani, J. Colloid Interface Sci., 2018, 513, 779–787 CrossRef CAS.
- B. Van de Voorde, M. Boulhout, F. Vermoortele, P. Horcajada, D. Cunha, J. S. Lee, J. S. Chang, E. Gibson, M. Daturi, J. C. Lavalley, A. Vimont, I. Beurroies and D. E. De Vos, J. Am. Chem. Soc., 2013, 135(26), 9849–9856 CrossRef CAS.
- N. A. Khan, J. W. Yoon, J. S. Chang and S. H. Jhung, Chem. Commun., 2016, 52(56), 8667–8670 RSC.
- J. Xiong, W. Zhu, H. Li, L. Yang, Y. Chao, P. Wu, S. Xun, W. Jiang, M. Zhang and H. Li, J. Mater. Chem. A, 2015, 3(24), 12738–12747 RSC.
- T. A. Saleh, K. O. Sulaiman, S. A. Al-Hammadi, H. Dafalla and G. I. Danmaliki, J. Cleaner Prod., 2017, 154, 401–412 CrossRef CAS.
- L. Ding, J. Wei, Z. Dai, Q. Guo and G. Yu, Int. J. Hydrogen Energy, 2016, 41(38), 16823–16834 CrossRef CAS.
- T. Mochizuki, M. Kubota, H. Matsuda and L. F. D'Elia Camacho, Fuel Process. Technol., 2016, 144, 164–169 CrossRef CAS.
- Y. Zhang, D. Duan, H. Lei, E. Villota and R. Ruan, Appl. Energy, 2019, 251, 113337 CrossRef CAS.
- U. Kumar, D. Goonetilleke, V. Gaikwad, J. C. Pramudita, R. K. Joshi, N. Sharma and V. Sahajwalla, ACS Sustainable Chem. Eng., 2019, 7(12), 10310–10322 CrossRef CAS.
- D. Chen, X. Chen, J. Sun, Z. Zheng and K. Fu, Bioresour. Technol., 2016, 216, 629–636 CrossRef CAS.
- G. S. dos Reis, E. C. Lima and C. H. Sampaio, J. Solid State Chem., 2018, 260, 106–116 CrossRef.
- G. S. dos Reis, C. H. Sampaio and E. C. Lima, Colloids Surf., A, 2016, 497, 304–315 CrossRef.
- D. R. Lima, A. Hosseini-Bandegharaei and P. S. Thue, Colloids Surf., A, 2019, 583, 123966 CrossRef CAS.
- H. Ullah, Q. Abbas, M. U. Ali, Amina, A. I. Cheema, B. Yousaf and J. Rinklebe, Chem. Eng. J., 2019, 373, 44–57 CrossRef CAS.
- Y. Zhang, Z. Xia, H. Huang and H. Chen, J. Anal. Appl. Pyrolysis, 2009, 84(1), 89–94 CrossRef CAS.
- E. Yagmur, Y. Gokce, S. Tekin, N. I. Semerci and Z. Aktas, Fuel, 2020, 267, 117232 CrossRef CAS.
- M. Ahmadi, M. Mohammadian, M. R. Khosravi-Nikou and A. Baghban, J. Hazard. Mater., 2019, 374, 129–139 CrossRef CAS.
- D. Li, J. Chen, Y. Fan, L. Deng, R. Shan, H. Chen, H. Yuan and Y. Chen, Energy Fuels, 2019, 33(9), 8927–8936 CrossRef.
- C. Deng and M. Zhu, Energy Fuels, 2020, 34(8), 9320–9327 CrossRef CAS.
- F. Liang, M. Lu, M. E. Birch, T. C. Keener and Z. Liu, J. Chromatogr. A, 2006, 1114(1), 145–153 CrossRef CAS.
- H.-H. T. Nguyen, D. iannel . Poster and W. Ball, Environ. Sci. Technol., 2007, 41(4), 1212–1217 CrossRef.
- J. Bu, G. Loh, C. G. Gwie, S. Dewiyanti, M. Tasrif and A. Borgna, Chem. Eng. J., 2011, 166(1), 207–217 CrossRef CAS.
- Y. Yang, H. Lu, P. Ying, Z. Jiang and C. Li, Carbon, 2007, 45(15), 3042–3044 CrossRef CAS.
- C. O. Ania and a. T. J. Bandosz, Langmuir, 2005, 21(17), 7752–7759 CrossRef CAS.
- Z. Shang, X. An, H. Zhang, M. Shen, F. Baker, Y. Liu, L. Liu, J. Yang, H. Cao, Q. Xu, H. Liu and Y. Ni, Carbon, 2020, 161, 62–70 CrossRef CAS.
- A. C. Lua and T. Yang, J. Colloid Interface Sci., 2004, 274(2), 594–601 CrossRef CAS.
- S. Y. Jia, Y. F. Zhang and Y. Liu, J. Hazard. Mater., 2013, 262, 589–597 CrossRef CAS.
- A. Srivastav and V. C. Srivastava, J. Hazard. Mater., 2009, 170, 1133–1140 CrossRef CAS.
- S. A. Ganiyu, K. Alhooshani and K. O. Sulaiman, Chem. Eng. J., 2016, 303, 489–500 CrossRef CAS.
- Al. Swat, A. A. Saleh and T. A. Ganiyu, J. Anal. Appl. Pyrolysis, 2017, 128, 246–256 CrossRef.
- A. A. Al. Swat, T. A. Saleh, S. A. Ganiyu, M. N. Siddiqui and K. R. Alhooshani, J. Anal. Appl. Pyrolysis, 2017, 128, 246–256 CrossRef.
- D. F. Caicedo, G. S. dos Reis and E. C. Lima, J. Environ. Chem. Eng., 2020, 8, 103875 CrossRef CAS.
- G. S. dos Reis, B. G. Cazacliu and C. R. Correa, J. Environ. Chem. Eng., 2020, 8, 103605 CrossRef CAS.
- Y. W. Shi, G. Z. Liu and L. Wang, Chem. Eng. J., 2015, 259, 771–778 CrossRef CAS.
- E. C. Lima, M. A. Adebayo and F. M. Machado, in Carbon Nanomaterials as Adsorbents for Environmental and Biological Applications, C. P. Bergmann and F. M. Machado, Springer International Publishing, 2015, ch. 3, pp. 33–69 Search PubMed.
- Q. Huo, J. Li, G. Liu, X. Qi, X. Zhang, Y. Ning, B. Zhang, Y. Fu and S. Liu, Chem. Eng. J., 2019, 362, 287–297 CrossRef CAS.
- X. M. a. C. S. Anning Zhou, J. Phys. Chem. B, 2006, 110(10), 4699–4707 CrossRef.
- Y. Sano, Appl. Catal., B, 2004, 49(4), 219–225 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07862j |
| ‡ These authors contributed to the work equally and should be regarded as co-first authors. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Weining Li‡a,
Bernard Wiafe Bineya,
Zhuo Lia,
Jiahua Shenb and
Zongxian Wanga
*a,
Weining Li‡a,
Bernard Wiafe Bineya,
Zhuo Lia,
Jiahua Shenb and
Zongxian Wanga