
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew zinc complexes derived from “self-adaptable” acyclic diiminodipyrromethanes as potent catalysts for the reduction of curing temperature of bisphenol-A/F benzoxazines†
Shiva K. Loke,
Eswar Pagadala,
S. Devaraju,
V. Srinivasadesikan and
Ravi K. Kottalanka *
*
Division of Chemistry, Department of Sciences and Humanities, Vignan's Foundation for Science Technology and Research (Deemed to be University), Vadlamudi, Guntur-522213, Andhra Pradesh, India. E-mail: ravikottalanka@gmail.com; Fax: +91 863 2344 707; Tel: +91 863 2344 700
First published on 1st October 2020
Abstract
The simple modification of the Schiff-base ligands often brings significant changes in the coordination properties of the metal-complexes, providing newer prospects for their unexplored applications. In this context, the present work utilized the “self-adaptable” acyclic diiminodipyrromethane Schiff's bases (2a and 2b) for the synthesis of their Zn-based complexes and explored their potential in the ring-opening polymerization of benzoxazines. The two zinc complexes of composition [Zn{(Ph)(CH3)C(2,6-iPr2C6H3–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–C4H2N)(2,6-iPr2C6H3–NCH–C4H2NH)}2] (3) and [ZnCl2{(Ph)(CH3)C(Ph3C–NHCH–C4H2N)2}] (4) were synthesized in good yields, and the structures were confirmed by single crystal X-ray diffraction (XRD). Later, zinc complexes (3 & 4) were used as catalysts to reduce the curing (ring-opening polymerization) temperature of benzoxazine monomers such as Bisphenol-A (BA-a) and Bisphenol-F (BF-a) benzoxazines. Dynamic scanning calorimetry (DSC) studies revealed that the on-set curing (Tp) temperatures were reasonably decreased upto 20% for the benzoxazines. Furthermore, the thermal stabilities of the polybenzoxazines (PBzs) derived in the presence of zinc catalysts (3 and 4) were compared with PBz obtained in the absence of catalyst under similar conditions. The thermal studies reveled that there is no significant changes in the initial degradation of polymers. However, the thermal stability in terms of char yields at 800 °C improved upto 10–21% for the bisphenol-A/F benzoxazines.
CH–C4H2N)(2,6-iPr2C6H3–NCH–C4H2NH)}2] (3) and [ZnCl2{(Ph)(CH3)C(Ph3C–NHCH–C4H2N)2}] (4) were synthesized in good yields, and the structures were confirmed by single crystal X-ray diffraction (XRD). Later, zinc complexes (3 & 4) were used as catalysts to reduce the curing (ring-opening polymerization) temperature of benzoxazine monomers such as Bisphenol-A (BA-a) and Bisphenol-F (BF-a) benzoxazines. Dynamic scanning calorimetry (DSC) studies revealed that the on-set curing (Tp) temperatures were reasonably decreased upto 20% for the benzoxazines. Furthermore, the thermal stabilities of the polybenzoxazines (PBzs) derived in the presence of zinc catalysts (3 and 4) were compared with PBz obtained in the absence of catalyst under similar conditions. The thermal studies reveled that there is no significant changes in the initial degradation of polymers. However, the thermal stability in terms of char yields at 800 °C improved upto 10–21% for the bisphenol-A/F benzoxazines.
Introduction
Polybenzoxazines (PBzs) are a new type of phenolic resin with good thermo-mechanical properties, which allows overcoming several shortcomings of conventional phenolic resins.1,2 Also, PBzs have numerous advantages including infinite molecular design, almost no shrinkage during curing, low dielectric constant (K), low moisture absorption, high heat and flame resistance, excellent thermal stability (high Tg, and high residual char), and good mechanical and dimensional stability.1,2 However, the major drawback of PBzs is that they need a high curing temperature for the polymerization process. Commonly, PBzs are developed by thermally induced ring opening polymerization of benzoxazines over 200 °C which limits the industrial application of PBzs. Thus, the reduction of curing temperature is highly enviable for industrial applications. Many organic compounds such as organic acids and bases have been studied to lower the ring-opening polymerization temperature of benzoxazines.3,4 However, the reduction of polymerization temperature achieved by these organic compounds is still substandard. On the other hand, Ishida et al. have demonstrated that Lewis acids such as metal halides and metal trifluoromethane sulfonates promote the polymerization of benzoxazine effectively.5 Though these Lewis acid catalysts are economical and potent, their miscibility in neat benzoxazines is limited. Takeshi Endo et al. proved that, acetylacetonato (acac) metal complexes of 4th period transition metals are highly efficient catalysts for polymerization of benzoxazines at low temperature without effecting thermal stability of obtained PBzs.6 This study indicates that, Lewis acids in complex structures would obviously increase both their stabilities and miscibility with monomers during the curing process and therefore, those metal complexes can be act as efficient catalysts for benzoxazine polymerization. Though, there are many Lewis acid catalysts reported in the literature, structurally fully characterized metal-complexes supported by acyclic Schiff's based ancillary ligands particularly for benzoxazine ring-opening polymerization with lowering of curing temperature were missing.In recent times, N-donor extended dipyrromethanes and their derivatives emerge as a new class of di-anionic (H2L) and tetra-anionic (H4L) Schiff base ligands with vast potential as ancillary ligands for the low-valent transition metals as well as for the alkali and alkaline-earth metals.7,8 The pioneering works in this area mainly include the extensive study on the syntheses of various structurally diversified metal complexes containing macrocyclic N-donor extended dipyrromethane Schiff base ligands and their utility in redox catalysis.9,10 However, very less attention has been paid towards the acyclic N-donor extended dipyrromethanes, formally known as diiminodipyrromethane Schiff bases.8b,i,j,11,12 The combination of both pyrrole and azomethine group of these ligands makes them unique due to the highly flexible coordination features. One of the factors that make these diiminodipyrromethane Schiff bases unique is that this set of ligands also displays pyrrole-imine to azafulvene-amine tautomerization (both single and double) under certain reaction conditions (Scheme 1), thus making them “self-adaptable”. Further, the ability of diiminodipyrromethane supported metal complexes to stabilize various charged species through hydrogen bonding interactions renders them quite unique.13
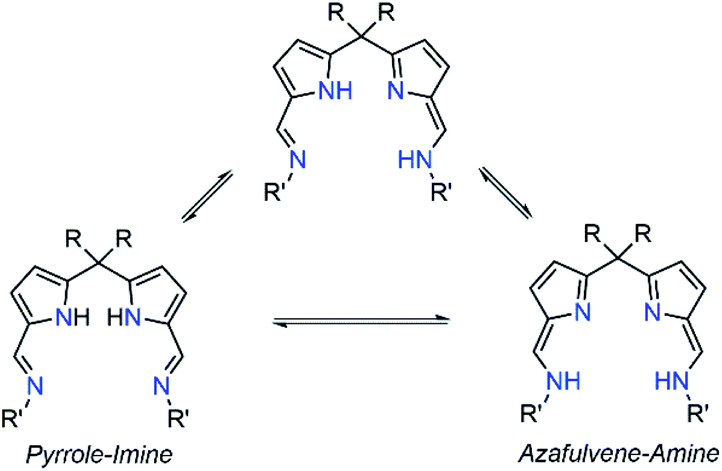 | ||
| Scheme 1 Possible pyrrole-imine to azafulvene-amine tautomerization (single and double). | ||
In this context, here we report the synthesis and structures of new acyclic N-donor extended dipyrromethanes Schiff base ligands with chemical formulae [(Ph)(CH3)C(2,6-iPr2C6H3–NCH–C4H2NH)2] (2a) and [(Ph)(CH3)C(Ph3C–NCH–C4H2NH)2] (2b) and corresponding zinc complexes of chemical compositions [Zn{(Ph)(CH3)C(2,6-iPr2C6H3–NCH–C4H2N)(2,6-iPr2C6H3–NCH–C4H2NH)}2] (3) and [ZnCl2{(Ph)(CH3)C(Ph3C–NHCH–C4H2N)2}] (4). The solid-state structures of acyclic Schiff base ligand (2a), zinc complexes (3 and 4) were analyzed in detail using single crystal X-ray diffraction (XRD) study and further characterizations were also carried out using various spectroscopic and analytical techniques. The density functional theory (DFT) calculations were performed to obtain more insight on the flexible coordination behaviour of the both ligands (2a and 2b). Further, we also report the curing behaviour of (ROP) of benzoxazines (BA-a and BF-a) using zinc complexes (3 and 4) as catalysts with the use of DSC analysis. The curing studies revealed that the both zinc complexes (3 and 4) are effective in reducing the curing (polymerization) temperature via coordination–insertion mechanism without affecting the polymer inherent property including thermal stability.
Results and discussion
Synthesis of acyclic diiminodipyrromethane Schiff bases (2a and 2b)
The acyclic N-donor extended dipyrromethane Schiff bases of composition [(Ph)(CH3)C(R–NCH–C4H2NH)2] (where R = 2,6-iPr2C6H3 for 2a and R = –CPh3 for 2b) were synthesized from easily assessable starting materials by following literature reports.8i,j,14 Dipyrromethane of composition [(Ph)(CH3)C(C4H3NH)2] (1) was obtained according to the literature procedure.14 Formylation at the meso-carbon position of dipyrromethane [(Ph)(CH3)C(C4H3NH)2] (1) via Vilsmeier–Haack reaction yielded mono-meso substituted dialdehyde [(Ph)(CH3)C{C4H2NH(CHO)}2] (2) in good yield and crystal structure of 2 was reported in our recent publication.15 The further treatment of 2 with 2,6-diisopropylaniline and triphenyl methylamine under standard Schiff base condensation method14 afforded acyclic N-donor extended dipyrromethane of compositions [(Ph)(CH3)C(2,6-iPr2C6H3–NCH–C4H2NH)2] (2a) and [(Ph)(CH3)C(Ph3C–NCH–C4H2NH)2] (2b) in very good yields (91% for 2a and 95% for 2b) (Scheme 2).
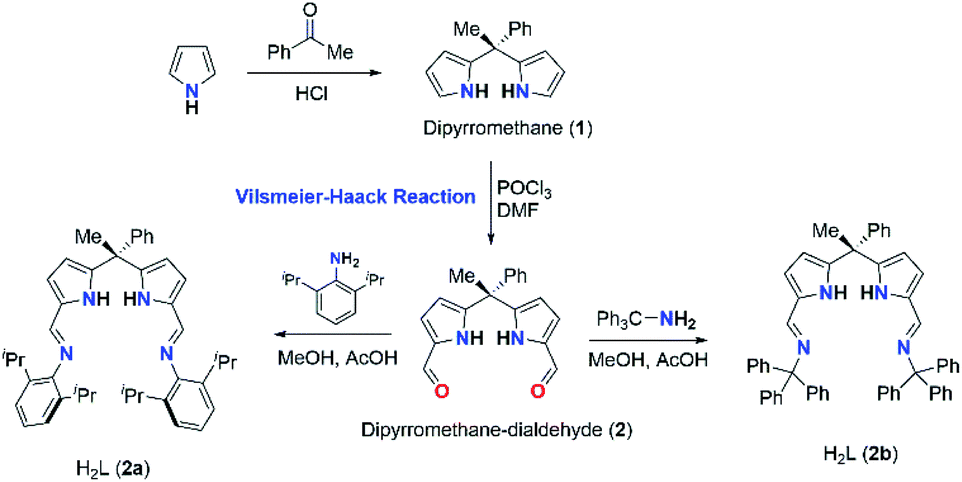 | ||
| Scheme 2 Synthesis of acyclic diiminodipyrromethanes (H2L) (2a and 2b). | ||
The both ligands were characterized by spectroscopic and analytical techniques. The crystals of suitable dimension for single crystal XRD analysis for acyclic diiminodipyrromethane (2a) were obtained after three days in ethyl acetate/hexane mixture. The solid-state structure of ligand (2a) was established by using single crystal XRD technique. The solid-state structure of acyclic Schiff base ligand (2a) is shown in Fig. S1 in the ESI† and the details of the structural parameters are given in Table 1 (see ESI†).
| Monomer+ | [% of Zn-Cat] | Ton-set (°C) | Tpeak (°C) | Tend (°C) |
|---|---|---|---|---|
| a DSC thermogram data were collected under N2 flow (20 mL min−1) and a heating rate of 10 °C min−1. | ||||
| BA-a | (Neat) | 165 | 218 | 278 |
| BA-a + [Zn(2a)2] | (3%) | 157 | 215 | 277 |
| (5%) | 143 | 200 | 258 | |
| (10%) | 133 | 195 | 252 | |
| BA-a + [Zn(2b-I)Cl2] | (3%) | 149 | 210 | 270 |
| (5%) | 144 | 207 | 265 | |
| (10%) | 136 | 199 | 250 | |
| BF-a | (Neat) | 162 | 217 | 278 |
| BF-a + [Zn(2a)2] | (3%) | 160 | 210 | 276 |
| (5%) | 139 | 198 | 267 | |
| (10%) | 132 | 191 | 252 | |
| BF-a + [Zn(2b-I)Cl2] | (3%) | 151 | 207 | 270 |
| (5%) | 148 | 206 | 273 | |
| (10%) | 142 | 198 | 253 | |
Synthesis of zinc catalysts (3 and 4)
The synthesis of two new zinc catalysts (3 and 4) were achieved by either direct zinc metalation using suitable Zn(II) agents or by salt metathesis route. The zinc metalation of acyclic N-donor extended dipyrromethane Schiff base ligand 2a was achieved using diethylzinc [ZnEt2] and H2L ligand 2a in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio in dry toluene at 80 °C under inert atmospheric conditions via alkane elimination route yielded a single-site, monometallic, non-helicate zinc complex of composition [Zn{(Ph)(CH3)C(2,6-iPr2C6H3–NCH–C4H2N)(2,6-iPr2C6H3–NCHC4H2NH)}2] (3) (Scheme 3) with good yield (80%). Though the alkane elimination method yields high purity of zinc complex 3, The title zinc complex was also synthesized via one-pot salt metathesis route under non-inert conditions to evade the usage of highly reactive and moisture sensitive diethylzinc reagent. The one-pot salt metathesis route involved the reaction between acyclic diiminodipyrromethane ligand 2a, alkali-metal bis(trimethylsilyl amide) [MN(SiMe3)2] (where M = Li or Na) and anhydrous ZnCl2 in 2:2:1 ration under non-inert atmospheric conditions at elevated temperature (Scheme 3).
:2 ratio in dry toluene at 80 °C under inert atmospheric conditions via alkane elimination route yielded a single-site, monometallic, non-helicate zinc complex of composition [Zn{(Ph)(CH3)C(2,6-iPr2C6H3–NCH–C4H2N)(2,6-iPr2C6H3–NCHC4H2NH)}2] (3) (Scheme 3) with good yield (80%). Though the alkane elimination method yields high purity of zinc complex 3, The title zinc complex was also synthesized via one-pot salt metathesis route under non-inert conditions to evade the usage of highly reactive and moisture sensitive diethylzinc reagent. The one-pot salt metathesis route involved the reaction between acyclic diiminodipyrromethane ligand 2a, alkali-metal bis(trimethylsilyl amide) [MN(SiMe3)2] (where M = Li or Na) and anhydrous ZnCl2 in 2:2:1 ration under non-inert atmospheric conditions at elevated temperature (Scheme 3).
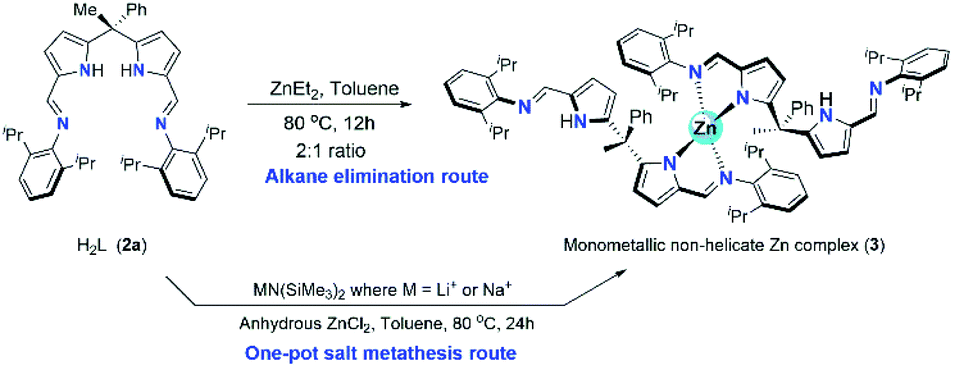 | ||
| Scheme 3 Synthesis of zinc complex 3. | ||
The formation of zinc complex 3 was confirmed by the single crystal XRD analysis and further characterized using spectroscopic/analytical techniques. The formation of zinc complex 3 is mainly due to the presence of sterically bulky Schiff base ligand 2a in the Zn(II) coordination sphere and due to the presence of phenyl and methyl groups at meso-carbon site of the ligand backbone which restricts the twisting around meso-carbon centre during the zinc metal coordination. Further the dangling nature of 2,6-iPr2C6H3 group attached to the imine-nitrogens of ligand moiety favours the more stable trans like confirmation. Therefore, the formation of single-site, monometallic, non-helicate can be successfully achieved. The crystals suitable for single crystal XRD analysis were obtained after 3 days from recrystallization of zinc complex 3 in toluene/n-hexane mixture at room temperature under ambient conditions. The single crystal X-ray diffraction analysis shows that the zinc complex 3 so obtained is crystalizes in monoclinic space group P21/c having 4 independent molecules in the unit cell. The details of the structural parameters are given in Table 1 in the ESI.† The solid-state structure of zinc complex 3 was shown in Fig. 1. In zinc complex 3, the central Zn(II) metal ion is four-fold coordinated by two mono-anionic diiminodipyrromethane ligands 2a and forms a distorted tetrahedral geometry around the Zn(II) cationic centre. Each ligand moiety chelated to the central metal ion through pyrrolide nitrogen (Npyr) and imine nitrogen (Nimine) atoms, thus forming a two five-membered metallacycles with bite angles of 85.38(6)° (for N1–Zn1–N2) and 85.02(6)° (for N5–Zn1–N6) respectively.
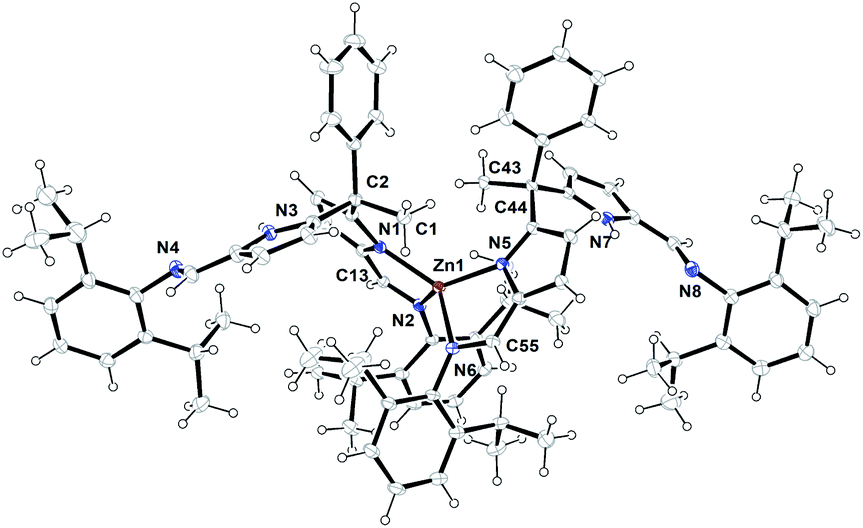 | ||
| Fig. 1 ORTEP diagram of zinc metal complex (3) with thermal displacement parameters drawn at the 30% probability level. Selected bond lengths [Å] and bond angles[°]: Zn1–N1 2.3015(16), Zn1–N2 2.0673(16), N1–C9 1.355(2), N1–C12 1.384(2), N2–C13 1.302(3), N2–C14 1.425(2), C12–C13 1.410(3), C11–C12 1.391(3), N3–C29 1.377(3), N3–C26 1.373(3), N4–C30 1.272(3), N4–C31 1.425(3); N5–Zn1–N2 129.54(6), N5–Zn1–N1 123.56(6), N5–Zn1–N6 85.02(6), N2–Zn1–N6 109.50(6), N1–Zn1–N2 85.38(6), N1–Zn1–N6 128.48(6). | ||
The typical bond distances and bond angles observed in zinc complex (3) were given in the Fig. 1. In case of zinc complex 3, the observed Zn–Npyr bond distances of 2.0305(16) Å (Zn1–N1) and 2.0203(16) Å (Zn1–N5) are slightly higher than the previously reported Zn1–Npyr distance of 1.978(3) Å in a bimetallic Zn2 helicate structures.11 This small discrepancy can be attributed to the sterically bulky 2,6-iPr2C6H3 groups attached to the imine nitrogen atom of the ligand 2a and also to the restricted rotation around the meso-carbon centre of ligand back bone due to the zinc metalation. The observed Zn1–Nimine bond distances 2.0673(16) Å (Zn1–N2) and 2.0690(16) Å (Zn1–N6) were found to be in good agreement with previously reported Zn–Nimine bond distance of 2.068(3) Å in the literature.11 The bond distances of imine –CN– group present in the zinc coordination sphere are a little elongated (C13–N2) 1.302(3) Å and (C55–N6) 1.293(3) Å in comparison to imine –CN– bond distance observed in uncoordinated ligand fragment (1.272(3) and 1.277(3) Å).
However, the observed imine –CN– bond distances corroborates well with previously reported literature.11,16a Though, the homoleptic zinc complex 3 possesses two mono-anionic diiminodipyrromethane moieties in the zinc coordination sphere, each ligand selectively coordinates to the central metal ion in a bidentate fashion through one of the iminopyrrolide fragment (particularly chelation via one pyrrolide nitrogen (Npyr) and one imine nitrogen (Nimine) center). The other iminopyrrolide fragments of each ligand moiety are oriented in such way that both nitrogen atoms (Npyr and Nimine) are dangling away from the metal ion (trans in Zn) and both C2 and C44 meso-carbon centres became asymmetric centres having a diastereotopic relationship between them. The similar kind of chelation behaviour was observed in the alkali metal complex of type [Na{κ2-(Ph3CNCH)2C4H2N}(THF)2] (trans in Na) in our earlier report.16a To the best of our knowledge, the crystal structure of zinc complex 3 is the first example of a single-site, monometallic non-helicate structure in the field of acyclic diiminodipyrromethane metal clusters.
In contrast to the complex 3, the zinc complex 4 was synthesized via direct reaction between diiminodipyrromethane Schiff base ligand 2b and anhydrous ZnCl2 in 1:1 ration in toluene at 80 °C temperature (Scheme 4). The main reason to follow different procedure for obtaining zinc complex (4) was, unlike acyclic diiminodipyrromethane Schiff base ligand 2a, the acyclic Schiff base ligand 2b undergoes double pyrrole-imine to azafulvene-amine tautomerization in the presence of ZnCl2 via the formation of presumably unstable zinc complex (5) and then the immediate structural rearrangement in the vicinity of hydrogen chloride leads to the formation of zinc complex (4) as an air-stable zinc coordination compound in a very good yield (Scheme 4).
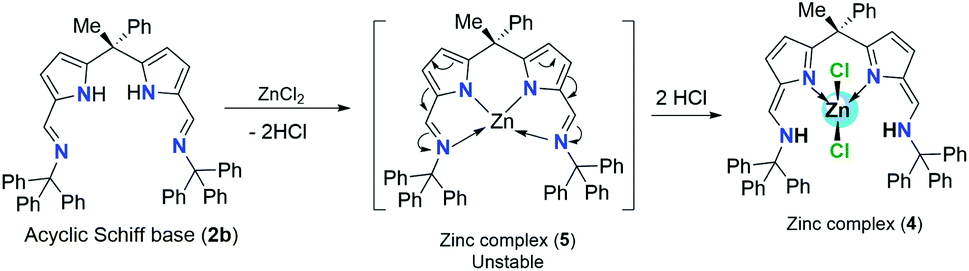 | ||
| Scheme 4 Pyrrole-imine to azafulvene-amine tautomerization in the presence of ZnCl2 and formation of intermediate zinc complex (5). | ||
Similar kind of tautomerization was also found in the four-coordinate metal halide and azide complexes of N-donor-extended dipyrromethanes [MX2(H2L)] formed between the tetradentate H2L diiminodipyrromethane ligand [(CH3)2C{tBu(Me)CHNCH–C4H2NH}2] and the transition metal halides MX2 (M = Fe, X = Br; M = Co, Zn, X = Cl).11 1H-NMR spectrum of zinc complex 4 showed a doublet resonance signal at δ 9.92–9.89 ppm can be best assigned to the N–H protons of Ph3CN-groups which are found to couple with azafulvene CC(H) with a coupling constant of 3JH–H = 12 Hz. A resonance signal observed at δ 7.44 ppm can be assigned to the azafulvene CC(H) protons. All other aromatic protons and pyrrole ring protons were obtained in the expected regions and fully supported the existence of zinc complex 4 even in the solution state as such. The crystals suitable for single crystal XRD analysis were obtained after recrystallization from hot toluene. The single crystal XRD analysis shows that the zinc complex 4 crystalizes in triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) having two independent molecules in the unit cell. The details of the structural parameters are given in table in the ESI.† The solid-state structure of zinc complex 4 was shown in Fig. 2. In the solid-state structure of zinc complex 4, the central Zn(II) metal ion is four-fold coordinated by two mono-anionic chlorine atoms and two pyrrolide nitrogen (Npyr) atoms of diiminodipyrromethane ligand (2b) and forms distorted tetrahedral geometry around the Zn(II) cationic centre. The ligand (2b) in its double pyrrole-imine to azafulvene-amine tautomeric form is chelated to the central metal ion in a bidentate fashion through chelation from two pyrrolide nitrogens (Npyr) rather than bidentate chelation from one pyrrolide nitrogen (Npyr) and one imine nitrogen (Nimine) atom as observed in the case of zinc complex 3. This kind of structural diversity was mainly due to the presence of highly sterically bulky triphenylmethyl (–CPh3) group attached to the imine-nitrogen centres which favoured the formation of amine-azafulvene tautomer at elevated temperatures. Also a six-membered metallacycle (N2–C9–C2–C3–N3–Zn1) with a bite angle of 97.73(9) Å (for N2–Zn1–N3) which is slightly higher than the bite angles 85.38(6)° (for N1–Zn1–N2) and 85.02(6)° (for N5–Zn1–N6) were observed for zinc complex 3. The Zn–Npyr bond distance of 1.979(2) Å (Zn1–N2 or Zn1–N3) was slightly smaller than the Zn–Npyr bond distance perceived in zinc complex 3 (2.0305(16) Å). This is presumably due to the sterically less hindered environment around the Zn(II) cationic centre in complex (4) compared to steric bulk in zinc complex (3). However, Zn–Npyr bond distance observed in zinc complex 4 was best fitted with reported Zn1–Npyr distance of 1.978(3) Å in bimetallic Zn2 helicate structures in the literature.11
having two independent molecules in the unit cell. The details of the structural parameters are given in table in the ESI.† The solid-state structure of zinc complex 4 was shown in Fig. 2. In the solid-state structure of zinc complex 4, the central Zn(II) metal ion is four-fold coordinated by two mono-anionic chlorine atoms and two pyrrolide nitrogen (Npyr) atoms of diiminodipyrromethane ligand (2b) and forms distorted tetrahedral geometry around the Zn(II) cationic centre. The ligand (2b) in its double pyrrole-imine to azafulvene-amine tautomeric form is chelated to the central metal ion in a bidentate fashion through chelation from two pyrrolide nitrogens (Npyr) rather than bidentate chelation from one pyrrolide nitrogen (Npyr) and one imine nitrogen (Nimine) atom as observed in the case of zinc complex 3. This kind of structural diversity was mainly due to the presence of highly sterically bulky triphenylmethyl (–CPh3) group attached to the imine-nitrogen centres which favoured the formation of amine-azafulvene tautomer at elevated temperatures. Also a six-membered metallacycle (N2–C9–C2–C3–N3–Zn1) with a bite angle of 97.73(9) Å (for N2–Zn1–N3) which is slightly higher than the bite angles 85.38(6)° (for N1–Zn1–N2) and 85.02(6)° (for N5–Zn1–N6) were observed for zinc complex 3. The Zn–Npyr bond distance of 1.979(2) Å (Zn1–N2 or Zn1–N3) was slightly smaller than the Zn–Npyr bond distance perceived in zinc complex 3 (2.0305(16) Å). This is presumably due to the sterically less hindered environment around the Zn(II) cationic centre in complex (4) compared to steric bulk in zinc complex (3). However, Zn–Npyr bond distance observed in zinc complex 4 was best fitted with reported Zn1–Npyr distance of 1.978(3) Å in bimetallic Zn2 helicate structures in the literature.11
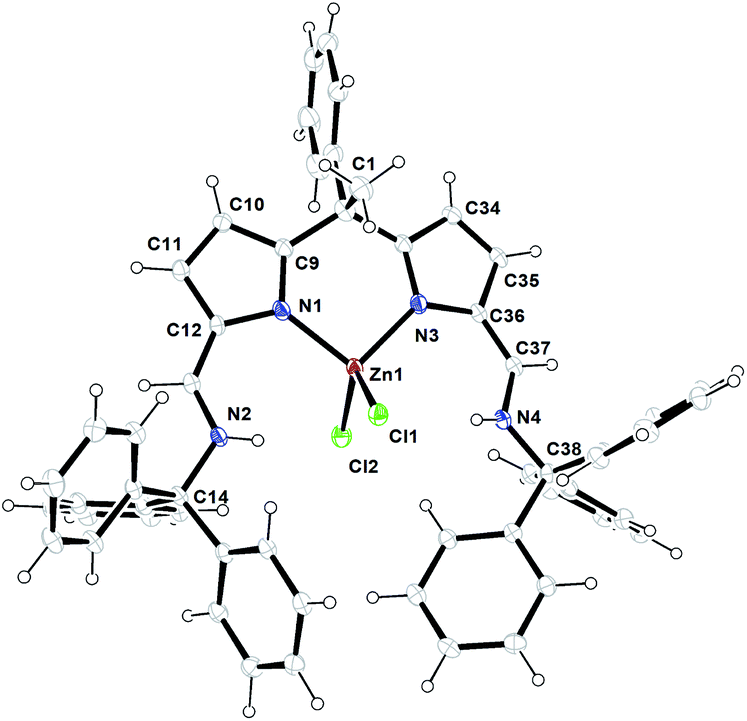 | ||
| Fig. 2 ORTEP diagram of zinc metal complex (4) with thermal displacement parameters drawn at the 30% probability level. Zn1–Cl1 2.2573(7), Zn1–Cl2 2.2301(8), Zn1–N1 1.979(2), N1–C9 1.347(3), N1–C12 1.392(3), C9–C10 1.414(4), C10–C11 1.366(4), C11–C12 1.416(4), C12–C13 1.383(4), N2–C13 1.347(3), N2–C14 1.392(3); N1–Zn1–N3 97.73(9), Cl1–Zn1–Cl2 113.57(3), N1–Zn1–Cl1 112.50(7), N1–Zn1–Cl2 109.54(7), N3–Zn1–Cl1 112.63(7), N3–Zn1–Cl2 109.77(7). | ||
DFT study
The Density Functional Theory (DFT) studies were performed for better understanding the flexible coordination behaviour of acyclic Schiff bases (2a and 2b) due to their self-adaptable nature during the complexation with metal centres. The computational calculations were employed at dispersion corrected ωB97XD17 and hybrid density functional B3LYP18 levels using 6-31g(d) basis set in the gas phase. All the computations were performed using Gaussian 09 (ref. 19) suite of program. Molecular geometries of acyclic Schiff base ligands (2a and 2b) in ground state were fully optimized in the gas phase using ωB97XD/6-31g(d) and B3LYP/6-31g(d) levels. As stated in the experimental results and discussion part, the Schiff base ligands 2a and 2b certainly undergoes double pyrrole-imine to azafulvene-amine tautomerization and hence having different molecular conformations namely 2a, 2a-I & 2b and 2b-I at elevated temperatures. The azafulvene-amine tautomerization of acyclic Schiff base (2a and 2b) to corresponding products (2a-I and 2b-I) was investigated using DFT.The transformation of ligands 2a and 2b are relatively more stable than 2a-I and 2b-I, respectively, which could be achieved through proton-transfer mechanism. The calculated energy barriers of transition states (TS-I & TS-II) and the related optimized structures were shown in Fig. 3. The transition states were confirmed with one imaginary frequency. The theoretical calculations reveal that the proton transfer requires activation energy of 23 kcal mol−1 (see TS-II) to form azafulvene-amine tautomer 2b-I from 2b endothermically and transformation of 2a into 2a-I shows 25.7 kcal mol−1 endothermically as well. Hence, we concluded that the activation energy of transition state TS-II is lower as compared to TS-I presumably due to the presence of Ph3C – which facilitates the proton transfer mechanism in 2b. To know the binding site of Zn, the electrostatic potential calculation was employed at B3LYP/6-31g(d) level of theory and the structures were depicted in Fig. S2 (ESI).†
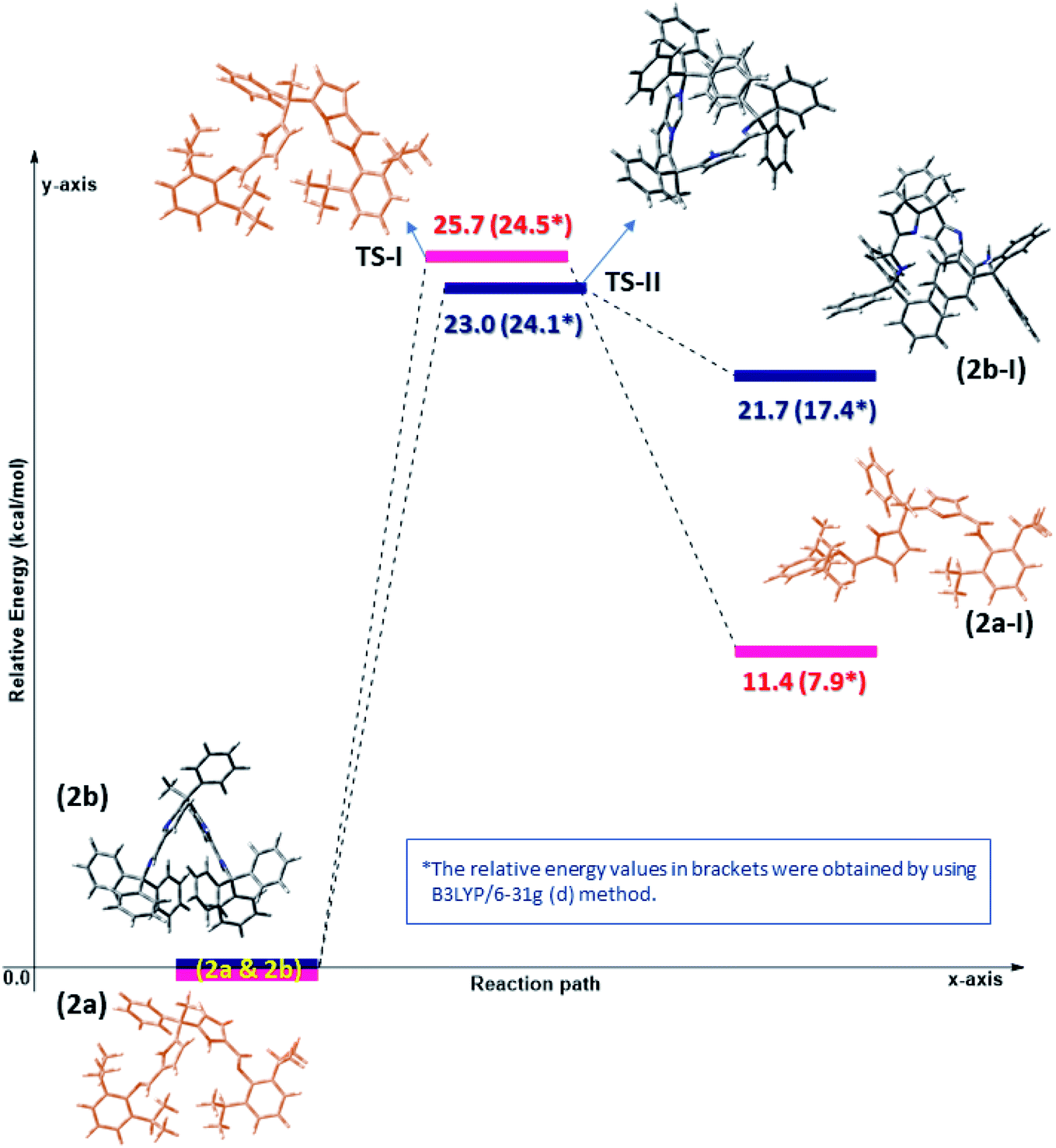 | ||
| Fig. 3 Computed potential energy surface for tautomeric forms of diiminodipyrromethane ligands (2a & 2b) in the gas phase at ωB97XD/6-31g(d) and B3LYP/6-31g(d) level. | ||
Catalytic study
The new zinc complexes (3 and 4) supported by acyclic diiminodipyrromethane Schiff base ligands (2a and 2b) were used as catalyst for study the ROP of benzoxazines such as BA-a and BF-a monomers with 10% catalyst (wt%) and polymerization process was examined with the help of DSC analysis. The sample was prepared by physical mixing (∼20 min; no solvent) of BA-a/BF-a monomer with Zn-Cat:(3 or 4) at room temperature to get homogeneous mixture followed by study of curing temperature profile of benzoxazine monomers by DSC from room temperature (RT) to 300 °C under N2 atmosphere. The curing temperature of the benzoxazines were screened by introducing 0%, 3%, 5% and 10% catalyst (Zn-Cat:3 or 4 wt%) in to benzoxazine monomers and checked their curing profiles (Scheme 5).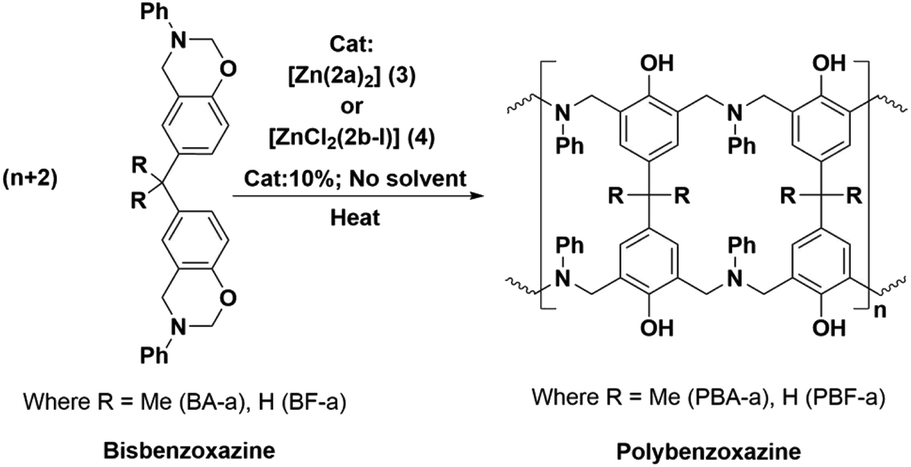 | ||
| Scheme 5 Typical ring-opening polymerization of BA-a and BF-a in the presence of Zn-Cat:(3 and 4). | ||
From the DSC analysis, under no-catalyst conditions, i.e. neat BA-a and BF-a benzoxazines showed maximum curing temperatures (Tpeak) 218 °C and 217 °C respectively. A slight modification in the curing temperature was observed when 3% and 5% catalyst loading was introduced (see Fig. S3 and S4 in the ESI†), whereas at 10% catalyst (optimized condition) showed significant decrease in the curing temperatures (Tpeak) such as 195 °C (Zn-Cat:3) & 199 °C (Zn-Cat:4) for BA-a and 191 °C (Zn-Cat:3) & 198 °C (Zn-Cat:4) for BF-a benzoxazines (see Fig. 4). According to DSC analysis, the two new zinc complexes (3 and 4) exhibited a clear catalytic activity and significant reduction in curing (ROP) temperatures. Typically, on-set curing temperature (Ton-set) values dropped for each case reasonably as much as 30–32 °C; maximum curing temperature (Tpeak) values dropped 19–26 °C and end curing temperatures (Tend) values dropped 25–28 °C (see Table 1). Further the successful ring opening polymerization of PBzs was confirmed by FT-IR (see Fig. S5 in ESI†). From the FT-IR spectra, the disappearance of peaks at 937 cm−1 and 1032 cm−1 confirms the ring opening of oxazine ring (C–O–C) of benzoxazines after polymerization.20
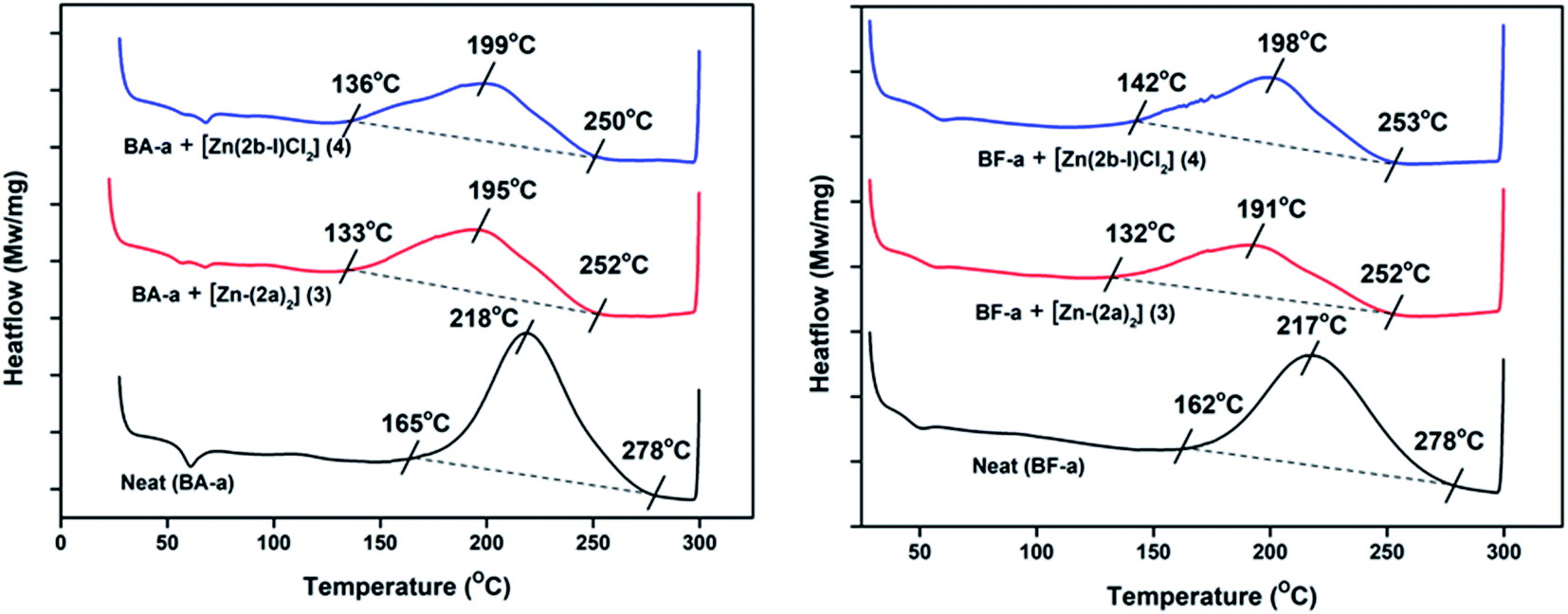 | ||
| Fig. 4 DSC curing profiles of BA-a and BF-a in the presence Zn catalysts (3 and 4) with 10% loading. | ||
Further, to check the thermal stability (Tg, degradation temperature, and char residue) of cured PBzs (0, 10% catalyst) were studied with the help of DSC and TGA analysis. Glass transition temperature (Tg) of the cured PBzs was checked with the DSC analysis and obtained Tg graph is shown in Fig. 5a. From the DSC graph (Fig. 5a), it was observed that in case of bisphenol-A based PBzs the glass transition temperature was slightly enhanced from 142 °C (for neat PBA-a polymer) to 144 °C in the presence of Zn-Cat:4. On the other hand, there is no considerable enhancement in the glass transition temperature (Tg = 148.6 °C) of bisphenol-F based PBz was observed (Fig. 5b). In addition, the thermal stability of PBzs (PBA-a and PBF-a) was studied with the help of TGA analysis from RT to 800 °C under inert conditions. The TGA thermogram results were shown in Fig. 5c and d, under no-catalyst condition the char yield for neat BA-a and BF-a PBzs were found to be 27.7% and 45.8% respectively. Whereas in the presence of Zn-catalysts (10 wt%), significant improvement in the char yield 37.1% (Zn-Cat:3) & 48.5% (Zn-Cat:4) for bisphenol-A PBz (Fig. 5c) and 48.3% (Zn-Cat:3) & 55.8% (Zn-Cat:4) for bisphenol-F PBz were observed (see Fig. 5d). These results indicate that, the structure, stability and steric bulk (nature of the ligand) on central metal ion plays major role on the ring-opening polymerization of benzoxazines. For instance, according to DSC analysis results, the Zn-Cat:3 is more effective than Zn-Cat:4 for lowering the curing temperature. Whereas on the other hand, the TGA analysis results indicates that, the Zn-Cat:4 is more efficient than Zn-Cat:3 for improving char yield. This discrepancy may be due to the structural diversity such as non-helical structure in the case of zinc complex 3 and double tautomerized zinc halide structure in the case of zinc complex 4. The presence of chloride ions in the zinc coordination sphere of Zn-complex 4, further influence the polymerization mechanism, curing temperature and char yield.
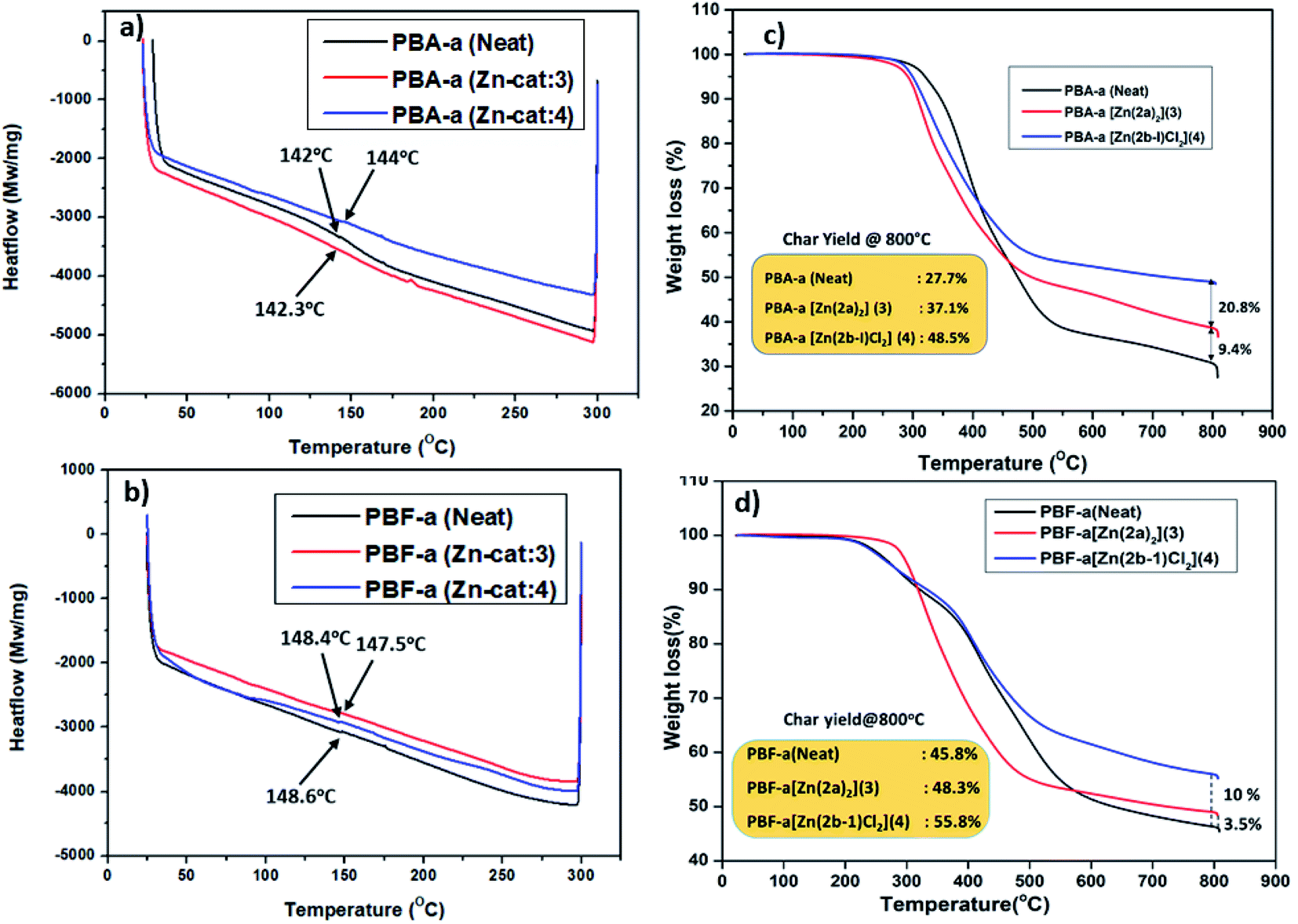 | ||
| Fig. 5 DSC thermograms (a and b) and TGA thermograms (c and d) of PBz's obtained from BA-a and BF-a monomers with and without catalysts (3 and 4). | ||
Plausible mechanism of [Zn(2a)2] (3) catalysed ROP of BA-a & BF-a
The plausible mechanism of [Zn(2a)2] (3) catalysed ring-opening polymerization of BA-a and BF-a was shown in Scheme 6. Under normal conditions, the zinc complex (3) is dormant to the polymerization process. However, as the BA-a/BF-a monomers are mixed with zinc catalyst (3) and supply of thermal energy, the tetra-coordinated zinc complex [Zn(2a)2] (3) undergoes structural transformation to form hexa-coordinated zinc complex [Zn(2a)(PBz)2] as an intermediate by leaving one of the bidentate ligand from the coordination sphere due to the “self-adaptable” nature of the coordinated acyclic diiminodipyrromethane Schiff's base ligand (2a) and due to the coordination of oxygen atom of oxazine ring of BA-a or BF-a to the central Zn2+ cationic centre. In zinc complex [Zn(2a)2] (3) the coordinated acyclic Schiff's base ligand “2a” acts as bi-dentate mono anionic ligand, whereas, in zinc complex [Zn(2a)(PBz)2] the ligand “2a” acts as tetra-dentate di-anionic ligand. Under these conditions, the active Zn2+ cationic centre may initiate the curing process by cleavage of C–O–C bond of the oxazine six-membered ring. The further addition of monomers to the active Zn2+ cationic centre promotes the coordination–insertion propagation steps that leads to the formation of PBzs as final product. As mentioned in the earlier, the cleavage of C–O–C bond during curing the process evidence by the disappearance of peaks at 937 cm−1 and 1032 cm−1 peaks in the IR spectrum of PBzs.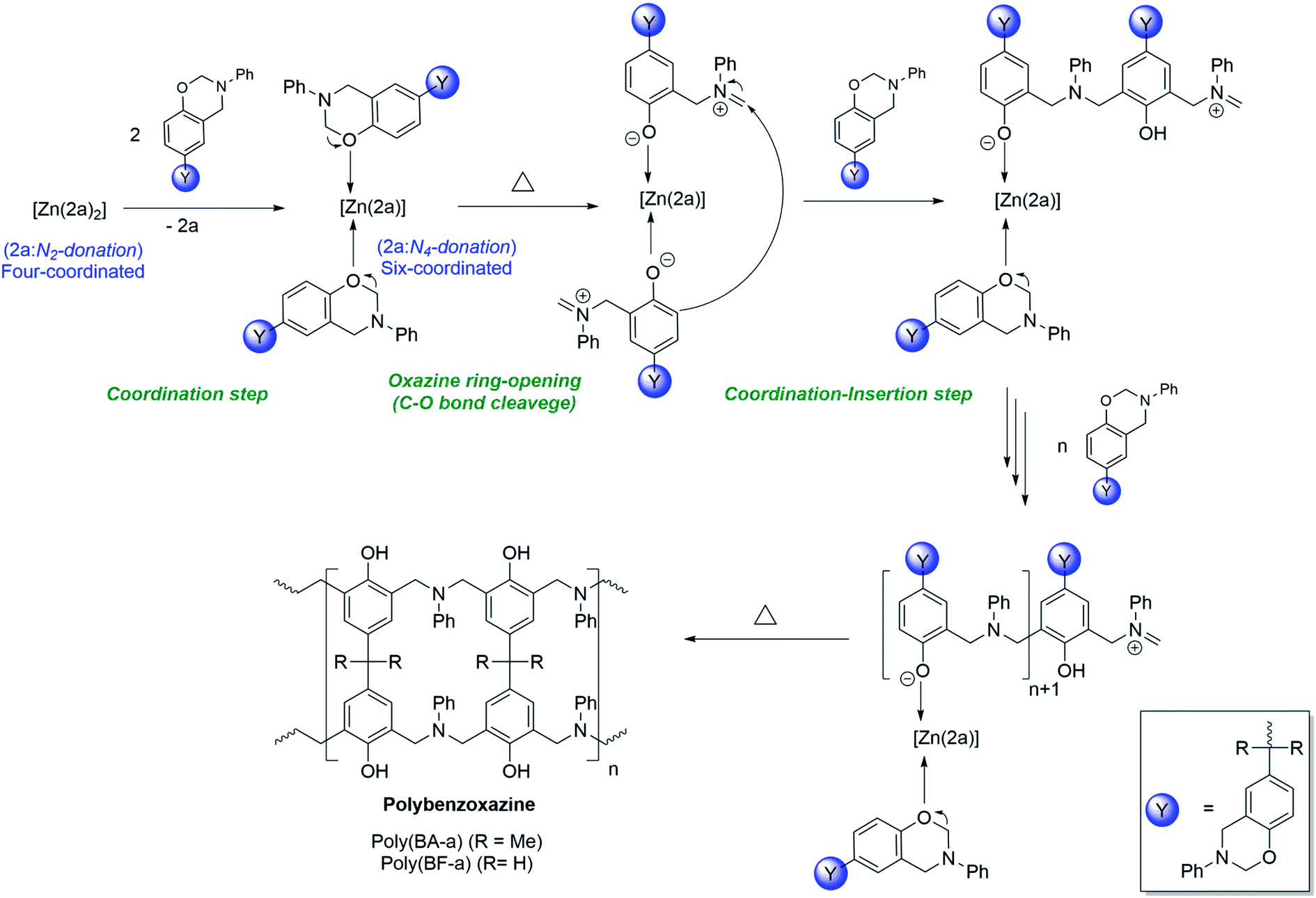 | ||
| Scheme 6 Plausible mechanism of [Zn(2a)2] (3) catalysed ring-opening polymerization of BA-a and BF-a. | ||
Plausible mechanism of [Zn(2b-I)Cl2] (4) catalysed ROP of BA-a & BF-a
Unlike zinc complex (3) the catalytic behaviour of zinc complex (4) can be best explained based on intra-structural transformation due to the azafulvene-amine to pyrrole-imine tautomerization in zinc complex (4) at higher temperatures. Under normal conditions the zinc complex (4) preferably exists in the form of [Zn(2b-I)Cl2], where “2b-I” acts as bidentate neutral ligand. However, at higher temperature, the zinc complex (4) can be transformed to zinc complex (5) [Zn(2b)] (where “2b” acts as tetradentate di-anionic ligand) and 2HCl as a biproduct. Hence, the curing (ROP) mechanism of BA-a or BF-a benzoxazines in the presence of zinc complex (4) can be anticipated via HCl mediated mechanism (Path-I) and Zn2+ cation mediated curing mechanism (Path-II) as shown in the Scheme 7. At higher temperatures, due to the intra-structural transformation, the obtained HCl can promote the polymerization process by forming hydroxy or amine salts with benzoxazine monomers as shown in the Scheme 7 (see Path-I). The further interaction of BA-a or BF-a monomers with hydroxy or amine slats lead the C–O–C bond cleavage and promotes the further polymerization process. Similar kind of mechanism was also proposed by Yagci et al. in the case of cyanuric chloride as potent catalyst for the reduction of curing temperature of benzoxazines.3a The curing process of benzoxazines in the presence of zinc complex (4) can also be explained via Path-II mechanism, which is likely to be mechanism anticipated in the zinc catalyst [Zn(2a)2] (3). According to this, the active form of Zinc complex (4) i.e. [Zn(2b)] (5) interacts with benzoxazine monomers and forms hexa-coordinated zinc complex [Zn(2b)] (5) as an intermediate species and followed by structural rearrangements via coordination–insertion mechanism lead the polymerization process till the end.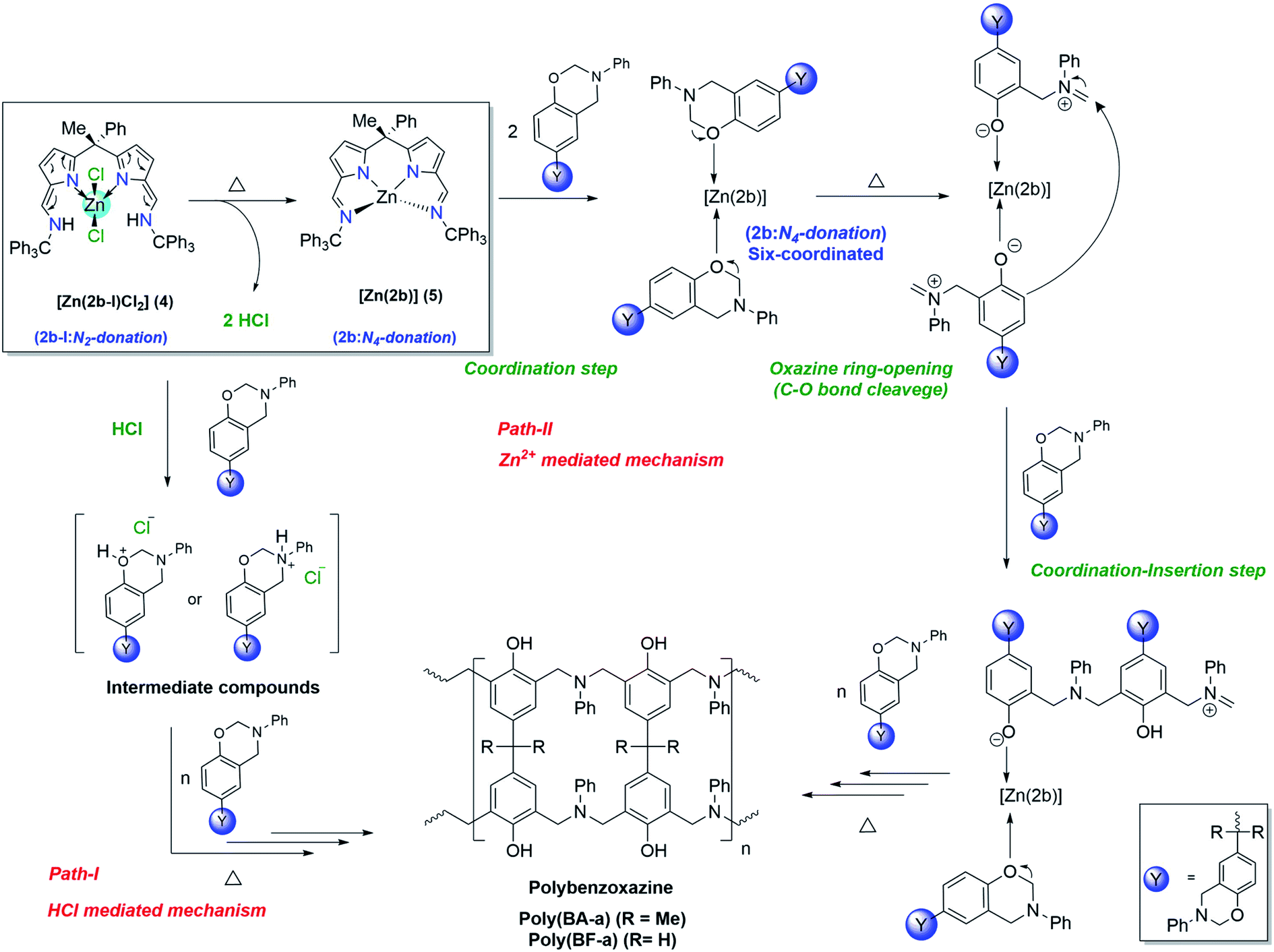 | ||
| Scheme 7 Plausible mechanism of [Zn(2b-I)Cl2] (4) catalysed ring-opening polymerization of BA-a and BF-a. | ||
Therefore, the present study proved that the zinc complexes (3 and 4) derived from acyclic diiminodipyrromethane Schiff bases are good catalysts for the ring-opening polymerization of benzoxazines. These catalysts are effective in reducing the curing temperature and improving the thermal stability (Tg, degradation temperature and char yield) of PBzs. However, the fine tuning of ligand architecture of diiminodipyrromethane Schiff's bases and modification in the Lewis acidity of the cationic metal centres will allow us to develop the more efficient catalysts for the benzoxazine ring-opening polymerization. Currently those studies were under investigation in our research group.
Experimental
General considerations
All manipulations of air-sensitive materials were performed with the rigorous exclusion of oxygen and moisture in flame dried Schlenk-type glassware either on a dual manifold Schlenk line, interfaced to a high vacuum (10−4 Torr) line. Hydrocarbon solvents (toluene and n-hexane) were distilled under nitrogen from LiAlH4 and stored under nitrogen atmosphere. HPLC grade methanol was purchased from Sigma Aldrich and it was further dried by using 3Å molecular and followed by distillation. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a VARIAN INOVA 400 spectrometer at Accu Analytical, Hyderabad, India. PerkinElmer Spectrum IR version 10.6.0 and Agilent Technologies FT-IR was used for FT-IR measurement fitted in the 450–4000 cm−1 range. Elemental analyses were performed on a TruSpec Micro CHNS 209-190. Curing behaviour and glass transition (Tg) temperature of benzoxazine monomers and PBzs were studied using differential scanning calorimetry (Hitachi DSC-7020) at a heating rate of 10 °C min−1 under nitrogen atmosphere. Thermal stability of the PBzs was carried out using thermo gravimetric analysis (Hitachi STA-7200) at the heating rate of 20 °C min−1 under nitrogen atmosphere.The starting materials like pyrrole, acetophenone, 2,6-diisopropylaniline, tritylamine, lithium bis(trimethylsilyl)amide, sodium bis(trimethylsilyl)amide, anhydrous ZnCl2 and diethylzinc (1 M solution) were purchased from Sigma Aldrich and used without further purification. 2,2′-(1-phenylethane-1,1-diyl)bis(1H-pyrrole) (1) and 5,5′-(1-phenylethane-1,1-diyl)bis(1H-pyrrole-2-carbaldehyde) (2) were reproduced by following literature reports.14,15 BA-a and BF-a benzoxazines were synthesized according to literature procudures.21
Synthesis of [(Ph)(CH3)C(2,6-iPr2C6H3–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CH–C4H2NH)2] (2a)
CH–C4H2NH)2] (2a)
To a dried methanolic solution (10 mL) of dipyrromethane-5,5′-dialdehyde (2) (2.0 g, 6.841 mmol), methanolic solution (10 mL) of 2,6-diisopropylaniline (5.45 g, 30.7 mmol) and a catalytic amount of glacial acetic acid (0.25 mL) were added under stirring at room temperature. The reaction mixture was stirred for another 12 h at room temperature. The solution was filtered and the solid was washed with cold methanol (5 mL) and then dried under high vacuum. Yield 3.80 g (91%). 1H NMR (400 MHz, CDCl3): δ 9.43–9.39 (d, J = 16 Hz, NH), 7.84 (s, 1H, NCH), 7.34–7.00 (m, 11H, ArH), 6.61–6.60 (d, 3-Pyr, J = 4 Hz, 1H), 6.15–6.14 (d, 4-Pyr, J = 4 Hz, 1H), 1.93 (s, CH3), 1.14–1.11 (m, 24H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 152.3 (NCH), 148.1 (ArC), 146.1 (ArC), 143.0 (ArC), 138.7 (ArC), 129.7 (PyrC), 128.3 (ArC), 127.0 (PyrC), 127.0 (ArC), 124.3 (ArC), 123.1 (ArC), 123.0 (ArC), 116.9 (PyrC), 109.4 (PyrC), 45.5 (meso-C), 27.7 (–C(Me)2), 23.6 (–C(CH3)2), 20.6 (CH3) ppm. FT-IR (selected frequencies, ν): 3410 (br, N–H), 3061 (w, ArC–H), 1618 (s, CN) cm−1. Elemental analysis: C42H50N4 (610.40): calcd C 82.58, H 8.25, N 9.17. Found C 82.45, H 8.22, N 9.13.
Synthesis of [(Ph)(CH3)C(Ph3C–NCH–C4H2NH)2] (2b)
To a dried methanolic solution (10 mL) of dipyrromethane-5,5′ dialdehyde (2) (2.0 g, 6.841 mmol), methanolic solution (10 mL) of tritylamine (3.548 g, 13.636 mmol) and a catalytic amount of glacial acetic acid (0.25 mL) were added under stirring. The reaction mixture was stirred for another 12 h at room temperature. The solution was filtered and the solid was washed with n-hexane (5 mL) and then dried under reduced pressure to afford the final product as an off-white powder. Yield: 4.00 g (95%). 1H NMR (400 MHz, CDCl3): δ 10.31 (s, NH), 9.26 (s, 1H, NCH), 7.59–7.02 (m, 35H, CPh3), 6.94–6.93 (d, pyr), 6.22–6.18 (d, pyr), 6.06–6.05 (d, pyr), 2.01 (s, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 150.36 (NCH), 147.79 (ArC), 145.86 (ArC), 145.33 (PyrC), 144.84 (PyrC), 132.77 (PyrC), 129.78 (ArC), 128.01 (ArC), 127.85 (ArC), 126.84 (ArC), 122.26 (ArC), 110.55(pyrC), 110.39 (PyrC), 55.43 (CPh3), 45.62 (meso-C), 20.94 (CH3) ppm. FT-IR (selected frequencies, ν): 3400 (br, N–H), 3059 (w, ArC-H), 1625 (s, CN) cm−1. Elemental Analysis: C56H46N4 (774.37): calcd C 86.79, H 5.98, N 7.23. Found C 86.74, H 5.94, N 7.22.
Synthesis of [Zn{(Ph)(CH3)C(2,6-iPr2C6H3–NCH–C4H2N)(2,6-iPr2C6H3–NCH–C4H2NH)}2] (3)
C–H), 7.87 (s, 2H, NC–H), 7.36–7.06 (m, 22H, ArH), 6.55–6.54 (d, 1H, Pyr, J = 4 Hz), 6.44–6.43 (d, 1H, Pyr, J = 4 Hz), 6.04–6.03 (d, 1H, Pyr, J = 4 Hz), 5.59–5.58 (d, 1H, Pyr, J = 4 Hz), 2.99–2.89 (m, 8H, –CH(Me2)), 1.83 (s, CH3), 1.28–1.26 (m, 48H, –CH(CH3)2) ppm; 13C NMR (100 MHz, CDCl3): δ 178.8 (Zn–NCH), 151.8 (NCH), 148.6 (ArC), 145.7 (ArC), 142.1 (ArC), 129.9 (ArC), 127.3 (ArC), 127.1 (ArC), 127.0 (ArC), 124.0 (ArC), 123.0 (ArC), 122.7 (ArC), 116.2 (ArC), 109.3 (ArC), 124.0 (PyrC), 123.0 (PyrC), 122.7 (PyrC), 116.2 (PyrC), 45.3 (–CH(Me)2), 28.3 (–CH(Me)2), 23.2 (CH3) ppm. FT-IR (selected frequencies, N–H, ν): 3415 (w, ArC-H), 1624 (s, CN) cm−1. Elemental analysis: C84H98ZnN8 (1282.72): calcd C 78.51, H 7.69, N 8.72. Found C 78.49, H 7.63, N 8.71.Synthesis of [ZnCl2{(Ph)(CH3)C(Ph3C–NHCH–C4H2N)2}] (4)
CH, J = 12 Hz), 7.44 (2H, NCH), 7.41–7.03 (m, 35H, ArH), 6.68 (d, Pyr, J = 4 Hz), 5.72 (d, Pyr, J = 4 Hz), 2.28 (s, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 179.2 (NCH), 150.5 (ArC), 150.4 (ArC), 148.9 (ArC), 146.2 (ArC), 146.0 (ArC), 145.2 (ArC), 130.0 (ArC), 127.0 (ArC), 126.9 (ArC), 126.8 (ArC), 121.8 (ArC), 115.1 (ArC), 110.6 (PyrC), 110.4 (PyrC), 109.4 (PyrC), 109.2 (PyrC), 28.5 (CH3) ppm. FT-IR (selected frequencies, ν): 3434 (w, ArC-H), 1633 (s, CN) cm−1. Elemental analysis: C56H46Cl2N4Zn (908.24): calcd C 73.81, H 5.09, Cl 7.78, N 6.15. Found C 73.77, H 5.05, Cl 7.75, N 6.14.X-ray crystallographic studies of acyclic Schiff base ligand (2a) and zinc complexes 3 and 4
Single crystals suitable for X-ray diffraction analysis of all the compounds (2a, 3 and 4) were obtained at room temperature (for 2a, and 3) and at −35 °C (for 4) under non-inert atmospheric conditions. Single crystals of suitable dimensions were mounted on a CryoLoop (Hampton Research Corp.) with a layer of light mineral oil and placed in a nitrogen stream at 298.15 K and all measurements were made on an Agilent Supernova X-calibur Eos CCD detector with graphite monochromatic Mo-Kα (0.71073 Å for 2a) and Cu-Kα (1.54184 Å for 3 and 4) radiation. Crystal data and structure refinement parameters are summarized in Table 1. The structures were solved by direct methods (SIR92)22 and refined on F2 by full-matrix least-squares methods; using SHELXL-97.23 Non-hydrogen atoms were anisotropically refined. H atoms were included in the refinement in calculated positions riding on their carrier atoms. The function minimized was [∑w(Fo2 − Fc2)2] (w = 1/[σ2(Fo2) + (aP)2 + bP]), where P = (max(Fo2,0) + 2Fc2)/3 with σ2(Fo2) from counting statistics. The functions R1 and wR2 were (∑||Fo| − |Fc||)/∑|Fo| and [∑w(Fo2 − Fc2)2/∑(wFo4)]1/2. The Ortep-3 program was used to draw the molecules. Crystallographic data (excluding structural factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1939817 (for 2a), 1900503 (for 3) and 1902678 (for 4).Conclusions
In this study, the synthesis of sterically hindered, acyclic diiminodipyrromethane Schiff base H2L ligands (2a and 2b) and its unusual zinc metal complexes (3 and 4) were well demonstrated. The stability and reactivity of all the compounds were further explained with the help of X-ray crystallography and DFT calculations. Among the ligands (2a and 2b), the diiminodipyrromethane Schiff base ligand 2b exhibits double pyrrole-imine to azafulvene-amine tautomerism at elevated temperatures due to the presence of bulky triphenylmethyl (–CPh3) groups on the imine nitrogen centres. Further, the present study towards the zinc metalation revealed the new coordination modes of acyclic diiminodipyrromethanes. In addition, both the zinc complexes (3 and 4) were screened for the ring-opening polymerization of BA-a and BF-a benzoxazines and they were proved to be efficient metal-based catalysts, In the presence of zinc-complexes, the on-set curing temperature was significantly decreased up to 20% and char yield improved up to 10–21%. Both zinc complexes were tolerance to moisture, highly active and harmless to the thermal stability of the obtained PBzs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the VFSTR Deemed to be University, Vadlamudi, Andhra Pradesh, India, under the scheme of seed grant (VFSTRU/Reg/A4/14/2017-18/278) for research faculty. Shiva and Eswar thank VFSTR for providing facilities and fellowships. Dr VS thank Prof. Shyi-Long Lee, Department of Chemistry and Biochemistry, National Chung Cheng University, Taiwan to provide the computational facilities to execute this work.Notes and references
- (a) A. Martos, M. Soto, H. Schäfer, K. Koschek, J. Marquet and R. M. Sebastián, Polymers, 2020, 12(2), 254 CrossRef CAS; (b) M. Arslan, React. Funct. Polym., 2019, 139, 9–16 CrossRef CAS; (c) M. Arslan, Turk. J. Chem., 2019, 43, 1472–1485 CrossRef CAS; (d) S. Devaraju, K. Krishnadevi, S. Sriharshitha and M. Alagar, J. Polym. Environ., 2019, 27, 141–147 CrossRef CAS; (e) L. Zhao, C. Zhao, C. Guo, Y. Li, S. Li, L. Sun, H. Li and D. Xiang, ACS Omega, 2019, 4(23), 20275–20284 CrossRef CAS; (f) Y. Liu, R. Yin, X. Yu and K. Zhang, Macromol. Chem. Phys., 2018, 1800291, 1–7 Search PubMed; (g) A. Hariharan, K. Srinivasan, C. Murthy and M. Alagar, New J. Chem., 2018, 42, 4067–4080 RSC; (h) M. Zeng, J. Chen, Q. Xu, Y. Huang, Z. Feng and Y. Gu, Polym. Chem., 2018, 9, 2913–2925 RSC; (i) S. Zhang, P. Yang, Y. Bai, T. Zhou, R. Zhu and Y. Gu, ACS Omega, 2017, 2(4), 1529–1534 CrossRef CAS; (j) M. Arslan, B. Kiskan and Y. Yagci, Sci. Rep., 2017, 7, 5207 CrossRef; (k) S. Li, C. Zhao, H. Gou, H. Li, Y. Li and D. Xiang, RSC Adv., 2017, 7, 55796–55806 RSC; (l) S. Rajasekar and N. Hari, High Perform. Polym., 2016, 1–13 Search PubMed.
- (a) N. Mantaranon, M. Kotaki, C. T. Lim and S. Chirachanchai, RSC Adv., 2016, 6, 91468–91476 RSC; (b) M. Z. Xu, K. Jia and X. B. Liu, eXPRESS Polym. Lett., 2015, 9(6), 567–581 CrossRef CAS; (c) G. Lligadas, A. Tüzün, J. C. Ronda, M. Galià and V. Cádiz, Polym. Chem., 2014, 5, 6636–6644 RSC; (d) A. D. Baranek, L. L. Kendrick, J. Narayanan, G. E. Tyson, S. Wand and D. L. Patton, Polym. Chem., 2012, 3, 2892–2900 RSC; (e) S. Sasaki, T. Ooya and T. Takeuchi, Polym. Chem., 2010, 1, 1684–1688 RSC; (f) H. C. Chang, H. T. Lin and C. H. Lin, Polym. Chem., 2012, 3, 970–978 RSC.
- (a) B. Akkus, B. Kiskan and Y. Yagci, Polym. Chem., 2020, 11, 1025–1032 RSC; (b) A. Rucigaj, B. Ali, M. Krajnc and U. Ṥebenik, eXPRESS Polym. Lett., 2015, 9(7), 647–657 CrossRef CAS; (c) A. Kocaarslan, B. Kiskan and Y. Yagci, Polymer, 2017, 122, 340–346 CrossRef CAS; (d) M. Arslan, B. Kiskan and Y. Yagci, Macromolecules, 2016, 49, 767–773 CrossRef CAS; (e) E. Semerci, B. Kiskan and Y. Yagci, Eur. Polym. J., 2015, 69, 636–641 CrossRef CAS; (f) S. Bektas, B. Kiskan, N. Orakdogen and Y. Yagci, Polymer, 2015, 75, 44–50 CrossRef CAS; (g) K. Zhang, Q. Zhuang, X. Liu, R. Cai, G. Yang and Z. Han, RSC Adv., 2013, 3, 5261–5270 RSC.
- (a) A. Sudo, H. Yamashita and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3631–3636 CrossRef CAS; (b) J. Wang, Y. Y. Z. Xu, Y. F. Fu and X. D. Liu, Sci. Rep., 2016, 6, 38584 CrossRef CAS; (c) J. Sun, W. Wei, Y. Xu, J. Qu, X. Liu and T. Endo, RSC Adv., 2015, 5, 19048–19057 RSC; (d) I. Gorodisher, R. J. DeVoe and R. J. Webb, Handb. Benzoxazine Resins, 2011, 211–234 CrossRef CAS; (e) J. Dunkers and H. Ishida, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1913–1921 CrossRef CAS.
- (a) Y. X. Wang and H. Ishida, Polymer, 1999, 40, 4563–4570 CrossRef CAS; (b) H. Ishida, US Pat., 6225440B1, 2001.
- A. Sudo, S. Hirayama and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2010, 48(2), 479–484 CrossRef CAS.
- (a) J. R. Pankhurst, S. Paul, Y. Zhu, C. K. Williams and J. B. Love, Dalton Trans., 2019, 48, 4887–4893 RSC; (b) J. R. Pankhurst, N. L. Bell, M. Zegke, L. N. Platts, C. A. Lamfsus, L. Maron, L. S. Natrajan, S. Sproules, P. L. Arnold and J. B. Love, Chem. Sci., 2017, 8, 108–116 RSC; (c) M. K. Deliomeroglu, V. M. Lynch and J. L. Sessler, Chem. Sci., 2016, 7, 3843–3850 RSC; (d) R. C. White, PhD thesis, The University of Edinburgh, 2014; (e) P. L. Arnold, G. M. Jones, S. O. Odoh, G. Schreckenbach, N. Magnani and J. B. Love, Nat. Chem., 2012, 4, 221–227 CrossRef CAS; (f) A. M. J. Devoille and J. B. Love, Dalton Trans., 2012, 41, 65–72 RSC; (g) A. M. J. Devoille, P. Richardson, N. L. Bill, J. L. Sessler and J. B. Love, Inorg. Chem., 2011, 50, 3116–3126 CrossRef CAS; (h) P. L. Arnold, D. Patel, A.-F. Pécharman, C. Wilson and J. B. Love, Dalton Trans., 2010, 39, 3501–3508 RSC.
- (a) P. L. Arnold, N. A. Potter, C. D. Carmichael, A. M. Z. Slawin, P. Roussel and J. B. Love, Chem. Commun., 2010, 46, 1833–1835 RSC; (b) S. D. Reid, C. Wilson, A. J. Blake and J. B. Love, Dalton Trans., 2010, 39, 418–425 RSC; (c) P. L. Arnold, N. A. Potter, N. Magnani, C. Apostolidis, J.-C. Griveau, E. Colineau, A. Morgenstern, R. Caciuffo and J. B. Love, Inorg. Chem., 2010, 49, 5341–5343 CrossRef CAS; (d) N. E. Borisova, M. D. Reshetova and Y. A. Ustynyuk, Chem. Rev., 2007, 107, 46–79 CrossRef CAS; (e) E. A. Katayev, K. Severin, R. Scopelliti and Y. A. Ustynyuk, Inorg. Chem., 2007, 46, 14–21 CrossRef; (f) E. A. Katayev, Y. A. Ustynyuk, V. M. Lynch and J. L. Sessler, Chem. Commun., 2006, 4682–4684 RSC; (g) P. L. Arnold, A. J. Blake, C. Wilson and J. B. Love, Inorg. Chem., 2004, 43, 8206–8208 CrossRef CAS; (h) J. M. Veauthier, W.-S. Cho, V. M. Lynch and J. L. Sessler, Inorg. Chem., 2004, 43(4), 1220–1592 CrossRef CAS; (i) S. D. Reid, A. J. Blake, W. Köckenberger, C. Wilson and J. B. Love, Dalton Trans., 2003, 4387–4388 RSC; (j) J. B. Love, A. J. Blake, C. Wilson, S. D. Reid, A. Novak and P. B. Hitchcock, Chem. Commun., 2003, 1682–1684 RSC.
- (a) G. Givaja, A. J. Blake, C. Wilson, M. Schröder and J. B. Love, Chem. Commun., 2003, 2508–2510 RSC; (b) J. Wang, X. Xiao, B. He, M. Jiang, C. Nie, Y.-W. Lin and L. Liao, Sens. Actuators, B, 2018, 262, 359–364 CrossRef CAS; (c) X.-J. Zheng, N. L. Bell, C. J. Stevens, Y.-X. Zhong, G. Schreckenbach, P. L. Arnold, J. B. Love and Q.-J. Pan, Dalton Trans., 2016, 45, 15910–15921 RSC; (d) N. L. Bell, P. L. Arnold and J. B. Love, Dalton Trans., 2016, 45, 15902–15909 RSC; (e) E. A. Connolly, J. W. Leeland and J. B. Love, Inorg. Chem., 2016, 55(2), 840–998 CrossRef CAS; (f) C. J. Stevens, A. Prescimone, F. Tuna, E. J. L. McInnes, S. Parsons, C. A. Morrison, P. L. Arnold and J. B. Love, Inorg. Chem., 2016, 55(1), 214–220 CrossRef CAS.
- (a) T. Cadenbach, J. R. Pankhurst, T. A. Hofmann, M. Curcio, P. L. Arnold and J. B. Love, Organometallics, 2015, 34(11), 2608–2706 CrossRef CAS; (b) P. L. Arnold, C. J. Stevens, J. H. Farnaby, M. G. Gardiner, G. S. Nichol and J. B. Love, J. Am. Chem. Soc., 2014, 136(29), 10218–10221 CrossRef CAS; (c) J. W. Leeland, C. Finn, B. Escuyer, H. Kawaguchi, G. S. Nichol, A. M. Z. Slawin and J. B. Love, Dalton Trans., 2012, 41, 13815–13831 RSC; (d) Q.-J. Pan, S. O. Odoh, G. Schreckenbach, P. L. Arnold and J. B. Love, Dalton Trans., 2012, 41, 8878–8885 RSC; (e) E. Askarizadeh, A. M. J. Devoille, D. M. Boghaei, A. M. Z. Slawin and J. B. Love, Inorg. Chem., 2009, 48, 7491–7500 CrossRef CAS.
- S. D. Reid, A. J. Blake, C. Wilson and J. B. Love, Inorg. Chem., 2006, 452, 636–643 CrossRef.
- S. D. Reid, C. Wilson, C. I. De Matteis and J. B. Love, Eur. J. Inorg. Chem., 2007, 33, 5286–5296 CrossRef.
- J. S. Hart, G. S. Nichol and J. B. Love, Dalton Trans., 2012, 41, 5785–5788 RSC.
- P. K. Muwal, A. Nayal, M. K. Jaiswal and P. S. Pandey, Tetrahedron Lett., 2018, 59, 29–32 CrossRef CAS.
- S. K. Loke, P. Eswar and R. K. Kottalanka, IUCrData, 2019, 4, 191660 CrossRef.
- (a) R. K. Kottalanka, A. Harinath, S. Rej and T. K. Panda, Dalton Trans., 2015, 44, 19865–19879 RSC; (b) R. K. Kottalanka, K. Naktode and T. K. Panda, Z. Anorg. Allg. Chem., 2014, 640, 114–117 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS; (b) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Peterssonet al., Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- M. Arslan, B. Kiskan and Y. Yagci, Polymers, 2018, 10, 239 CrossRef.
- (a) M. Selvi, S. Devaraju and M. Alagar, Polym. Bull., 2019, 76, 3785–3801 CrossRef CAS; (b) M. R. Vengatesan, S. Devaraju, K. Dinakaran and M. Alagar, J. Mater. Chem., 2012, 22, 7559–7566 RSC.
- A. Altomare, M. C. Burla, G. Camalli, G. Cascarano, C. Giacovazzo, A. Gualiardi and G. Polidori, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR and DSC analysis of ROP studies of Bzs; crystallographic information of 2a, 3 and 4; CCDC: 1939817 (2a), 1900503 (3) and 1902678 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra07837a |
| This journal is © The Royal Society of Chemistry 2020 |