
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Confinement of a Au–N-heterocyclic carbene in a Pd6L12 metal–organic cage†
Lihua Zeng‡
,
Shujian Sun‡,
Zhang-Wen Wei ,
Yu Xin,
Liping Liu and
Jianyong Zhang*
,
Yu Xin,
Liping Liu and
Jianyong Zhang*
MOE Laboratory of Polymeric Composite and Functional Materials, School of Materials Science and Engineering, School of Chemistry, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: zhjyong@mail.sysu.edu.cn
First published on 27th October 2020
Abstract
A Au(I)–N-heterocyclic-carbene (NHC)-edged Pd6L12 molecular metal–organic cage is assembled from a Au(I)–NHC-based bipyridyl bent ligand and Pd2+. The octahedral cage structure is unambiguously established by NMR, electrospray ionization-mass spectrometry and single crystal X-ray crystallography. The electrochemical behaviour was analyzed by cyclic voltammetry. The octahedral cage has a central cavity for guest binding, and is capable of encapsulating PF6− and BF4− anions within the cavity.
Introduction
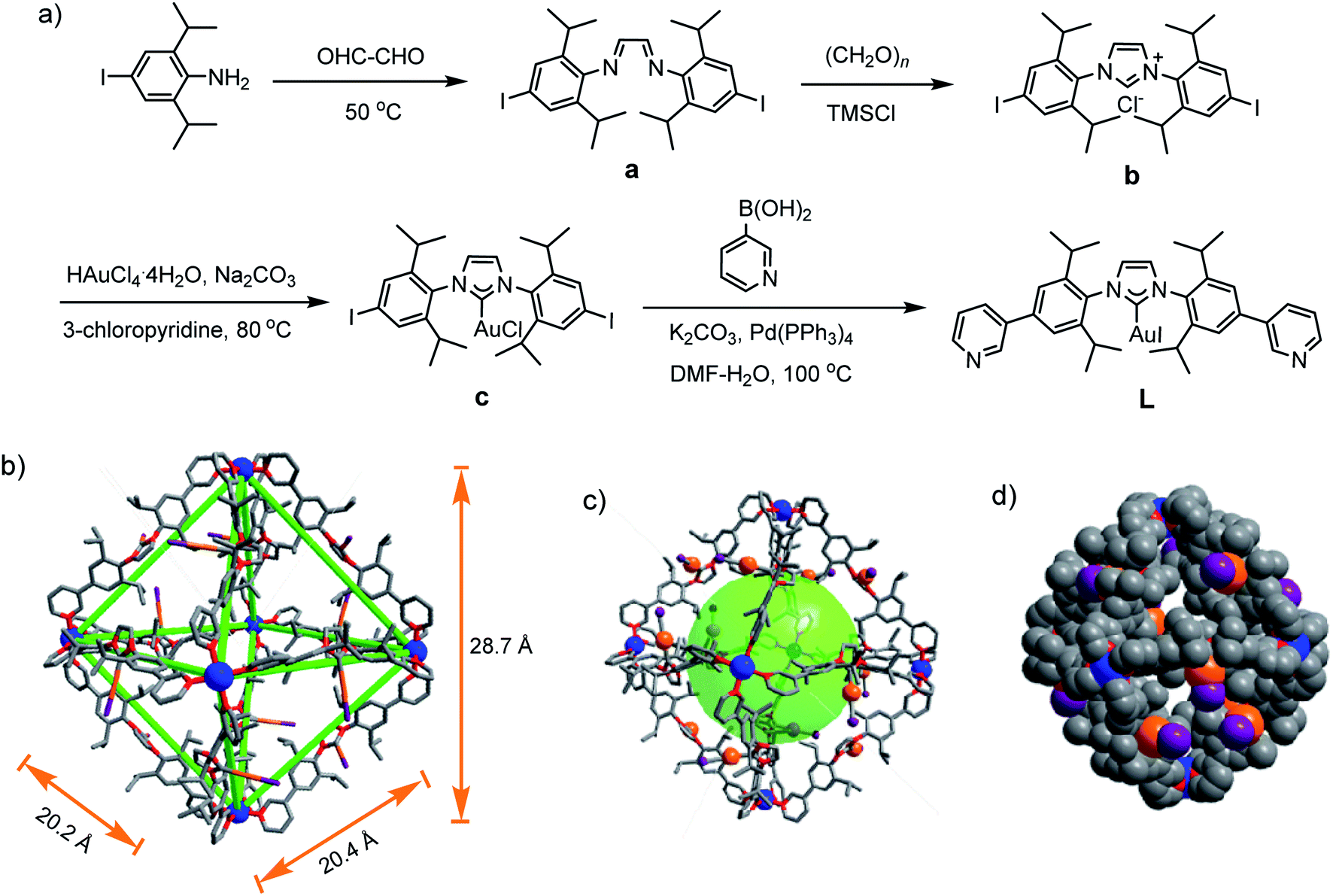
The self-assembly of metal–organic cages (MOCs)1–3 has been investigated for diverse potential applications, such as catalysis,4 sensing,5,6 molecular storage and sequestration,7 drug delivery,8 and so on. For MOCs the shape, cavity size, and ligand can influence their guest binding and transformation capability.9–11 N-heterocyclic-carbenes (NHCs) are a class of electron-donating ligands,12 and have been applied in catalyzing various organic transformations.13,14 Discrete assemblies based on NHC ligands including molecular metallocycles and cages have received increasing attention15–18 because they may induce reactivity change, selectivity and product distribution variation.19,20 However, few molecular cages with well-defined cavities are reported in the literature.21–23 Remarkably, Nitschke et al. reported M4L6 (M = Zn(II), Cd(II)) cages with Au(I)–NHC-based ligand.23 In our effort to build NHC-based cages with large cavity, we wish to report herein that a Pd6L12 metal–organic cage containing twelve Au(I)–NHC centres is assembled from a rigid, bent N-heterocyclic-carbene-based bispyridyl ligand and palladium(II) ions.Results and discussion
For the self-assembly of Pd6L12 cage, bis(pyridyl)-functionalised Au(I)–NHC ligand L was designed and synthesized (Fig. 1a). First diiodo-functionalised imidazolium chloride (b) was synthesized from bis-Schiff's compound a24 and paraformaldehyde via a ring-closing step in the presence of chlorotrimethylsilane. Then Au(I)–NHC compound c25 was synthesized by the reaction of imidazolium salt b with HAuCl4·4H2O and 3-chloropyridine in the presence of base (Na2CO3). Subsequently, L was synthesized by Suzuki–Miyaura cross-coupling reaction between c and 3-pyridylboronic acid (Fig. S1–S10†). 1H NMR shows that the imidazolium C–H resonance appeared at δ 10.34 for b and disappeared for L. 13C NMR spectra show the resonance of the imidazolium C–H carbon at 139.60 ppm for b and the resonance of the metallated carbene-carbon atoms at 185.55 ppm. The coordination cage Pd6L12 was successfully assembled by heating a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of L and Pd(NO3)2·2H2O in DMSO at 70 °C for 6 h. 1H NMR indicates that the quantitative formation of Pd6L12 (12 NO3− anions are omitted for clarity, the same hereinafter) (Fig. 2 and S11†). The cage is highly symmetric. Compared with the free ligand L the original sharp signals of pyridine moieties turn into broad peaks and shift downfield, for example the signals at 9.05, 8.64 and 8.26 ppm for the Hi, Hh and Hc pyridyl protons are downfield-shifted to 9.74, 9.09 and 8.62 ppm, respectively, which is ascribed to the coordination of palladium(II) ions to the ligand.
:1 mixture of L and Pd(NO3)2·2H2O in DMSO at 70 °C for 6 h. 1H NMR indicates that the quantitative formation of Pd6L12 (12 NO3− anions are omitted for clarity, the same hereinafter) (Fig. 2 and S11†). The cage is highly symmetric. Compared with the free ligand L the original sharp signals of pyridine moieties turn into broad peaks and shift downfield, for example the signals at 9.05, 8.64 and 8.26 ppm for the Hi, Hh and Hc pyridyl protons are downfield-shifted to 9.74, 9.09 and 8.62 ppm, respectively, which is ascribed to the coordination of palladium(II) ions to the ligand.
 | ||
| Fig. 1 (a) Synthetic route of L, single crystal X-ray structures of Pd6L12, (b) sticks, (c) ball-and-stick model, (d) space-filling model. Anions, solvent molecules and hydrogen atoms are omitted for clarity. Color coding: C: gray; N: red; I: purple; Au: orange; Pd: blue. | ||
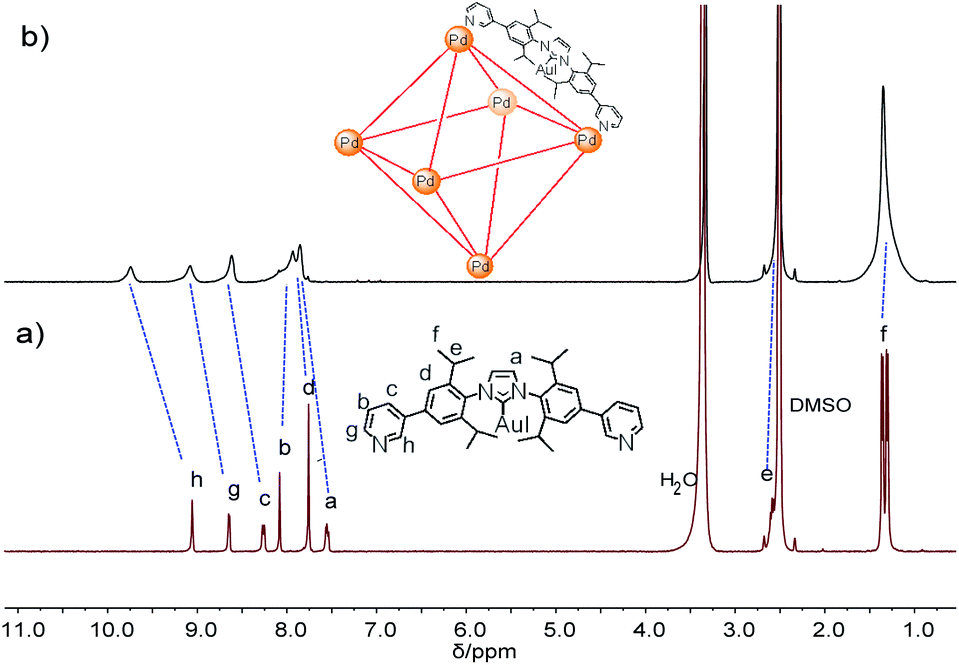 | ||
| Fig. 2 1H NMR spectra of (a) L and (b) Pd6L12 in DMSO-d6 (400 MHz, 298 K). | ||
Gratifyingly, single crystals of Pd6L12 (NO3− salt) suitable for X-ray diffraction analysis were obtained over one month by slow diffusion of ethyl acetate vapour into a solution of Pd6L12 in DMSO (Table S1†). The crystals crystallize in the trigonal space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) . The cubic symmetric unit is composed of six Pd(II) metal centres linked by twelve L ligands, forming the octahedron edges (Fig. 1b). Nevertheless, nitrate ions and solvent molecules could not be reasonably located in this highly disordered structure. The bend angle between pyridine rings and central Au–NHC ring is 174.1°. Two pyridyl donors on the same ligands adopt syn-conformation. The bond angles Ccarbene–Au–Itrans range from 176 to 179° are in fact close to linearity (C18–Au1–I1 176.1(11)°) and the Au1–C18 bond length of 1.96(3) Å, which is close to those of the reported (NHC)gold(I) complexes.26 Each Pd(II) has a square planar geometry with Pd–N bond distances of 1.98(2)–2.03(2) Å and the angle between the two 3-pyridyl coordinating motifs in L is 88–93°, within the normal range for those reported analogous pyridine-based ligand assembled Pd6L12 complexes.27,28 The cavity size is approximately 20.2 × 20.4 × 28.7 Å3, which is defined by the six Pd(II) ions. The opposing Pd(II)–Pd(II) distance is 28.7 Å and adjacent Pd–Pd distances are 20.2–20.4 Å. One of the two diisopropyl groups on each ligand points to the cavity, and the C–C distance of diisopropyl groups on opposite ligands is 16.3 Å.
. The cubic symmetric unit is composed of six Pd(II) metal centres linked by twelve L ligands, forming the octahedron edges (Fig. 1b). Nevertheless, nitrate ions and solvent molecules could not be reasonably located in this highly disordered structure. The bend angle between pyridine rings and central Au–NHC ring is 174.1°. Two pyridyl donors on the same ligands adopt syn-conformation. The bond angles Ccarbene–Au–Itrans range from 176 to 179° are in fact close to linearity (C18–Au1–I1 176.1(11)°) and the Au1–C18 bond length of 1.96(3) Å, which is close to those of the reported (NHC)gold(I) complexes.26 Each Pd(II) has a square planar geometry with Pd–N bond distances of 1.98(2)–2.03(2) Å and the angle between the two 3-pyridyl coordinating motifs in L is 88–93°, within the normal range for those reported analogous pyridine-based ligand assembled Pd6L12 complexes.27,28 The cavity size is approximately 20.2 × 20.4 × 28.7 Å3, which is defined by the six Pd(II) ions. The opposing Pd(II)–Pd(II) distance is 28.7 Å and adjacent Pd–Pd distances are 20.2–20.4 Å. One of the two diisopropyl groups on each ligand points to the cavity, and the C–C distance of diisopropyl groups on opposite ligands is 16.3 Å.
Further evidence for Pd6L12 was provided by 1H–1H homonuclear correlation spectroscopy (COSY) and 1H–1H nuclear overhauser effect spectroscopy (NOESY), which both reveal important cross peaks between the two observed sets of NMR signals (e.g. Hc–Hb and Hc–Hh) (Fig. S12 and S13†). In addition, diffusion-ordered NMR spectroscopy (DOSY) shows the selective formation of a single species (Fig. S14†). The same diffusion coefficient at D = 5.75 × 10−11 m2 s−1 corresponds to the dynamic radius of 19.0 Å according to the Stokes–Einstein equation.28 Further structural evidence was given by electrospray ionization-mass spectrometry (ESI-MS) (Fig. 3). After anion exchange of NO3− for PF6− ions in DMSO solution, a series of prominent peaks with continuous charge states of [M–(PF6−)n]n+ (n = 4–8) were detected for Pd6L12. The isotopic distribution patterns of each peak agreed well with the simulated patterns.
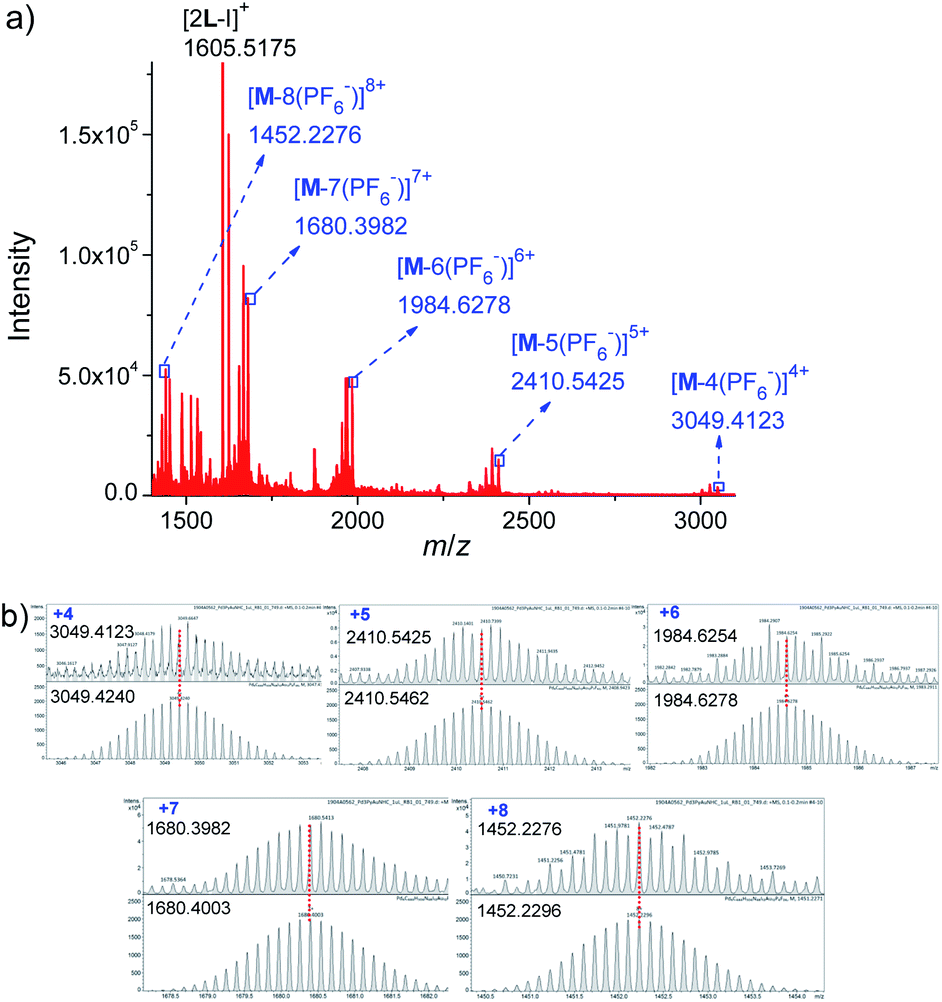 | ||
| Fig. 3 (a) ESI-TOF-MS spectrum of Pd6L12 (M) (PF6− as counterion) and (b) measured (top) and calculated (bottom) isotope patterns for various charge states observed for [Pd6L12]12+·12PF6−. | ||
The chemical composition of L and Pd6L12 were characterized by Fourier transform infrared spectroscopy (FT-IR) (Fig. S15†). The absorption of Pd6L12 at 1632 cm−1 corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N in-ring stretching in the pyridine rings shifts to lower energy compare with L at 1638 cm−1, which is attributed to the coordination of L to Pd2+ via the nitrogen atom. Both L and Pd6L12 exhibit characteristic C–Ncarbene bands typically at 1467 and 1443 cm−1.29 The strong absorption band at approximately 1384 cm−1 is attributed to the NO3− ions for Pd6L12.
N in-ring stretching in the pyridine rings shifts to lower energy compare with L at 1638 cm−1, which is attributed to the coordination of L to Pd2+ via the nitrogen atom. Both L and Pd6L12 exhibit characteristic C–Ncarbene bands typically at 1467 and 1443 cm−1.29 The strong absorption band at approximately 1384 cm−1 is attributed to the NO3− ions for Pd6L12.
To investigate the electrochemical behaviour, cyclic voltammetry analyses were carried out in the potential range from −2.0 to +2.0 V at a scan rate of 50 mV s−1 in a solution of tetrabutylammonium hexafluorophosphate (TBAPF6) in dry DMSO (0.10 mol L−1) as a supporting electrolyte on glassy carbon electrode (3 mm in diameter) (Fig. 4). L gave a reduction wave at a potential of −1.068 V, indicating the reduction of Au(I) to metallic Au(0) corresponding to previously reported Au(I)–NHC analogues.30,31 The analogous process was significantly shifted to a more anodic potential at −0.890 V for Pd6L12. In addition to the reduction processes, a single irreversible oxidation peak was also observed at 0.812 V for Pd6L12, positively shifted by 0.287 V from L (+0.525 V), which is consistent with the oxidation processes of I− to I2 on clean glassy carbon surface.32,33 It is worth noting that a new irreversible cathodic potential appeared at −0.732 V, attributed to the reduction process of NO3− to NO2− within the cage,34 perhaps owing to the change of ionic status in the cavity after coordination.35 The reduction current for Pd6L12 were acquired at scan rates of 50–200 mV s−1, showing that two reduction waves are located below the scan rates of 100 mV s−1 (Fig. S16†). With the increase in the sweep rate, the two waves merge into one broad peak, suggesting that the redox process takes place under simple diffusion-control for the cage.36
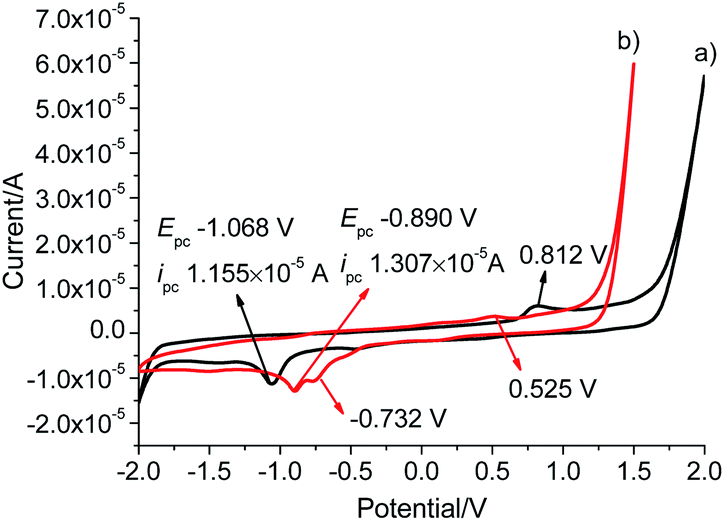 | ||
| Fig. 4 Cyclic voltammograms of (a) L and (b) Pd6L12 in DMSO solution at a scan rate of 50 mV s−1, results are reported versus Ag wire. | ||
The resulting cage structure of Pd6L12 was further investigated to explore its encapsulation of anions. The original nitrate anions are difficult to detect by NMR, so anion exchange of nitrate with other anions were performed. Various anions (20 equiv., excess) were added to a DMSO solution of Pd6L12 (0.75 mmol L−1) and the mixture was allowed to react for 4 h at room temperature, then diethyl ether was added to precipitate out pale yellow solid prior to the acquisition of NMR spectroscopy. After the introduction of NaPF6 31P NMR spectroscopy shows that phosphorus resonances from PF6− in DMSO appear expected sharp septet, with the main peak at δ −144.19 ppm with coupling constant of JPF = 711.2 Hz,37 while bound PF6− anions display the multiplets centred on δ −142.70 ppm, shifted by Δδ = 1.49 ppm with JPF = 719.3 Hz to lower field (Fig. 5a and S17†). Moreover, 19F NMR shows that one new set of peaks at −65.19 ppm (JFP = 725.7 Hz) are downshifted with free PF6− (−70.15 ppm, JFP = 710.6 Hz) (Fig. 5b and S18†). Additionally for 1H NMR Hi proton resonance inside the central cavity is upfield shifted by 0.70 ppm (Fig. S19†), while other proton resonances are essentially unaffected. These results demonstrate that two types of PF6− anions are present in the solution, some are encapsulated within the cavity, and others are free in the solution.
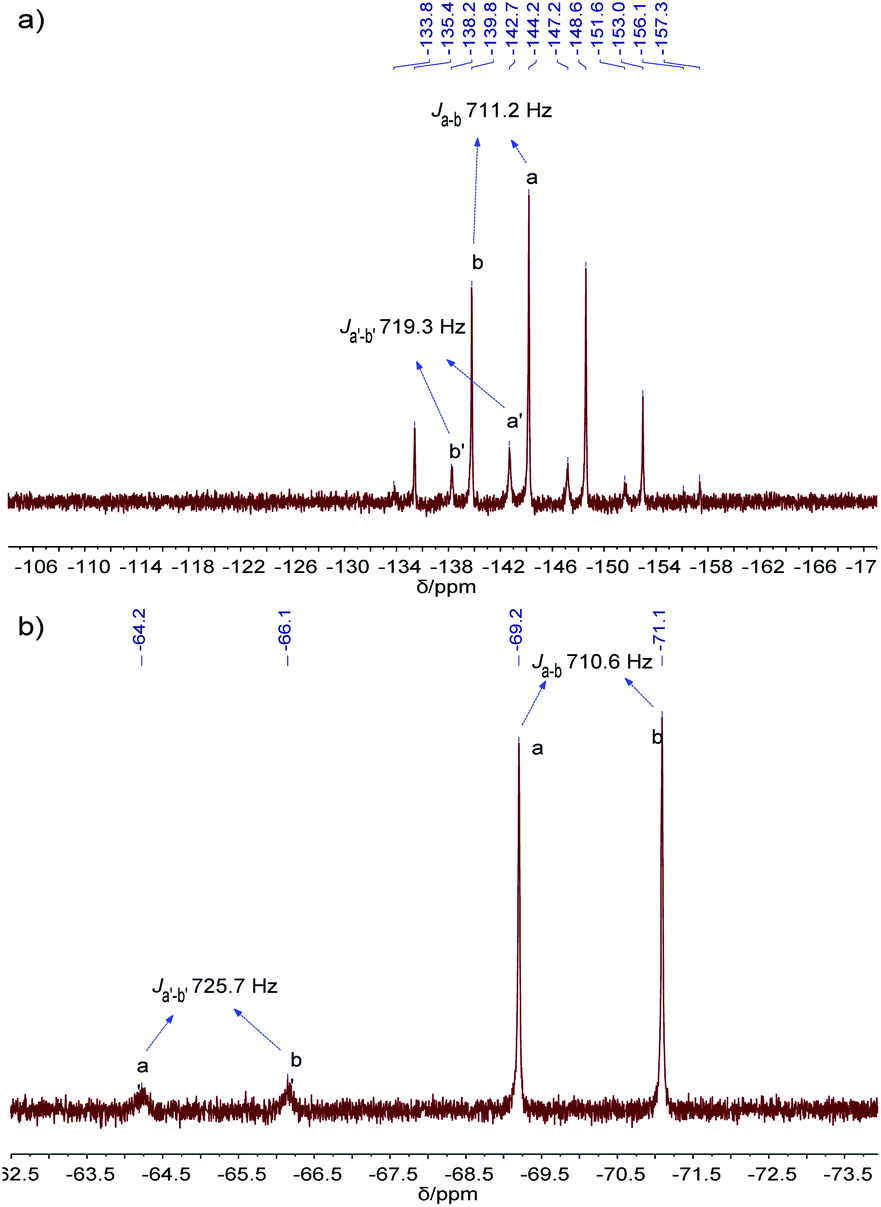 | ||
| Fig. 5 (a) 31P NMR (162 MHz, 298 K), and (b) 19F NMR (376 MHz, 298 K) spectra of PF6−@Pd6L12 in DMSO-d6. | ||
BF4− anions can also be encapsulated inside the cavity. With the addition of KBF4 to Pd6L12, 19F NMR shows two signals, one strong signal at −148.27 ppm corresponding well with the peak of free BF4− in DMSO-d6 (−148.3 ppm), and the other new signal at −144.1 ppm (Fig. S20 and S21†). 11B NMR show that a new signal at 2.79 ppm is also assigned to bound BF4− in the cavity (Fig. S22 and S23†).38,39 Simultaneously 1H NMR reveals that the proton resonance of Hi inside the central cavity is shifted upfield (Δδ = 0.30 ppm) compared to Pd6L12 (Fig. S19†). These results suggest that some BF4− anions are bound within the cavity of Pd6L12. However, addition of OTf− to Pd6L12 causes neither new signals nor notable shifts, and 19F NMR gives only one sharp signal corresponding to free OTf−, suggesting that the nitrate ions in the cage was unaffected by OTf− (Fig. S19 and S24†).
Conclusions
In summary, we have successfully synthesized Au(I)–NHC bispyridine ligand (L) and Au(I)–NHC-edged Pd6L12 coordination cage has been assembled from L and Pd2+. The cage Pd6L12 has been fully characterized by NMR, diffusion-ordered NMR spectroscopy, FT-IR, mass spectrometry and cyclic voltammogram. The single crystal X-ray diffraction confirms unequivocally the octahedral structure of Pd6L12. The octahedral cage has central cavity for guest binding, and has been shown to be capable of encapsulating PF6− and BF4− anions within the cavity. It paves a way to build NHC-based metal–organic container molecules with large cavity for subsequent guest binding and transformation.Experimental
Synthesis of L
A mixture of c (689.1 mg, 0.715 mmol), 3-pyridylboronic acid (357.5 mg, 2.86 mmol), anhydrous potassium carbonate (988.1 mg, 7.15 mmol), tetrakistriphenylphosphine palladium(0) (0.00715 mmol, 8.3 mg) in 18 mL degassed DMF–H2O (v/v = 5:1) was stirred at 100 °C for 24 h under argon atmosphere, the progress of the reaction was monitored by TLC. After completion of the reaction, the solvent was removed and the residue was dispersed in water and extracted with CH2Cl2. The CH2Cl2 solution was washed with water and brine, dried over anhydrous MgSO4, filtered and concentrated in vacuo. Purification by column chromatography on silica gel (ethyl acetate:hexane 1:1) to give the product as white powder which was further purified by washing with hexane (464 mg, 75%). 1H NMR (400 MHz, DMSO-d6): δ 9.05 (s, 2H), 8.64 (s, 2H), 8.27–8.22 (m, 2H), 8.08 (s, 2H), 7.76 (s, 4H), 7.57–7.53 (m, 2H), 2.59 (dt, J = 14.0, 6.9 Hz, 4H), 1.36 (d, J = 6.8 Hz, 12H), 1.30 (d, J = 6.9 Hz, 12H). 13C NMR (101 MHz, CDCl3): δ 185.55, 149.02, 148.43, 146.49, 140.41, 136.25, 134.79, 133.79, 123.65, 123.29, 123.14, 29.04, 24.48, 24.06. Q-TOF-MS(ESI+) (m/z): calcd. for [M + H]+ [C37H43N4AuI + H]+ 867.2198, found 867.2206 and calcd for [M–AuI + H]+ [C41H44N2O2 + H]+ 543.3488, found 543.3471.
Synthesis of Pd6L12
L (174.0 mg, 0.2 mmol) and Pd(NO3)2·2H2O (27.0 mg, 0.1 mmol) were dissolved in DMSO (10.0 mL) were mixed. Each time 1 mL of the mixture was taken and stirred at 70 °C for 6 h. The quantitative formation of Pd6L12 was confirmed by 1H NMR and 1H DOSY NMR. The addition of diethyl ether to the yellow homogeneous solution precipitated the product, and it was collected by filtration, washed with diethyl ether, and dried in vacuo to give a pale yellow solid (13.8 mg, 70%). 1H NMR (500 MHz, DMSO-d6): δ 9.74 (s, 2H), 9.08 (s, 2H), 8.60 (s, 2H), 7.88 (d, J = 39.1 Hz, 8H), 2.55 (s, 4H), 1.35 (s, 24H). 1H DOSY NMR (400 MHz, DMSO-d6): D = 7.16 × 10−11 m2 s−1. Pd6L12 (PF6− salt) was prepared by adding excess NaPF6 to the DMSO solution of Pd6L12. Electrospray ionization mass spectra were recorded for Pd6L12 (PF6− salt). m/z calcd for C444H504N48Au12I12P8F48Pd6 [M–4(PF6)]4+ 3049.4240, found 3049.4123; m/z calcd for C444H504N48Au12I12P7F42Pd6 [M–5(PF6)]5+ 2410.5462, found 2410.5425; m/z calcd for C444H504N48Au12I12P6F36Pd6 [M–6(PF6)]6+ 1984.6278, found 1984.6254; m/z calcd for C444H504N48Au12I12P5F30Pd6 [M–7(PF6)]7+ 1680.4003, found 1680.3982; m/z calcd for C444H504N48Au12I12P4F24Pd6 [M–8(PF6)]8+ 1452.2296, found 1452.2276; m/z calcd for C74H84N8Au2I [2L–I]+ 1605.5190, found 1605.5175.X-ray crystallography for Pd6L12
Quality single crystals were obtained by the slow diffusion of ethyl acetate vapour into a DMSO solution of Pd6L12 (NO3− salt) to give colourless crystals of Pd6L12 over one month. A crystal was picked (0.2 × 0.2 × 0.2 mm3) and coated in paratone oil, attached to a glass silk which was inserted in a stainless steel stick, then quickly transferred to the Agilent Technologies SuperNova X-ray diffractometer with the Enhance X-ray Source of Cu Kα radiation (λ = 1.54184 Å) using the ω–ϕ scan technique. Data collection were measured at 150.00 (10) K. The unit cell parameters were solved by direct methods and the unit cell parameters refined against all data by anisotropic full-matrix least-squares methods on F2 with the SHELXL program.40 Hydrogen atoms were calculated in ideal positions (riding model). All nitrate ions and solvent molecules could not be reasonably located because of highly disordered structures, and were removed by PLATON/SQUEEZE routine.41Crystallographic data for Pd6L12: C74H84Au2I2N8Pd FW = 1839.62, trigonal, R, a = 29.8768 (16) Å, b = 29.8768 (16) Å, c = 74.341 (3) Å, α = 90°, β = 90°, γ = 120°, V = 57468 (7) Å3, Z = 18, T = 150.00 (10) K, λ = 1.54184 Å, ρcalcd = 0.957 mg m−3, μ = 9.349 mm−1, 13205 reflections were collected (7693 were unique) for 6.856 < 2θ < 79.938, R(int) = 0.0372, R1 = 0.0888, wR2 = 0.2668 [I ≥ 2σ(I)], R1 = 0.1033, wR2 = 0.2821 (all data) for 725 parameters and 825 restraints, GOF = 1.059. Selected bond lengths and bond angles are presented in Table S1.† The crystal structure was submitted to the Cambridge Structural Database under the CCDC number 2013514.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the NSF of Guangdong Province (2019A1515010710) for financial support.Notes and references
- M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC.
- M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., 2013, 125, 1358–1362 CrossRef.
- O. Chepelin, J. Ujma, X. Wu, A. M. Z. Slawin, M. B. Pitak, S. J. Coles, J. Michel, A. C. Jones, P. E. Barran and P. J. Lusby, J. Am. Chem. Soc., 2012, 134, 19334–19337 CrossRef CAS.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS.
- D. P. August, G. S. Nichol and P. J. Lusby, Angew. Chem., Int. Ed., 2016, 55, 15022–15026 CrossRef CAS.
- H. Lee, D. Kim, H. Oh and O.-S. Jung, Chem. Commun., 2020, 56, 2841–2844 RSC.
- M. B. Duriska, S. M. Neville, J. Lu, S. S. Iremonger, J. F. Boas, C. J. Kepert and S. R. Batten, Angew. Chem., Int. Ed., 2009, 48, 8919–8922 CrossRef CAS.
- W. Cullen, S. Turega, C. A. Hunter and M. D. Ward, Chem. Sci., 2015, 6, 625–631 RSC.
- S. Löffler, J. Lübben, A. Wuttke, R. A. Mata, M. John, B. Dittrich and G. H. Clever, Chem. Sci., 2016, 7, 4676–4684 RSC.
- D. R. Martir, A. Pizzolante, D. Escudero, D. Jacquemin, S. L. Warriner and E. Zysman-Colman, ACS Appl. Energy Mater., 2018, 1, 2971–2978 CrossRef CAS.
- Y. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649–2652 CrossRef CAS.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS.
- Z. Guo, N. R. Song, J. H. Moon, M. Kim, E. J. Jun, J. Choi, J. Y. Lee, C. W. Bielawski, J. L. Sessler and J. Yoon, J. Am. Chem. Soc., 2012, 134, 17846–17849 CrossRef CAS.
- D. Zhang, H. Yang, A. Martinez, K. Jamieson, J.-P. Dutasta and G. Gao, Chem.–Eur. J., 2014, 20, 17161–17167 CrossRef CAS.
- M.-M. Gan, J.-Q. Liu, L. Zhang, Y.-Y. Wang, F. E. Hahn and Y.-F. Han, Chem. Rev., 2018, 118, 9587–9641 CrossRef CAS.
- C. Mejuto, G. Guisado-Barrios, D. Gusev and E. Peris, Chem. Commun., 2015, 51, 13914–13917 RSC.
- A. Rit, T. Pape and F. E. Hahn, J. Am. Chem. Soc., 2010, 132, 4572–4573 CrossRef CAS.
- N. Sinha, F. Roelfes, A. Hepp, C. Mejuto, E. Peris and F. E. Hahn, Organometallics, 2014, 33, 6898–6904 CrossRef CAS.
- S. Roland, J. M. Suarez and M. Sollogoub, Chem.–Eur. J., 2018, 24, 12464–12473 CrossRef CAS.
- A. C. H. Jans, X. Caumes and J. N. H. Reek, ChemCatChem, 2019, 11, 287–297 CrossRef CAS.
- P. J. Altmann and A. Pöthig, Angew. Chem., Int. Ed., 2017, 56, 15733–15736 CrossRef CAS.
- G. Xu, S. Leloux, P. Zhang, J. M. Suárez, Y. Zhang, E. Derat, M. Ménand, O. Bistri-Aslanoff, S. Roland, T. Leyssens, O. Riant and M. Sollogoub, Angew. Chem., Int. Ed., 2020, 59, 7591–7597 CrossRef CAS.
- W. J. Ramsay, J. A. Foster, K. L. Moore, T. K. Ronson, R. J. Mirgalet, D. A. Jefferson and J. R. Nitschke, Chem. Sci., 2015, 6, 7326–7331 RSC.
- H. Yang, G. Li, Z. Ma, J. Chao and Z. Guo, J. Catal., 2010, 276, 123–133 CrossRef CAS.
- W. Wang, A. Zheng, P. Zhao, C. Xia and F. Li, ACS Catal., 2014, 4, 321–327 CrossRef CAS.
- M. Pažický, A. Loos, M. J. Ferreira, D. Serra, N. Vinokurov, F. Rominger, C. Jäkel, A. S. K. Hashmi and M. Limbach, Organometallics, 2010, 29, 4448–4458 CrossRef.
- K. Suzuki, M. Tominaga, M. Kawano and M. Fujita, Chem. Commun., 2009, 1638–1640 RSC.
- C. Gütz, R. Hovorka, C. Klein, Q.-Q. Jiang, C. Bannwarth, M. Engeser, C. Schmuck, W. Assenmacher, W. Mader, F. Topić, K. Rissanen, S. Grimme and A. Lützen, Angew. Chem., Int. Ed., 2014, 53, 1693–1698 CrossRef.
- D. Rendón-Nava, D. Mendoza-Espinosa, G. E. Negrón-Silva, J. L. Téllez-Arreola, A. Martínez-Torres, A. Valdez-Calderón and S. González-Montiel, New J. Chem., 2017, 41, 2013–2019 RSC.
- L. Aldous, D. S. Silvester, C. Villagrán, W. R. Pitner, R. G. Compton, M. C. Lagunas and C. Hardacre, New J. Chem., 2006, 30, 1576–1583 RSC.
- F. J.-B. dit Dominique, H. Gornitzka, A. Sournia-Saquet and C. Hemmert, Dalton Trans., 2009, 340–352 RSC.
- H. V. Huynh, S. Guo and W. Wu, Organometallics, 2013, 32, 4591–4600 CrossRef CAS.
- S. G. Bratsch, J. Phys. Chem., 1989, 18, 1–21 CAS.
- L. Szpyrkowicz, S. Daniele, M. Radaelli and S. Specchia, Appl. Catal., B, 2006, 66, 40–50 CrossRef CAS.
- F. Jiang, N. Wang, Z. Du, J. Wang, Z. Lan and R. Yang, Chem.–Asian J., 2012, 7, 2230–2234 CrossRef CAS.
- T. S. Varley, M. Hirani, G. Harrison and K. B. Holt, Faraday Discuss., 2014, 172, 349–364 RSC.
- T. Endo, H. Murata, M. Imanari, N. Mizushima, H. Seki, S. Sen and K. Nishikawa, J. Phys. Chem. B, 2013, 117, 326–332 CrossRef CAS.
- B. E. Aroussi, L. Guénée, P. Pal and J. Hamacek, Inorg. Chem., 2011, 50, 8588–8597 CrossRef.
- M. D. Wise, J. J. Holstein, P. Pattison, C. Besnard, E. Solari, R. Scopelliti, G. Bricogne and K. Severin, Chem. Sci., 2015, 6, 1004–1010 RSC.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental details, spectra and structure data. CCDC 2013514. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra07509d |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2020 |