
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe-based metal–organic frameworks as heterogeneous catalysts for highly efficient degradation of wastewater in plasma/Fenton-like systems†
Xumei Tao *a,
Xinjie Yuana,
Liang Huangb,
Shuyong Shangc and
Dongyan Xua
*a,
Xinjie Yuana,
Liang Huangb,
Shuyong Shangc and
Dongyan Xua
aState Key Laboratory Base for Eco-Chemical Engineering, College of Chemical Engineering, Qingdao University of Science and Technology, Qingdao 266042, Shandong, China. E-mail: qiqitxm_2002@sina.com.cn
bCollege of Electromechanical Engineering, Qingdao University of Science and Technology, Qingdao 266042, Shandong, China
cDepartment of Science, Technology and Discipline Construction, Chengdu Normal University, Chengdu 611130, Sichuan, China
First published on 2nd October 2020
Abstract
Fe-based metal organic frameworks (Fe-MOFs) were successfully synthesized with the dielectric barrier discharge (DBD) plasma method and FeSO4·7H2O as the Fe precursor. Fe-MOFs were used as Fenton-like catalysts in DBD plasma/Fenton-like technology to treat wastewater, which addressed the issues with iron solubility. Since the valence state of iron will affect the catalytic performance, the Fe precursor FeSO4·7H2O was added to regulate the valence state and adjust the catalytic performance by improving the availability of active sites. The influences of discharge voltage, catalyst addition amount, H2O2 addition amount and pH on the degradation efficiency of methyl orange (MO) were systematically examined. Through free radical capture experiments, the reaction mechanism of the plasma/Fenton-like catalytic degradation process was deduced primarily as the coordinated oxidation process of hydroxyl radicals (·OH), photo-generated holes (h+) and superoxide radicals (·O2−). The reusability experiments proved that the catalyst was stable and reusable. The possible degradation pathways were proposed based on the identification of intermediate products generated in the degradation process by liquid chromatography-mass spectrometry (LC-MS) analyses.
1 Introduction
Fenton oxidation technology is considered a promising method for the treatment of toxic organic pollutants in wastewater because of its advantages such as strong oxidation potential, mild reaction conditions, and easy operation.1,2 However, the Fenton process possesses the disadvantages of a narrow pH range, low H2O2 utilization rate, and sludge production, which limits its large-scale application.3,4Plasma, a novel technology, has attracted wide attention in recent years.5,6 In order to degrade wastewater more effectively and rapidly, researchers have studied the synergy of plasma technology and Fenton technology. During the process of plasma degradation of wastewater, active substances such as H2O2 and ·OH are generated.7 When an appropriate amount of Fe2+-containing substance is added, a Fenton system is formed, and a large amount of ·OH is generated after the reaction, which increases the amount of active substance, thereby accelerating the degradation of organic matter.8 Plasma technologies applied for the plasma/Fenton process include glow discharge (GDP) plasma,9,10 high voltage pulsed discharge plasma (PDP),11 gliding arc discharge (GAP) plasma12,13 and DBD plasma.14 Hu et al.9 studied the synergistic effect of the GDP/Fenton system and found that GDP can be significantly enhanced by the addition of iron ions as both Fenton catalysts and flocculants. Dai et al.11 studied high voltage PDP combined with the Fenton reaction with both ferrous ion (Fe2+) salts and a carbon steel grounded electrode as iron sources for the degradation of bisphenol A (BPA). Plasma/Fenton treatment induced an increased rate of BPA removal. Slamani et al.12 initiated the Fenton process by GAD plasma for the degradation of paracetamol (PCM) in water. Combining Fenton with GAD discharge induced a rapid degradation of PCM and gave a high degree of mineralization. Marković et al.14 studied the treatment of ibuprofen with homogeneous catalyst FeSO4 in the DBD/Fenton system, in which the degradation efficiency was improved. The above results showed that the degradation efficiency was promoted under the plasma/Fenton system, but the degradation efficiency was improved at the expense of electrode loss or iron ion erosion.15 DBD, with features of high safety, uniform discharge and long electrode life,16 has not been combined with Fenton technology using heterogeneous catalysts to treat wastewater. Therefore, it is of great significance to develop new heterogeneous catalysts with low cost, high activity, good stability and environment friendliness.
Fe-MOFs, which lose less iron ions in solution, have received increasing attention as Fenton-like catalysts. Fe-MOFs can provide iron active sites useful for the heterogeneous Fenton reaction with different iron environments, enabling the advantages of homogeneous iron complexes over a broader range of pH.17–19 More interestingly, Fe-MOFs prepared with DBD plasma showed improved adsorption properties20 and Fenton properties,21 as demonstrated in our previous work. Also, the valence of an iron ion in Fe-MOFs affects the catalytic performance. Li et al.22 synthesized a series of Fe-MOFs by controlling the composition of mixed valence Fe2+ and Fe3+ precursors with dual functions, namely, high adsorption capacity for toluene and good photocatalytic degradation performance. In addition, Fe-MOFs could be recycled and reused several times for the transformation without a significant degradation in catalytic activity.23
In this work, Fe-MOFs using FeSO4·7H2O as the iron precursor were synthesized by DBD plasma and used as heterogeneous catalysts to degrade wastewater in a DBD plasma/Fenton-like system. The effects of different TA/Fe molar ratios and reaction conditions on the catalytic degradation of MO were studied. Through capture agent experiments and LC-MS analysis, the reaction mechanism and possible degradation paths of the degradation process were deduced.
2 Material and methods
2.1 Preparation of Fe-MOFs by DBD plasma
The DBD plasma reaction device for preparing Fe-MOFs is shown in Fig. 1. Briefly, FeSO4·7H2O (2.87 g) and trimesic acid (TA) (0.73 g) (TA/Fe source molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) were added into 150 mL of N,N-dimethylformamide (DMF) and 150 mL of absolute ethanol, and stirred until completely dissolved. Then, the mixed solution was pumped into the DBD reactor and discharged for a certain time. After cooling naturally to room temperature, the supernatant liquid was poured out and the dark orange solid product was washed with DMF and ethanol several times for each. Then, it was dried at 160 °C for 4 h to obtain the product, which was labelled as Fe-MOFs.
:3) were added into 150 mL of N,N-dimethylformamide (DMF) and 150 mL of absolute ethanol, and stirred until completely dissolved. Then, the mixed solution was pumped into the DBD reactor and discharged for a certain time. After cooling naturally to room temperature, the supernatant liquid was poured out and the dark orange solid product was washed with DMF and ethanol several times for each. Then, it was dried at 160 °C for 4 h to obtain the product, which was labelled as Fe-MOFs.
 | ||
| Fig. 1 Fe-MOFs preparation device diagram. | ||
As shown in Fig. 1, a stainless steel mesh served as the high voltage electrode while a stainless steel rod served as the low voltage electrode. A voltage generator (CTP-2000K, Nanjing Suman Electronics Co, Ltd) was used to generate non-thermal plasma. The voltage was measured by an oscilloscope (UTD2012CEX, UNI-T Co, Ltd). The solution was first driven into the inner tube of the reactor through a peristaltic pump; after filling the inner tube, the solution formed a wall-mounted liquid film on the outer surface of the inner tube and overflowed to the outer tube. After discharging for a certain time, Fe-MOFs were synthesized.
2.2 Materials characterization
X-ray diffraction (XRD) was carried out by a Japanese Rigaku D/max-rA type X-ray diffractometer with Cu Kα radiation operated at 40 kV and 150 mA. XRD patterns were scanned in the 2θ range of 3–80° at a scan rate of 10° min−1 (step size 0.02° s−1). Scanning Electron Microscopy (SEM) was conducted with JSM-6700F to characterize the surface morphology of the samples. Energy Dispersive Spectroscopy (EDS) was used for the qualitative detection of Fe and O in the Fe-MOFs. The transmission electron microscopy (TEM) images were acquired using a JEOL (JEM-2100, Japan) operating at 200 kV. The BET specific surface areas were measured by N2 adsorption at 196 °C using an ASAP 2020 apparatus. The X-ray photoelectron spectra (XPS) were collected with an ESCALAB 250xi spectrometer, monochromatic radiation from the Al Kα radiation (150 W), and the Al Kα monochromatic line (1486.6 eV) was adopted for the high-resolution analysis. The intermediates and reaction products of MO decontamination were analyzed using LC-MS (WATWES-e2695 A0360F, America).2.3 Plasma/Fenton-like process
The plasma/Fenton-like experimental device is shown in Fig. S1.† The reactor was similar to that for preparing Fe-MOFs. Typically, MO and H2O2 were dissolved in water at room temperature. A certain amount of Fe-MOFs and H2O2 was added to 200 mL of MO solution (200 mg L−1) in a beaker. The air pump played a role in preventing catalyst precipitation and participating effectively in the reaction. Magnetic stirring was set at 500 rpm. Samples were taken at set intervals using a 1 mL pipette, then diluted and centrifuged at 3000 rpm for 5 min. Then, the absorbance of the supernatant was measured with a UV-visible spectrophotometer at 463 nm. The degradation rate of the MO solution was defined as eqn (1-1).| Degradation rate (%) = (1 − C/C0) × 100% | (1-1) |
Finally, in order to compare the synergistic degradation effect, DBD alone and Fenton alone reactions were conducted under the same conditions.
3 Results and discussion
3.1 Characterization analysis of Fe-MOFs
:1 had a small number of impurity peaks, which might be caused by underutilization in the case of excessive acid. With increasing TA/Fe molar ratios, the amount of acid was relatively reduced, which could then be effectively utilized and thus be more stable.
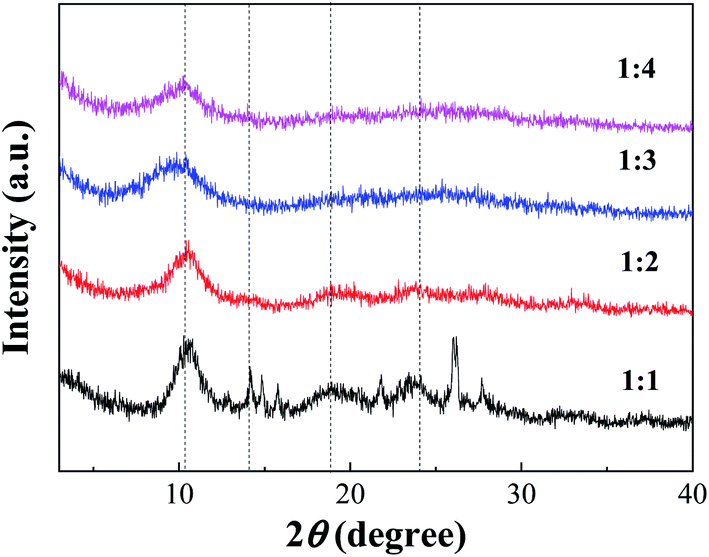 | ||
| Fig. 2 XRD of Fe-MOFs with different TA/Fe molar ratios. | ||
According to the XRD patterns, the average particle diameters of Fe-MOFs with different TA/Fe molar ratios were calculated. As shown in Table S1,† the estimation error of the average particle diameter was usually ± 0.5 nm.26 It can be seen that the particle size of the Fe-MOFs with a TA/Fe molar ratio of 1:3 was the smallest. When the catalyst particle size is smaller, the specific surface area may be larger and more active sites may be exposed; thus, the corresponding catalytic activity may be higher.
:1, it showed a large area of aggregation with irregular shapes and different particle sizes. With increasing TA/Fe molar ratios, the morphology appeared spherical and became more and more regular, and the particle size also tended to be more uniform. When the TA/Fe molar ratio was 1:3, the morphology was flower-like, and the ratio of width to length was small, which provided a larger surface area and increased number of active sites. SEM analyses revealed that the TA/Fe molar ratio had a noticeable effect on the morphology of the as-synthesized products.25
 | ||
| Fig. 3 SEM images of Fe-MOFs with different TA/Fe molar ratios (a–d) 1:1; (e–h) 1:2; (i–l) 1:3; (m–p) 1:4. | ||
:3 contained the most Fe. However, when the TA/Fe molar ratio was 1:4, the percentage of Fe decreased. The possible reason was that although the TA/Fe molar ratio increased, the Fe source effectively participating in the reaction was relatively reduced and the activity was reduced, which was consistent with the law when studying the preparation conditions.During the plasma/Fenton-like degradation of MO, the concentrations of Fe2+ and total Fe ions in the solution were measured, as shown in Fig. S2.† The results showed that the concentration of free Fe ions was 0.10–1.26 μg mL−1, which only accounted for 0.04–0.30% of the total amount of iron added. It was verified that the material could effectively fix Fe ions and prevent Fe ion loss during the reaction.
:3, the width to length ratio of the sample was small, which was consistent with the SEM results.
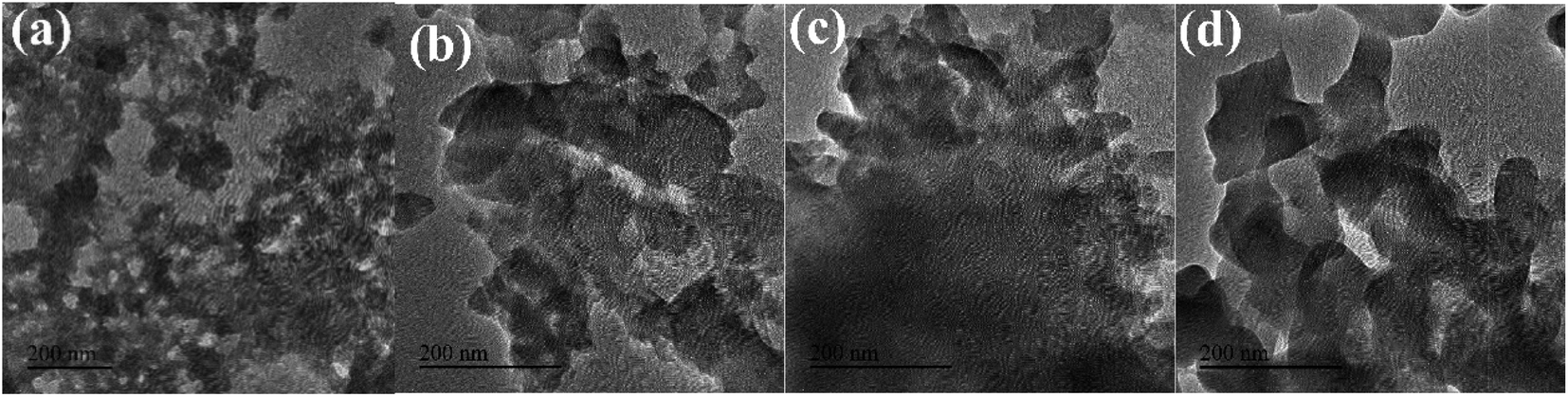 | ||
| Fig. 4 TEM images of Fe-MOFs with different TA/Fe molar ratios (a) 1:1; (b) 1:2; (c) 1:3; (d) 1:4. | ||
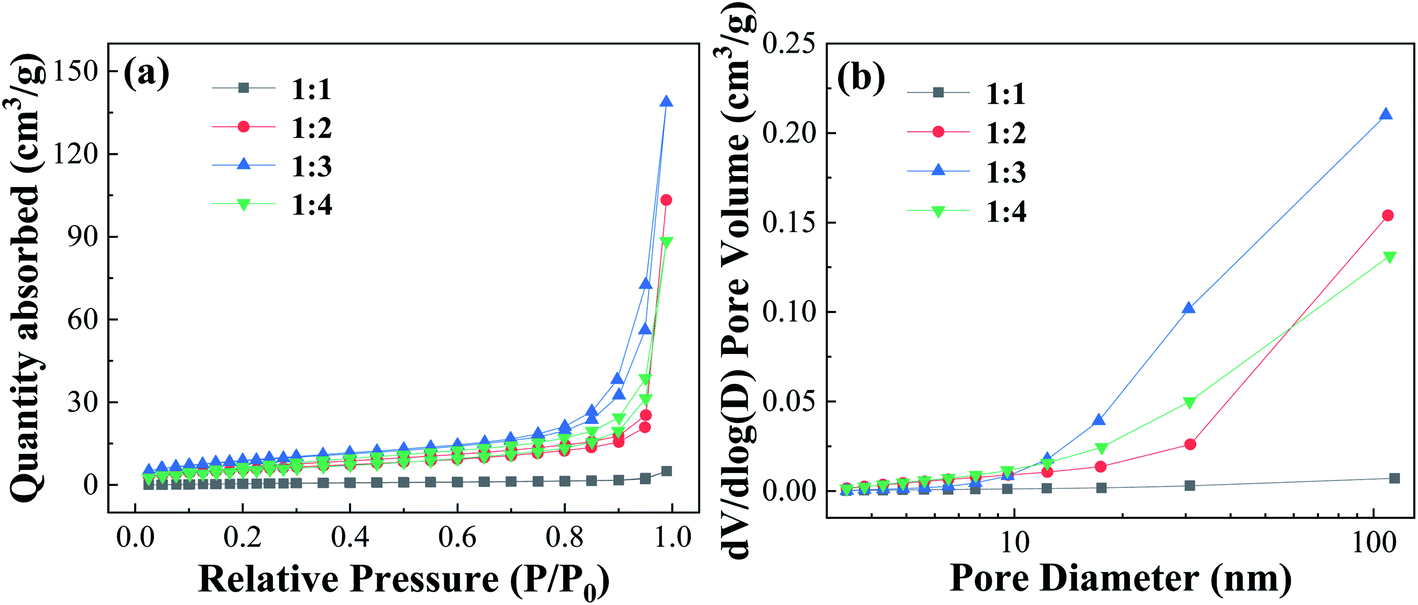 | ||
| Fig. 5 Nitrogen adsorption–desorption isotherms curves and pore size distribution of Fe-MOFs with different TA/Fe molar ratios. | ||
Table S3† shows the surface and pore properties of Fe-MOFs. With increasing TA/Fe molar ratios, the values of specific surface area, average pore diameter, and pore volume of Fe-MOFs increased. When the TA/Fe molar ratio was 1:3, the maximum specific surface area was obtained, which was 32.021 m2 g−1. Due to the nature of the structure, the amount of MO molecules adsorbed on the inner surface increased, which was convenient for adsorption and thus the catalytic reaction was accelerated. Furthermore, a large specific surface area not only results in more active sites, but also increases the contact area of the reaction, thereby accelerating the reaction rate. When the TA/Fe molar ratio was 1:4, the specific surface area, average pore size and pore volume of Fe-MOFs decreased, which may be caused by the differences in morphology and structure. This was consistent with the SEM analysis results.
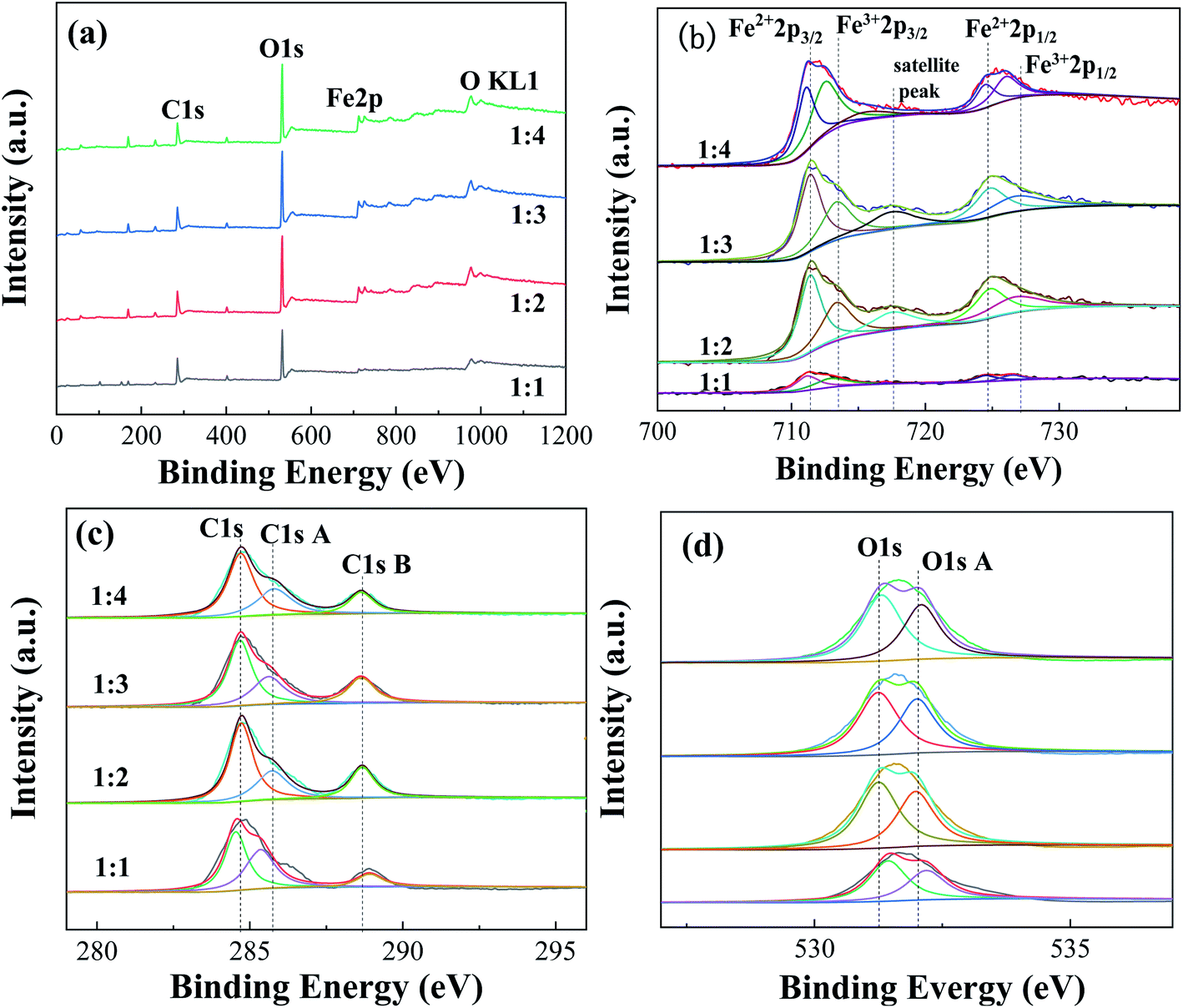 | ||
| Fig. 6 XPS spectra of Fe-MOFs with different TA/Fe molar ratios (a) survey spectrum; (b) Fe 2p; (c) C 1s; (d) O 1s. | ||
Table S5† presents the proportions of different valence iron in Fe-MOFs. The ratio of Fe2+/Fe3+ and the relative proportion of oxygen vacancies in Fe-MOFs were estimated from the relative area of the deconvolution peak. When the TA/Fe molar ratio was 1:3, the ratio of Fe2+/Fe3+ and [O]s was significantly higher than that of the other samples, which presented more oxygen vacancies and was very beneficial to the Fenton catalytic degradation process. Fe-MOFs can drive the formation of more ·OH and ·OOH based on eqn (1) and (2). Then, MO is degraded by the abundant ·OH and ·OOH. e− and h+ are produced under ultraviolet light (eqn (3)). The Fe3+ is easily reduced to Fe2+ by absorbing e− (eqn (4)), which enables the continuous formation of ·OH.22 In addition, surface oxygen vacancies are generated during the reduction of Fe3+ to Fe2+, which would also affect the catalytic performance. Fe-MOFs with a TA/Fe molar ratio of 1:3 had the highest content of Fe2+/Fe3+ and oxygen vacancies. Furthermore, the content of Fe2+ and Fe3+ in Fe-MOFs with a TA/Fe molar ratio of 1:1 was higher, because the area formed by the satellite peak was small and the proportion was relatively small, so the proportion of Fe2+ and Fe3+ was relatively large. This was consistent with the XRD and XPS results.
| Fe2+ + H2O2 → Fe3+ + ·OH + OH− | (1) |
| Fe3+ + H2O2 → Fe2+ + ·OOH + H+ | (2) |
| hv → h+ + e− | (3) |
| Fe3+ + e− → Fe2+ | (4) |
3.2 Plasma/Fenton-like process of Fe-MOFs
Fig. S3† shows a comparison of the performance of plasma alone, Fenton alone, and the plasma/Fenton-like system. When the discharge voltage was 14.5 kV, the addition amount of Fe-MOFs with a TA/Fe molar ratio of 1:3 was 1 g L−1 and the addition amount of H2O2 was 2.0 mL L−1, it can be found that the plasma/Fenton-like catalysis was better than plasma and Fenton alone.
Fig. S4† shows the effect of different reaction conditions on the performance of Fe-MOFs. From Fig. S4 (a),† it can be seen that 200 mg L−1 MO was completely degraded within 8 min, and 350 mg L−1 MO can reach 96% degradation within 12 min. With the increase of MO concentration, the degradation rate was correspondingly slower. It can be seen that Fe-MOFs could not only process low-concentration MO solutions, but also have a beneficial effect on MO in higher concentrations. Solutions with higher initial organic concentrations needed longer treatment times to achieve a given degradation degree.35 Fig. S4(b)† showed that the degradation rate increased with the increase of discharge voltage. Nevertheless, energy consumption needs to be considered. It can be seen from Fig. S4(c)† that when the optimal amount was added, the degradation efficiency was the fastest.36 The concentration of H2O2 played a crucial role in deciding the overall efficiency of the degradation process. However, the operating oxidant dosage must be carefully identified. The presence of H2O2 was harmful to many of the organisms and would affect the overall degradation efficiency significantly, hence the excess amount was not recommended. Moreover, the negative effect of H2O2 was the scavenging of generated ·OH, which occurred at high quantities of H2O2.35 Therefore, the amount of H2O2 should be reduced as much as possible to ensure efficiency. According to Fig. S4(e),† the pH of 3 was beneficial for the Fenton reaction because pH significantly affects the solubility and oxidation state of Fe.37 The kinetics of MO degradation in the experiments were studied.38–40 The kinetic behavior of MO degradation was fit by a pseudo-first-order model, as highlighted in the inset panel of Fig. S4.† It was found that the maximum pseudo-first-order rate constant was 0.669 min−1 (R2 = 0.991).
Fig. S5† shows the recycling experiment results of Fe-MOFs prepared in the conditions of discharge voltage of 14.5 kV, reactant concentration of 12 g L−1, TA/Fe molar ratio of 1:3 and discharge time of 100 min, which showed the best performance. The samples still maintained more than 89% degradation efficiency for MO after six cycles. A comparison of the degradation capacity between the DBD plasma/Fenton system and other systems is shown in Table S6.† Fe-MOFs synthesized by DBD plasma had great advantages in the DBD plasma/Fenton treatment of MO.
3.3 Reaction mechanism of plasma/Fenton-like process
In order to identify the role of various active species on MO degradation in the plasma/Fenton-like process and to discuss the reaction mechanism, tert-butanol (TBA), ammonium oxalate (AO) and benzoquinone (BQ) were used as capture agents of ·OH, h+ and ·O2−, respectively. As shown in Fig. S6,† after the addition of the capture agents, the reaction efficiency was reduced because the capture agents consumed the active species in the reaction system that contributed to the degradation of MO. It proved that active species including ·OH, h+ and ·O2− were generated during the reaction and played a vital role in the degradation process of MO.For the Fenton process, Fe2+ catalyzed the decomposition of H2O2, which resulted in the generation of ·OH radicals.17 The generated Fe3+ could be reduced by reaction with excess H2O2 to form Fe2+ again and more radicals such as ·O2−; it could also attack organic contaminants.36 For the DBD plasma process, energetic electrons exist that collide with other molecules such as O2 and H2O to produce various active species including ·OH and ·O2−, in addition to some active molecules such as O3 and H2O2. Furthermore, h+ and photo-generated electrons (e−) were generated on the surface of Fe-MOFs and the catalytic redox reaction occurred to produce H2O2 and dissolved oxygen. This residual H2O2 could be utilized through the Fenton reaction to produce more ·OH,41 which further enhanced the degradation of MO. When the plasma reaction was combined with the Fenton reaction, the amount of active substances increased, thus the degradation of organic matter was accelerated. The degradation of MO was finally achieved by the redox reactions of the active species.
The reaction intermediates of MO in the plasma/Fenton-like process formed during the first 8 min were analyzed by LC-MS. The obtained mass spectra are provided in Fig. S7.† The detected intermediates with different mass-to-charge (m/z) ratios and molecular structures are summarized in Table S7.† A degradation mechanism (Scheme 1) was proposed based on the species detected in this study and previous literature. Before degradation, the peak of m/z = 304.07 was the parent molecule of MO after losing Na+ due to dissolution in aqueous solution. The intermediate with a m/z of 290.06 was formed due to the demethylation, followed by deamination to generate azobenzesulfonic acid (m/z = 261.03). Then, it was decomposed into sulfanilic acid (m/z = 172.00) and aniline (m/z = 93.05) (Path 1). Sulfanilic acid was methylated and deaminated to form phenylmethylamine (m/z = 107.07) and then converted to aniline. Azobenzesulfonic acid can also be decomposed to 4-diazenyl-benzene (m/z = 106.05) and p-hydroxy-benzenesulfonic acid (m/z = 172.99) (Path 2). P-hydroxybenzenesulfonic acid was oxidized to benzenesulfonic acid (m/z = 156.99) and nitrobenzene (m/z = 138.0197), which was then oxidized to phenol (m/z = 110.03), while 4-diazenyl benzene was converted to aniline. The further oxidation of hydroquinone and aniline resulted in the aromatic ring opening and formation of short-linear aliphatic carboxylic acids. Finally, the aliphatic carboxylic acids can directly convert into CO2 and H2O.
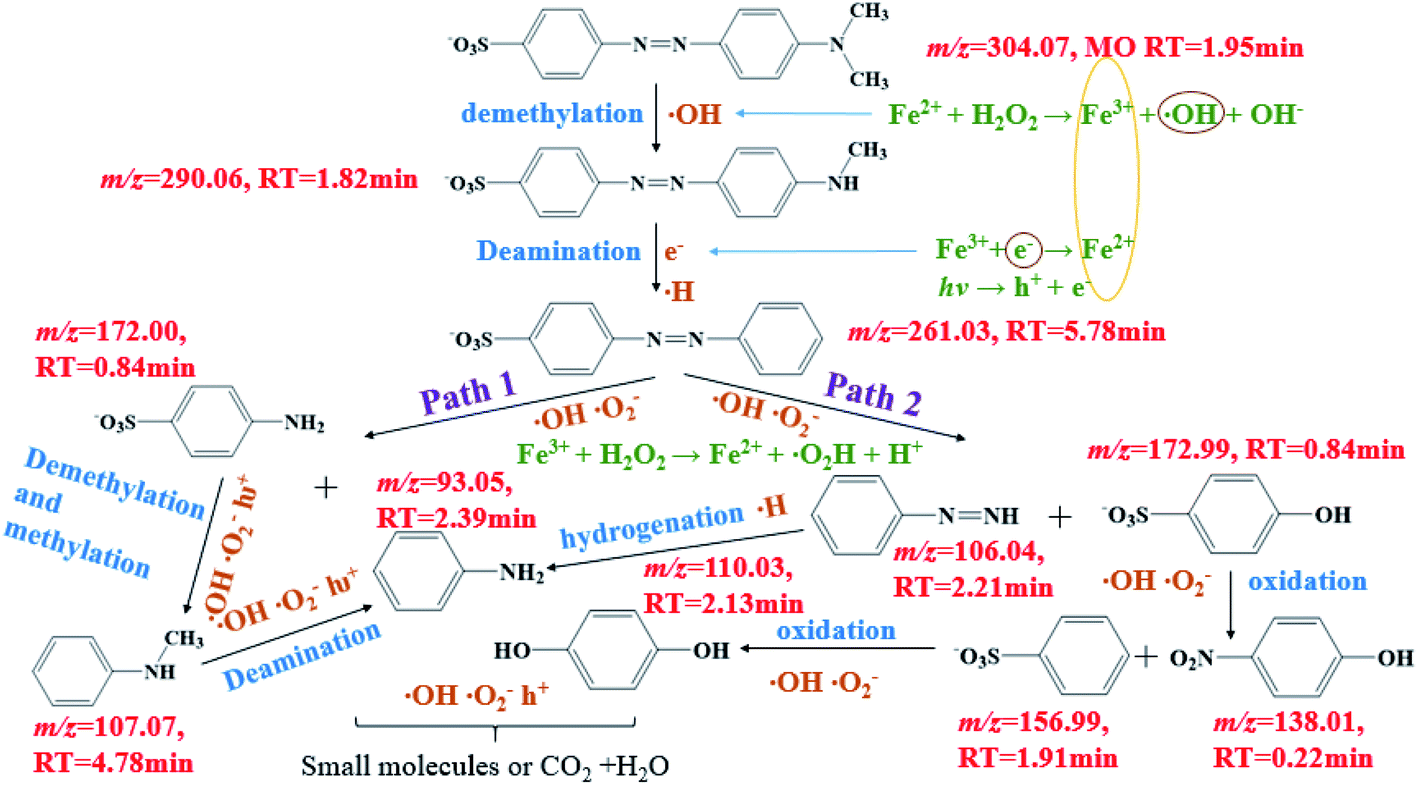 | ||
| Scheme 1 Proposed degradation pathways of MO in the plasma/Fenton-like system. | ||
4 Conclusions
Fe-MOFs using FeSO4·7H2O as an iron source were successfully synthesized by DBD plasma and applied for plasma/Fenton-like technology as heterogeneous catalysts. The synergy of DBD plasma and Fenton induced an increased rate of MO removal. The results showed that Fe-MOFs had a stable crystal structure, increased specific surface area and pore size, and thus, more iron-based active sites. In addition, the valence of iron in the synthesized Fe-MOFs affected the catalytic performance. At a catalyst addition amount of 1 g L−1, H2O2 addition amount of 2.0 mL L−1, discharge voltage of 13.7 kV, and pH of 3, the degradation rate of 200 mL (200 mg L−1) MO reached 96.4% at 8 min. Through free radical capture experiments, the reaction mechanism of the plasma/Fenton-like catalytic degradation process was deduced mainly as the coordinated oxidation process of ·OH, h+ and ·O2−. Collaborative technology improves the efficiency of wastewater treatment, and also provides new ideas for the study of pollutant degradation. More importantly, as Fenton-like catalysts, Fe-MOFs exhibited excellent durability in six cycles of degradation, which proved that the catalyst was stable and reusable. In addition, the degradation intermediates of MO were identified using LC-MS, and possible transformation pathways were proposed.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Talent Fund of Shandong Collaborative Innovation Center of Eco-Chemical Engineering from Qingdao University of Science and Technology [XTCXQN23]. The authors would like to acknowledge the support of Mengzhen Wang for useful discussions and help.References
- M. Malakootian, A. Shahesmaeili, M. Faraji, H. Amiri and S. S. Martinez, Process Saf. Environ. Prot., 2020, 134, 292–307, DOI:10.1016/j.psep.2019.12.004.
- M. H. Zhang, H. Dong, L. Zhao, D. X. Wang and D. Meng, Sci. Total Environ., 2019, 670, 110–121, DOI:10.1016/j.scitotenv.2019.03.180.
- L. J. Di, H. Yang, T. Xian, X. Q. Liu and X. J. Chen, Nanomaterials, 2019, 9, 399, DOI:10.3390/nano9030399.
- H. Wang, M. Jing, Y. Wu, W. L. Chen and Y. Ran, J. Hazard. Mater., 2018, 353, 53–61, DOI:10.1016/j.jhazmat.2018.03.053.
- S. Ma, K. Kim, S. Chun, S. Y. Moon and Y. C. Hong, Chemosphere, 2020, 243, 125377, DOI:10.1016/j.chemosphere.2019.125377.
- G. S. Oehrlein and S. Hamaguchi, Plasma Sources Sci. Technol., 2018, 27, 1–21, DOI:10.1088/1361-6595/aaa86c.
- A. Fahmy, J. Friedrich, F. Poncin-Epaillard and D. Debarnot, Thin Solid Films, 2016, 616, 339–347, DOI:10.1016/j.tsf.2016.08.045.
- F. Alaa, E. Z. Adham, A. M. Saeed, A. Z. Sayed, M. A. Ezz El-Arab and H. A. Shehata, Chemosphere, 2018, 210, 102–109, DOI:10.1016/j.chemosphere.2018.06.176.
- P. Hu, Y. K. Liu, B. Jiang, X. Zheng, J. T. Zheng and M. B. Wu, Ind. Eng. Chem. Res., 2015, 54, 8277–8286, DOI:10.1021/acs.iecr.5b01012.
- A. Khataee, S. Bozorg, S. Khorram, M. Mehrangiz Fathinia, Y. Hanifehpour and S. W. Joo, Ind. Eng. Chem. Res., 2013, 52, 18225–18233, DOI:10.1021/ie403283n.
- F. Dai, X. R. Fan, G. R. Stratton, C. L. Bellona, T. M. Holsen, B. S. Crimmins, X. Y. Xia and S. M. Thagard, J. Hazard. Mater., 2016, 308, 419–429, DOI:10.1016/j.jhazmat.2016.01.068.
- S. Slamani, F. Abdelmalek, M. R. Redouane and A. Addou, J. Photochem. Photobiol., A, 2018, 359, 1–10, DOI:10.1016/j.jphotochem.2018.03.032.
- C. M. Du, L. L. Zhang, J. Wang, C. R. Zhang, H. X. Li and Y. Xiong, Plasma Chem. Plasma Process., 2010, 30, 855–871, DOI:10.1007/s11090-010-9249-0.
- M. Marković, M. Jović, D. Stanković, V. Kovačević, G. Roglić, G. Gordana Gojgić-Cvijović and D. Manojlović, Sci. Total Environ., 2015, 505, 1148–1155, DOI:10.1016/j.scitotenv.2014.11.017.
- J. L. Wang and L. J. Xu, Crit. Rev. Environ. Sci. Technol., 2012, 42, 251–325, DOI:10.1080/10643389.2010.507698.
- C. L. Chang and T. S. Lin, Plasma Chem. Plasma Process., 2005, 25, 227–243, DOI:10.1007/s11090-004-3034-x.
- R. Thür, N. V. Velthovena, S. Slootmaekers, J. Diddena, R. Verbekea, S. Smoldersa, M. Dickmannb, W. Eggerc, D. D. Vosa and I. F. J. Vankelecom, J. Membr. Sci., 2019, 576, 78–87, DOI:10.1016/j.memsci.2019.01.016.
- A. Shrivastav, S. Sundriyal, P. Goel, H. Kaur, S. K. Tuteja, K. Vikrant, K. H. Kim, U. K. Tiwari and A. Deep, Coord. Chem. Rev., 2019, 393, 48–78, DOI:10.1016/j.ccr.2019.05.006.
- F. Martínez, P. Leo, G. Orcajo, M. Díaz-Garcíab, M. Sanchez-Sanchezb and G. Calleja, Catal. Today, 2018, 313, 6–11, DOI:10.1016/j.cattod.2017.10.002.
- X. M. Tao, C. Sun, Y. Y. Han, L. Huang and D. Y. Xu, CrystEngComm, 2019, 21, 1–24, 10.1039/C9CE00015A.
- X. M. Tao, C. Sun, L. Huang, Y. Y. Han and D. Y. Xu, RSC Adv., 2019, 9, 6379–6386, 10.1039/c8ra09211g.
- P. Li, S. Kim, J. Jin, D. H. Chun and J. H. Park, Appl. Catal., B, 2020, 263, 118284, DOI:10.1016/j.apcatb.2019.118284.
- T. D. Le, K. D. Nguyen, V. T. Nguyen, T. Truong and N. T. S. Phan, J. Catal., 2016, 333, 94–101, DOI:10.1016/j.jcat.2015.10.026.
- G. Q. Song, Z. Q. Wang, L. Wang, G. R. Li, M. J. Huang and F. X. Yin, Chin. J. Catal., 2014, 35, 185–195, DOI:10.1016/S1872-2067(12)60729-3.
- D. Z. Chen, S. S. Chen, Y. J. Jiang, S. S. Xie, H. Y. Quan, L. Hua, X. B. Luo and L. Guo, RSC Adv., 2017, 7, 49024–49030, 10.1039/c7ra09234b.
- V. M. Kariuki, I. Y. azgan, A. Akgul, A. Kowal, A. Omowunmi, M. Parlinska and O. A. Sadik, Environ. Sci.: Nano, 2015, 2, 518–527, 10.1039/C5EN00053J.
- R. Wang, Y. Gong, H. J. Xu, S. Y. Wei and D. Y. Wu, Chin. J. Inorg. Anal. Chem., 2018, 34, 906–916, DOI:10.11862/CJIC.2018.110.
- M. Anbia, V. Hoseini and S. Sheykhi, J. Ind. Eng. Chem., 2012, 18, 1149–1152, DOI:10.1016/j.jiec.2012.01.014.
- N. Torres, J. Galicia, Y. Plasencia, A. Cano, F. Echevarría, L. F. Desdin-Garcia and E. Reguera, Colloids Surf., A, 2018, 549, 138–146, DOI:10.1016/j.colsurfa.2018.04.016.
- Y. J. Li, G. S. Hou, J. Yang, J. Xie, X. L. Yuan, H. Yang and M. M. Wang, RSC Adv., 2016, 6, 16395–16403, 10.1039/c5ra24915e.
- X. H. Li, W. L. Guo, Z. H. Liu, R. Q. Wang and H. Liu, Appl. Surf. Sci., 2016, 369, 130–136, DOI:10.1016/j.apsusc.2016.02.037.
- F. M. Zhang, Y. Jin, J. Shi, W. D. Zhu and M. S. El-Shall, Chem. Eng. J., 2015, 269, 236–244, DOI:10.1016/j.cej.2015.01.092.
- B. J. Zhu, X. Y. Yu, Y. Jia, F. M. Peng, B. Sun, M. Y. Zhang, T. Luo, J. H. Liu and X. J. Huang, J. Phys. Chem. C, 2012, 116, 8601–8607, DOI:10.1021/jp212514a.
- T. A. Vu, G. H. Le, C. D. Dao, L. Q. Dang, K. T. Nguyen, K. Quang, Q. K. Nguyen, P. T. Dang, H. T. K. Tran, Q. T. Duong, V. Tuyen, T. V. Nguyen and G. D. Lee, RSC Adv., 2015, 5, 5261–5268, 10.1039/c4ra12326c.
- L. Labiadh, M. A. Oturan, M. Panizza, N. B. Hamadi and S. Ammar, J. Hazard. Mater., 2015, 297, 34–41, DOI:10.1016/j.jhazmat.2015.04.062.
- A. Babuponnusami and K. Muthukumar, J. Ind. Eng. Chem., 2014, 2, 557–572, DOI:10.1016/j.jece.2013.10.011.
- F. C. Moreira, R. A. R. Boaventura, E. Brillas and V. J. P. Vilar, Appl. Catal., B, 2016, 202, 217–261, DOI:10.1016/j.apcatb.2016.08.037.
- S. Ammar, M. A. Oturan, L. Labiadh, A. Guersalli, R. Abdelhedi, N. Oturan and E. Brillas, Water Res., 2015, 74, 77–87, DOI:10.1016/j.watres.2015.02.006.
- S. Midassi, A. Bedoui and N. Bensalah, Chemosphere, 2020, 260, 127558, DOI:10.1016/j.chemosphere.2020.127558.
- Z. Q. Yang, H. L. Chen, J. H. Wang, Q. F. Yuan, F. Wang and B. H. Zhou, J. Environ. Chem. Eng., 2020, 8, 104057, DOI:10.1016/j.jece.2020.104057.
- X. L. Hao, M. H. Zhou, Y. Zhang and L. C. Lei, Plasma Chem. Plasma Process., 2006, 26, 455–468, DOI:10.1007/s11090-006-9028-0.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07402k |
| This journal is © The Royal Society of Chemistry 2020 |