
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermodynamics of selective serotonin reuptake inhibitors partitioning into 1,2-dioleoyl-sn-glycero-3-phosphocholine bilayers†
Dat T. N. Ngo ad,
Trinh Q. Nguyen‡
ad,
Hieu K. Huynhb and
Trang T. Nguyen*cd
ad,
Trinh Q. Nguyen‡
ad,
Hieu K. Huynhb and
Trang T. Nguyen*cd
aDepartment of Biotechnology, International University, Block 6, Linh Trung Ward, Thu Duc District, Ho Chi Minh City, Vietnam
bUniversity of Medicine and Pharmacy at Ho Chi Minh City, 217 Hong Bang, Ward 11, District 5, Ho Chi Minh City, Vietnam
cDepartment of Chemical Engineering, International University, Block 6, Linh Trung Ward, Thu Duc District, Ho Chi Minh City, Vietnam. E-mail: nttrang@hcmiu.edu.vn
dVietnam National University, Ho Chi Minh City, Vietnam
First published on 27th October 2020
Abstract
Knowledge of thermodynamics of lipid membrane partitioning of amphiphilic drugs as well as their binding site within the membrane are of great relevance not only for understanding the drugs' pharmacology but also for the development and optimization of more potent drugs. In this study, the interaction between two representatives of selective serotonin reuptake inhibitors, including paroxetine and sertraline, and large unilamellar vesicles (LUVs) composed of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) was investigated by second derivative spectrophotometry and Fourier transform infrared spectroscopy (FTIR) to determine the driving force of the drug partitioning across lipid membranes. It was found that temperature increase from 25 to 42 °C greatly enhanced the partitioning of paroxetine and sertraline into DOPC LUVs, and sertraline intercalated into the lipid vesicles to a greater extent than paroxetine in the temperature range examined. The partitioning of both drugs into DOPC LUVs was a spontaneous, endothermic and entropy-driven process. FTIR measurements suggested that sertraline could penetrate deeply into the acyl tails of DOPC LUVs as shown by the considerable shifts in the lipid's CH2 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching modes induced by the drug. Paroxetine, however, could reside closer to the head groups of the lipid since its presence caused a larger shift in the PO2− bands of DOPC LUVs. The findings reported here provide valuable insights into the influence of small molecules' chemical structure on their molecular interaction with the lipid bilayer namely their possible binding sites within the lipid bilayer and their thermodynamics profiles of partitioning, which could benefit rational drug design and drug delivery systems.
O stretching modes induced by the drug. Paroxetine, however, could reside closer to the head groups of the lipid since its presence caused a larger shift in the PO2− bands of DOPC LUVs. The findings reported here provide valuable insights into the influence of small molecules' chemical structure on their molecular interaction with the lipid bilayer namely their possible binding sites within the lipid bilayer and their thermodynamics profiles of partitioning, which could benefit rational drug design and drug delivery systems.
1. Introduction
Most of the drugs available on the market target transmembrane proteins.1 As a result, the ability of a drug molecule to cross the cell membrane and interact with lipid bilayers is a critical benchmark in drug design, development and optimization.2 Additionally, the thermodynamics of drug–lipid membrane interaction not only offers essential information regarding the force that drives a drug across lipid membranes but also reveals its pharmacological behavior, thus it has been of great research interest.3–10Selective serotonin reuptake inhibitors (SSRIs) have been considered as the first-line medicines for the treatment of depression and other mental disorders11 since they alleviate the symptoms of depression by raising serotonin level. When SSRIs are administered, they inhibit the neuronal uptake pump for serotonin by binding to the serotonin transporter, and thus prevent the re-uptake of serotonin.12,13 To reach their targets, SSRIs must partition into cell membranes, as a result, any interactions between SSRIs and lipid membranes are crucial in understanding the mechanism of action of the drugs. SSRIs have been found to not only accumulate in the cell membranes14,15 but also alter the physical structure and properties of lipid bilayers upon their partitioning.16–19 It was previously reported that fluoxetine, a common SSRI, partitioned to a greater extent into liquid-crystalline 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) bilayers than into solid-gel 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) bilayers and the membrane partitioning of fluoxetine increased with temperature in which the lipid phase changed from solid-gel to liquid-crystalline.16 Furthermore, fluoxetine structurally disordered both DOPC and DPPC bilayers upon its partitioning.16 The disordering effect of fluoxetine upon its partitioning into 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and DPPC bilayers was also observed.19
Although SSRIs–lipid membrane interactions have been subjected to several studies, the thermodynamics of SSRIs partitioning into lipid membranes, especially the Gibbs free energy, enthalpy and entropy change of partitioning, has not been described. Moreover, the force that drives the partitioning of SSRIs into lipid bilayer has not yet been elucidated. Additionally, previous studies on SSRIs–lipid membrane interaction were only carried out with fluoxetine as the representative drug16,17,19 while little attention has been paid to paroxetine (PAX) and sertraline (SER) in spite of their wide usage in the treatment of depression.11,20,21
As cationic amphiphilic drugs, PAX and SER are defined by two main physicochemical attributes: a hydrophobic ring system and a basic, nitrogen-containing group.22 In PAX, the hydrophobic ring system is composed of a piperidine bearing 1,3-benzodioxol-5-yloxymethyl and 4-fluorophenyl substituents at positions 3 and 4, respectively, while the nonpolar moiety of SER is tetralin which has methylamino and 3,4-dichlorophenyl substituents at positions 1 and 4, respectively. The nitrogen-containing group of PAX is the piperidine group, while that of SER is the methylamino group, both of which are secondary amine. Additionally, the halogen moiety on the hydrophobic ring system of the two drugs also differs, in which PAX has one fluorine atom while SER possesses two chlorine atoms. Due to the heterogeneity in their chemical structures, the physicochemical properties of PAX and SER, such as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P and total polar surface area (TPSA), are also different, which lead to the dissimilarity in pharmacokinetic and pharmacologic action between PAX and SER.21,23–26 The chemical structures of PAX and SER were depicted in Fig. 1.
P and total polar surface area (TPSA), are also different, which lead to the dissimilarity in pharmacokinetic and pharmacologic action between PAX and SER.21,23–26 The chemical structures of PAX and SER were depicted in Fig. 1.
 | ||
| Fig. 1 Chemical structures of paroxetine, sertraline and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC). | ||
In this study, thermodynamics of the two SSRIs, PAX and SER, partitioning into lipid membranes, were characterized in an attempt to provide a more comprehensive understanding of SSRIs–lipid membrane interactions. Large unilamellar vesicles (LUVs) composed of DOPC were chosen in the present work as a model of lipid membranes. With a cis double bond between the C9 and C10 in each chain (see Fig. 1), DOPC exists in the liquid-crystalline state at a temperature above its Tm (−16.5 °C27), mimicking well the fluid cell membranes, and thus has been commonly used as a model lipid for probing drug–lipid membrane interactions.18,28–31
Thermodynamics of membrane partitioning of PAX and SER was determined by measuring DOPC liposome/water partition coefficients (Kp) of the drugs at varying temperatures using second derivative spectrophotometry, an effective method in measurement of drug partitioning owing to its ability to eliminate the light scattering from lipid vesicles.16,17,32,33 Gibbs energy (ΔG), enthalpy (ΔG) and entropy (ΔS) change of the drug partitioning were then extracted from Van't Hoff figure in which logarithms of Kp values were plotted against inversed temperature. Along with partition coefficient determination, the conformational changes in DOPC bilayer were investigated by FTIR to provide complementary information regarding the thermodynamics profiles of SSRIs-DOPC LUVs interaction. Particularly, the ordering degree of the lipid acyl chains before and after the drug partitioning can be revealed by changes in the location of the CH2 stretching bands34 and the hydration state of the interfacial regions can be obtained by frequency shifts in the CO and PO2− bands of the lipid head groups.35
2. Methods
Paroxetine (PAX) and sertraline (SER) were obtained from Sigma Aldrich (USA). 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) in chloroform was purchased from Avanti Polar Lipids (USA) and used without further purification. The stock solution of lipid was made at a concentration of 20 mg mL−1. HEPES (4-(2-hydroxyethyl)-1-piperazine-ethanesulfonic acid) buffer was obtained from Sigma Aldrich (USA). All liquid suspensions were prepared with 10 mM HEPES buffer containing 50 mM NaCl, at pH 7.4.2.1 Preparation of large unilamellar vesicles
Chloroform in the lipid solution was removed by evaporation using a stream of nitrogen. The residual amount of chloroform was further removed under vacuum for at least 8 h at room temperature. The dried lipid film was then suspended with HEPES buffer at pH 7.4 and sonicated at room temperature. The lipid suspensions were subjected to 5 cycles of a cooling–heating process. Large unilamellar vesicles (LUVs) were produced by extruding the lipid suspensions through a polycarbonate membrane of 0.1 μm pore size (Whatman Inc., USA) using a mini-extruder (Avanti Polar Lipids, USA).2.2 Preparation of drug–liposome mixtures
Sample solutions were prepared by adding the drug solution at a fixed concentration (0.50 mM for PAX and 0.045 mM for SER) to different concentrations of the lipid suspensions. Reference samples were prepared identically but without the drugs. All drug–liposome mixtures were vortexed for 1 min and then incubated at 25 °C, 32 °C, 37 °C or 42 °C for 30 min before being subjected to absorbance measurements.2.3 Collection of absorption spectra and second derivative of absorption spectra
The absorption spectrum of each sample was collected using a microcell cuvette with a chamber volume of 700 μL by Agilent Cary 60 UV-Vis spectrophotometer (Agilent, USA) with a temperature regulated cell holder set at the given temperature. The spectral window was from 205 nm to 330 nm for PAX and from 205 nm to 260 nm for SER with a wavelength interval (Δλ) of 1 nm. The second derivative spectra were calculated using Origin (Origin Lab, WA, USA) based on the Savitzky–Golay method in which the second-order polynomial convolution of 20 points was employed.2.4 Determination of partition coefficient
The partition coefficient (Kp) of a drug between the lipid phase and the water phase is defined as:
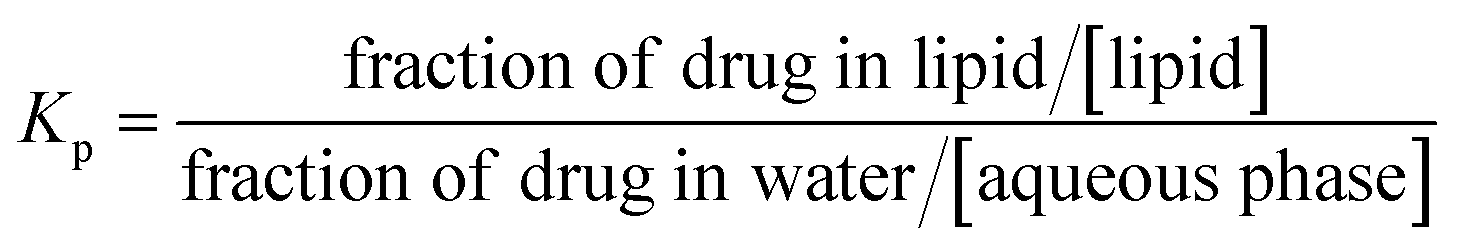 | (1) |
The fraction of the bound drug is defined as ΔD/ΔDmax, where ΔD is the derivative intensity difference between absorption in the presence and absence of lipid vesicles and is directly proportional to the drug concentration in the lipid phase.
| ΔD = D − D0 | (2) |
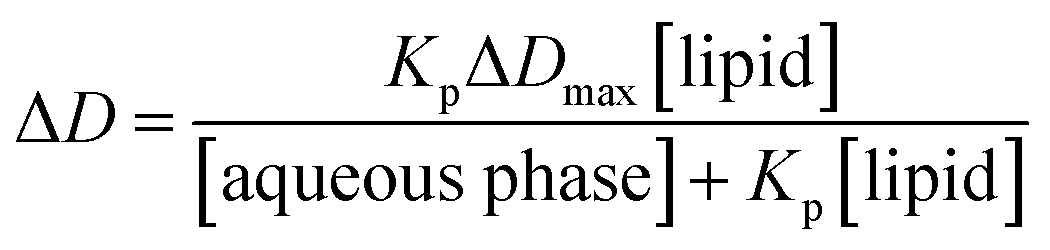 | (3) |
Non-linear least square fitting of eqn (3) with the experimental values of ΔD and [lipid] for a selected drug concentration can be performed in order to obtain Kp. In principle, ΔD value at any wavelength can be used to determine Kp.36,37 In practice, ΔD values at the wavelength that give the smallest standard deviation in the calculation of Kp have been frequently used. In our case, the Kp values at 219 nm exhibited smaller errors as compared to those calculated at other wavelengths; thus, this wavelength was chosen for the Kp calculation of both PAX and SER.
2.5 Determination of thermodynamic parameters
Thermodynamics of the partitioning of a drug from the aqueous phase into the lipid phase can be determined by the relationship between changes in Gibbs free energy ΔG and logarithm of Kp:|
ΔG = −RTlnKp
| (4) |
Gibbs free energy (ΔG) can be represented as the change in enthalpy (ΔH) minus the change in entropy (ΔS) at a certain temperature T:
| ΔG = ΔH − T × ΔS | (5) |
Substituting eqn (4) into eqn (5), Van't Hoff equation is established:
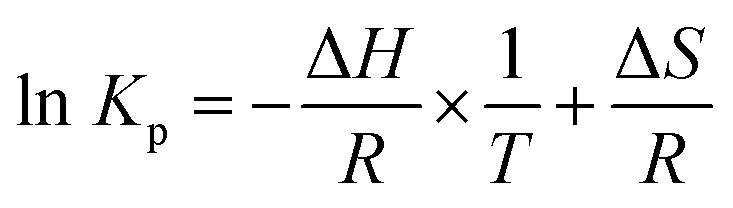 | (6) |
Kp values against 1/T.
2.6 ATR-FTIR measurement
The SSRIs at a concentration of 2 mg mL−1 in HEPES buffer (pH 7.4) were added into DOPC LUVs to obtain mixtures of DOPC: drug = 85:15 (mol%). The drug–lipid mixtures were then incubated for 30 min at 25 °C prior to IR measurements. The Tensor 27 FTIR spectrometer (Bruker Corporation, USA) with the scan range of 4000–1000 cm−1, the spectral resolution of 2 cm−1 and an average of 64 scans was used to acquire the spectra. The structural changes within the lipid bilayers could be reflected by the frequency shifts in the CH2 stretching modes of the acyl chain (2800–3000 cm−1), the CO stretching region (1700–1760 cm−1) and the PO2− stretching bands of the lipid head group (1150–1270 cm−1). The FTIR spectra were background subtracted, baseline corrected and normalized against to the maximum intensity of the CH2 band. Gaussian mathematical transformation on PeakFit v4.12 (Systat Software Inc., USA) was used to perform the Fourier deconvolution in order to reveal the component peaks.38 The peak deconvolution of the CH2, CO and PO2− regions in the FTIR spectra was performed first by self-location of the peak assignments with the full width at half maximum kept at a constant for each stretching mode and the heights left as a free parameter. The peak positions, intensities and the Gaussian shapes were then altered until the best fits of the spectral shapes were obtained.
3. Results
3.1 Absorption and second derivative spectra of SSRIs
The absorption spectra of PAX and SER with the presence of varying concentrations of DOPC LUVs at 25, 32, 37 and 42 °C were collected. The absorption spectra of PAX and SER at 25 °C were shown in Fig. 2A and 3A, respectively as representative examples. The absorption spectra in Fig. 2A and 3A were obtained by subtracting the reference (pure lipid) spectra from the sample (mixture of drug and lipid) spectra that contain the same lipid concentration. It is noted that the concentrations of PAX (0.50 mM) and SER (0.045 mM) in this work complied with the Beer's Law for absorption and the choice of drug concentration should not affect its partition coefficient into the lipid bilayer.2,39–41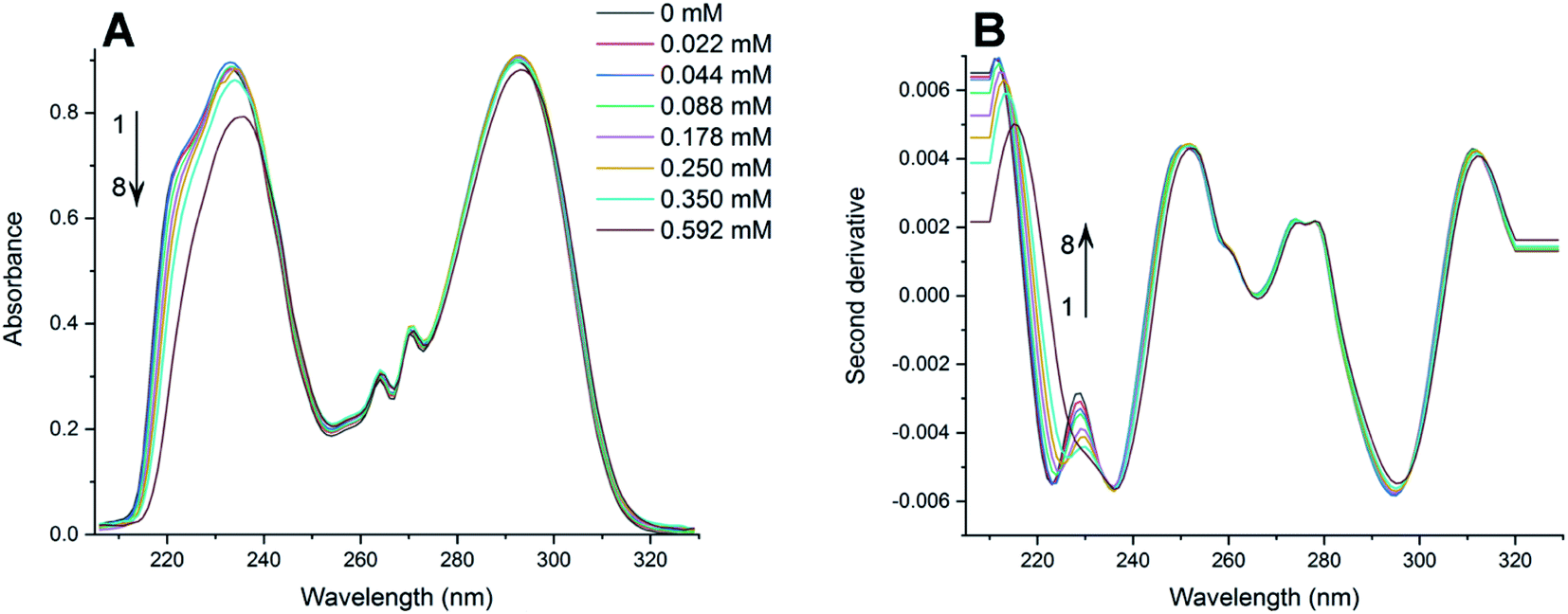 | ||
| Fig. 2 (A) Absorption spectra of PAX in HEPES buffer (pH 7.4, 25 °C) containing various amounts of DOPC LUVs (mM): (1) 0; (2) 0.022; (3) 0.044; (4) 0.088; (5) 0.178; (6) 0.250; (7) 0.350; (8) 0.592. (B) Second derivative spectra of PAX calculated from the absorption spectra (A). | ||
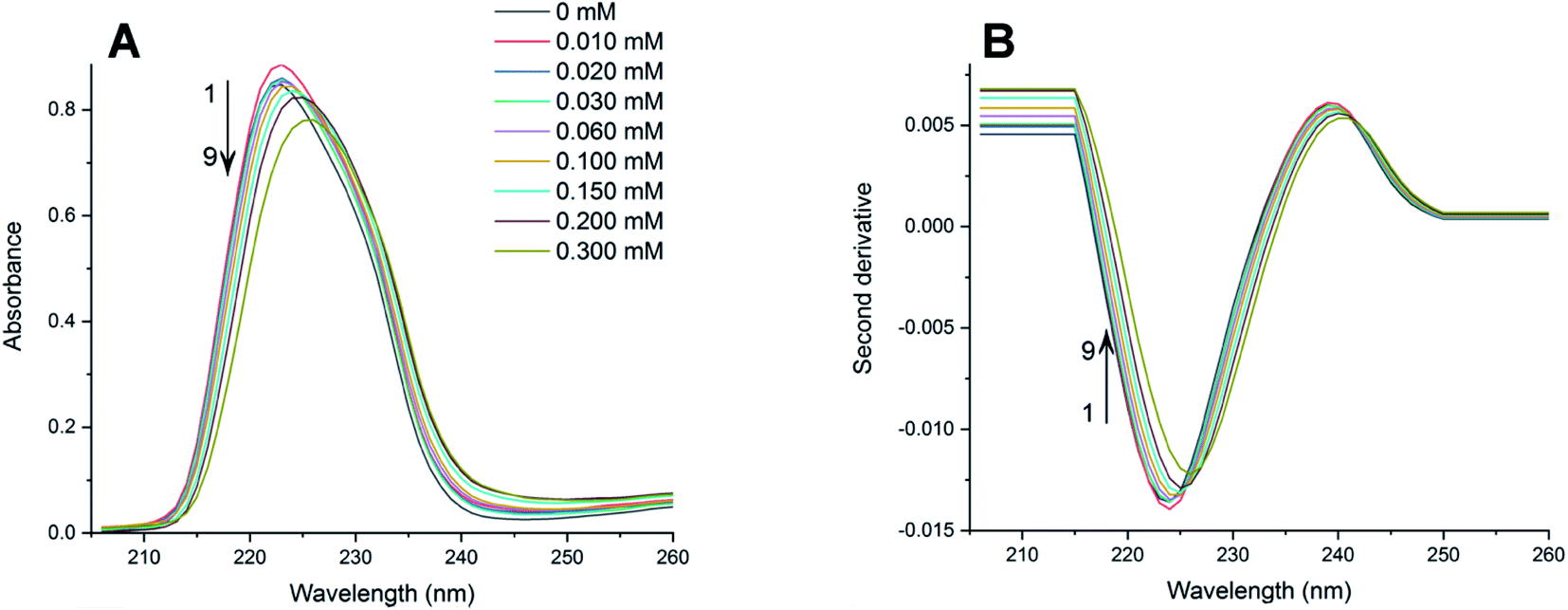 | ||
| Fig. 3 (A) Absorption spectra of SER in HEPES buffer (pH 7.4, 25 °C) containing various amounts of DOPC LUVs (mM): (1) 0; (2) 0.010; (3) 0.020; (4) 0.030; (5) 0.060; (6) 0.100; (7) 0.150; (8) 0.200; (9) 0.300. (B) Second derivative spectra of SER calculated from the absorption spectra (A). | ||
Increasing the concentration of DOPC LUVs resulted in a reduction of intensity (hypochromic effect) and a deviation in absorption maxima (λmax) of PAX from 232 nm to longer wavelength (bathochromic effect). SER also exhibited the same hypochromic and bathochromic effects, from 223 nm to longer wavelength, upon the introduction of increasing the lipid concentration. A hypochromic effect marks a significant interaction between a drug and lipid bilayers.42,43 A bathochromic effect occurs when the polarity of the environment surrounding a drug decreases.44,45 These phenomena indicated that both PAX and SER partitioned into DOPC LUVs.8,46 As a result of light scattering coming from the lipid vesicles, no isosbestic point was detected in the absorption spectra of either drug. In order to eliminate the background signals and to improve the resolution of overlapped signals between the aqueous phase and the lipid phase, the second derivative was then applied to the absorption data. The second derivative spectra calculated from the absorption spectra in Fig. 2A and 3A were depicted in Fig. 2B and 3B, respectively. For both PAX and SER, the minima of the second derivatives increased in intensity and shifted toward longer wavelengths with increasing the lipid concentration. Moreover, in the second derivative spectra, PAX exhibited two isosbestic points at 235 nm and 297 nm, while those of SER were located at 227 nm and 242 nm. The presence of these isosbestic points indicated that the background signal arisen from light scattering was removed in the second derivative spectra8,47 and suggested that PAX and SER equilibrated in two phases, the lipid phase and the aqueous phase.48
3.2 Partition coefficients of SSRIs in DOPC LUVs
The Kp values of PAX and SER into DOPC LUVs were obtained by the nonlinear least-squares fitting, as described in the experimental section. The ΔD values were calculated as the differences in the second derivative of absorption between spectrum 1 and the remaining spectra in Fig. 2B and 3B at a wavelength of 219 nm.ΔD/ΔDmax values, which denote the fractions of either PAX or SER partitioned into DOPC LUVs, were plotted versus the lipid concentration and shown in Fig. 4. Solid lines represented the theoretical curves calculated from eqn (3). The calculated Kp values of PAX and SER partitioned into DOPC LUVs were listed in Table 1.
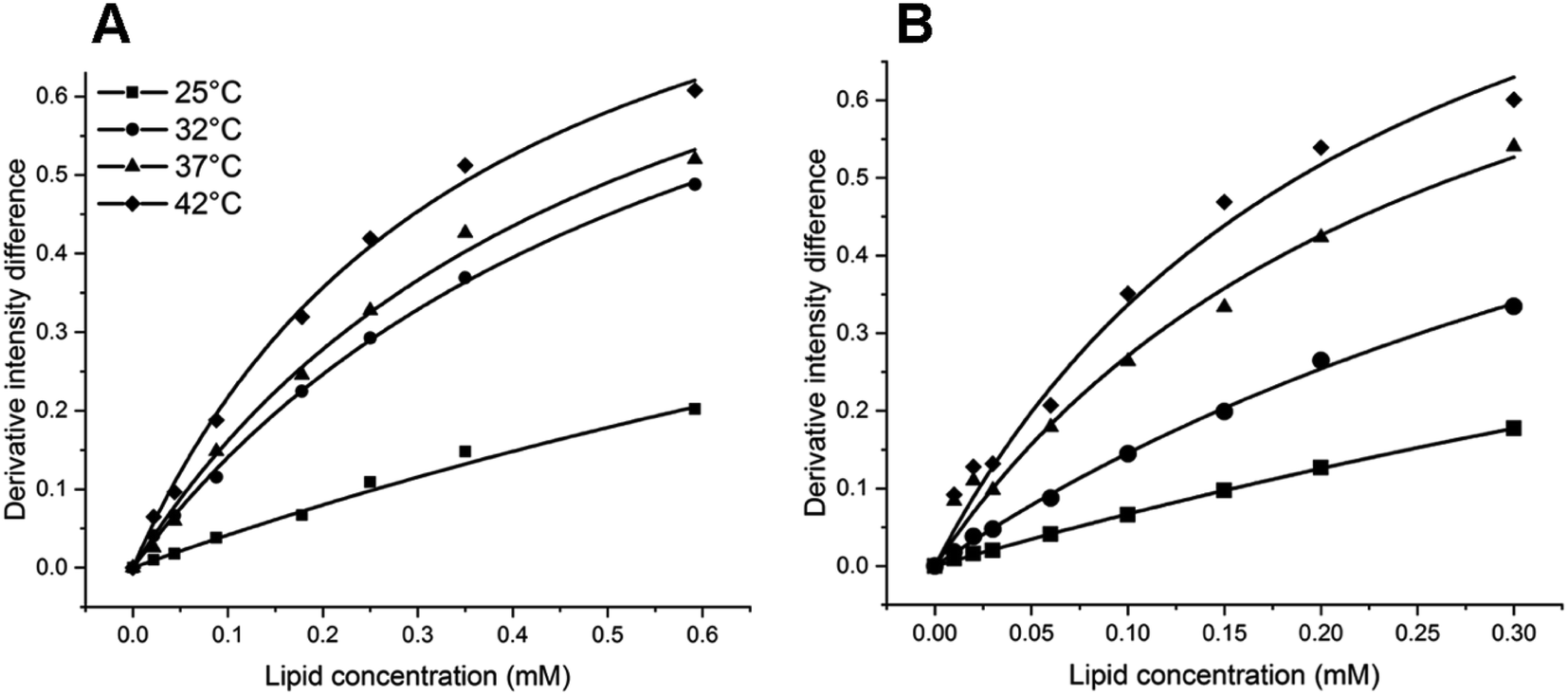 | ||
| Fig. 4 Derivative intensity difference value (ΔD/ΔDmax) of (A) PAX and (B) SER in DOPC LUVs as a function of lipid concentration (mM) at 25 °C (filled squares), 32 °C (filled circles), 37 °C (filled triangles) and 42 °C (filled diamonds). The solid lines show the theoretical curves calculated from eqn (3) using the experimental values. | ||
Both PAX and SER partitioned into DOPC vesicles to a greater extent with temperature, as demonstrated by the increase in their Kp values. When the temperature increased from 25 to 32 °C, the Kp values of PAX and SER increased 2.7-fold and 2.3-fold, respectively. The increases in the Kp values of PAX and SER were 1.2-fold and 2.0-fold from 32 to 37 °C, and 1.4-fold and 1.2-fold from 37 to 42 °C, respectively. Furthermore, the Kp values of SER were consistently higher than those of PAX across the temperature range examined.
3.3 Thermodynamics of SSRIs' partitioning into DOPC LUVs
The Van't Hoff plots for PAX and SER partitioning into DOPC LUVs were shown in Fig. 5 and the calculated thermodynamics parameters were listed in Table 2. The negative values of the free energy change for PAX and SER (ΔGw→l) indicated that the transfer process of the drugs from the aqueous phase into DOPC LUVs was energetically favorable. The Van't Hoff plots for both PAX and SER exhibited negative slopes, i.e., positive enthalpy changes (ΔHw→l), revealing that the partitioning of these drugs into the lipid bilayers was an endothermic process. Additionally, both ΔHw→l and ΔSw→l were positive, indicating that entropy mainly drives the incorporation of the drugs into the lipid bilayers.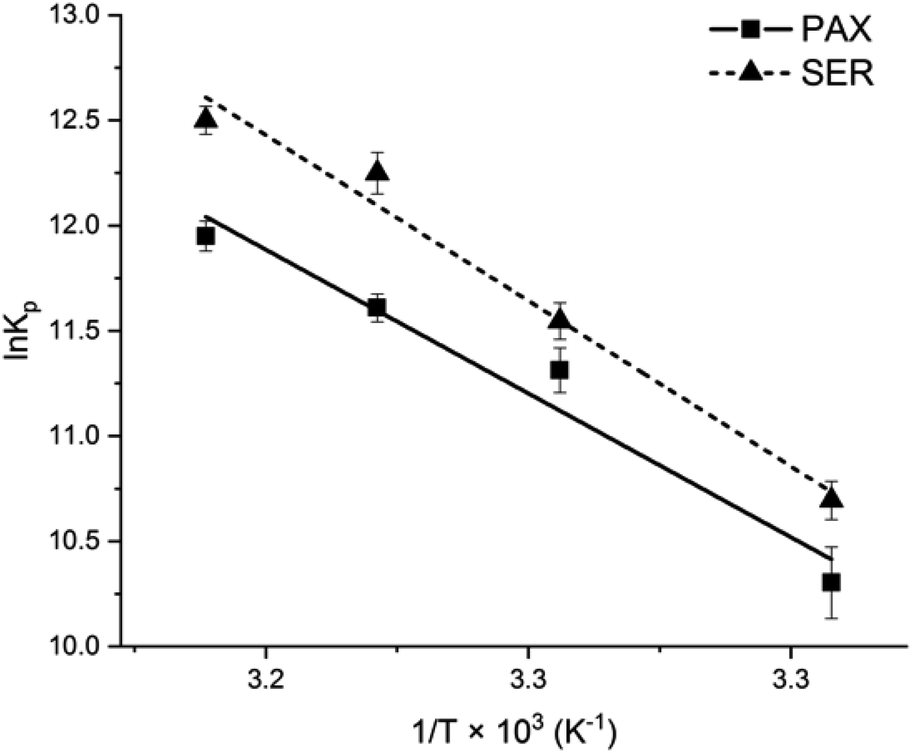 | ||
| Fig. 5 Van't Hoff plot of PAX (filled squares) and SER (filled triangles) partitioning into DOPC LUVs. | ||
3.4 Structural changes of DOPC induced by SSRIs partitioning
The structural changes of DOPC LUVs upon the SSRIs partitioning were examined by FTIR at 25 °C to provide a better comprehension of the drugs–lipid membrane interaction. A mathematical addition of the pure DOPC spectrum to the spectrum of either PAX or SER was conducted to validate the perturbation of the SSRIs on DOPC bilayers. The resultant addition spectra were different from the drug–lipid mixture spectra in terms of shape and vibrational frequency assignments, confirming the interaction of SSRIs–lipid bilayers (see the ESI†).The normalized FTIR spectra of DOPC LUVs with and without PAX or SER in the CH2, CO and PO2− regions were shown in Fig. 6. The precision of the IR instrument employed in this work is better than 0.01 cm−1 at a resolution of 2 cm−1; thus, a frequency shift of 1 cm−1 for the stretching vibrational band would be significant.16,49,50
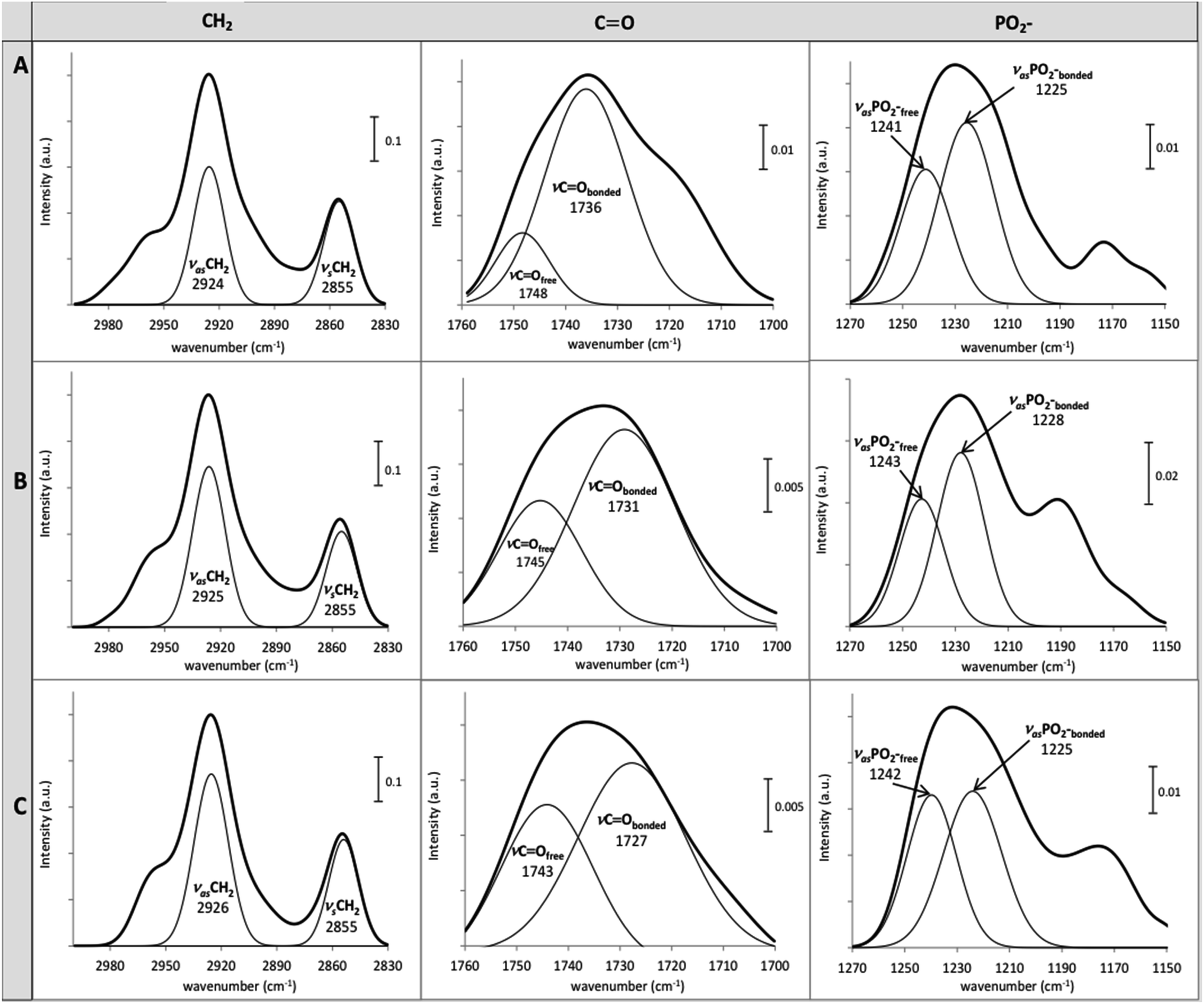 | ||
| Fig. 6 Normalized FTIR spectra of (A) pure DOPC LUVs and those with (B) PAX, (C) SER in HEPES buffer (pH 7.4 and 25 °C), in the regions of CH2, CO and PO2−. The displayed frequencies were obtained from the deconvolution of the spectra. | ||
The CH2 symmetric (νsCH2) and asymmetric stretch (νasCH2) frequencies, located around 2850 cm−1 and 2920 cm−1, respectively, are related to changes of the trans/gauche isomerization of the lipid acyl chain.51 The νsCH2 and νasCH2 of DOPC LUVs, were resided at 2855 cm−1 and 2924 cm−1, respectively (Fig. 6). The partitioning of PAX and SER induced an increment of gauche conformers in the lipid acyl chain indicated by the upward shift of 1 and 2 cm−1 of the νasCH2, respectively. This reflects the disordered motion of DOPC's acyl chains caused by the presence of the drugs.
The partitioning of the SSRIs into DOPC LUVs also substantially perturbed the interfacial regions of DOPC LUVs as revealed by changes in the location of the ester carbonyl νCO stretching modes which appeared near 1730 cm−1 and 1740 cm−1 for the hydrogen bonded CO (νCObonded) and free CO (νCOfree), respectively.52 Both PAX and SER provoked considerable downward shifts of the νCO bands of DOPC LUVs and the shift caused by SER was more pronounced than that by PAX. Particularly, SER induced a downshift of 9 cm−1 and 5 cm−1 while PAX induced a downshift of 5 cm−1 and 3 cm−1, in the νCObonded and νCOfree, respectively (see Fig. 6). Such decreases in the νCO frequencies of DOPC LUVs in the presence of the drugs indicated the increase in hydrogen bonding between the drug molecules and the ester carbonyl group of the lipid.
Conformational changes in DOPC LUVs upon the partitioning of the SSRIs can be further elucidated with regard to the lipid head group's hydration state. The hydration state of the head group can be reflected by the spectral characteristics of asymmetric νasPO2− stretching (1220–1270 cm−1).34 The νasPO2− bands of DOPC LUVs were deconvoluted into fully hydrated (νasPO2−bonded) and free PO2− (νasPO2−free) at 1225 cm−1 and 1241 cm−1, respectively. The frequency of the lipid νasPO2−bonded remained unchanged in the presence of SER but increased by 3 cm−1 upon the partitioning of PAX. The νasPO2free was upshifted by 2 cm−1 and 1 cm−1 with the presence of PAX and SER, respectively. All these upward shifts signified an increase in the dehydration of the lipid head group upon the drug partitioning. In particular, the dehydration effect caused by PAX was more pronounced than that by SER, denoting this drug could replace more water molecules from the lipid head group.
4. Discussion
In this study, the thermodynamics of membrane partitioning of the two common SSRIs, namely PAX and SER, was characterized to provide a better understanding of SSRIs–lipid membrane interaction. The Kp values of PAX and SER into DOPC LUVs were measured at varying temperatures by second derivative spectrophotometry and the conformational changes in the lipid vesicles induced by the drug incorporation were also determined using ATR-FTIR.As the temperature increased from 25 to 42 °C, DOPC became more fluid, facilitating the partitioning of PAX and SER into the lipid bilayer, which lead to an increase in the Kp values for both drugs. Additionally, the partitioning of PAX and SER into the lipid bilayer was a spontaneous process as indicated by the negative ΔG. The increase in temperature raised the kinetic energy of both the drug and the lipid molecules and promoted the rate of which the collision between them occurs. An increased chance of collision between the drug molecules and the lipid bilayer could largely promote the interaction and thus contributed to the greater partitioning. The increase in Kp according to the rise in temperature is in agreement with other works in which fluoxetine, another SSRI, was found to partition to a greater extent into DOPC LUVs as temperature increased from 25 to 37 °C16 while haloperidol, a dopamine antagonist, was reported to incorporate more strongly into DOPC vesicles as temperature increased from 10 to 40 °C.53
SER was found to partition more strongly into DOPC LUVs as compared to PAX at all temperatures studied. This could be related to the more lipophilic property of SER than that of PAX, indicated by its higher logP value (logP = 4.30 (ref. 54)) compared to the latter (logP = 3.9555). Besides, the lower polarity of SER could further contribute to its partitioning behavior. The polarity of a small molecule can be represented by its topological polar surface area (TPSA), which is the surface sum of all polar atoms. Molecules with low polarity tend to have a small TPSA and thus partitions more robustly into the membrane, while those with a large TPSA are likely to have surface interaction with the lipid's head group.56 Additionally, those with a TPSA of greater than 140 Å2 could be poor at permeating cell membranes, while those with a TPSA of less than 90 Å2 can effectively penetrate the blood–brain barrier.57,58 Both SER and PAX could freely diffuse into and interact with the lipid membranes as they have a TPSA of 12.0 Å259 and 39.7 Å2,60 respectively. Since the TPSA of SER is much smaller than that of PAX, SER penetrated into DOPC LUVs to a greater extent while PAX interacted more strongly with the lipid head group at the water–bilayer interface, as affirmed by the frequency shifts in the CH2, ester carbonyl and phosphate regions of DOPC LUVs in the presence of the drugs.
The partitioning process of PAX and SER into DOPC LUVs followed the classical hydrophobic effect, i.e., entropy-driven with positive ΔHw→l and ΔSw→l.61–63 The net value of ΔHw→l has its origin from two sources: the energy required to form the cavities within the lipid bilayer and the energy released during the drug–lipid interaction.7,64 The contribution to ΔHw→l due to the cavities formed within the lipid bilayer was positive as energy is expended to disrupt the lipid packing of the bilayer. On the other hand, the negative contribution to ΔHw→l originated from the exothermic process of drugs–lipid interactions such as the formation of hydrogen bonds between the drugs and the ester carbonyl group of the lipid (as revealed by the downward shift of the νCO frequencies upon the drug partitioning), and the hydrophobic/van der Waals interactions between the nonpolar moieties of the drugs and the nonpolar acyl chains of DOPC. Since SER is more lipophilic (i.e., having a higher value of logP) and possibly formed more hydrogen bonds with the carbonyl region of DOPC LUVs (i.e., having a larger shift in the lipid's νCO frequencies), it is expected that the enthalpy loss (ΔHw→l < 0) due to its interaction with DOPC bilayer would be larger than that of PAX, resulting a smaller value of the overall net ΔHw→l. However, the net ΔHw→l value (84.83 ± 1.20 kJ mol−1) of the partitioning of SER is higher than that of PAX (74.74 ± 1.26 kJ mol−1), revealing that the enthalpy increase (ΔHw→l > 0) by disruption of the lipid packing to accommodate the drugs predominates and largely contributes to the overall ΔHw→l. Furthermore, the thermodynamic parameters showed that the transfer processes of PAX and SER from the aqueous phase into DOPC LUVs were associated with positive values of entropy change (ΔSw→l > 0), which could be attributed to the disordering of the lipid acyl chains as observed by the upward shift in the νasCH2 of DOPC LUVs by 1 and 2 cm−1 in the presence of PAX and SER, respectively. The disordering effect on DOPC's acyl chains induced by SER was more pronounced as shown by the larger νasCH2 shift, suggesting that SER could penetrate more deeply into the lipid bilayer. The disordering effect of another SSRI, fluoxetine, on the lipid acyl chains was also reported previously.16,17,19 The increase in the ΔSw→l could also be related to the displacement of water molecules surrounding the lipid's interfacial region as the SSRIs transfer from the aqueous phase to the lipid bilayer. This explanation was affirmed by the downward shift of the νasPO2−free of DOPC LUVs, which signifies the increase in dehydration state of the lipid head group upon the drugs' incorporation. Altogether, it could be inferred that SER penetrated more deeply into the lipid's acyl chains and disrupted the ordering of the lipid to a greater extent while PAX interacted more strongly with the lipid head group and accumulated in the interfacial region.
Overall, the partitioning of PAX and SER into liquid-crystalline DOPC LUVs was a spontaneous, endothermic process driven mainly by entropy, as shown by negative ΔGw→l, positive ΔHw→l and ΔSw→l. Such partitioning behavior can be attributed to the classical hydrophobic effect.61–63 The partitioning of amphiphilic molecules into lipid bilayer can be ascribed to either of two main effects: classical or nonclassical hydrophobic effect.62,63 To distinguish between the classical and nonclassical hydrophobic effect, the free energy of transfer of the amphiphilic molecule to the bilayer is taken into account. The classical hydrophobic effect is dominated by a large positive entropy change (i.e., entropy-driven), while the nonclassical hydrophobic effect is dictated by a large negative enthalpy change (i.e., enthalpy-driven).61–63 The partitioning of several cationic amphiphilic drugs into fluid-phase lipid membranes was also found to follow the classical hydrophobic effect such as anti-malarials (mefloquine, 4-anilinoquinoline derivatives),65,66 beta-blockers (propranolol, alprenolol, bupranolol),67 antibiotic (azithromycin)68 and antipsychotics (promethazine, trifluoperazine, trimeprazine).69
The implement of ATR-FTIR in complementary with second derivative spectrophotometry is effective in the determination of the molecular interaction a drug may have with a model lipid bilayer. Although SER partitioned into DOPC bilayer to a greater extent than PAX as indicated by the Kp values, the influence of PAX on the phosphate bands of the lipid head group was larger than that of SER. Particularly, the νasPO2−free was upshifted by 2 cm−1 in the presence of PAX while this shift was 1 cm−1 with SER. Furthermore, the νasPO2−bonded remained unchanged in the presence of SER but increased by 3 cm−1 upon the partitioning of PAX. Therefore, if we based our conclusion purely from the Kp values, we would have inaccurately deduced that SER largely affects all three regions of the lipid (CH2, CO, PO2−) and missed out important information that in fact, SER strongly affects the CH2 and CO regions while PAX strongly affects the PO2− region. Electrostatic interaction may be one of the forces that contributed to the partitioning of these SSRIs. Initially, the unbound drug in the aqueous phase could randomly collide with the lipid bilayer. When this happens, the amine moiety of either drug could be protonated and electrostatically interacted with the PO2− region of the lipid head group. As the drug moved toward the hydrophobic core of the bilayer, hydrophobic interaction between the piperidine ring of PAX or the tametraline group of SER with the acyl chain took over. Additionally, the presence of the polarizable atoms in the structure of the two SSRIs also contributed to the different degree of their partitioning. PAX has one fluorine atom and two oxygen atoms while SER has two chlorine atoms. The discrepancy in polarizable atoms results in PAX being much more polar than SER. Such a difference was reflected by the three-times larger TPSA of PAX as compared to that of SER. Consequently, the much smaller polar surface area of SER allowed it to permeate the lipid bilayer to a greater extent. All things considered, due to the dissimilarities in the chemical structure of the two SSRIs, their possible binding sites within the lipid bilayer were also different such that PAX could be situated mainly on the headgroup of the bilayer while SER could reside primarily near the hydrophobic core.
5. Conclusions
We have investigated the interaction of PAX and SER with DOPC LUVs from the thermodynamics aspect by second derivative spectrophotometry and FTIR. The partition coefficients of PAX and SER into DOPC LUVs were determined at four different temperatures: 25, 32, 37 and 42 °C. The IR spectra of DOPC LUVs with and without the drugs were also collected. It was found that temperature increase from 25 to 42 °C facilitates the partitioning of PAX and SER into DOPC LUVs, and SER partitioned more strongly into the lipid bilayer than PAX did. The thermodynamics profiles of PAX and SER indicated that their partitioning processes into DOPC LUVs are energetically favorable, endothermic and follow the classical hydrophobic effect (i.e., entropy-driven). Despite having the same thermodynamics profiles, PAX and SER differ in their influence on the lipid conformation upon their partitioning, and notably, their possible binding sites within the lipid bilayer. In particular, PAX possibly bound to the phosphate group of the lipid bilayer while SER could bind to the carbonyl moiety of the lipid bilayer. The differences in the degree of lipid membrane partitioning and possible binding sites could also be related to the heterogeneity in their polarizable atoms, which ultimately could affect the pharmacokinetic and pharmacologic properties of PAX and SER.From these findings, it might be inferred that the partitioning of other antidepressant drugs in the SSRI class into fluid lipid bilayer could also conform to the classical hydrophobic effect. Therefore, any modification in the chemical structure of the SSRIs that modulates their hydrophobicity could lead to significant variations in the degree of membrane partitioning. Additionally, alteration of the SSRIs' functional groups, most notably the polarizable atoms such as the halogen moiety, could largely influence their binding sites within the lipid bilayer. This information could be beneficial to rational drug design, optimization and drug delivery system.
Abbreviations
| DOPC | 1,2-Dioleoyl-sn-glycero-3-phosphocholine |
| DPPC | 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine |
| DMPC | 1,2-Dimyristoyl-sn-glycero-3-phosphocholine |
| LUV | Large unilamellar vesicle |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazine-ethanesulfonic acid |
| SSRI | Selective serotonin reuptake inhibitor |
| PAX | Paroxetine |
| SER | Sertraline |
| ATR-FTIR | Attenuated total reflectance-Fourier-transform infrared spectroscopy |
| Tm | Melting temperature |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper is dedicated to the memory of Trinh Q. Nguyen who passed away on February 21st, 2020. The authors thank Uyen P. N. Dao and Vy Y. T. Le for their technical assistance. This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 108.99-2018.325.References
- J. P. Overington, B. Al-Lazikani and A. L. Hopkins, Nat. Rev. Drug Discovery, 2006, 5, 993–996 CrossRef CAS.
- C. Matos, J. L. C. Lima, S. Reis, A. Lopes and M. Bastos, Biophys. J., 2004, 86, 946–954 CrossRef CAS.
- N. C. Garbett and J. B. Chaires, Expert Opin. Drug Discovery, 2012, 7, 299–314 CrossRef CAS.
- G. A. Holdgate, Expert Opin. Drug Discovery, 2007, 2, 1103–1114 CrossRef CAS.
- A. D. Fearon and G. Y. Stokes, J. Phys. Chem. B, 2017, 121, 10508–10518 CrossRef CAS.
- E. S. Rowe, F. Zhang, T. W. Leung, J. S. Parr and P. T. Guy, Biochemistry, 1998, 37, 2430–2440 CrossRef CAS.
- H. R. Lozano and F. Martínez, Braz. J. Pharm. Sci., 2006, 42, 601–613 CAS.
- S. Takegami, K. Kitamura, T. Kitade, A. Kitagawa and K. Kawamura, Chem. Pharm. Bull., 2003, 51, 1056–1059 CrossRef CAS.
- M. Arrowsmith, J. Hadgraft and I. W. Kellaway, Biochim. Biophys. Acta, Lipids Lipid Metab., 1983, 750, 149–156 CrossRef CAS.
- B. G. Akinoglu, M. Gheith and F. Severcan, J. Mol. Struct., 2001, 565, 281–285 CrossRef.
- F. R. Walker, Neuropharmacology, 2013, 67, 304–317 CrossRef CAS.
- P. Benfield, R. C. Heel and S. P. Lewis, Drugs, 1986, 32, 481–508 CrossRef CAS.
- J. Mazella, O. Pétrault, G. Lucas, E. Deval, S. Béraud-Dufour, C. Gandin, M. El-Yacoubi, C. Widmann, A. Guyon and E. Chevet, PLoS Biol., 2010, 8, 819 CrossRef.
- J. Chen, D. Korostyshevsky, S. Lee and E. O. Perlstein, PLoS One, 2012, 7, 1–13 Search PubMed.
- S. J. Erb, J. M. Schappi and M. M. Rasenick, J. Biol. Chem., 2016, 291, 19725–19733 CrossRef CAS.
- V. T. Pham, T. Q. Nguyen, U. P. Dao and T. T. Nguyen, Spectrochim. Acta, Part A, 2018, 191, 50–61 CrossRef CAS.
- T. T. T. Do, U. P. N. Dao, H. T. Bui and T. T. Nguyen, Chem. Phys. Lipids, 2017, 207(Part A), 10–23 CrossRef CAS.
- R. Kapoor, T. A. Peyear, R. E. Koeppe and O. S. Andersen, J. Gen. Physiol., 2019, 151, 342–356 CrossRef CAS.
- F. Momo, S. Fabris and R. Stevanato, Biophys. Chem., 2005, 118, 15–21 CrossRef CAS.
- R. M. Hirschfeld, J. Clin. Psychiatry, 1999, 60, 326–335 CrossRef CAS.
- C. Sanchez, E. H. Reines and S. A. Montgomery, Int. Clin. Psychopharmacol., 2014, 29, 185–196 CrossRef.
- M. M. Rainey, D. Korostyshevsky, S. Lee and E. O. Perlstein, Genetics, 2010, 185, 1221–1233 CrossRef CAS.
- P. A. Marken and J. S. Munro, Prim. Care Companion J. Clin. Psychiatry, 2000, 02, 205–210 CrossRef.
- A. M. Koenig and M. E. Thase, Pol. Arch. Intern. Med., 2009, 119, 478–486 CAS.
- P. C. A. Kam and G. W. M. Chang, Anaesthesia, 2004, 52, 982–988 CrossRef.
- H. G. Nurnberg, P. M. Thompson and P. L. Hensley, J. Clin. Psychiatry, 1999, 60, 574–579 CrossRef CAS.
- A. S. Ulrich, M. Sami and A. Watts, Biochim. Biophys. Acta, Biomembr., 1994, 1191, 225–230 CrossRef CAS.
- A. S. Klymchenko and R. Kreder, Chem. Biol., 2014, 21, 97–113 CrossRef CAS.
- J. B. Larsen, C. Kennard, S. L. Pedersen, K. J. Jensen, M. J. Uline, N. S. Hatzakis and D. Stamou, Biophys. J., 2017, 113, 1269–1279 CrossRef CAS.
- A. S. Reddy, D. T. Warshaviak and M. Chachisvilis, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 2271–2281 CrossRef CAS.
- C. Peetla, A. Stine and V. Labhasetwar, Mol. Pharm., 2009, 6, 1264–1276 CrossRef CAS.
- K. Kitamura, N. Imayoshi, T. Goto, H. Shiro, T. Mano and Y. Nakai, Anal. Chim. Acta, 1995, 304, 101–106 CrossRef CAS.
- K. Kitamura, T. Goto and T. Kitade, Talanta, 1998, 46, 1433–1438 CrossRef CAS.
- D. C. Lee and D. Chapman, Biosci. Rep., 1986, 6, 235–256 CrossRef CAS.
- J. M. Boggs, Biochim. Biophys. Acta, Rev. Biomembr., 1987, 906, 353–404 CrossRef CAS.
- S. Takegami, K. Kitamura, T. Funakoshi and T. Kitade, Chem. Pharm. Bull., 2008, 56, 663–667 CrossRef CAS.
- A. A. Omran, K. Kitamura, S. Takegami, T. Kitade, A.-A. Y. El-Sayed, M. H. Mohamed and M. Abdel-Mottaleb, Chem. Pharm. Bull., 2002, 50, 312–315 CrossRef CAS.
- A. L. Stancik and E. B. Brauns, Vib. Spectrosc., 2008, 47, 66–69 CrossRef CAS.
- M. Luxnat and H.-J. Galla, Biochim. Biophys. Acta, Biomembr., 1986, 856, 274–282 CrossRef CAS.
- C. Rodrigues, P. Gameiro, S. Reis, J. L. F. C. Lima and B. de Castro, Anal. Chim. Acta, 2001, 428, 103–109 CrossRef CAS.
- G. D. Eytan, R. Regev, G. Oren and Y. G. Assaraf, J. Biol. Chem., 1996, 271, 12897–12902 CrossRef CAS.
- H. S. Schwartz and P. M. Kanter, Eur. J. Cancer, 1965, 15, 923–928 CrossRef.
- W. Werner, J. Baumgart, G. Burckhardt, W. F. Fleck, K. Geller, W. Gutsche, H. Hanschmann, A. Messerschmidt, W. Römer, D. Tresselt and G. Löber, Biophys. Chem., 1990, 35, 271–285 CrossRef CAS.
- R. Welti, L. J. Mullikin, T. Yoshimura and G. M. Helmkamp, Biochemistry, 1984, 23, 6086–6091 CrossRef CAS.
- A. Pola, K. Michalak, A. Burliga, N. Motohashi and M. Kawase, Eur. J. Pharm. Sci., 2004, 21, 421–427 CrossRef CAS.
- M. Luxnat, H. J. Müller and H. J. Galla, Biochem. J., 1984, 224, 1023 CrossRef CAS.
- A. A. Omran, K. Kitamura, S. Takegami, A.-A. Y. El-Sayed and M. Abdel-Mottaleb, J. Pharm. Biomed. Anal., 2001, 25, 319–324 CrossRef CAS.
- K. A. Connors, in Binding Constants: The Measurement of Molecular Complex Stability, Wiley-Interscience, 1987, pp. 141–147 Search PubMed.
- W. Cong, Q. Liu, Q. Liang, Y. Wang and G. Luo, Biophys. Chem., 2009, 143, 154–160 CrossRef CAS.
- K. Cieslik-Boczula, J. Szwed, A. Jaszczyszyn, K. Gasiorowski and A. Koll, J. Phys. Chem. B, 2009, 113, 15495–15502 CrossRef CAS.
- K. Cieślik-Boczula and A. Koll, Biophys. Chem., 2009, 140, 51–56 CrossRef.
- A. Blume, W. Hübner and G. Messner, Biochemistry, 1988, 27, 8239–8249 CrossRef CAS.
- A. B. Sarmento, M. C. P. Lima and C. R. Olivejra, J. Pharm. Pharmacol., 1993, 45, 601–605 CrossRef CAS.
- K. Tihanyi, B. Noszal, K. Takacs-Novak and K. Deak, Med. Chem., 2006, 2, 385–389 CrossRef.
- N. Agrawal, Open Anal. Chem. J., 2013, 7, 1–5 CrossRef CAS.
- G. M. M. El Maghraby, A. C. Williams and B. W. Barry, Int. J. Pharm., 2005, 292, 179–185 CrossRef CAS.
- H. Pajouhesh and G. R. Lenz, Neurotherapeutics, 2005, 2, 541–553 CrossRef.
- S. A. Hitchcock and L. D. Pennington, J. Med. Chem., 2006, 49, 7559–7583 CrossRef CAS.
- I. Bytheway, M. G. Darley and P. L. A. Popelier, ChemMedChem, 2008, 3, 445–453 CrossRef CAS.
- R. Longhi, S. Corbioli, S. Fontana, F. Vinco, S. Braggio, L. Helmdach, J. Schiller and H. Boriss, Drug Metab. Dispos., 2011, 39, 312–321 CrossRef CAS.
- M. Fernández-Vidal, S. H. White and A. S. Ladokhin, J. Membr. Biol., 2010, 239, 5–14 CrossRef.
- J. Seelig and P. Ganz, Biochemistry, 2002, 30, 9354–9359 CrossRef.
- W. C. Wimley and S. H. White, Biochemistry, 2002, 32, 6307–6312 CrossRef.
- C. P. Mora and F. Martínez, J. Chem. Eng. Data, 2007, 52, 1933–1940 CrossRef CAS.
- M.-L. Go and T.-L. Ngiam, Chem. Pharm. Bull., 1997, 45, 2055–2060 CrossRef CAS.
- M.-L. Go, T.-L. Ngiam and J. A. Rogers, Chem. Pharm. Bull., 1995, 43, 289–294 CrossRef CAS.
- G. V. Betageri and J. A. Rogers, Int. J. Pharm., 1987, 36, 165–173 CrossRef CAS.
- N. Fa, S. Ronkart, A. Schanck, M. Deleu, A. Gaigneaux, E. Goormaghtigh and M. P. Mingeot-Leclercq, Chem. Phys. Lipids, 2006, 144, 108–116 CrossRef CAS.
- A. M. S. Ahmed, F. H. Farah and I. W. Kellaway, Pharm. Res., 1985, 02, 119–124 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Figures show the mathematical addition of the pure DOPC spectrum to the spectrum of either PAX or SER. See DOI: 10.1039/d0ra07367a |
| ‡ Deceased. |
| This journal is © The Royal Society of Chemistry 2020 |