
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of tough, anisotropic, chemical-crosslinker-free poly(vinyl alcohol) nanofibrous cryogels via electrospinning†
Yoshiyasu Nagakawaab,
Mikiya Katob,
Shin-ichiro Suyebc and
Satoshi Fujita *bc
*bc
aBiotechnology Group, Tokyo Metropolitan Industrial Technology Research Institute, 2-4-10, Aomi, Koto-ku, Tokyo 135-0064, Japan
bDepartment of Frontier Fiber Technology and Sciences, Graduate School of Engineering University of Fukui, 3-9-1, Bunkyo, Fukui, 910-8507, Japan. E-mail: fujitas@u-fukui.ac.jp
cLife Science Innovation Center, University of Fukui, 3-9-1, Bunkyo, Fukui, 910-8507, Japan
First published on 15th October 2020
Abstract
PVA hydrogels with anisotropic structures have many biomedical applications; however, the hydrophilicity of PVA nanofibers degrades their mechanical properties, and the residual unreacted chemical crosslinkers are disadvantageous for medical use. Therefore, maintaining the stability of aqueous solutions without using crosslinkers is essential while synthesizing electrospun anisotropic PVA nanofibers. Herein, we developed a novel fabrication method for synthesizing tough, anisotropic, and chemical-crosslinker-free nanofibrous cryogels composed of poly(vinyl alcohol) (PVA) and glycerol (Gly) via electrospinning in conjunction with freeze–thawing treatment. Wide-angle X-ray diffraction, attenuated total reflection Fourier-transform infrared spectroscopy, and differential scanning calorimetry analysis revealed an enhanced crystallinity of the PVA and hydrogen bonds in the PVA/Gly nanofibers after freeze–thawing, thereby leading to improved stability of the PVA/Gly nanofiber in water. The scanning electron microscopy observation and tensile tests revealed that the addition of Gly improved both the orientation and the mechanical properties. The values of the toughness parallel and vertical to the fiber axis direction were 4.20 ± 0.63 MPa and 2.17 ± 0.27 MPa, respectively, thus revealing the anisotropy of this mechanical property. The PVA/Gly nanofibrous cryogel consisted of physically crosslinked biocompatible materials featuring toughness and mechanical anisotropy, which are favorable for medical applications including tissue engineering.
Introduction
Hydrogels are three-dimensional, hydrophilic, polymeric networks capable of retaining large amounts of aqueous solutions; they are utilized in various industries such as agriculture and food chemistry,1 hygiene products,2–4 cosmetics,3 and biomedical applications due to their excellent biocompatibility, which is based on their high water absorption and retention.4,5 Hydrogel structures exhibiting anisotropic properties are essential for biomedical applications.Extracellular matrices (ECMs) contain hydrogels composed of biopolymers with anisotropic structures that endow the ECMs with high strength and anisotropic mechanical properties.6–8 Anisotropic structures impact cell behavior and function.9–13 The method for controlling cellular microenvironments that mimic three-dimensional natural tissues and organs is extremely favorable for use as a cell scaffold in regenerative medicine and tissue engineering.14–16 Furthermore, anisotropic hydrogels are also desirable for medical devices, e.g., wound dressing,17 vascular applications,18,19 self-inflating tissue expanders,20 and self-expandable biliary stents.21 However, the hydrogels fabricated usually have isotropic structures, which would negatively impact their applicability to these devices. Therefore, a fabrication method for constructing anisotropic hydrogels with excellent biocompatibility is highly required.
Poly(vinyl alcohol) (PVA), due to its biocompatibility, has numerous biomedical applications, including wound dressing,22,23 contact lenses,24 and implants for various tissues and organs such as cartilage,25 vascular access,26 stent for biliary drainage,21 ligament,27 and tissue engineering applications.28 The fabrication of PVA hydrogels with anisotropic structures using electrospinning has been investigated due to its small fiber diameter, high surface-to-volume ratio, and controllable porous structure and shape. However, the hydrophilicity of PVA nanofibers destabilizes the aqueous solution and subsequently decreases their mechanical properties, thereby limiting their suitability for biomedical applications.29 Chemical crosslinking methods form permanent, irreversible covalent bonds between the polymer chains in the PVA nanofibers, thus improving their mechanical properties and increasing their stability in aqueous solutions.30 However, the leach of residual unreacted chemical crosslinkers has a distinct undesirable effect for use in biomedical applications.31,32 Therefore, fabrication methods for synthesizing electrospun anisotropic PVA nanofibers while maintaining the stability of aqueous solutions without the use of crosslinkers are urgently required.
PVA can be physically crosslinked via cryogelation methods such as the freeze–thawing (FT).33 This gelation method addresses the toxicity because it does not require a crosslinker and is thus appropriate for biomedical applications.31,32 PVA polymer networks form inter- and intramolecular hydrogen bonds as well as crystallites during the freezing process, where PVA chains are aggregated by ice crystal formation.32 However, when PVA nanofibers are fabricated via electrospinning, insufficient physical crosslink formation in the PVA polymer networks may occur due to the low water content in the electrospun fibers. The incorporation of an additive agent is considered an effective method for reinforcing electrospun PVA nanofibers, thus promoting crystallite formation. The incorporation of cellulose nanocrystals (CNCs) reinforces the hydrogen bonding formation between PVA and CNCs.34 Recently, Shi et al. reported that glycerol (Gly) promotes hydrogen bonding with PVA chains, thus resulting in gelation while maintaining the water stability and high mechanical property.35 Accordingly, we hypothesized that the fabrication of electrospun PVA nanofibers in conjunction with Gly would promote crystal formation with hydrogen bonds between the Gly and PVA chains, thereby facilitating the fabrication of nanofibers without chemical crosslinkers.
Herein, we investigated the fabrication condition of PVA/Gly electrospun hydrogel nanofibers and its characterization. PVA/Gly hydrogel nanofibers can be fabricated via the electrospinning method by mixing PVA and Gly using water as a solvent. This method is advantageous in that it enables the PVA/Gly hydrogel nanofibers to maintain good stability in water and mechanical properties superior to those of PVA nanofibers without Gly. Wide-angle X-ray diffraction (WAXD), attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR), and differential scanning calorimetry (DSC) indicated that reinforcement of hydrogen bonding and the crystallinity between PVA and Gly contributed to the stability in water. The fabrication of electrospun PVA/Gly is simple and enables formation without chemical crosslinkers, thus making it applicable for various biomedical applications.
Experimental
Fabrication of nanofibers
PVA (Mw 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–98000, saponification degree 99+%, Sigma-Aldrich, St. Louis, USA) and Gly (Nacalai Tesque, Kyoto, Japan) were purchased and used without further purification. The electrospinning solutions were prepared by dissolving PVA powder in distilled water under magnetic stirring at 100 °C for 3 h to obtain concentrations ranging from 4–15% (w/v). Subsequently, Gly was added to the solution at concentrations of 5, 10, and 15% (v/v). The samples were designated as x-PVA/y-Gly, where x and y are the concentrations of PVA and Gly in the solutions, respectively.
000–98000, saponification degree 99+%, Sigma-Aldrich, St. Louis, USA) and Gly (Nacalai Tesque, Kyoto, Japan) were purchased and used without further purification. The electrospinning solutions were prepared by dissolving PVA powder in distilled water under magnetic stirring at 100 °C for 3 h to obtain concentrations ranging from 4–15% (w/v). Subsequently, Gly was added to the solution at concentrations of 5, 10, and 15% (v/v). The samples were designated as x-PVA/y-Gly, where x and y are the concentrations of PVA and Gly in the solutions, respectively.
The fabrication process of the nanofibers is shown in Scheme 1. The PVA and PVA/Gly nanofibers were electrospun on aluminum foil using a commercial electrospinning setup (NANON, MECC, Fukuoka, Japan), which consisted of a syringe pump, a high-voltage power supply, and a rotating collector placed in a closed chamber. The conditions for the electrospinning process were as follows: an applied electric field of 2.5 kV cm−1, a flow rate of 0.5 mL s−1 for both solutions, a collector rotation speed of 2000 rpm (linear velocity of 13.2 m s−1), a distance of 10 cm between the needle and rotating collector to ensure that the fibers were aligned, and a spinning duration of 3 h. The samples were designated as PVA as-spun and PVA/Gly as-spun nanofibers. The PVA as-spun and PVA/Gly as-spun nanofibers were subjected to FT treatment involving three cycles of freezing (−30 °C for 24 h) and subsequent thawing (25 °C for 24 h in a desiccator), thereby producing PVA FT and PVA/Gly FT nanofibers.
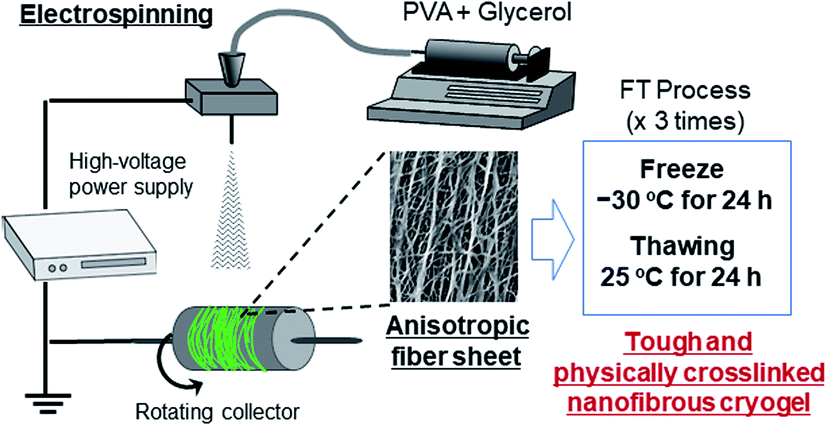 | ||
| Scheme 1 Schematic representation of the fabrication of PVA nanofibrous cryogels. | ||
Scanning electron microscopy (SEM)
The nanofiber samples were dehydrated by sequentially and gently immersing in serially diluted ethanol solutions (50, 60, 70, 80, 90, 95, and 100%) for 10 min and t-butyl alcohol three times for 10 min each. The samples were then lyophilized overnight using a freeze dryer (FDU-830, EYELA, Tokyo, Japan). The samples were then sputtered with Pt/Pd using an ion sputter (MSP-1S, Vacuum Device Inc., Ibaraki, Japan) for 120 s and imaged using SEM (S-2600HS, Hitachi, Ibaraki, Japan) at an accelerating voltage of 12 kV. The diameter of the nanofibers was examined at ten randomly selected areas of the scaffolds using an image processing software (http://fiji.sc; ImageJ, NIH, USA).Water stability test
The water stability of the nanofibers was evaluated by comparing the morphologies before and after immersion in water. The morphologies of the nanofibers collected in the basal plate were observed using SEM with JSM-6390MM (JEOL, Tokyo, Japan) at an acceleration of 15 kV. Subsequently, the samples were immersed in distilled water for 24 h. After immersion in water, the nanofibers were dried and their morphologies were reobserved under the same condition.Swelling tests
The water absorbability of the nanofibers was evaluated by immersing in distilled water for 24 h. The SR of the nanofibers was calculated as follows:| SR = Ws/Wd × 100 |
Wide-angle X-ray diffraction (WAXD) measurement
The WAXD measurements were performed on the nanofibers with an Ultima IV (Rigaku, Tokyo, Japan) using Ni-filtered Cu Kα rays at 40 kV and 20 mA, scanning at 1 deg per min in the 2θ range of 10–60° at a resolution of 0.05°. Three scans were accumulated.Differential scanning calorimetry (DSC) measurement
DSC was performed using DSC-60Plus (Shimadzu, Kyoto, Japan), using an aluminum pan as a reference. Specimens weighing 2–4 mg were sealed inside the non-hermetic aluminum pans and heated at a scanning rate of 10 °C min−1 from 30 °C to 300 °C under a flowing nitrogen atmosphere. The glass transition temperature (Tg), enthalpy relaxation, recrystallization temperature (Tc), and melting point (Tm) were recorded.Attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy
The ATR-FTIR spectra of the nanofibers were measured with a Nicolet 6700 system (Thermo Scientific, Waltham, USA). The measurements were performed for wavenumbers ranging from 4000–500 cm−1 at a resolution of 4 cm−1 using KBr detector with 32 scans.Analysis of fiber orientation
The orientation of nanofibers was quantified based on a second-order parameter, which was calculated from the power spectrum of the Fourier transform of the images using an image processing software (ImageJ; plugin: directionality). The second-order parameter in a 2D plane, S, was defined as follows:| S = 2(cos2θ) − 1 = (cos2θ) |
2θ) is the average value of cos2θ.36,37
Tensile tests
The mechanical property was investigated on the nanofiber sheet using KES-01-SH (Katotec, Kyoto, Japan) at a test speed of 0.001 mm s−1. The nanofibers were strained in the parallel or vertical direction of the nanofiber sheets. The elastic moduli of the nanofibers were calculated from the slope of the stress–strain curves in their linear regions. The tensile strengths and elongations at break were obtained from the elongation points of the samples. The toughness values of the nanofibers were calculated by integrating the stress in the strain ranging from the start to the elongation point.Results
Effect of Gly concentration on the fabrication condition for PVA/Gly nanofibers
The spinnability of 15% PVA solution with 5–15% Gly was investigated to optimize the concentration of Gly to be added. Numerous beads were observed in the nanofibers containing 5% Gly (Fig. 1a). In contrast, smooth PVA/Gly hydrogel nanofibers could be fabricated using 10% Gly (Fig. 1b). Conversely, it was difficult to form fibers at 15% Gly due to the increase in viscosity and the formation of droplets (Fig. 1c). The diameters of the PVA/Gly nanofibers increased as the Gly concentration increased, but the uniformity of the fiber diameter appeared to decrease at 5 or 15% Gly. Based on the SEM analyses, the average values and standard deviations of the fiber diameters for the 15-PVA/5-Gly, 15-PVA/10-Gly, and 15-PVA/15-Gly nanofibers were 743 ± 373 nm, 800 ± 255 nm, and 1144 ± 459 nm, respectively (Fig. 1d–f). Consequently, the remaining experiments were conducted on the 15-PVA/10-Gly nanofibers. For comparison, nanofibers were also fabricated using 15% PVA solution without the addition of Gly (hereinafter PVA fiber). The diameter of the PVA fiber was estimated to be 322 ± 60.9 nm.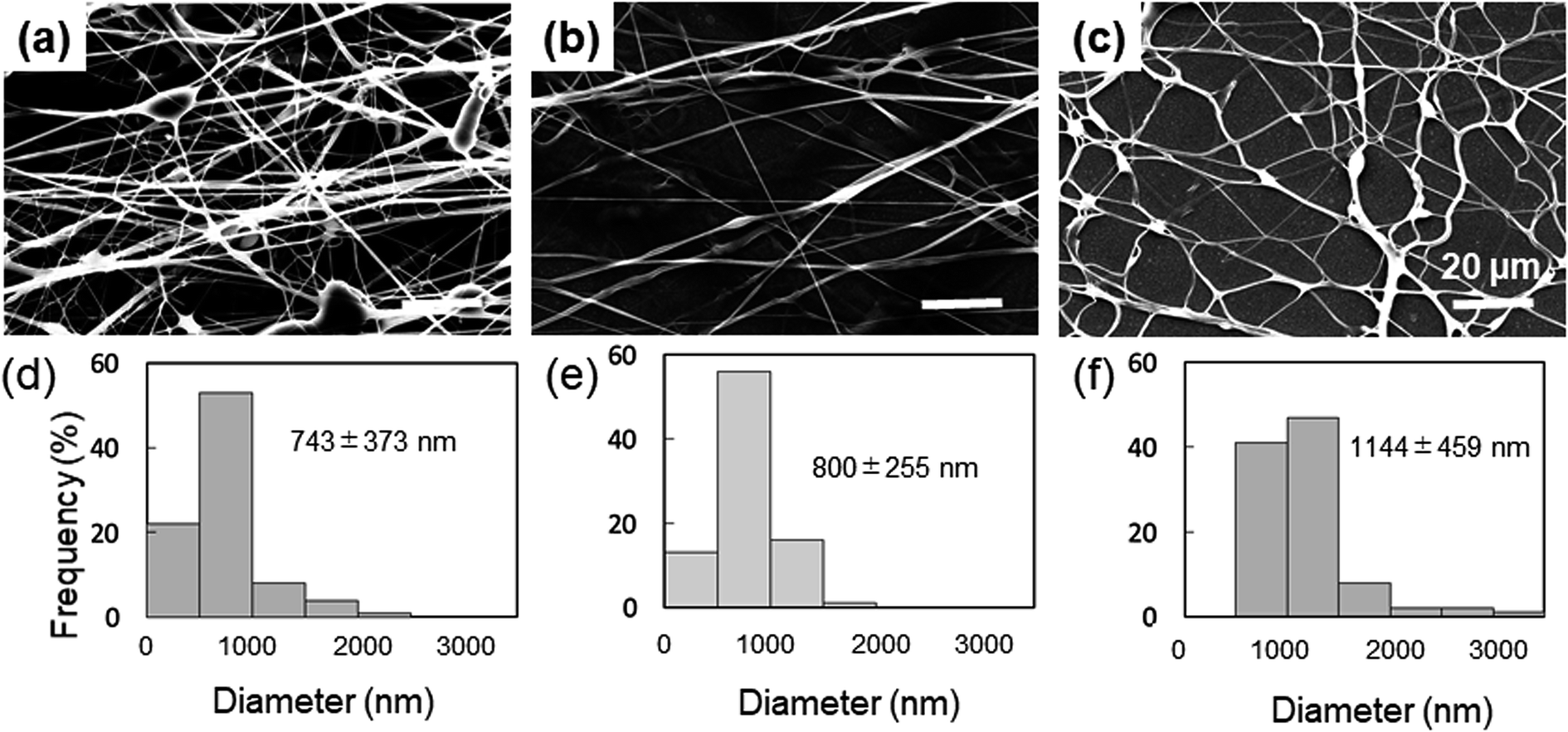 | ||
| Fig. 1 Morphologies of 15-PVA/5-Gly, 15-PVA/10-Gly, and 15-PVA/15-Gly nanofibers. SEM images (a–c) and distributions of the fiber diameter (d–f) of the 15-PVA/5-Gly, 15-PVA/10-Gly, and 15-PVA/15-Gly nanofibers, respectively. | ||
Morphology of the fabricated nanofibers
The electrospun nanofibers were processed with the FT treatment to promote crosslinking. The fibers were then hydrated by immersing into water for 24 h. The morphologies of the PVA and PVA/Gly nanofibers before and after hydration are shown in Fig. 2. The fiber structures prior to hydration were observed for various nanofibers before and after the FT treatment (Fig. 2a–d). Comparing the PVA and PVA/Gly as-spun nanofibers (Fig. 2a and c, respectively) with the corresponding PVA and PVA/Gly FT nanofibers (Fig. 2b and d, respectively) reveals that the FT treatment did not change their morphologies. On the other hand, after hydration the changes in the morphologies varied significantly depending on the nanofibers (Fig. 2e–h). The PVA as-spun nanofiber almost dissolved after swelling in water, resulting in the formation of a film-like structure (Fig. 2e). The PVA FT nanofiber was also dissolved, but a part of the fibrous morphology remained (Fig. 2f). On the other hand, the network structure of the PVA/Gly as-spun nanofibers was maintained, but the nanofibers adhered to each other after swelling (Fig. 2g). In contrast, the fiber structure was maintained in the PVA/Gly FT nanofibers, even after swelling (diameter: 970 ± 250 nm), indicating that the FT treatment promoted crosslinking in the nanofibers (Fig. 2h). These results indicated that FT treatment and Gly addition contribute to the stability in water and PVA-based hydrogel nanofibers were obtained. | ||
| Fig. 2 Morphologies of the PVA and PVA/Gly nanofibers before and after swelling. SEM images of (a) PVA as-spun, (b) PVA FT, (c) PVA/Gly as-spun, and (d) PVA/Gly FT nanofibers before swelling. SEM images of (e) PVA as-spun, (f) PVA FT, (g) PVA/Gly as-spun, and (h) PVA/Gly FT nanofibers after swelling in water for one day. | ||
Chemical characterization of hydrogel nanofibers
The swelling ratio (SR) decreased when the FT treatment was conducted for both the PVA and PVA/Gly nanofibers (Fig. 3). The SR of the PVA as-spun nanofibers (1200 ± 160%) decreased to 780 ± 60% (PVA FT nanofibers) after FT treatment. On the other hand, the SR of the PVA/Gly as-spun nanofibers (540 ± 10) decreased slightly after FT treatment (500 ± 20), indicating that the decrease in rate due to FT treatment was significantly smaller than that of the PVA nanofibers.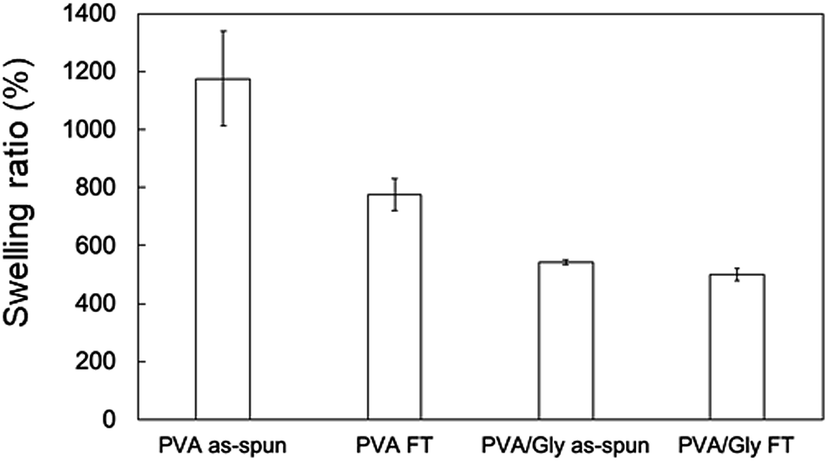 | ||
| Fig. 3 Swelling ratios of the nanofibers. Data are presented as the mean ± SD (n = 3). | ||
WAXD measurements were performed to evaluate the crystallinity of the PVA and PVA/Gly nanofibers. Fig. 4 depicts the X-ray diffraction (XRD) patterns of the PVA as-spun, PVA FT, PVA/Gly as-spun, and PVA/Gly FT nanofibers. A sharp crystalline reflection at 19.5° (2θ) corresponding to [10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ] and [101] was observed for all the nanofibers, which correlates with existing reports.38,39 The crystallinities of the nanofibers were estimated based on the results of the XRD analysis. The crystallinity of the PVA as-spun nanofiber was estimated at 33%, and increased to 41% after FT treatment. The increase in crystallinity was also observed in the PVA/Gly as-spun (43%) and PVA/Gly FT nanofibers (46%).
] and [101] was observed for all the nanofibers, which correlates with existing reports.38,39 The crystallinities of the nanofibers were estimated based on the results of the XRD analysis. The crystallinity of the PVA as-spun nanofiber was estimated at 33%, and increased to 41% after FT treatment. The increase in crystallinity was also observed in the PVA/Gly as-spun (43%) and PVA/Gly FT nanofibers (46%).
 | ||
| Fig. 4 WAXD patterns of (a) PVA as-spun and PVA FT nanofibers, and (b) PVA/Gly as-spun and PVA/Gly FT nanofibers. | ||
The DSC profiles of the nanofibers are shown in Fig. 5. Endothermic peaks derived from the nanofiber structure were observed in the range of 30–80 °C for all samples. Tm was observed at 225 °C for both the PVA as-spun and PVA FT nanofibers (Fig. 5a). The thermogram of neat Gly was also measured and the Tm was observed at 250 °C, whereas the Tm values of the PVA/Gly as-spun and PVA/Gly FT nanofibers were 263 °C and 271 °C, respectively, indicating that the addition of Gly in the PVA nanofibers led to an increase in the Tm (Fig. 5b).
 | ||
| Fig. 5 DSC profiles of (a) PVA as-spun and PVA FT nanofibers, and (b) PVA/Gly as-spun, PVA/Gly FT nanofibers, and Gly. | ||
ATR-FTIR analysis was performed to examine the effects of FT on the PVA and PVA/Gly nanofibers. The characteristic peaks of PVA were identified at 1094 cm−1 (CO stretching) and 3490 cm−1 (symmetric and asymmetric OH stretching) in all samples (Fig. 6a and b). However, peaks were observed at 1140 cm−1 (CO stretching) in the PVA FT, PVA/Gly as-spun, and PVA/Gly FT nanofibers except for the PVA as-spun nanofibers (Fig. 6c and d). The peak at 1140 cm−1 is due to the hydrogen crystallization bonding process of PVA.40 The crystallinity (χ) can be calculated as follows:41
| χ = −13.1 + 89.5(A1144/A1094) |
 | ||
| Fig. 6 ATR-FTIR spectra of the (a) PVA as-spun and PVA FT nanofibers, and (b) PVA/Gly as-spun, PVA/Gly FT nanofibers, and Gly. The figures in parts (c) and (d) are magnifications of (a) and (b). | ||
Evaluation of mechanical property
The anisotropic mechanical property of the nanofibers was evaluated by fabricating the samples in sheets; the SEM images and analyses of the fiber orientation are shown in Fig. 7. To facilitate comparison, the morphology of the PVA fiber is also shown in Fig. 7a and b. The orientation index (S) of the PVA/Gly nanofibers (0.500 ± 0.030) was higher than that of the PVA nanofibers (0.211 ± 0.035). The mechanical properties of the sheet-shaped PVA/Gly and PVA nanofibers were tested parallel and vertical to the fiber axis (Fig. 8 and Table 1). The typical stress–strain curves of the PVA and PVA/Gly nanofibers at parallel and vertical directions are shown in Fig. 8a and b, and their elastic moduli, tensile strengths, and elongation at break are summarized in Table 1. | ||
| Fig. 7 Morphologies of 15-PVA and 15-PVA/10-Gly nanofibers. SEM images (a) and (c) and orientation (b) and (d) of the 15-PVA and 15-PVA/10-Gly hydrogel nanofibers, respectively. | ||
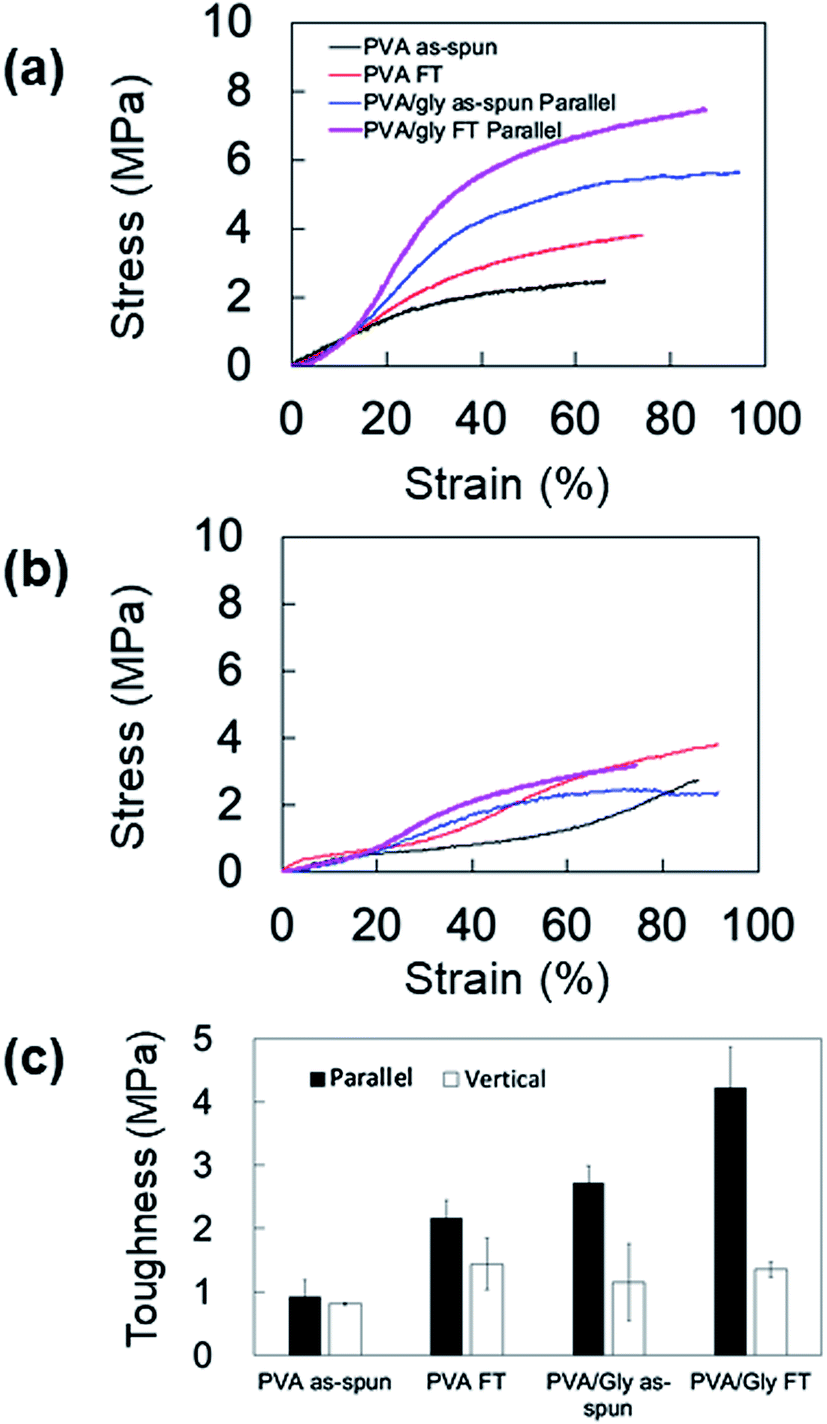 | ||
| Fig. 8 Mechanical properties of the PVA and PVA/Gly nanofibers in the parallel and vertical directions. Typical stress–strain curves of the PVA and PVA/Gly nanofibers in (a) parallel and (b) vertical directions. (c) Comparisons of the toughness values in the parallel and vertical directions for the PVA and PVA/Gly nanofibers. | ||
| Elastic modulus (MPa) | Tensile strength (MPa) | Elongation at break (%) | ||
|---|---|---|---|---|
| PVA as-spun | Parallel | 8.4 ± 0.24 | 2.4 ± 0.46 | 66 ± 4.7 |
| Vertical | 4.4 ± 0.76 | 2.7 ± 0.17 | 77 ± 2.0 | |
| PVA FT | Parallel | 13 ± 0.37 | 3.7 ± 0.14 | 81 ± 1.0 |
| Vertical | 11 ± 0.74 | 3.8 ± 0.69 | 73 ± 2.9 | |
| PVA/Gly as-spun | Parallel | 8.8 ± 0.27 | 5.0 ± 0.66 | 93 ± 2.6 |
| Vertical | 5.8 ± 1.2 | 2.7 ± 0.52 | 83 ± 2.4 | |
| PVA/Gly FT | Parallel | 11 ± 0.86 | 6.8 ± 0.85 | 83 ± 3.2 |
| Vertical | 7.2 ± 0.59 | 3.2 ± 0.20 | 78 ± 2.4 |
The FT treatment increased the elastic moduli and tensile strengths for both the PVA and PVA/Gly nanofibers parallel to the fiber axis. The elastic modulus and tensile strength of PVA as-spun nanofibers were 8.4 ± 0.24 MPa and 2.4 ± 0.46 MPa, respectively. After the FT treatment, the elastic modulus and tensile strength increased to 13 ± 0.37 MPa and 3.7 ± 0.14 MPa, respectively. Comparable results were obtained in the PVA/Gly nanofibers: the elastic modulus (8.8 ± 0.27 MPa) and tear strength (5.0 ± 0.66 MPa) of the PVA as-spun nanofibers increased to 11 ± 0.86 MPa and 6.8 ± 0.85 MPa after the FT treatment. In particular, the tear strengths of the PVA/Gly nanofibers were significantly higher than those of the PVA nanofibers (Fig. 8a and Table 1). The value of the elongation at break for the PVA as-spun nanofibers was 66 ± 4.7%, which increased to 81 ± 1.0% after FT treatment. Conversely, the value of the elongation at break for the PVA/Gly as-spun nanofibers (93 ± 2.6) decreased after the FT treatment (83 ± 3.2).
To evaluate the anisotropic mechanical property of the nanofibers, the samples were tested vertical to the fiber axis (Fig. 8b and Table 1). Similar to the results of samples tested in a parallel direction to the fiber axis, the elastic moduli and tensile strengths for both the PVA and PVA/Gly nanofibers at vertical directions increased after the FT treatment (Table 1). However, the elastic moduli of samples tested in the vertical direction were smaller compared to those in the parallel direction. In particular, the tensile strengths of the PVA/Gly as-spun and FT nanofibers vertical to the fiber axis were significantly smaller than those parallel to the fiber axis, whereas slight differences in the tensile strengths were observed in the PVA nanofibers for both the parallel and vertical directions. These results indicate that the mechanical property of PVA/Gly nanofibers indicated anisotropy.
Mechanical anisotropy was further confirmed by comparing the toughness values of the PVA and PVA/Gly nanofibers at parallel and vertical directions (Fig. 8c). The FT treatment increased the toughness of both the PVA and PVA/Gly nanofibers. The toughness of the PVA/Gly as-spun nanofiber (2.7 ± 0.27 MPa) was slightly higher than that of the PVA FT nanofiber (2.2 ± 0.27 MPa) even prior to FT treatment, as observed in those of tear strengths (Table 1). Moreover, the toughness of the PVA/Gly FT nanofiber (4.23 ± 0.63 MPa) was approximately two times higher than that of the PVA FT nanofiber. By contrast, there were slight differences in the toughness values of the PVA and PVA/Gly nanofibers in the vertical direction. These results clearly demonstrated the mechanical anisotropy of the PVA/Gly nanofibers.
Discussion
The study is aimed at investigating tough, toxic-crosslinker-free PVA hydrogel nanofibers with anisotropic mechanical properties. Hydrogels without chemical crosslinkers are appropriate for use in biomedical devices because there is no risk of toxicity arising from leaching of unreacted chemical crosslinkers. Additionally, hydrogels with weak mechanical properties are limited in terms of their biomedical applicability.42 Furthermore, mechanical anisotropy is desirable for various medical applications, as described in the above section. Electrospinning is a suitable method for controlling the anisotropic structure of nanofibers. Moreover, Gly is not only biologically safe but also promotes hydrogen bonding. Thus, we have hypothesized that tough anisotropic nanofibers can be fabricated via electrospinning in conjunction with Gly addition and FT treatment.In preliminary experiments, we examined dimethyl sulfoxide (DMSO) and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) as solvents for electrospinning PVA nanofibers, as these solvents have higher and lower boiling points than water, respectively. However, fabricating nanofibers with DMSO was difficult, probably because of its high boiling point (189 °C), resulting in a membrane-like formation on the rotating collector. An HFIP solution with 4% PVA formed nanofibers, but the diameter of PVA/HFIP nanofibers exceeded the nanometer scale (Fig. S1†). Furthermore, the ATR spectra of the electrospun fibers exhibited a peak at 1180 cm−1 (C–F stretching, Fig. S2†), indicating the presence of residual HFIP. Thus, this solvent is inappropriate for biomedical applications because HFIP induces toxicity. Thus, we used water as a solvent in subsequent experiments.
The distance between the needle and rotating collector affects the spinnability and morphology of PVA nanofibers. When the distance was below 10 cm, insufficient spinnability was observed, and the fiber diameters were on the order of micrometers. On the other hand, a needle-rotating collector distance above 10 cm decreased both the fiber diameters and orientation. Accordingly, to obtain PVA nanofibers with an anisotropic structure, we fabricated the PVA nanofibers at a needle-rotating collector distance of 10 cm.
We also optimized the Gly concentration in the 15% PVA solution. The Gly content in the PVA solution impacted the fabrication condition for the PVA/Gly hydrogel nanofibers (Fig. 1). Uniform fiber diameters and oriented PVA/Gly hydrogel nanofibers can be fabricated with 10% Gly (Fig. 1b, 7c and d), but were not achievable with 5 or 15% Gly, possibly due to the changes in both the viscosity and volatility of the PVA/Gly solution (Fig. 1a and c). The amount of Gly introduced into the PVA solution could potentially impact the increase in the PVA/Gly fiber diameter. In fact, the diameters of the PVA/Gly nanofibers increased as the Gly concentration increased (Fig. 1d–f). Furthermore, compared to PVA nanofibers, the addition of Gly presumably contributed to the increase in the orientation index (S) for PVA/Gly nanofibers because the Gly addition reduces the volatility of the solution. The reduced volatility makes it easier to receive the traction force from the rotating collector and decrease the wind effect. Conversely, the thinner diameter of the PVA nanofibers is easily influenced by the wind effect of the collector, resulting in a lower orientation value than that of the PVA/Gly nanofibers (Fig. 7).
The Gly addition also impacts the crystal formation on the PVA/Gly hydrogel nanofibers. The peak shift of the XRD patterns was not observed for both the PVA and PVA/Gly hydrogel nanofibers (Fig. 4). However, the effect of FT treatment on the formation of PVA/Gly crystallites was significantly smaller than that of the PVA nanofibers, indicating that Gly addition would contribute to increased crystal formation for the nanofibers. This result is inconsistent with that previously reported by Shi et al.: the crystallinity of the PVA hydrogel decreases with the Gly addition to the hydrogel.35 However, it explains the difference in the amount of Gly added to the PVA hydrogel. Based on the report by Shi et al., Gly was added to the 15% PVA solution at a maximum ratio of 1:2 using water/Gly as a solvent; the amount of Gly was larger than that of PVA. On the other hand, only 10% Gly was added to the PVA solution (v/v) in this study. This is indicative that a small amount of Gly increases the crystallinity of the PVA nanofibers. Water could be considered to be almost evaporated, whereas Gly completely remained in the fiber after electrospinning. Therefore, the weight percentage of Gly in 15-PVA/10-Gly nanofibers was estimated to be 40%.
The crystallite formation and hydrogen bonding in the PVA/Gly nanofibers, which were facilitated by Gly, were also confirmed by the ATR spectra. Similar to the results of the WAXD measurements, the estimation of crystallinity using ATR spectra indicated that a slight change in the crystal formation was observed in the PVA/Gly hydrogel nanofibers compared with that in the PVA nanofibers. An ATR peak at 1140 cm−1 was observed in the PVA/Gly as-spun nanofibers but not in the PVA as-spun nanofibers (Fig. 6c and d), suggesting that the crystal formation and hydrogen bonding in the OH between PVA and Gly formed by the end of spinning. In addition, the OH peak of the PVA/Gly as-spun nanofibers was observed at 3342 cm−1 and its peak shifted to 3330 cm−1 after FT treatment, suggesting that the FT treatment facilitated hydrogen bonding. The peak at 1140 cm−1 can be assigned to C–O stretching due to the formation of hydrogen bonds in microcrystals.40 Moreover, the results of the DSC also suggest enhanced hydrogen bonding. The Tms of PVA and Gly were observed to be 225 and 250 °C, respectively. On the other hand, the Tm of the PVA/Gly nanofibers was 263 °C, which increased slightly to 270 °C after the FT treatment (Fig. 5b). These results suggest that Gly addition enhances the hydrogen bonding in the crystal structure on the PVA/Gly nanofibers.
The enhanced crystal formation and hydrogen bonding of the PVA/Gly hydrogel nanofibers improved the stability in water. This was confirmed by morphology observations before and after swelling and swelling tests (Fig. 2 and 3). The swelling ratios of the PVA (1200 ± 160%) and PVA/Gly (540 ± 10%) as-spun nanofibers were higher than those of the PVA FT (780 ± 60%) and PVA/Gly (500 ± 20%) nanofibers, but a small decrease was observed in the PVA/Gly hydrogel nanofibers compared to the PVA nanofibers. These results appeared consistent with the crystallinity values estimated by the results of the WAXD and ATR (Fig. 4 and 6). However, the morphology of the PVA FT in water was not observed (Fig. 2f), even after FT treatment. Meanwhile, the swelling value of the PVA/Gly nanofibers was smaller than that of the PVA nanofibers, indicating that the water stability of the PVA/Gly nanofibers was higher than that of the PVA nanofibers. There was a slight difference in the crystallinity between the PVA FT and PVA/Gly FT nanofibers. Further, only the PVA/Gly FT nanofibers exhibited stability in water (Fig. 2f and h). The water stability observed in the PVA/Gly FT nanofibers was probably due the enhancement of crystallite formation as well as the hydrogen bonding between PVA and Gly. Although all PVA/Gly FT nanofibers exhibited stability in water, they swelled slightly, resulting in the loss of their original structure. Nonetheless, simple methods that avoid the use of chemical crosslinkers may further stabilize the PVA/Gly nanofibers in water, e.g., by increasing the FT cycles and/or drying time.40,43
The anisotropic mechanical property of the PVA/Gly nanofibers was identified by the tensile tests (Fig. 8). The elastic modulus and tensile strength of the PVA nanofibers increased after FT treatment in the parallel direction. Similar results were observed in the PVA/Gly hydrogel nanofibers and the tensile strengths were higher than those of the PVA nanofibers owing to the larger diameters and the higher orientation index (S) (Fig. 7). There was a slight difference in the elastic moduli between the PVA and PVA/Gly nanofibers because these values were estimated by the early linear region of the stress–strain curves. The mechanical properties of the PVA/Gly nanofibers tested in the parallel direction demonstrated different trends compared with those in the vertical direction. The elastic moduli, tear strengths, and toughness of the PVA/Gly nanofibers in the parallel direction were significantly higher than those in the vertical direction (Fig. 8 and Table 1). In particular, the tensile strength of the PVA/Gly nanofibers in the parallel direction was significantly higher than that in the vertical direction, whereas the tear strength of the PVA nanofibers was almost the same in both the parallel and vertical directions (Table 1). These results can be explained by the value of the orientation index (S). The value of the orientation index for the PVA nanofibers (0.211 ± 0.035) was lower than that for the PVA/Gly nanofibers (0.500 ± 0.030). Thus, a slight difference was observed in terms of the tensile strengths in both the parallel and vertical directions. On the other hand, the higher degree of orientation of the PVA/Gly nanofibers would lead to anisotropic mechanical properties, resulting in a decrease of elastic modulus and tensile strength in the vertical direction. Comparable trends were also confirmed by the toughness values of the PVA and PVA/Gly nanofibers (Fig. 8c). The toughness of the PVA/Gly FT nanofibers was approximately two times higher than that of the PVA FT nanofibers because of the larger diameter and higher orientation index (S). Consequently, these results indicate that the Gly addition in conjunction with FT treatment would improve the mechanical property and anisotropic structure of PVA nanofibers.
Herein, we developed a method for fabricating PVA nanofibers with water stability and mechanical anisotropy by adding Gly. PVA/Gly hydrogel nanofibers can be fabricated by a physically crosslinking method that does not use toxic chemical crosslinkers, thereby rendering the nanofibers biologically safe. Thus, it can be used in various biomedical applications such as implantation medical devices or regenerative medicine. The electrospinning method enables the fabrication of nanofibers with various shapes such as tubules, bulk samples, and sheets, which are applicable to multiple types of medical devices. For instance, nanofibers with conduit shapes can be potentially utilized as base polymers of biliary stents because of its anisotropic structure, which would lead to anisotropic swelling and mechanical properties.21 Hollow fibers also can be fabricated in conjunction with the core–shell nanofiber fabrication method,44,45 which is applicable for use in fluid removal or membrane plasmapheresis. Furthermore, PVA hydrogels with antibacterial property can be fabricated while maintaining the suppressed release of antibacterial reagents;23 these hydrogels may be used in gauze and surgical suture. In the future, we intend to optimize the properties of the PVA/Gly nanofibers based on the properties required for some applications.
Conclusions
We developed PVA/Gly hydrogel nanofibers fabricated by the electrospinning method with water stability and anisotropic mechanical properties. The amount of Gly was the predominant factor for fabricating PVA/Gly hydrogel nanofibers. The Gly addition in the hydrogel nanofibers enhanced the formation of hydrogen bonds in the crystalline structure of PVA, leading to stability in water. Furthermore, the PVA/Gly hydrogel nanofibers exhibited mechanical anisotropy and a higher orientation than the neat PVA nanofibers. PVA/Gly hydrogel nanofibers are physically crosslinked, comprising biocompatible materials. These hydrogel nanofibers are potentially applicable to medical applications and tissue engineering.Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Ni, M. Liu, S. Lu, L. Xie and Y. Wang, J. Agric. Food Chem., 2011, 59, 10169–10175 CrossRef CAS.
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS.
- S. H. Aswathy, U. Narendrakumar and I. Manjubala, Heliyon, 2020, 6, e03719 CrossRef CAS.
- E. Caló and V. V. Khutoryanskiy, Eur. Polym. J., 2015, 65, 252–267 CrossRef.
- C. R. Lynch, P. P. D. Kondiah, Y. E. Choonara, L. C. du Toit, N. Ally and V. Pillay, Front. Bioeng. Biotechnol, 2020, 8, 228 CrossRef.
- A. Gautieri, S. Vesentini, A. Redaelli and M. J. Buehler, Nano Lett., 2011, 11, 757–766 CrossRef CAS.
- M. A. Meyers, P. Y. Chen, M. I. Lopez, Y. Seki and A. Y. Lin, J. Mech. Behav. Biomed. Mater., 2011, 4, 626–657 CrossRef.
- Y. Ohyabu, S. Yunoki, H. Hatayama and Y. Teranishi, Int. J. Biol. Macromol., 2013, 62, 296–303 CrossRef CAS.
- R. McBeath, D. M. Pirone, C. M. Nelson, K. Bhadriraju and C. S. Chen, Dev. Cell, 2004, 6, 483–495 CrossRef CAS.
- D. E. Discher, P. Janmey and Y.-l. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS.
- T. Lecuit and P.-F. Lenne, Nat. Rev. Mol. Cell Biol., 2007, 8, 633–644 CrossRef CAS.
- K. N. Dahl, A. J. Ribeiro and J. Lammerding, Circ. Res., 2008, 102, 1307–1318 CrossRef CAS.
- A. D. Doyle and K. M. Yamada, Exp. Cell Res., 2016, 343, 60–66 CrossRef CAS.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS.
- M. Miron-Mendoza, V. Koppaka, C. Zhou and W. M. Petroll, Exp. Cell Res., 2013, 319, 2470–2480 CrossRef CAS.
- M. Verhulsel, M. Vignes, S. Descroix, L. Malaquin, D. M. Vignjevic and J.-L. Viovy, Biomaterials, 2014, 35, 1816–1832 CrossRef CAS.
- S. D. Hudson, J. L. Hutter, M.-P. Nieh, J. Pencer, L. E. Millon and W. Wan, J. Chem. Phys., 2009, 130, 034903 CrossRef.
- L. Millon, H. Mohammadi and W. Wan, J. Biomed. Mater. Res., Part B, 2006, 79, 305–311 CrossRef CAS.
- L. E. Millon, M.-P. Nieh, J. L. Hutter and W. Wan, Macromolecules, 2007, 40, 3655–3662 CrossRef CAS.
- M. Swan, D. Bucknall, T. Goodacre and J. Czernuszka, Acta Biomater., 2011, 7, 1126–1132 CrossRef CAS.
- Y. Nagakawa, S. Fujita, S. Yunoki, T. Tsuchiya, S. Suye and T. Itoi, J. Appl. Polym. Sci., 2019, 48851 Search PubMed.
- M. Kokabi, M. Sirousazar and Z. M. Hassan, Eur. Polym. J., 2007, 43, 773–781 CrossRef CAS.
- S. Yunoki, M. Kohta, Y. Ohyabu, M. Sekiguchi, T. Kubo and T. Iwasaki, J. Appl. Polym. Sci., 2014, 131, 40456 CrossRef.
- S.-H. Hyon, W.-I. Cha, Y. Ikada, M. Kita, Y. Ogura and Y. Honda, J. Biomater. Sci., Polym. Ed., 1994, 5, 397–406 CrossRef CAS.
- M. I. Baker, S. P. Walsh, Z. Schwartz and B. D. Boyan, J. Biomed. Mater. Res., Part B, 2012, 100, 1451–1457 CrossRef.
- J. Negishi, K. Nam, T. Kimura, Y. Hashimoto, S. Funamoto, T. Higami, T. Fujisato and A. Kishida, J. Biomed. Mater. Res., Part B, 2014, 102, 1426–1433 CrossRef.
- D. Moreau, A. Villain, M. Bachy, H. Proudhon, D. N. Ku, D. Hannouche, H. Petite and L. Corté, J. Mater. Sci.: Mater. Med., 2017, 28, 114 CrossRef.
- Y. Hou, W. Xie, K. Achazi, J. L. Cuellar-Camacho, M. F. Melzig, W. Chen and R. Haag, Acta Biomater., 2018, 77, 28–37 CrossRef CAS.
- M. Miraftab, A. N. Saifullah and A. Çay, J. Mater. Sci., 2015, 50, 1943–1957 CrossRef CAS.
- C. Tang, C. D. Saquing, J. R. Harding and S. A. Khan, Macromolecules, 2010, 43, 630–637 CrossRef CAS.
- C. M. Hassan and N. A. Peppas, in Biopolymers PVA Hydrogels, Anionic Polymerisation Nanocomposites, Springer, 2000, pp. 37–65 Search PubMed.
- W. Wan, A. D. Bannerman, L. Yang and H. Mak, in Polymeric Cryogels, Springer, 2014, pp. 283–321 Search PubMed.
- C. M. Hassan and N. A. Peppas, Macromolecules, 2000, 33, 2472–2479 CrossRef CAS.
- S. Huan, L. Bai, W. Cheng and G. Han, Polymer, 2016, 92, 25–35 CrossRef CAS.
- S. Shi, X. Peng, T. Liu, Y.-N. Chen, C. He and H. Wang, Polymer, 2017, 111, 168–176 CrossRef CAS.
- M. Reffay, L. Petitjean, S. Coscoy, E. Grasland-Mongrain, F. Amblard, A. Buguin and P. Silberzan, Biophys. J., 2011, 100, 2566–2575 CrossRef CAS.
- O. Batnyam, H. Shimizu, K. Saito, T. Ishida, S. Suye and S. Fujita, RSC Adv., 2015, 5, 80357–80364 RSC.
- R. Ricciardi, F. Auriemma, C. De Rosa and F. Lauprêtre, Macromolecules, 2004, 37, 1921–1927 CrossRef CAS.
- R. Ricciardi, F. Auriemma, C. Gaillet, C. De Rosa and F. Lauprêtre, Macromolecules, 2004, 37, 9510–9516 CrossRef CAS.
- E. Otsuka and A. Suzuki, J. Appl. Polym. Sci., 2009, 114, 10–16 CrossRef CAS.
- O. Tretinnikov and S. Zagorskaya, J. Appl. Spectrosc., 2012, 79, 521–526 CrossRef CAS.
- H. Kamata, Y. Akagi, Y. Kayasuga-Kariya, U.-i. Chung and T. Sakai, Science, 2014, 343, 873–875 CrossRef CAS.
- T. Fukumori and T. Nakaoki, J. Appl. Polym. Sci., 2014, 131, 40578 CrossRef.
- Y. Wakuda, S. Nishimoto, S. Suye and S. Fujita, Sci. Rep., 2018, 8, 1–10 CrossRef CAS.
- S. Fujita, Y. Wakuda, M. Matsumura and S. Suye, J. Mater. Chem. B, 2019, 7, 6556–6563 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07322a |
| This journal is © The Royal Society of Chemistry 2020 |