
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and in vitro characterization of valsartan-loaded ethyl cellulose and poly(methyl methacrylate) nanoparticles†
Eszter Hajba-Horvátha,
Emese Birób,
Mirella Mirankóa,
Andrea Fodor-Kardosab,
László Trifb and
Tivadar Feczkó *ab
*ab
aResearch Institute of Biomolecular and Chemical Engineering, Faculty of Engineering, University of Pannonia, Egyetem u. 10., H-8200, Veszprém, Hungary. E-mail: hajba-horvath@mukki.richem.hu; miranko@mukki.richem.hu
bInstitute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, Magyar Tudósok Körútja 2., H-1117, Budapest, Hungary. E-mail: emese.biro@gmail.com; kardos@mukki.richem.hu; trif.laszlo@ttk.hu; tivadar.feczko@gmail.com
First published on 9th December 2020
Abstract
Valsartan is an antihypertensive drug used primarily orally, however, due to its hydrophobic nature it has got low bio-availability thus requiring higher dosage/frequency and causing more side effects. The aim of our work was to prepare valsartan-loaded nanoparticles by using ethyl cellulose and poly(methyl methacrylate) polymers which can be administered orally and to investigate the preparation conditions and their significance as potential drug carriers for valsartan delivery by in vitro release studies. Ethyl cellulose and poly(methyl methacrylate) polymers were used for the preparation of nanoparticles by single emulsion-solvent evaporation technique. The formation of drug-loaded nanoparticles was designed by experimental design for size and encapsulation efficiency, in addition the prepared nanosuspensions were nano spray dried in order to gain a powder form that is easy to handle and store. Both of the nano spray dried formulations had an amorphous structure in contrast to the pure drug according to differential scanning calorimetry and X-ray diffraction analysis, which can be advantageous in drug absorption. The originally processed ethyl cellulose-valsartan nanoparticles increased the solubility of the drug in the model intestinal medium, while poly(methyl methacrylate)-valsartan nanoparticles enabled substantially prolonged drug release. The release kinetics of both types of nanoparticles could be described by the Weibull model.
1. Introduction
Nanotechnology is one of the most promising and fastest developing fields in medical and pharmaceutical applications. The use of nanoparticles is increasingly spreading in these areas due to their advantageous properties which make them extremely effective in drug delivery systems. When developing a new diagnostic or therapeutic agent, the most important product requirements are specific drug targeting and delivery, biocompatibility, biodegradability, enhanced bioavailability, and minimized toxicity and side effects.1 Drug loaded nanoparticles offer a great opportunity to fulfil these requirements. The size is a very important feature of the nanoparticles which has a great effect on their behaviour in biological organisms. According to a commonly used definition, the size of nanoparticles ranges from 10 to 1000 nm.2 The reason they are attractive for medical purposes is that these particles have relatively large surface to mass ratio, moreover, they can be characterized by the ability to bind and deliver several compounds.3 They also provide a great possibility for increasing the drug solubility and bioavailability.4 Wide range of compounds can be used for drug encapsulation in nanoparticles, such as lipids, chitosan, lactic acid, polymers, carbon or metals.3 Also several methods have been developed for engineering nanoparticles.5 The most commonly used are nanoprecipitation,6 single7 and double emulsion-solvent evaporation method,8 spray-drying, solvent diffusion method, coacervation phase separation method, polymerization and impinging jet.9 It is possible to control the particle characteristics via changing the process parameters such as drug to polymer ratio, presence and amount of different emulsifiers, temperature, organic to water phase ratio, flow rate, stirring rate, etc. The drug release is determined by the diffusion of a drug molecule through the carrier matrix.10 Alternatively, the degradation or swelling of the carrier matrix or the cleavage of drug–polymer linkage can control the drug release rate from the nanoparticles. Encapsulating the drug in different polymers can enhance the solubility and bioavailability.Valsartan is an orally active antihypertensive drug which causes reduction in blood pressure due to selective inhibition of angiotensin II receptor type 1.11 Beside its primary oral application, also transdermal administration was investigated for the treatment of hypertension.12 Further application of valsartan can be the treatment of chronic diabetic wounds.13 The examined drug can be characterized with a hydrophobic nature with only 0.021 mg ml−1 solubility in water.14 The drug is mostly absorbed in the small and large intestine.15 Oral administration of such lipophilic active ingredients is impeded by the barriers of the gastrointestinal tract (GIT) because of the poor solubility and permeability through the mucus membrane.16 This fact leads to relatively low bioavailability which is only 23–25% for valsartan.14 Despite of the difficulties of oral administration in the case of these therapeutic agents, oral tablet form is the most popular among patients due to its convenience. Therefore nearly 50% of the available drugs on the global market can be found in oral tablet form.17
Valsartan bioavailability was increased using various carrier systems, such as crystalline and spray-dried drug18 or precipitated active agent attached to protamine-functionalized montmorillonite,19 adsorption on mesoporous silica nanoparticles20,21 or graphene oxide.22 Some types of nanocarriers have already been developed, e.g. lipid-based23 gelatin-oleic acid24 or chitosan nanoparticles.25 However, to our knowledge synthetic biocompatible polymeric nanoparticles have not been used so far for valsartan microencapsulation. Ethyl cellulose and poly(methyl methacrylate) are promising drug carrier polymers with potential oral application.26,27 Both ethyl cellulose and poly(methyl methacrylate) nanoparticles were found to be safe in cytotoxicity studies in human cell lines.28,29 The WHO suggests ethyl cellulose as a food additive with negligible toxicity and low hazard to health, which showed no adverse reactions up to 5000 mg per kg per day for 3 month administration through oral route.30 Moreover, the daily repeated dose oral toxicity studies of ethyl cellulose31 and poly(methyl methacrylate)32,33 nanoparticles in rats did not show any treatment-related abnormal behavioural traits, mortality or toxicity in histopathological studies in the sub-acute period. The aim of our study was to prepare and investigate valsartan-loaded nanoparticles by two polymers which showed promising physical and chemical features in preliminary preparation study and can be administered orally. Further goal was to study their significance as potential drug carriers for valsartan delivery by in vitro release studies. Experimental parameters were determined by Box–Behnken design in order to control the particle size and the encapsulation efficiency. Emulsification was applied for the preparation of the nanoparticles containing ethyl cellulose and poly(methyl methacrylate) encapsulating polymers. According to the principle of this microencapsulation technique7 the oil phase containing the dissolved drug and polymer was dispersed in the water phase, then the organic solvent was evaporated.34 Nano spray-drying was applied to produce solid products from the prepared nanosuspensions to take advantage of solid-state properties.35 Thus, with further formulation of the nanoparticles, easy-to-store and easy-to-handle products were formed. The chemical and physical properties of the obtained nanoparticles were examined and in vitro drug release studies were carried out. The selected polymers could accelerate and slow the liberation of therapeutics.
2. Experimental
2.1. Materials
Standard gift sample of valsartan was provided by EGIS pharmaceutical company, Hungary. Ethyl cellulose (EC-4, viscosity: 4 mPa s, 5 wt% in 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 toluene/ethyl alcohol, 25 °C) was a kind gift from Dow Chemical Company (Dow Wolff Cellulosics GmbH, Bomlitz, Germany). Poly(methyl methacrylate) (PMMA, Mw = 35000) was purchased from Reanal Ltd., Hungary, polyvinyl alcohol (PVA, 87–90% hydrolyzed, MW = 30000–70000) was purchased from Sigma Aldrich. Dichloromethane (DCM) (Carlo Erba Reagents SAS.) and ethyl acetate (EtOAc) were purchased from Scharlab Hungary Ltd., HCl was purchased from Sigma Aldrich. Na2HPO4·2H2O, and NaH2PO4·2H2O were purchased from Reanal Ltd., Hungary. Cellulose ester dialysis membrane (Spectra Por 131420 Biotech-Grade CE Dialysis Tubing, 100000 MWCO) was obtained from Thermo Fischer Scientific (Waltham, MA).
:20 toluene/ethyl alcohol, 25 °C) was a kind gift from Dow Chemical Company (Dow Wolff Cellulosics GmbH, Bomlitz, Germany). Poly(methyl methacrylate) (PMMA, Mw = 35000) was purchased from Reanal Ltd., Hungary, polyvinyl alcohol (PVA, 87–90% hydrolyzed, MW = 30000–70000) was purchased from Sigma Aldrich. Dichloromethane (DCM) (Carlo Erba Reagents SAS.) and ethyl acetate (EtOAc) were purchased from Scharlab Hungary Ltd., HCl was purchased from Sigma Aldrich. Na2HPO4·2H2O, and NaH2PO4·2H2O were purchased from Reanal Ltd., Hungary. Cellulose ester dialysis membrane (Spectra Por 131420 Biotech-Grade CE Dialysis Tubing, 100000 MWCO) was obtained from Thermo Fischer Scientific (Waltham, MA).
2.2. Preparation of valsartan-loaded ethyl cellulose and poly(methyl methacrylate) nanoparticles
2.3. Nano spray-drying of the obtained nanosuspensions
The scaled up suspensions of ethyl cellulose-valsartan and poly(methyl methacrylate)-valsartan nanoparticles containing PVA emulsifier were nano spray dried without any pre-treatment in order to bring the nanoformulas into a more easily treatable form. For this purpose, a Büchi Nano Spray Dryer B-90 (Büchi Labortechnik AG, Flawil, Switzerland) was used. The experimental conditions of the spray-drying process were briefly the following: spray mesh size: 7 μm, pump: 60%, drying air flow rate: 90 l min−1, pressure: 26 hPa. The inlet temperature was 100 °C (except for PMV4, where it was 120 °C). The solid products were examined with different analytical methods (TG-DSC, XRD, SEM), their particle size was also determined and in vitro drug release studies were carried out.2.4. Determination of particle size and size distribution
Particle size and size distribution of the freshly prepared nanosuspensions was determined by dynamic light scattering (DLS) method using Malvern Zetasizer ZS (Malvern Instruments, Malvern, UK) at 25 °C. Five parallel size measurements were carried out for each sample. The analysis of the spray dried products were carried out by laser diffraction method using Malvern Mastersizer 2000 (Malvern Instruments, Malvern, UK) after dispersing the particles in cyclohexane containing 1% soy lecithin emulsifier. After spray drying the solid product was redispersed in corresponding amount of deionized water in the case of ECV54 and PMV4 and the hydrodynamic size analysis was achieved again by Malvern Zetasizer ZS (Malvern Instruments, Malvern, UK) under the same circumstances.2.5. S/TEM imaging of the nanoparticles
The morphology of the nanoparticles in the nanosuspensions was determined by transmission electron microscopy (TEM). In order to prepare the samples for the analysis, 1 ml of five-fold diluted suspension per sample was centrifuged (15000 rpm, 30 min or 10 min, Hermle Z216 MK centrifuge) at 25 °C then washed with 1 ml deionized water. After washing, the centrifugation and washing were repeated. The supernatant was discarded at each step of the process. Samples for TEM analysis were prepared by depositing a drop of aqueous suspension of sedimented particles on copper TEM grids covered by continuous carbon amorphous support film. TEM analyses were performed using a Talos F200X G2 instrument (Thermo Fisher), operated at 200 kV accelerating voltage, equipped with a field-emission gun and a four-detector Super-X energy-dispersive X-ray spectrometer, and capable of working in both conventional TEM and scanning transmission (STEM) modes. In our study STEM high-angle annular dark-field (HAADF) images were collected to visualize the particles of the ethyl cellulose and poly(methyl methacrylate) nanosuspensions. We applied low beam current and fast image acquisition to avoid beam damage of the particles. In the case of the spray-dried samples, scanning electron microscope (FEI Apreo scanning electron microscope, Thermo-Fisher, Waltham, MA) was used to determine the morphology of the nanoparticles. The spray dried samples were used without further sample preparation and they were mounted on carbon stubs and they were imaged at 2 to 20 kV.
2.6. X-ray powder diffraction (XRD) analysis
XRD analysis (XRD, Philips PW 3710) was carried out for ECV54 and PMV4 spray dried samples which were produced from nanoparticle suspensions in order to determine the structure of the products. The raw materials used for the nanoparticle production were also analysed.2.7. Determination of encapsulation efficiency
The drug loading and encapsulation efficiency were investigated measuring the non-encapsulated drug in the supernatant after centrifugation of the nanosuspensions. The samples which were prepared on the basis of the experimental design, were ultracentrifuged (40000 rpm, 10 min, Beckman Coulter Optima Max Ultracentrifuge) in order to separate the non-encapsulated drug. In the case of ethyl cellulose-valsartan nanosuspensions the supernatant was diluted to be within the detectable linear calibration range (5–60 μg ml−1). The absorbance of solutions was measured spectrophotometrically with a T80+ UV/Vis spectrophotometer (PG Instruments Ltd., Leicestershire, UK) at 250 nm. For the determination of the non-encapsulated drug in poly(methyl methacrylate)-valsartan nanoparticles a previously validated HPLC-UV (Young Lin YL 9100, YL Instruments Co., Ltd., Gyeonggi-do, Korea) method was used with the following measurement parameters: column: c-18, 5 μm 4.6 × 150 mm, mobile phase: MeOH/w-30–90 gradient, flow rate: 1 ml min−1, temperature: 35 °C, detection wavelength: 250 nm. The experimental non-encapsulated drug was quantified using the peak area of each sample at retention time of 8.27 min. In this method the supernatant samples were diluted to be within the 5–100 μg ml−1 concentration range.
2.8. Thermogravimetric and differential scanning calorimetric (TG-DSC) measurements
Thermal behavior of samples was investigated by a Setaram LabsysEvo (Lyon, France) TG-DSC system, in flowing (50 ml min−1) high purity (99.999%) argon atmosphere. Samples, without any sample preparation, were weighed directly into 100 μl Al crucibles (the reference cell was empty) and heated from 25 °C to 300 °C temperature interval, with a heating rate of 10 °C min−1. The obtained data was baseline corrected and further processed with the thermoanalyzer's processing software (Calisto Processing, ver.2.08). The thermal analyzer (both the temperature scale and calorimetric sensitivity) was calibrated by a multipoint calibration method, in which seven different certified reference materials were used to cover the thermal analyzer's entire operating temperature range.2.9. In vitro drug release studies
The in vitro release study was performed with a dialysis bag method under sink conditions mimicking the gastric and intestinal pH.43 Briefly, 30 mg of spray dried nanosuspensions (ECV55 and PMV33) containing 7.0 mg drug were weighed, redispersed in 4 ml 0.1 M HCl and filled into a dialysis bag. The bag was put in 400 ml glass beaker containing 196 ml 0.1 N HCl and was magnetically stirred (IKA RCT, Staufen, Germany) at 150 rpm for 2 hours. The bag containing the nanoparticle suspension was placed in another 400 ml beaker including 196 ml of phosphate buffer solution at pH 6.8, where it has been stirred (IKA RCT, Staufen, Germany) at 150 rpm for 4 hours. To prepare the buffer of the desired pH, proper amount of 0.1 M Na2HPO4 and NaH2PO4·2H2O stock solutions were mixed. The system was tempered at 37 °C and stirred at 150 rpm using an IKA RCT magnetic stirrer (Staufen, Germany) during the whole experimental period. At time periods of every 30 min, 1 ml of sample was taken from the medium, and they were analysed by UV-Vis spectrophotometry (Shimadzu UV-1800, Shimadzu, Japan) as described above. The detectable linear calibration range was 2.5–40 μg ml−1. The experiments were repeated three times.2.10. Drug dissolution and release kinetics
Several models are applied to characterize the dissolution profile and release of drugs. Most commonly used equations are zero order- and first order kinetics, Higuchi, Korsmeyer–Peppas, Hixson–Crowell, Ritger–Peppas, power law, Brazel–Peppas, Baker–Lonsdale, Hopfenberg, Weibull and Peppas–Sahlin kinetic correlations.44 Release kinetics of valsartan-loaded formulations have already been described by zero order, Higuchi and Weibull models.45,46 In our study the application of zero order- and first order kinetics, Higuchi, Korsmeyer–Peppas and Weibull models were tested, firstly, in their linearized form. Under acidic condition, the low release rate can be described by the simplest zero order kinetics. At pH 6.8 the active agent has got substantial solubility in phosphate buffer, hence, the most frequently used first order kinetic model, the Higuchi model (eqn (1)) was investigated at first, which is generally suitable to describe the dissolution of soluble drugs from matrix-structured nanoparticles. This model was originally created for the description of dissolution of active agents of slightly and very soluble drugs from solid and semi-solid matrices. It can be also applied for non-swelling polymers:| f = KHt1/2 | (1) |
The Korsmeyer–Peppas model is a developed Higuchi model (eqn (2) and (3)) using a comprehensive semi-empiric equation for polymer containing systems:
| f = kdtn | (2) |
|
logf = logK + nlogt
| (3) |
The Weibull model (eqn (4) and (5)) is an empiric relationship that has not got kinetic model basis, nevertheless, it can be used for the comparison of drug release from different matrices:
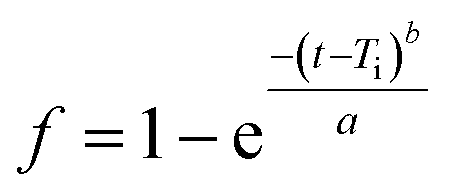 | (4) |
|
log[−ln(1 − f)] = blog(t − Ti) − loga
| (5) |
The form parameter (b) characterizes the type of curve:
(1) b = 1 (exponential)
(2) b > 1 (sigmoid, with ascendant curvature delimited by an inflection point)
(3) b < 1 (parabolic, displaying high initial slope and a consistent exponential character).
In eqn (4), the scale parameter, a defines the process time scale. The location parameter, Ti gives the time lag before the actual onset of the dissolution process. Fitting the lines, the curve shape can be determined by their correlation coefficients (R2), which approaches the release profile most exactly. The original form (eqn (4)) of the model has also been fitted by non-linear iteration. To describe the release profiles, our recently applied triphasic release model,47 developed by Lucero-Acuña et al. (eqn (5)),48 has also been examined.
2.11. Experimental design
Pre-formulation studies were performed with both type of polymer-valsartan nanoparticles in order to evaluate the effect of the experimental conditions on the particle size and encapsulation efficiency. On the basis of the results of these experiments three variables were selected, which were found to mostly influence the mentioned result parameters. Thus three factor Box–Behnken fractional factorial experimental design was performed with the help of STATISTICA® software42 using polymer, drug and emulsifier concentration as independent factors. By determining the three different levels for each parameter (the lowest, mean and highest value) the program generated 15 runs for both cases. This reduced significantly the number of experiments followed from different combinations of the selected independent factors and 3 repetitions for the combination of the mean values. The main advantage of using this experimental design was to obtain adequate amount of useful information with the reduction of necessary experimental work.After performing all the experiments according to the experimental design this software was also used for the evaluation of the results. The analysis made it possible to define the independent parameters which had statistically significant effect on the dependent parameters, such as particle size and encapsulation efficiency. According to the Student's t-test, P value less than 0.05 (P < 0.05) was considered to indicate a statistically significant effect.
3. Results and discussion
3.1. Variation of size
After testing of different methods, single emulsion-solvent evaporation method was found to be the most applicable procedure to prepare valsartan-loaded ethyl cellulose and poly(methyl methacrylate) nanoparticles. In preliminary experiments the processing range of variables was determined. The volumes of oil and water phase were kept constant.49,50 The highest values of drug and polymer concentrations (40 and 80 mg in 3 ml solvent, respectively) were selected, because at higher concentrations formation of aggregates was observed. The minimum concentrations were determined considering the fact that the valsartan has got increased solubility in PVA solution, thus the encapsulation efficiency would be substantially decreased, if the initial drug concentration is too low. The particles were prepared on the basis of the experimental parameters determined by using STATISTICA® software.42 The average hydrodynamic diameter of the obtained ethyl cellulose-valsartan nanoparticles ranged from 160 nm to 213 nm (see Table 1, see in ESI†), their polydispersity was 0.047–0.082, well below 0.1, which means that they can be characterized by a narrow size distribution in the case of each parameter combination. Typical size distribution of the ethyl cellulose-valsartan nanoparticles is shown in Fig. 1 (marked as ECV54). As it can be seen in Fig. S1 (in ESI),† the calculated and measured particle size data gave a very good match.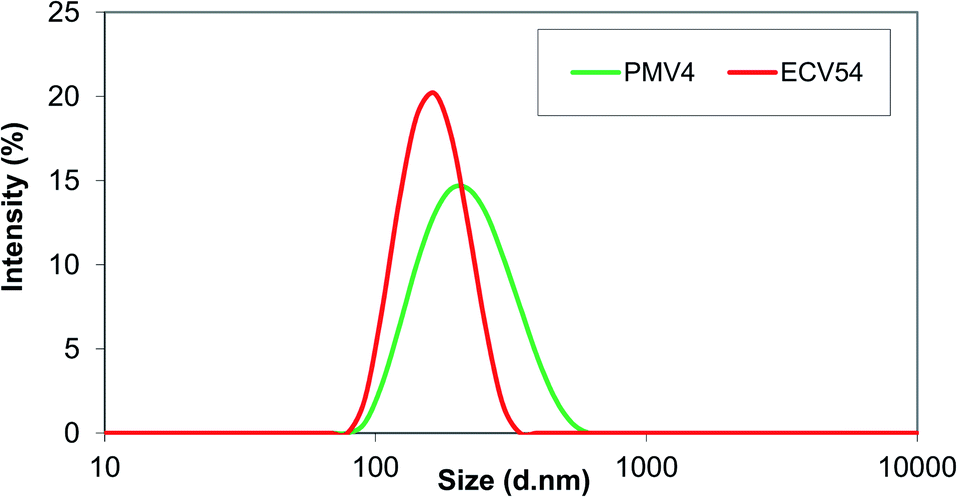 | ||
| Fig. 1 Particle size distribution of poly(methyl methacrylate)-valsartan (PMV4) and ethyl cellulose-valsartan (ECV54) nanoparticles. | ||
Statistical analysis was carried out on the measured particle size data in order to determine the influences of the independent process variables on the particle size. As a result of the analysis the effects of the parameters were characterized by the Pareto-chart (Fig. S2, ESI†). It can be concluded that in the preparation process of ethyl cellulose-valsartan nanoparticles the emulsifier (PVA) concentration (linear (L) and quadratic (Q) effect) and ethyl cellulose concentration (linear (L) effect) had significant effect on the particle size, while the influence of valsartan concentration was found not to be significant. Therefore, the particle size can be reduced by increasing the PVA concentration and decreasing the polymer concentration (Fig. S3 in ESI†).
As Table 2 (see in ESI†) shows, 195 nm–221 nm average particle size range was experimentally determined for the prepared poly(methyl methacrylate)-valsartan nanoparticles. Their polydispersity ranged from 0.050–0.118, which was below 0.1 for most of the parameter combinations, which represents wider size distribution than that obtained using ethyl cellulose for valsartan entrapment, nevertheless, it was still adequately monodisperse (Fig. 1, marked as PMV4). Similarly to the ethyl cellulose-valsartan nanoparticles the calculated and measured particle size data showed a very good correlation (Fig. S4 in ESI†).
It was found in the case of poly(methyl methacrylate)-valsartan nanoparticles that the PVA concentration (linear (L) and quadratic (Q) effect), the polymer concentration (linear (L) effect) and the active ingredient concentration affected significantly the particle size (Fig. S5 in ESI†). The particle size could be reduced by increasing the emulsifier concentration, and decreasing the valsartan and poly(methyl methacrylate) concentration (Fig. S6–S8 in ESI†).
TEM images also supported the results of size measurements, and they indicated formation of spherical nanoparticles (Fig. 2).
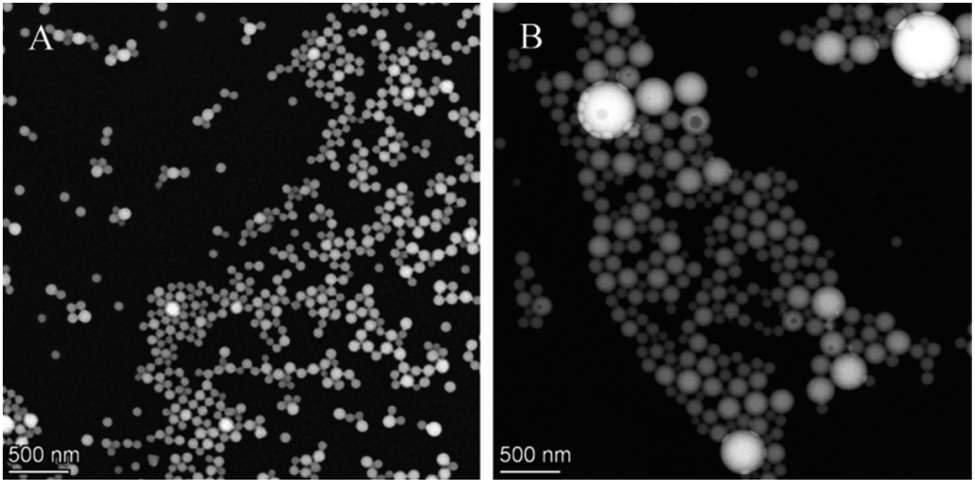 | ||
| Fig. 2 TEM images of ethyl cellulose- (A) and poly(methyl methacrylate)-valsartan (B) nanoparticles. | ||
3.2. Variation of encapsulation efficiency
Besides the effect of the studied experimental parameters on the average hydrodynamic diameter, the encapsulation efficiency was also examined as a function of polymer-, active ingredient- and emulsifier concentration. Table 3 (see in ESI†) summarizes the combinations of the examined factors according to the experimental setup and the encapsulation efficiency data which were calculated on the basis of measurements of supernatant samples for ethyl cellulose-loaded valsartan nanoparticles. The encapsulation efficiency ranged from 84.1% to 94.7% and the calculated and measured data correlated well (Fig. S9 in ESI†). As a result of the performed statistical analysis on the data set, it was found that PVA concentration (linear (L) and quadratic (Q) effect), valsartan concentration (linear (L) effect) had statistically significant effect on the encapsulation process (Fig. S10 in ESI†). The encapsulation efficiency could be enhanced by increasing the valsartan- and polymer concentration and decreasing the PVA concentration (Fig. S11–S13 in ESI†) as it was expected.The encapsulation efficiency was also determined for valsartan-loaded poly(methyl methacrylate) nanoparticles on the basis of the measurements of the supernatant samples after centrifugation. The encapsulation efficiency results ranged from 84.0% to 93.8% (Table S4 in ESI†). It can be stated on the basis of the correlation of the calculated and experimentally determined data that they showed good correlation (Fig. S14 in ESI†). As a result of the statistical analysis of the measured data in the three factor Box–Behnken fractional factorial experimental design, PVA-, valsartan- and the polymer concentration were identified as statistically significant factors on the encapsulation efficiency during formation of poly(methyl methacrylate)-valsartan nanoparticles with linear effect in each case (Fig. S15 in ESI†). The fitted surface of the studied variables was also performed in order to demonstrate these effects. Corresponding to the expectations the encapsulation efficiency could be improved by increasing the valsartan- and polymer concentration and decreasing the PVA concentration (Fig. S16–S18 in ESI†) similarly to the case with the ethyl cellulose encapsulating polymer. However, the decrease of the emulsifier concentration leads to the increase of the particle size, by changing the experimental parameters, despite the relatively low PVA concentration small particle size and at the same time high encapsulation efficiency were achieved for both polymer in our system.
Regarding the emulsifier concentration, our results harmonize with the observations reported in the literature. Sadoun et al.51 also found that the higher surfactant concentration decreased the encapsulation efficiency in a similar Box–Behnken experimental design. In their study the amount of poly(lactic acid) (PLA) and poly(ε-caprolactone) (PCL) as well as aqueous phase volume were examined as independent factors besides hydroxypropyl methylcellulose (HPMC) surfactant concentration. It was also stated, that aqueous phase volume decreased the encapsulation efficiency of valsartan while the polymer amount (PLA or PCL) had no significant effect on this parameter. As it is mentioned in the cited work,51 similar effect of PVA on encapsulation efficiency was reported by Yan et al.52 The explanation of this occurrence was the enhanced solubility of the drug according to Kumar et al.53 who examined the solubility of naproxen sodium. In contrast to the results found by Sadoun et al.,51 the amount of the two encapsulating polymers influenced the encapsulation efficiency significantly. The encapsulation efficiency achieved by Sadoun et al.51 was substantially lower (from 21.36 ± 1.29% to 57.90 ± 3.87%) compared to ours which could be the result of the different structure of their microparticles from our nanoparticles, the applied polymer characters, nevertheless, the significantly higher volume (40–80 ml) of outer water phase with similar drug concentration in their study also contributed definitely to the reduction of entrapped drug, since the drug dissolution in water is not negligible especially in the presence of emulsifiers.
3.3. Nano spray dried composites
According to the results of the particle size analysis by laser diffraction method, the particles, which were formed by spray drying of the poly(methyl methacrylate)-valsartan nanosuspensions, were 1–10 μm in size, while the particles from the ethyl cellulose-valsartan suspension ranged from 1 μm to 5 μm (Fig. 3).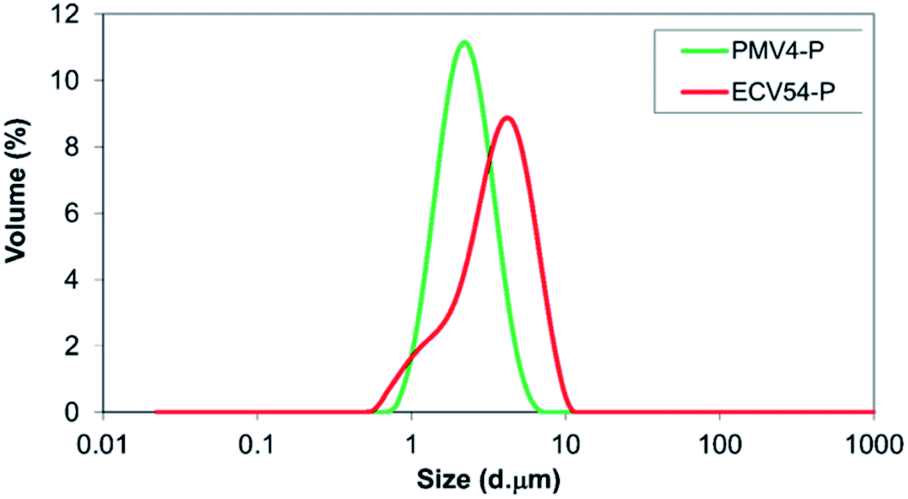 | ||
| Fig. 3 Particle size distribution of nano spray dried suspension of poly(methyl methacrylate-valsartan) (PMV4-P) and ethyl cellulose-valsartan (ECV54-P) nanoparticles. | ||
As it can be seen in the scanning electron microscope (SEM) images of the spray dried samples, the PVA emulsifier with the spheroid nanoparticles forms porous particles of some micrometer in size (Fig. 4).
 | ||
| Fig. 4 SEM images of nano spray dried ethyl cellulose- (A) and poly(methyl methacrylate)-valsartan (B) nanoparticles. | ||
After resuspending the spray-dried nanoparticles in MilliQ water, different behaviour was observed in the case of the two studied polymers. During the examination of ethyl cellulose-valsartan particles, aggregation process occurred, which could not be eliminated completely despite ultrasonication (Fig. 5). On the other hand, the poly(methyl methacrylate)-valsartan nanoparticles could be easily resuspended even without ultrasonication. After resuspending them the original size distribution was determined (Fig. 6). Resuspendability of the spray dried samples is strongly influenced by the interaction between the particles of the powder product and the emulsifier. During the nano spray drying process, the nanometer size particles of the prepared emulsions are dried together with the water phase, containing PVA as emulsifier. When the micrometer size particles of the spray dried powder are resuspended with water, the emulsifier in the composite particles should be available for the water in order to get suspensions with homogeneous distribution again and with the original particle size. In the case of the nanosuspensions prepared with the two encapsulating polymers, respectively, the same experimental parameters were applied during the spray drying. Under the same conditions, the original particle size of the nanosuspensions plays an important role in the drying process. Due to the smaller size of the ethyl cellulose-valsartan nanoparticles, they can be characterised by a greater diffusion coefficient, hence, they move faster in the spray dried droplets compared to the poly(methyl methacrylate)-valsartan nanoparticles. During spray drying process, particle formation is determined by droplet evaporation rate and diffusional motion of the solutes. The ratio between these two processes is expressed by the Peclet number.54 Our system can be characterized by a low Peclet number due to the relatively low inlet temperature which decreases the evaporation rate. In this case the fast diffusion motion of the small ethyl cellulose-valsartan nanoparticles dominates against the velocity of the receding droplet surface. Under these conditions the droplets undergo shrinking while the solutes and the solid particles migrate to the centre of the droplets. After saturation or supersaturation is reached, dense and solid particles are produced. The emulsifier in the composite particles with such structure is less available for the water therefore the particles become hardly resuspendable, which can result in the aggregation of the particles. In our system this could happen with the spray dried ethyl cellulose-valsartan particles while the poly(methyl methacrylate)-valsartan nanoparticles with greater size could be easily resuspended due to the better availability of the emulsifier for the water.
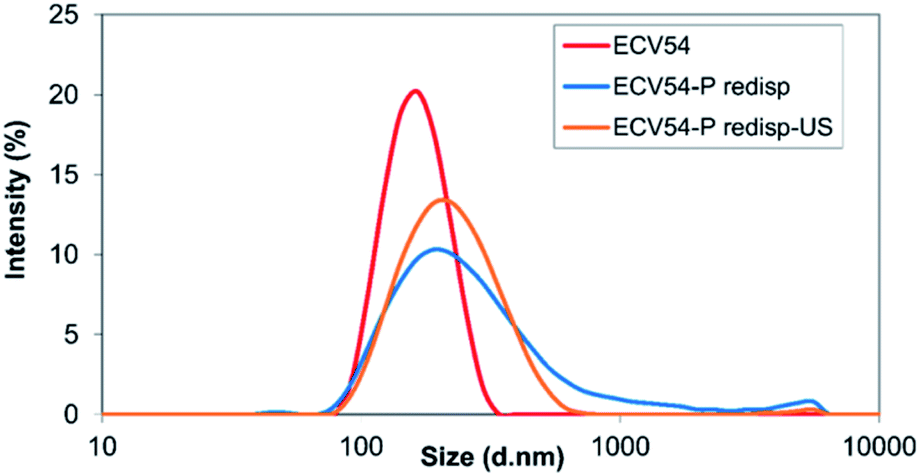 | ||
| Fig. 5 Size distribution of ethyl cellulose-valsartan nanosuspension (ECV54), its spray dried and resuspended suspension (ECV54-P redisp) and the spray dried subsequently redispersed suspension using sonication (ECV54-P redisp-US). | ||
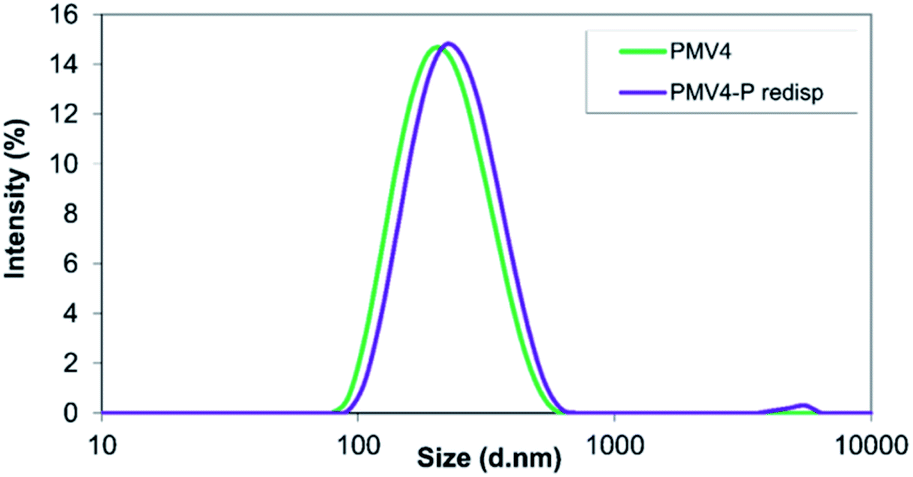 | ||
| Fig. 6 Size distribution of poly(methyl methacrylate)-valsartan nanosuspension (PMV4) and of the spray-dried and resuspended suspension (PMV4-P redisp). | ||
3.4. TG-DSC and XRD analysis
Differential scanning calorimetry (Fig. 7) and thermogravimetry (Fig. 8) were performed on the nano spray dried samples. The spray dried poly(methyl methacrylate)-valsartan and ethyl cellulose-valsartan nanoparticles seemed to be similar in their heat flow and thermogravimetric properties. TG-DSC analysis indicated significant amount of PVA and its effect since the melting endotherm of the emulsifier could be detected at approximately 190 °C in the case of both spray dried samples. The results of these experiments suggest the amorphous structure of the active ingredient in both of the samples prepared by different polymers. The effect of the encapsulating polymers from a termic point of view could be negligible despite the fact that they represented similar mass to the PVA as a consequence of their similar concentration related to the spray dried nanosuspensions.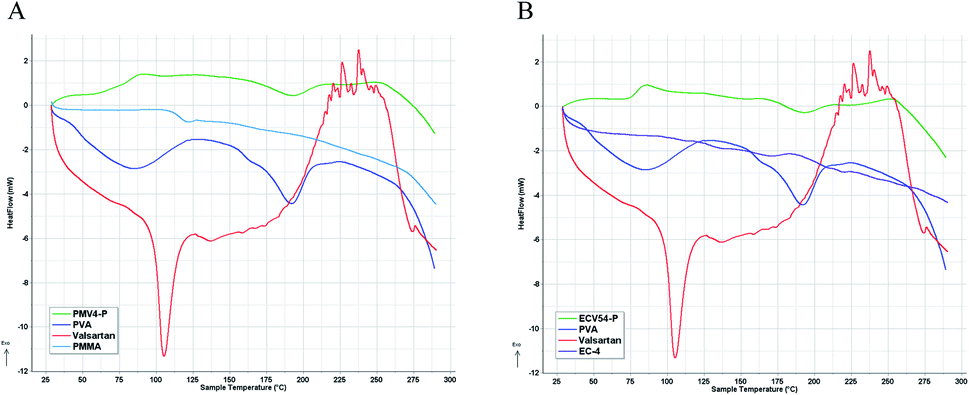 | ||
| Fig. 7 DSC diagrams of PVA emulsifier, valsartan, and the polymers as well as that of nano spray dried suspension of poly(methyl methacrylate) (PMMA)-valsartan (PMV4-P, (A)) and ethyl cellulose (EC-4)-valsartan (ECV54-P, (B)) nanoparticles. | ||
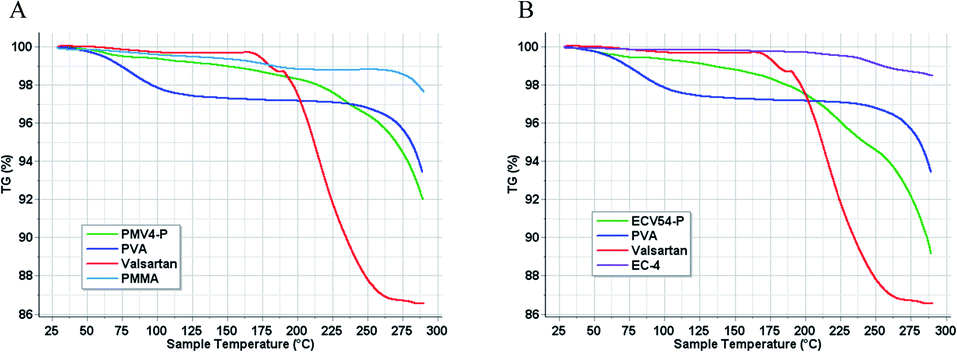 | ||
| Fig. 8 TG diagrams of PVA emulsifier, valsartan, and the polymers as well as that of nano spray dried suspension of poly(methyl methacrylate) (PMMA)-valsartan (PMV4-P, (A)) and ethyl cellulose (EC-4)-valsartan (ECV54-P, (B)) nanoparticles. | ||
To prove the results of DSC investigation, XRD analysis was done, which showed amorphous structure for both nano spray dried samples which were produced from nanoparticle suspensions (Fig. 9). However, the valsartan showed crystalline and amorphous structure as well, both of the nano spray dried nanosuspensions had exclusively amorphous character.
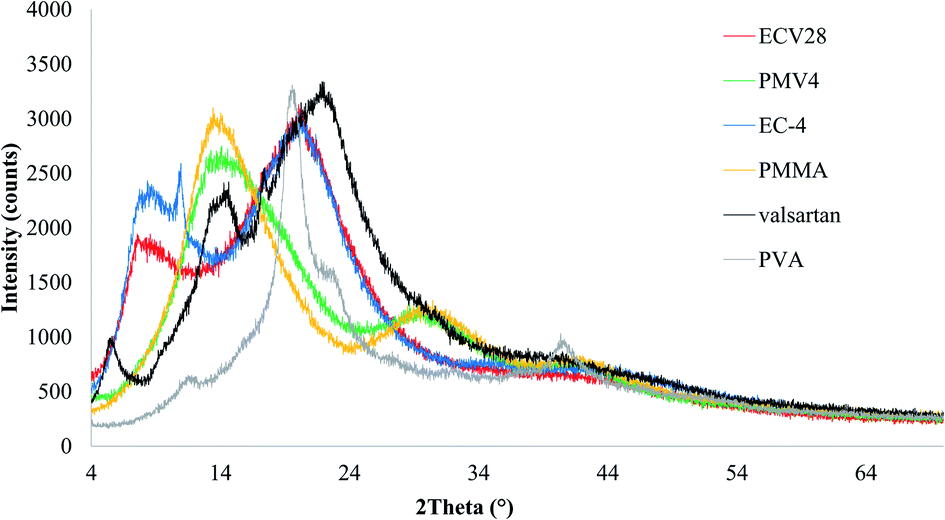 | ||
| Fig. 9 XRD results of nano spray dried samples from ethyl cellulose (EC-4)-valsartan (ECV28, red line) and poly(methyl methacrylate) (PMMA)-valsartan (PMV4, green line) nanoparticle suspensions and that of the raw materials: EC-4 (blue line), PMMA (yellow line), valsartan (black line), PVA (grey line). | ||
3.5. In vitro drug release studies
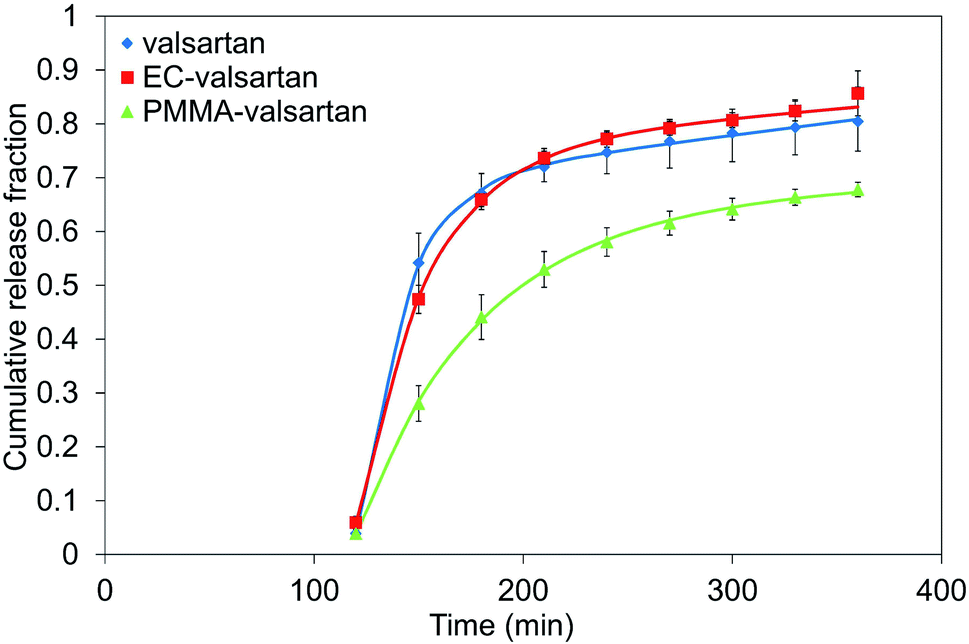
The cumulative drug release (%) was determined for gastric and intestinal pH at each sampling interval. In order to evaluate the effect of drug encapsulation into the nanoparticles and considering the effect of membrane diffusion, an in vitro release study was performed with the non-encapsulated drug as well. The used amount of valsartan was the same as in the examined spray dried samples. Microencapsulation by poly(methyl methacrylate) polymer resulted in considerably sustained drug release from the nanoparticles with incomplete release, however, the valsartan-loaded ethyl cellulose nanoparticles enabled faster dissolution (Fig. 10) than the pure drug. A similar trend was observed by Streubel et al.55 by studying a multiparticulate, floating drug delivery system. They found that the drug release increased in the following rank order of encapsulating polymers: PMMA < EC < Eudragit RS. They also concluded that the type of the polymer significantly affected the drug release rate. Regarding the effect of the different polymers on the release profile, Sadoun et al.51 conducted a dissolution study of valsartan-loaded PLA and PCL microparticles in a phosphate buffered saline at pH = 6.8, where a rapid release of the non-encapsulated valsartan was observed, and both encapsulating polymer resulted in sustained drug release from the drug-loaded particles. In our study, due to the low solubility of the active ingredient at acidic pH, the drug release was below 6% from both nanoparticulate samples and also the free drug during the first two hours of the study in 0.1 N HCl. The drug release from the nanoparticles increased significantly right after placing the dialysis membrane into the phosphate buffer solution at pH 6.8. 47.4% drug release was observed at the end of the first time interval (30 min) in this medium for ethyl cellulose-valsartan nanoparticles (ECV55) and 28.1% for the poly(methyl methacrylate)-valsartan nanoparticles (PMV33) for the same interval. At the first sampling point at pH 6.8, ethyl cellulose-valsartan nanoparticles provided lower release than the dissolution of free drug (54.2%), however, later on the liberation from this nanoparticle exceeded that from the free drug. At the end of the whole study period 85.7% and 67.8% of the encapsulated drug was released from ECV55 and PMV33, respectively, while 80.5% of the free drug was dissolved. Recently, Hamed56 showed the strong dependency of valsartan dissolution on pH and buffer capacity of the applied media. In that study the valsartan dissolution enhanced as a result of growing pH and buffer capacity. This important finding emphasizes the importance of release media and its necessary consideration when release data are compared.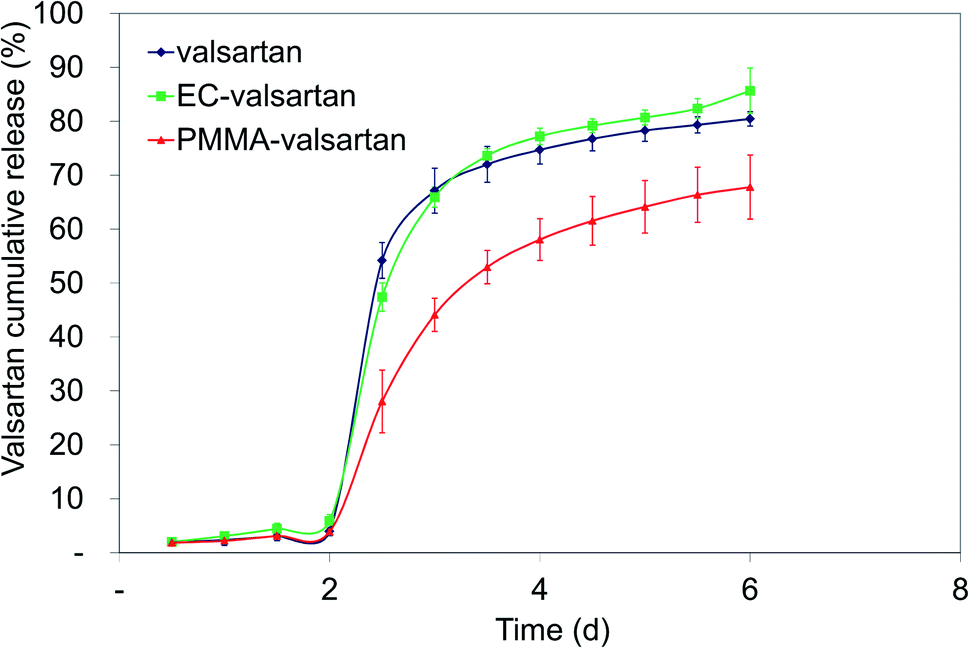 | ||
| Fig. 10 In vitro release profile of valsartan from ethyl cellulose-valsartan nanoparticles in 0.1 N HCl for 2 h and in phosphate buffer solution at pH 6.8 for further 4 h. | ||
However, sustained release from ethyl cellulose particles is reported in the literature57,58 with other drugs, similarly to the type of the encapsulating polymer, the particle formation is also influenced by the active ingredient. The quickly dissolving fraction of the drug in the case of our valsartan-loaded ethyl cellulose nanoparticles can be composed of the non-encapsulated valsartan and the drug present on the surface of the nanoparticles. They had smaller size compared to the poly(methyl methacrylate)-valsartan nanoparticles which resulted in larger surface of the particles, where greater amount of the drug could be quickly dissolved. The release profile of our poly(methyl methacrylate)-valsartan nanoparticles was similar to the tablet form having the smallest coat thickness (B5) prepared and studied by Sokar et al.59 They investigated 5 different tablet composition and concluded that B5 composition provided the most beneficial release behaviour. The biphasic way of the drug release from poly(methyl methacrylate) particles with an incomplete release is also mentioned in the review paper of Bettencourt et al.60 This can be explained by the structural properties of the polymer which cause less mobility of the active ingredient, therefore it leads to slow and incomplete release. A possible way to improve the release rate is to synthesize functionalized PMMA microspheres or to formulate PMMA composites with hydrophilic polymers in order to increase the hydrophilicity of the polymer. The sustained drug release from our poly(methyl methacrylate)-valsartan nanoparticles is indicative of a diffusion-controlled process, which is also supported by the experience of Streubel et al.55 Baek et al.61 examined the dissolution profile of spray died emulsions at pH 1.2 and pH 6.8 and they found a significant increase of valsartan release from the redispersible dry emulsions compared to the pure material. Most of the above mentioned findings reported in the literature concern to oil-in-oil emulsion systems with different conditions for particle formation and drug release. The dissolution of the encapsulated drug is strongly influenced by the pore size of the particles, the dissolution medium and the interfacial tension which all differ from the parameters of our oil-in-water system. The drug release in different systems is controlled by different processes, therefore, in our study considerably sustained drug release and also elevated dissolution could be achieved from valsartan-loaded nanoparticles depending on the type of used biocompatible encapsulating polymers.
3.6. Release kinetics
In the acidic medium the dissolution is very low, and zero order kinetics can sufficiently characterize the change (Fig. 11, Table 1), hence it is not reasonable to analyse a more complicated model. | ||
| Fig. 11 Zero order kinetics of valsartan, ethyl cellulose-valsartan- (EC-valsartan) and poly(methyl methacrylate)-valsartan (PMMA-valsartan) nanoparticles in the acidic medium. | ||
| Zero order | First order | Higuchi | Korsmeyer–Peppas | Weibull | |
|---|---|---|---|---|---|
| pH | 1.2 | 6.8 | 6.8 | 6.8 | 6.8 |
| R2 | R2 | R2 | R2 | R2 | |
|---|---|---|---|---|---|
| Valsartan | 0.976 | 0.885 | 0.838 | 0.840 | 0.998 |
| EC-valsartan | 0.995 | 0.910 | 0.825 | 0.808 | 0.986 |
| PMMA-valsartan | 0.972 | 0.920 | 0.888 | 0.849 | 0.992 |
At pH 6.8 some models applied in the area of drug delivery have been investigated to describe the concentration changes. To decide whether the model can be suitable for the purpose, their linearized forms have been plotted in the first approach. First order kinetics, Higuchi and Korsmeyer–Peppas models were found to be unsuitable to characterize the valsartan release kinetics in the used medium (Table 1).
The benefit of Weibull model is its suitability for linearization. Fig. 12 shows the curves fitted using Weibull model, while it can be concluded from R2 values in Table 1 that its modelled values are in very good agreement with the measured ones. Since the ‘b’ values are smaller than 1, parabolic curve was fitted to the measured points (eqn (5)). Ti parameter represents the time shift that shows values also for earlier time points related to the first measured result (pH = 6.8, 150 min). For Ti parameter 135 min was chosen.
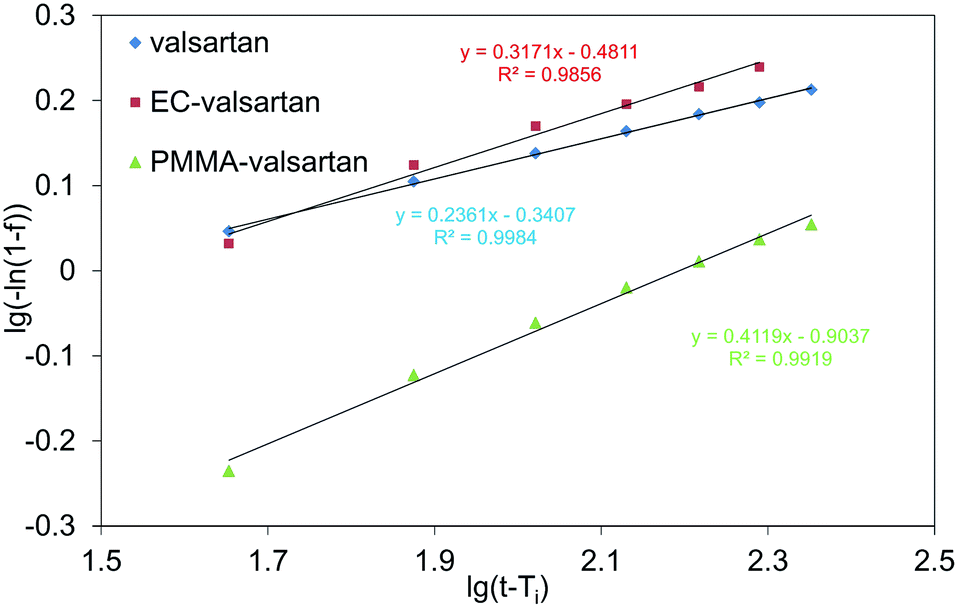 | ||
| Fig. 12 Weibull model at Ti = 135 min for valsartan, ethyl cellulose-valsartan- (EC-valsartan) and poly(methyl methacrylate)-valsartan (PMMA-valsartan) nanoparticles. | ||
To eliminate the inaccuracy of linearization, non-linear fitting was also used. Ti parameter can be fitted during non-linear fitting (Table 2). The modelled values of linear and non-linear fitting are compared in Fig. 13. The extension and direction of model bias depends on the function shape. a, b and Ti parameters for linear- and non-linear fitting are collected in Table 2.
| Linear fitting | Non-linear fitting | |||||
|---|---|---|---|---|---|---|
| Ti (chosen value), min | a | b | Ti, min | a | b | |
| Valsartan | 135 | 2.19 | 0.24 | 147.7 | 1.82 | 0.20 |
| EC-valsartan | 135 | 3.03 | 0.32 | 144.7 | 2.49 | 0.28 |
| PMMA-valsartan | 135 | 8.01 | 0.41 | 142.5 | 6.49 | 0.37 |
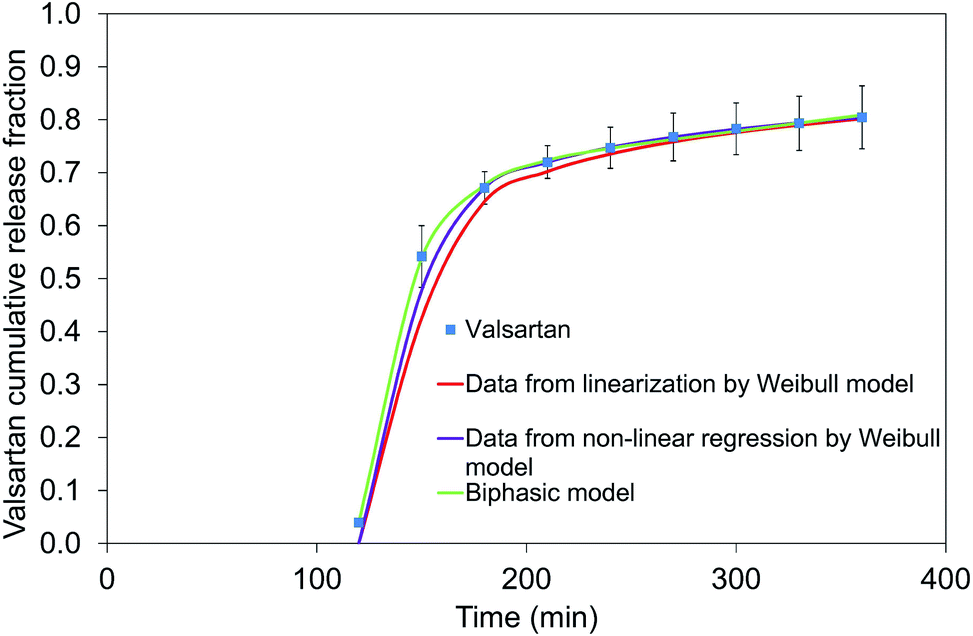 | ||
| Fig. 13 Linear and non-linear regression of Weibull model and the biphasic model for valsartan dissolution at pH = 6.8. | ||
The Weibull model is an empirical relationship and a mathematical equation for the description of a given curve shape, hence, one definite mechanism cannot be deduced from it. Consequently, the triphasic model47 fitting has been used to gain data regarding the mechanism.
In the triphasic model the first, second and third term describe the initial quick dissolution, release due to polymer degradation and slow diffusion, respectively. In our valsartan containing systems sigmoid curve of polymer degradation cannot be observed, thus the concentration change can be characterized by a biphasic profile composed of the terms of quick dissolution and diffusion.
In Fig. 13 all of the three relationships approach the result values quite good, however, the optimal correlation resulted from the biphasic model. Fig. 14 shows the dissolution profiles of free drug and release of valsartan-loaded nanoparticles fitted by the biphasic model. Biphasic model suggests a mechanisms in which the drug adsorbed on the surface is dissolved in the so-called initial burst, then, the drug diffusing from the polymer matrix diffuses through the dialysis membrane into the outer phase. Table 3 shows the parameters calculated from the biphasic model. kd is the kinetic constant that includes the diffusion from polymer and dialysis membrane as well.
 | ||
| Fig. 14 Biphasic model of valsartan, ethyl cellulose-valsartan- (EC-valsartan) and poly(methyl methacrylate)-valsartan (PMMA-valsartan at pH = 6.8.). | ||
| θb | kb | kd | n | dt | |
|---|---|---|---|---|---|
| Valsartan | 0.69 | 0.045 | 4.65 × 10−4 | 1.012 | 118.7 |
| EC-valsartan | 0.75 | 0.029 | 3.01 × 10−4 | 1.011 | 117.3 |
| PMMA-valsartan | 0.64 | 0.017 | 1.56 × 10−4 | 1.011 | 116.4 |
In the scientific literature generally the tablet with accelerated dissolution is targeted, which is compared to the commercially available Diovan. In the release of Diovan tablet, at pH 1.2 10% and 40% valsartan dissolve after 30 min and 120 min, respectively.62 At pH 6.8 the dissolution of valsartan is naturally more accelerated, it changes in 15 min and 45 min between 80% and 99%.63,64 These published formulations provide faster release related to our nano spray dried nanosuspension, however, our product cannot be considered as a ready-to-use drug, but a promising pre-formulation that may be functionalized to prepare a targeted drug delivery system.
4. Conclusions
Valsartan-loaded ethyl cellulose and poly(methyl methacrylate) polymeric nanoparticles were prepared by single emulsion-solvent evaporation technique. The nanoparticle preparation was studied by Box–Behnken experimental design, and resulted in average size smaller than 213 nm (ethyl cellulose-valsartan) and 221 nm (poly(methyl methacrylate)-valsartan-valsartan) accompanied with low polydispersity for both nanotherapeutics. The maximized encapsulation efficiencies were as high as 94.7% and 93.8%, respectively. The nanoparticle suspensions were formulated to easy-to-handle and storable powder form using nano spray-drying. Both of the spray-dried drug forms had amorphous character despite of the partly crystalline raw drug. Amorphous form is generally more beneficial concerning the absorption of drug. The dried poly(methyl methacrylate)-valsartan could be readily redispersed in water, however, ethyl cellulose-valsartan nanoparticles showed some aggregation after resuspension. Consequently, the size of the obtained poly(methyl methacrylate)-valsartan nanoparticles is more suitable to achieve the favourable characteristics of a spray dried product. The significance of prepared nanoparticles as potential valsartan carriers was also tested by in vitro release studies. It was found that ethyl cellulose-valsartan nanoparticles increased the solubility of the drug in the model intestinal medium which enhanced the bioavailability of the poorly water soluble drug. Poly(methyl methacrylate)-valsartan nanoparticles enabled substantially prolonged drug release. The release kinetics of valsartan from both type of nanoparticles could be described by Weibull model and a biphasic release pattern. Our in vitro results are showing that the nanoparticles structurally changed the active agent, which could be advantageous in the intestinal absorption, therefore both encapsulating polymers have potential significance in valsartan delivery depending on the potential future applications. In order to clarify their role in the delivery of this antihypertensive, the complicated enzymatic processes and the cross-reactions in the human body should be taken into account, therefore in vivo studies are necessary to clarify the real performance of the nanotherapeutics, which will be the next step of the drug development work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to acknowledge the National Competitiveness and Excellence Program, Hungary for providing funding in frame of GINOP-2.2.1-15-2016-00023 and the TKP2020-IKA-07 project financed under the 2020-4.1.1-TKP2020 Thematic Excellence Programme by the National Research, Development and Innovation Fund of Hungary. S/TEM studies were performed at the electron microscopy laboratory of the University of Pannonia, established using grant no. GINOP-2.3.3-15-2016-0009 from the European Structural and Investments Funds and the Hungarian Government. The authors are especially grateful to the EGIS Pharmaceuticals PLC for their partnership and they thank Péter Pekker and Miklós Jakab for the TEM/SEM imaging of the nanoparticles and Éva Kristóf-Makó for the X-ray powder diffraction analysis.References
- R. Ran, Q. Sun, T. Baby, D. Wibowo, A. P. J. Middelberg and C. X. Zhao, Chem. Eng. Sci., 2017, 169, 78 CrossRef CAS.
- H. J. Jeon, Y. I. Jeong, M. K. Jang, Y. H. Park and J. W. Nah, Int. J. Pharm., 2000, 207, 99 CrossRef CAS.
- W. H. Jong and P. De Borm, Int. J. Nanomed., 2008, 3, 133 CrossRef.
- C. Ding and Z. Li, Mater. Sci. Eng., C, 2017, 76, 1440 CrossRef CAS.
- C. I. C. Crucho and M. T. Barros, Mater. Sci. Eng., C, 2017, 80, 771 CrossRef CAS.
- I. D. Styliari, V. Taresco, A. Theophilus, A. Cameron, M. Garnett and C. Laughton, RSC Adv., 2020, 10, 19521 RSC.
- T. Feczkó, A. Piiper, T. Pleli, C. Schmithals, D. Denk, S. Hehlgans, F. Rödel, T. J. Vogl and M. G. Wacker, Pharmaceutics, 2019, 11, 489 CrossRef.
- T. Feczkó, J. Tóth and J. Gyenis, Colloids Surf., A, 2008, 319, 188 CrossRef.
- R. Baber, L. Mazzei, N. T. K. Thanh and A. J. Gavriilidis, J. Flow Chem., 2016, 6, 268 CrossRef CAS.
- J. H. Lee and Y. Yeo, Chem. Eng. Sci., 2015, 125, 75 CrossRef CAS.
- P. Gupta and A. Mahajan, RSC Adv., 2015, 5, 26686 RSC.
- A. Ahad, M. Aqil and A. Ali, Int. J. Biol. Macromol., 2014, 64, 144 CrossRef CAS.
- P. Abadir, S. Hosseini, M. Faghih, A. Ansari, F. Lay, B. Smith, A. Beselman, D. Vuong, A. Berger, J. Tian, D. Rini, K. Keenahan, J. Budman, T. Inagami, N. Fedarko, G. Marti, J. Harmon and J. Walston, J. Invest. Dermatol., 2018, 138, 434 CrossRef CAS.
- S. Agrawal and M. Kasturi, World J. Pharm. Res., 2016, 5, 833 CAS.
- X. Lin, H. Jiang and J. Zhou, J. China Pharm. Univ., 2012, 43, 130 CAS.
- N. Siddiqui, H. Asif, C. M. Lakshita, A. Shamsher, M. Moloy and S. B. Parminder, J. Appl. Pharm. Sci., 2011, 01, 12 Search PubMed.
- P. P. Desai, A. A. Date and V. B. Patravale, Drug Discovery Today: Technol., 2012, 9, e87 CrossRef CAS.
- Q. Ma, H. Sun, E. Che, X. Zheng, T. Y. Jiang, C. S. Sun and S. L. Wang, Int. J. Pharm., 2013, 441, 75 CrossRef CAS.
- A. Kumar, P. Davern, B. K. Hodnett and S. P. Hudson, Colloids Surf., B, 2019, 175, 554 CrossRef CAS.
- P. B. Santosh and R. Neeraj, Asian J. Pharm., 2016, 10, S86 CAS.
- N. Biswas, Eur. J. Pharm. Sci., 2017, 99, 152 CrossRef CAS.
- I. P. de Sousa, K. Buttenhauser, W. Suchaoin, A. Partenhauser, M. Perrone, B. Matuszczak and A. Bernkop-Schnurch, Int. J. Pharm., 2016, 509, 360 CrossRef.
- M. Aslam, M. Aqil, A. Ahad, A. K. Najmi, Y. Sultana and A. Ali, J. Mol. Liq., 2016, 219, 897 CrossRef CAS.
- H. L. T. Phuong, T. D. T. Thao and B. J. Lee, Int. J. Pharm., 2013, 455, 235 CrossRef.
- T. Niaz, S. Shabbir, S. Manzoor, A. Rehman, A. Rahman, H. Nasir and M. Imran, Carbohydr. Polym., 2016, 142, 268 CrossRef CAS.
- O. A. Adeleke, Int. J. Pharm., 2019, 100023 CAS.
- F. Ahangaran, A. H. Navarchian and F. Picchioni, J. Appl. Polym. Sci., 2019, 136, 48039 CrossRef.
- S. Duarah, R. D. Durai and V. B. Narayanan, Drug Delivery Transl. Res., 2017, 7, 750 CrossRef CAS.
- P. E. Feuser, L. S. Bubniak, C. N. Bodack, A. Valério, M. C. S. Silva, E. Ricci-Júnior, C. Sayer and P. H. H. de Araújo, J. Nanosci. Nanotechnol., 2016, 16, 7669 CrossRef CAS.
- C. C. DeMerlis, D. R. Schoneker and J. F. Borzelleca, Food Chem. Toxicol., 2005, 43, 1355 CrossRef CAS.
- S. S. Bachhav, V. D. Dighe and P. V. Devarajan, Mol. Pharm., 2018, 15, 4434 CrossRef CAS.
- U. M. D. Lekshmi, G. Poovi, N. Kishore and P. N. Reddy, Int. J. Pharm., 2010, 396, 194 CrossRef.
- U. M. D. Lekshmi, N. Kishore and P. N. Reddy, J. Biomed. Nanotechnol., 2011, 7, 578 CrossRef CAS.
- S. Desgouilles, C. Vauthier, D. Bazile, J. Vacus, J. L. Grossiord, M. Veillard and P. Couvreur, Langmuir, 2003, 19, 9504 CrossRef CAS.
- K. Glaubitt, M. Ricci and S. Giovagnoli, AAPS PharmSciTech, 2019, 20, 19 CrossRef.
- K. Saravana Kumar, P. Jayachandra Reddy and K. B. Chandra Sekhar, J. Pharm. Res., 2011, 4, 3943 Search PubMed.
- P. B. O'Donnell and J. W. Mcginity, Adv. Drug Delivery Rev., 1997, 28, 25 CrossRef.
- A. Sannino, C. Demitri and M. Madaghiele, Materials, 2009, 2, 353 CrossRef CAS.
- A. N. Generalova, S. V. Sizova, V. A. Oleinikov, V. P. Zubov, M. V. Artemyev, L. Spernath, A. Kamyshny and S. Magdassi, Colloids Surf., A, 2009, 342, 59 CrossRef CAS.
- R. Dhivya, J. Ranjani, J. Rajendhran, J. Mayandi and J. Annaraj, Mater. Sci. Eng., C, 2018, 82, 182 CrossRef CAS.
- A. Sahu, P. Solanki and S. Mitra, Int. J. Nanomed., 2018, 13, 101 CrossRef CAS.
- STATISTICA (Version 13.4.0.14), StatSoft Inc., Tulsa, OK, 1984–2018 Search PubMed.
- H. Ünal, N. Öztürk and E. Bilensoy, Beilstein J. Org. Chem., 2015, 11, 204 CrossRef.
- M. L. Bruschi, Mathematical models of drug release, in Strategies to Modify the Drug Release from Pharmaceutical Systems, Woodhead Publishing, 2015, p. 63 Search PubMed.
- N. Chella, B. Daravath and D. Kumar, Eur. J. Drug Metab. Pharmacokinet., 2016, 41, 517 CrossRef CAS.
- H. Meda, D. Rao, H. Chowdary and M. B. Tej, Curr. Pharm. Anal., 2020, 10, 3748 Search PubMed.
- G. Babos, J. Rydz, M. Kawalec, M. Klim, A. Fodor-Kardos, L. Trif and T. Feczkó, Int. J. Mol. Sci., 2020, 21, E7312 CrossRef.
- A. Lucero-Acuña, C. Gutiérrez-Valenzuela, R. Esquivel and R. Guzmán-Zamudio, RSC Adv., 2019, 9, 8728 RSC.
- L. O. Beniteza, J. M. Castagninia, M. C. Añónb and P. R. Salgado, LWT--Food Sci. Technol., 2020, 118, 108809 CrossRef.
- D. J. McClements, Food emulsions – principles, practices, and techniques, CRC Press Taylor & Francis Group, USA, 2016 Search PubMed.
- O. Sadoun, F. Rezgui and C. G'Sell, Mater. Sci. Eng., C, 2018, 90, 189 CrossRef CAS.
- Y. D. Yan, J. H. Sung, K. K. Kim, D. W. Kim, J. O. Kim, B. J. Lee, C. S. Yong and H. G. Choi, Int. J. Pharm., 2012, 422, 202 CrossRef CAS.
- S. M. Kumar, M. J. N. Chandrasekar, R. Gopinath, R. Srinivasan, N. J. Nanjan and B. Suresh, Drug Delivery, 2007, 14, 163 CrossRef CAS.
- J. Vicente, J. Pinto, J. Menezes and F. Gaspar, Powder Technol., 2013, 247, 1 CrossRef CAS.
- A. Streubel, J. Siepmann and R. Bodmeier, Int. J. Pharm., 2002, 241, 279 CrossRef CAS.
- R. Hamed, Pharm. Dev. Technol., 2018, 23, 1168 CrossRef CAS.
- V. R. Malipeddi, R. Awasthi and K. Dua, Interv. Med. Appl. Sci., 2016, 8, 60 Search PubMed.
- M. Jelvehgari, D. Hassanzadeh, F. Kiafar, B. D. Loveymi and S. Amiri, Iran. J. Pharm. Res., 2011, 10, 457 CAS.
- M. Sokar, A. Hanafy, A. Elkamel and S. El-gamal, Indian J. Pharm. Sci., 2016, 77, 470 CrossRef.
- A. Bettencourt and A. J. Almeida, J. Microencapsulation, 2012, 29, 353 CrossRef CAS.
- I. H. Baek, J. S. Kim, E. S. Haa, G. H. Chooa, W. Choc, S. J. Hwang and M. S. Kim, Int. J. Biol. Macromol., 2014, 69, 222 CrossRef CAS.
- P. S. Rajinikanth, Y. Suyu and S. Garg, International Scholarly and Scientific Research & Innovation, 2012, 6, 12 Search PubMed.
- S. Revathi, S. K. Moulali and M. D. Dhanaraju, Der Pharm. Lett., 2015, 7, 315 Search PubMed.
- S. C. Dinda, D. P. Pattanayak and U. L. Narayan, Int. J. Pharm. Sci. Nanotechnol., 2011, 6, 44 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07218d |
| This journal is © The Royal Society of Chemistry 2020 |