
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative analysis of hydrogen and chalcogen bonds in two pyrimidine-5-carbonitrile derivatives, potential DHFR inhibitors: an integrated crystallographic and theoretical study†
Lamya H. Al-Wahaibia,
Kushumita Chakrabortyb,
Nora H. Al-Shaalan a,
Mohamed Yehya Annavi Syed Majeedb,
Olivier Blacquec,
Aamal A. Al-Mutairid,
Ali A. El-Emame,
M. Judith Percinof and
Subbiah Thamotharan*b
a,
Mohamed Yehya Annavi Syed Majeedb,
Olivier Blacquec,
Aamal A. Al-Mutairid,
Ali A. El-Emame,
M. Judith Percinof and
Subbiah Thamotharan*b
aDepartment of Chemistry, College of Sciences, Princess Nourah Bint Abdulrahman University, Riyadh 11671, Saudi Arabia
bBiomolecular Crystallography Laboratory, Department of Bioinformatics, School of Chemical and Biotechnology, SASTRA Deemed University, Thanjavur-613401, India. E-mail: thamu@scbt.sastra.edu
cDepartment of Chemistry, University of Zürich, Winterthurerstrasse 190, 8057 Zürich, Switzerland
dDepartment of Chemistry, College of Sciences, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh 11671, Saudi Arabia
eDepartment of Medicinal Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
fUnidad de Polímeros y Electrónica Orgánica, Instituto de Ciencias, Benemérita Universidad Autónoma de Puebla, Val3-Ecocampus Valsequillo, Independencia O2 Sur 50, San Pedro Zacachimalpa, Puebla-C.P.72960, Mexico
First published on 6th October 2020
Abstract
Two potential bioactive pyrimidine-5-carbonitrile derivatives have been synthesized and characterized by spectroscopic techniques (1H and 13C-NMR) and the three dimensional structures were elucidated by single crystal X-ray diffraction at low temperature (160 K). In both structures, the molecular conformation is locked by an intramolecular C–H⋯C interaction involving the cyano and CH of the thiophene and phenyl rings. The intermolecular interactions were analyzed in a qualitative manner based on the Hirshfeld surface and 2D-fingerprint plots. The results suggest that the phenyl and thiophene moieties have an effect on the crystal packing. For instance, the chalcogen bonds are only preferred in the thiophene derivative. However, both structures uses a common N–H⋯O hydrogen bond motif. Moreover, the structures of 1 and 2 display 1D isostructurality and molecular chains stabilize by intermolecular N–H⋯O and N–H⋯N hydrogen bonds. The nature and extent of different non-covalent interactions were further characterized by the topological parameters derived from the quantum theory of atoms-in-molecules approach. This analysis indicates that apart from N–H⋯O hydrogen bonds, other non-covalent interactions are closed-shell in nature. A strong and linear N–H⋯O hydrogen bond shows intermediate bonding character between shared and closed-shell interactions. The molecular docking analysis suggests that both compounds display potential inhibitory effect against the dihydrofolate reductase (DHFR) enzyme from humans and Staphylococcus aureus.
Introduction
Pyrimidine and its related derivatives have long been known as efficient chemotherapeutic agents.1 The chemotherapeutic potency of pyrimidine-based drugs is attributed to their inhibitory effect on the biosynthesis of vital enzymes responsible for nucleic acids such as thymidylate synthetase (TSase), thymidine phosphorylase (TPase), dihydrofolate reductase (DHFR) and reverse transcriptase (RTase). Several pyrimidine-based drugs are currently used as efficient anticancer2–6 and antiviral agents against human immunodeficiency viruses (HIV),7–10 hepatitis B virus (HBV),11,12 herpes simplex virus (HSV)13 and SARS-CoV virus.14 In addition, several pyrimidine-based drugs are currently used as antibacterial agents. Trimethoprim was early discovered as a potent drug mainly in the treatment of urinary tract infections acting by inhibition of dihydrofolate reductase (DHFR).15 The next generations of DHFR inhibitors including brodimoprim,16 epiroprim17 and iclaprim18 were developed as highly potent antibacterial drugs for the treatment of severe respiratory tract infections. DHFR inhibitors bind bacterial DHFR 105 times tighter than it does to vertebrate DHFR. By virtue of their mechanism of action, antibacterial DHFR inhibitors exert their activity through blocking synthesis of DNA, RNA and proteins, thereby arresting bacterial cell growth.19,20 Pyrimidine-based DHFR inhibitors are also used as potent antiprotozoal agents for the treatment of malaria,21,22 trypanosomiasis23 and leishmaniasis.24 It has been reported that the barbituric acid, a pyrimidine heterocylic nucleus, was used as a molecular recognition species by non-covalent interactions especially hydrogen bonds or via coordination to metal ions. Using this molecular recognition approach, different types of receptors that belong to organic, inorganic and organometallic have been developed for barbituric acid.25In view of the diverse applications of the pyrimidine derivatives, we have synthesized two pyrimidine-5-carbonitrile derivatives and crystal structures of these compounds have been studied in detail in the present investigation. The reported compounds vary with respect to the thiophene and phenyl substituents. A CSD (Cambridge Structural Database; CSD Version 5.41, November 2019; update 1 March 2020) search26 was conducted using the compound 1 as a template in which substituents at 2 and 4 positions kept ‘X’ (any) and this search yielded 8 hits (csd refcodes: GAHTIS, ILEREY, MUTGOY, MUTGUE, OKUWEY, SEDTAY, XAYLUF and ZAPQEO). In all these structures, a strong and directional N–H⋯O hydrogen bonded R22(8) synthon is formed between amino and carbonyl groups of the pyrimidine ring except in 2-amino-4-(3,4-dimethyoxyphenyl)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile N,N-dimethylformamide solvate (csd refcode: SEDTAY27). This self-assembled R22(8) synthon was disrupted due to the presence of N,N-dimethylformamide solvate. However, the carbonyl group of the solvate has participated in a N–H⋯O bond with amino group of the pyrimidine moiety.
Among 8 hits, 3 structures (6-oxo-4-propyl-2-(propylthio)-1,6-dihydropyrimidine-5-carbonitrile; csd refcode: ILEREY,28 2-[(4-fluorobenzyl)sulfanyl]-4-(2-methylpropyl)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile; csd refcode: OKUWEY29 and 2-[(2-methoxyethyl)sulfanyl]-4-(2-methylpropyl)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile; csd refcode: ZAPQEO30) were found to be closely related structures to the title compounds. In these 3 structures, the S atom is present at the 2nd position of the pyrimidine ring. In addition to hydrogen bond donors and acceptors, two chalcogen atoms (O and S) also present in these 3 structures and in the title compounds. The chalcogen bond (ChB) is one of the types of non-covalent interactions and has gained great interest in studying the role of such a bond in different areas including crystal engineering, materials science and biological sciences etc. The chalcogen bond is defined as R–Ch⋯A, where Ch is the chalcogen bond donor (electron-poor) and A is the chalcogen bond acceptor (electron-rich).31 The lone pair carbonyl and π system could act as chalcogen acceptors in the title compounds and their closely related structures. Therefore, a detailed investigation has been performed to study the nature and strength of chalcogen bond in the title compounds in addition to conserved and self-assembled R22(8) synthon mediated by the N–H⋯O hydrogen bond in pyrimidine-5-carbonitrile containing compounds.
To gain qualitative and quantitative insights into intra- and intermolecular interactions, we performed different analyses, including the Hirshfeld surface, 2D-fingerprint plots, energy frameworks, PIXEL energy, and Bader's quantum theory of atoms-in-molecules (QTAIM).32 The first four criteria ((a) topology, (b) electron density, (c) Laplacian of electron density and (d) mutual penetration of the hydrogen and the acceptor atom) of Koch–Popelier33 have been used to characterize the nature and strength of non-covalent interactions as previously reported.34,35 In addition, molecular docking study of the title pyrimidine-5-carbonitrile derivatives as DHFR inhibitors is reported.
Materials and methods
Synthesis and crystallization
The pyrimidine-5-carbonitrile derivatives 1 and 2 were synthesized starting with the appropriate aldehydes A via condensation with thiourea B and ethyl cyanoacetate C, in ethanol, in the presence of anhydrous potassium to yield the intermediates 6-(thiophen-2-yl)-2-thiouracil-5-carbonitrile D36 and 6-phenyl-2-thiouracil-5-carbonitrile E,37 which were reacted with bromoethane in the presence of potassium carbonate to yield the target compounds 1 and 2 (Scheme 1).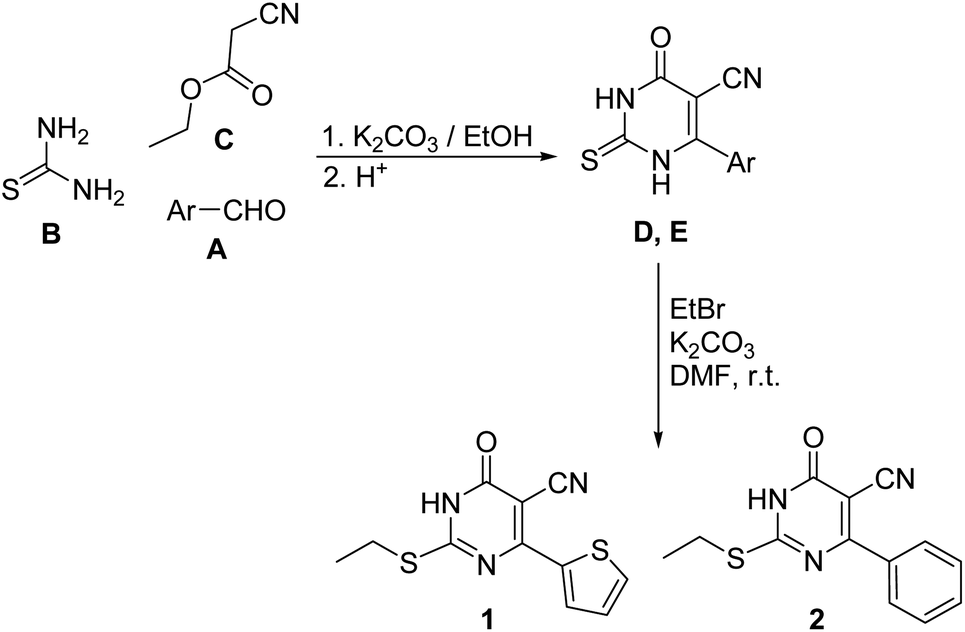 | ||
| Scheme 1 The synthetic pathway for compounds 1 and 2. | ||
Bromoethane (1.1 g m, 0.01 mol) and anhydrous potassium carbonate (1.38 g, 0.01 mol) were added to a solution of the appropriate 2-thiouracil-5-carbonitrile D or E (0.01 mol) in DMF (10 mL) and the mixture was stirred at room temperature for 12 hours. Water (15 mL) was the added to the reaction with stirred for further 30 minutes. The precipitated crude product was filtered, washed with cold water, dried and crystallized from aqueous ethanol. The pure single crystals of compounds 1 and 2 were obtained by slow evaporation of ethanolic solution at room temperature.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 177.60 (C-6). FT-IR (in cm−1) (NH: 3451, thiophene CH: 3028, aliphatic CH: 2841, CN: 2218, CO: 1651, CN: 1522).O), 177.54 (C-6). FT-IR (in cm−1) (NH: 3450, aromatic CH: 3018, aliphatic CH: 2849, CN: 2219, CO: 1657, CN: 1534). The FT-IR spectra for compounds 1 and 2 are given in the ESI.†
O), 177.60 (C-6). FT-IR (in cm−1) (NH: 3451, thiophene CH: 3028, aliphatic CH: 2841, CN: 2218, CO: 1651, CN: 1522).O), 177.54 (C-6). FT-IR (in cm−1) (NH: 3450, aromatic CH: 3018, aliphatic CH: 2849, CN: 2219, CO: 1657, CN: 1534). The FT-IR spectra for compounds 1 and 2 are given in the ESI.†Single crystal X-ray diffraction
X-ray intensity data were measured at 160 K from single crystals of 1 and 2 on a Rigaku OD XtaLAB Synergy, Dualflex, Pilatus 200 K diffractometer using a single wavelength X-ray source (Cu Kα radiation: λ = 1.54184 Å). Pre-experiment, data collection, data reduction and analytical absorption correction38 were performed with the program suite CrysAlisPro (Version 1.171.40.39a, Rigaku Oxford Diffraction, 2018).Using Olex2,39 the structure was solved with the SHELXT structure solution program40 and the structural refinement was performed with the SHELXL 2018/3 program package.41 In 2, the phenyl ring was disordered over two sets of positions with site-occupancy factors of 0.501(3) and 0.499(3). For both structures, the position of amine H atom was located from a difference Fourier map and refined freely along with its isotropic temperature factors. The methyl H atoms were constrained to an ideal geometry with C–H = 0.98 Å and Uiso(H) = 1.5Ueq(C), but were allowed to rotate freely about the C–C bond. The remaining H atoms were fixed by a riding model approach with C–H = 0.95–0.99 Å and Uiso(H) = 1.2Ueq(C). PLATON was used to check the result of the X-ray analysis.42Details of theoretical calculations
For all the density functional theory (DFT) calculation, the M06-2X functional43 and cc-pVTZ basis set44 was used with the incorporation of Grimme's dispersion correction (D3).45 The geometry optimization carried out using the refined models of 1 and 2 (major disordered component) with Gaussian 09 program.46 Further, the optimized structures were subjected to vibrational frequency calculation in order to confirm the energy minima on the potential energy surface.The H involving bond lengths were adjusted to their typical neutron diffraction values (C–H = 1.083 Å and N–H = 1.009 Å). After resetting the bond lengths, the resultant molecular geometries were used for the following analysis: (i) qualitative analysis of intermolecular interactions using Hirshfeld surface47 and 2D-fingerprint plots48 with the program CrystalExplorer-17.5 (ref. 49) and (ii) intermolecular interaction energies (Etot) for dimers of 1 and 2 using PIXELC module of CLP program package.50–53 The energy frameworks of these two structures (for 2, the major disordered component was considered) were performed using CrystalExplorer with B3LYP/6-31G(d,p) level of theory.54 For the PIXELC calculation, the electron density for the monomer of 1 and 2 (both major and minor disordered components) has been obtained separately at the MP2/6-31G** level of theory using the Gaussian 09 program.
The topological analysis of the intra- and intermolecular interactions was carried out using AIMALL package.55 For this calculation, the wave functions were calculated at normalized crystal structure geometries with M06-2X-D3/cc-pVTZ level of theory. The dissociation energy (De) for different non-covalent interactions was obtained by EML formula.56 Various topological properties such as ρ(r), ∇2ρ(r), V(r), G(r) and H(r) were computed for intra and intermolecular interactions at their bond critical points (BCPs). Further, the wave functions and cube files were used as input for the molecular electrostatic potential surfaces of 1 and 2. These surfaces were mapped over the electron density at 0.001 au using WFA-SAS suite.57
Molecular docking analysis
The 3D structures of dihydrofolate reductase (DHFR) enzyme from two different species (Homo sapiens; PDB ID: 2W3A and Staphylococcus aureus; PBD ID: 2W9H) were used in the present study to investigate the anticancer and antibacterial activities of the title pyrimidine derivatives utilizing an in silico molecular docking approach. The docking calculation was performed using the Schrödinger suite 2019-4 (Schrödinger, LLC, New York, NY, 2019). For docking calculation, the protein structure was prepared using the protein prep wizard module with default settings. Similarly, the title compounds and control inhibitor (trimethoprim) were prepared using the liprep module with OPLS3e force field. The grid box was constructed based on the position of the trimethoprim inhibitor complexed with human and bacterial enzymes. The glide XP docking58 was performed to predict the best binding pose and assess inhibitory potential using docking scores.Results and discussion
The present work probes the role of two substituents (thiophene and phenyl moieties) at the 4th position of the pyrimidine ring on the intermolecular interactions, crystal packing, evaluation of non-covalent interactions, energy frameworks and inhibitory potential against DHFR enzyme from human and methicillin resistant Staphylococcus aureus. Further, a detailed evaluation of the intermolecular interactions using the QTAIM approach is presented.Description of crystal structures of 1 and 2
Single crystal data and refinement parameters for 1 and 2 are presented in Table 1. Compound 1 {systematic name: 2-(ethylthio)-1,6-dihydro-6-oxo-4-(thiophen-2-yl)pyrimidine-5-carbonitrile} crystallizes in the monoclinic system with the space group P21/n. Compound 2 {systematic name: 2-(ethylthio)-1,6-dihydro-6-oxo-4-phenylpyrimidine-5-carbonitrile} crystallizes in the triclinic system with the centrosymmetric P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. Thermal ellipsoidal plots of compounds 1 and 2 are shown in Fig. 1. As mentioned in the experimental section, the phenyl ring of 2 was disordered over two orientations with nearly equal site occupancy factors. The thioethyl moiety is nearly coplanar with the pyrimidine ring in both 1 and 2. The dihedral angle formed between the mean planes of these groups being 7.28° in 1 and 6.37° in 2. It should be noted that the phenyl ring (27.45–28.20° in minor and major disordered components) is more twisted than the thiophene ring (5.51°) with respect to the mean plane of the pyrimidine ring.
space group. Thermal ellipsoidal plots of compounds 1 and 2 are shown in Fig. 1. As mentioned in the experimental section, the phenyl ring of 2 was disordered over two orientations with nearly equal site occupancy factors. The thioethyl moiety is nearly coplanar with the pyrimidine ring in both 1 and 2. The dihedral angle formed between the mean planes of these groups being 7.28° in 1 and 6.37° in 2. It should be noted that the phenyl ring (27.45–28.20° in minor and major disordered components) is more twisted than the thiophene ring (5.51°) with respect to the mean plane of the pyrimidine ring.
| 1 | 2 | |
|---|---|---|
| Crystal data | ||
| Chemical formula | C11H9N3OS | C13H11N3OS |
| Mr | 263.33 | 257.31 |
| Crystal system, space group | Monoclinic, P21/n | Triclinic, P |
| Temperature (K) | 160 (1) | 160 (1) |
| a, b, c (Å) | 14.5546 (2), 4.9907 (5), 17.1813 (3) | 4.6908 (2), 9.2376 (3), 14.6613 (3) |
| α, β, γ (°) | 90, 113.268 (2), 90 | 94.308 (2), 93.766 (2), 100.924 (3) |
| V (Å3) | 1146.50 (12) | 619.95 (4) |
| Z | 4 | 2 |
| Radiation type | Cu Kα (λ = 1.54184) | Cu Kα (λ = 1.54184) |
| μ (mm−1) | 4.10 | 2.25 |
| ρCalc (g cm−3) | 1.526 | 1.378 |
| Crystal size (mm3) | 0.09 × 0.09 × 0.03 | 0.13 × 0.03 × 0.02 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Data collection | ||
| Diffractometer | XtalLab Synergy, Dualflex, Pilatus 200K diffractometer | XtalLab Synergy, Dualflex, Pilatus 200K diffractometer |
| Absorption correction | Analytical | Analytical |
| Tmin, Tmax | 0.745, 0.908 | 0.848, 0.960 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 11447, 2336, 2147 |
13068, 2524, 2219 |
| Rint | 0.027 | 0.037 |
| (sinθ/λ)max (Å−1) |
0.625 | 0.625 |
|
||
| Refinement | ||
| R[F2 > 2σ(F2)], wR(F2), S | 0.025, 0.069, 1.06 | 0.033, 0.089, 1.02 |
| No. of reflections | 2336 | 2524 |
| No. of parameters | 159 | 205 |
| H-atom treatment | H-atom treated by a mixture of independent and constrained refinement | H-atom treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.29, −0.23 | 0.30, −0.31 |
| CCDC no. | 2022218 | 2022219 |
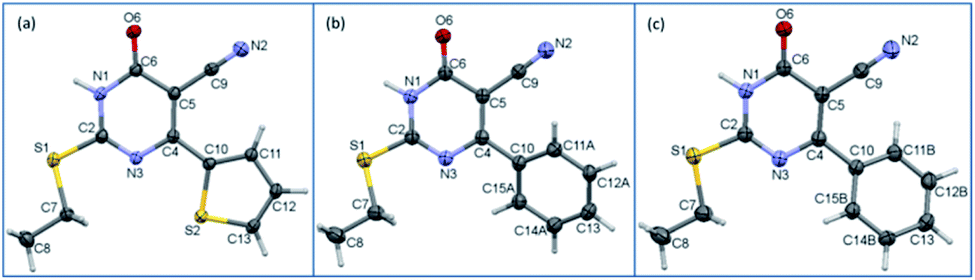 | ||
| Fig. 1 ORTEP diagrams of compounds (a) 1, (b) major disordered component of 2 and (c) minor disordered component of 2 drawn at the 50% probability level. Atom-numbering scheme is given. | ||
The structural superimposition of X-ray structures of 1 and 2 is given in Fig. 1. As can be seen from Fig. 1, the phenyl ring is slightly twisted compared to thiophene ring. The optimized structures of 1 and 2 are in close agreement with their corresponding X-ray structure geometry.
Intramolecular interactions
The molecular conformation of 1 and 2 is stabilized by an intramolecular C–H⋯C interaction involving one of the protons (H11) of the thiophene ring and the C9 atom of cyano group. A similar intramolecular interaction has also been observed in many cyano containing organic compounds reported earlier from our group.59–61 This intramolecular interaction was further examined in both X-ray and optimized structures using QTAIM analysis. In the X-ray geometry of 1, the bond path is established between H and cyano carbon (hence it is a C–H⋯C interaction) whereas in the corresponding optimized structure, the bond path is shifted to cyano N from cyano carbon (hence it is a C–H⋯N interaction). The molecular graphs showing the intramolecular interaction in X-ray and optimized structures of 1 and 2 are depicted in Fig. 2. The topological parameters for these interactions are summarized in Table S1.† From the analysis, we note that the intramolecular interaction observed in X-ray and optimized structures of 1 and 2 shows van der Waals type nature (based on KP-4 rule) except C–H⋯N interaction in the optimized structure of 1. This intramolecular C–H⋯N interaction displays hydrogen bonding character. Moreover, intramolecular C–H⋯C interaction is slightly stronger in the X-ray structure of 2 compared to 1 and the optimized structure of 2. It is of interest to note that the intramolecular C–H⋯C interaction is absent as evident from the topological analysis in related structures in which aliphatic groups are present at the 2nd position of the pyrimidine nucleus.28–30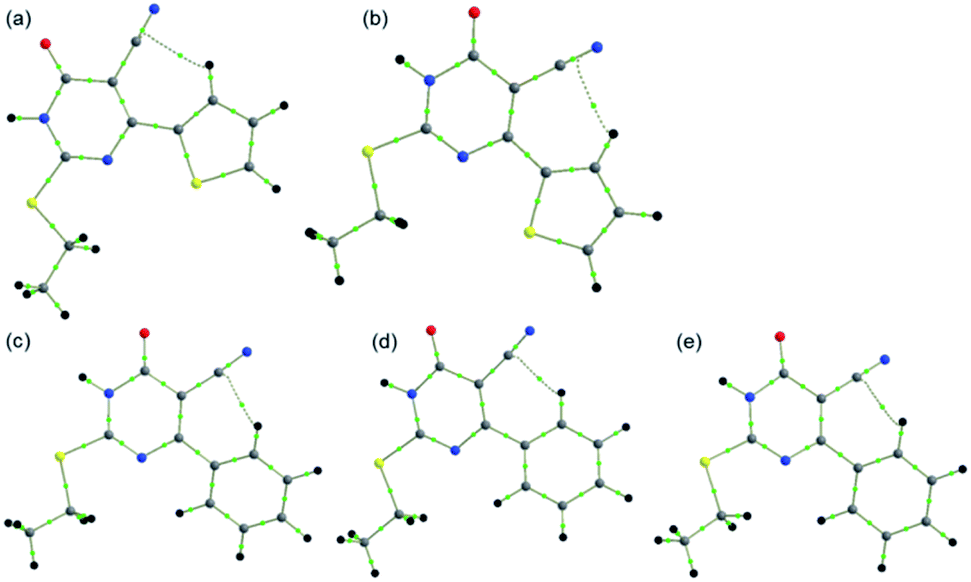 | ||
| Fig. 2 Molecular graphs showing intramolecular interactions in (a) X-ray structure of 1 (b) optimized structure of 1, (c) X-ray structure of 2 (major disordered component), (d) X-ray structure of 2 (minor disordered component) and (e) optimized structure of 2. | ||
Hirshfeld surface (HS) and 2D-fingerprint plots
In 1, the highly directional N1–H1⋯O6 hydrogen-bonded dimer is showing bright red areas on the HS, whereas the intermolecular C7–H72⋯N2 interaction and S2⋯S2 (lp⋯lp) contact are also visible on the HS with pale red marks. In 2, two intermolecular hydrogen bonding interactions (N1–H1⋯O6 and C8–H81⋯N2) are visible on the HS and large red areas appeared over the phenyl ring due to disorder. Fig. 3 highlights the short intermolecular interactions observed in 1 and 2 and these features suggest that these interactions play an essential role in the stabilization of crystal structure.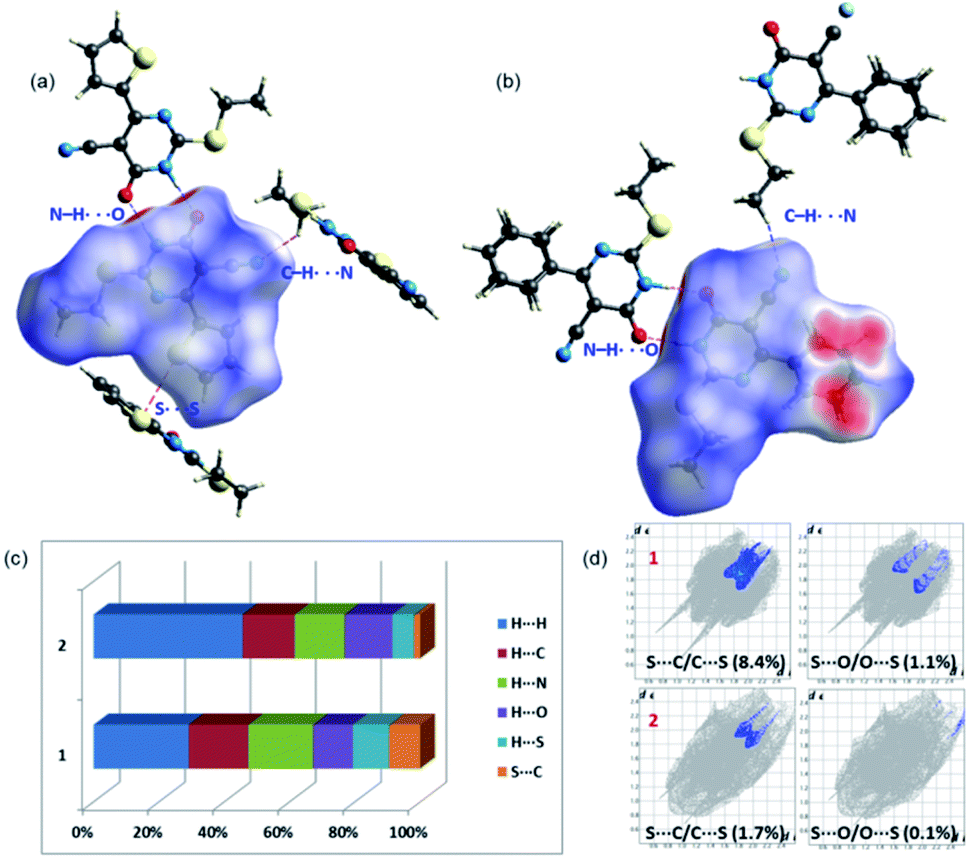 | ||
| Fig. 3 Short intermolecular interactions visualized using Hirshfeld surfaces mapped over dnorm for (a) 1 and (b) 2 and (c) the relative contribution of different intermolecular interactions observed in 1 and 2 (d) the relative contribution of two chalcogen contacts (S⋯C/O) in 1 and 2. | ||
To explain the effect of thiophene and phenyl substituents on the intermolecular interactions observed in the solid state, we obtained 2D-fingerprint plots (2D-FP) (Fig. S1†). The result shows that the intermolecular H⋯H contacts have a significant contribution to both structures. The intermolecular H⋯N/C/O interactions provide substantial contributions toward crystal packing and the relative contributions of these contacts are comparable in 1 and 2. However, there is a substantial increase of H⋯H (16.2%) contacts in 2 compared to 1 due to the presence of phenyl substituent and its disorder. Further, the intermolecular S⋯C(π) contact described as a chalcogen bond, is reduced in 2 by 6.6% towards the crystal packing compared to 1. It is worthy to note that the 3.6% increase of S⋯S contacts to the total HS area due to the presence of additional S atom in 1 and its participation in the intermolecular interaction. The corresponding contact appeared around 3.4 Å in the FP plot and this contact contributes about 1.0% towards the crystal packing of a related structure.29
It should be pointed out that the chalcogen bond (S⋯O) contributes 1.1% to the total HS area in 1 and the corresponding interaction is found to be 0.1%. In one of the closely related structures, the corresponding contact contributes about 1.7% to the crystal packing.30 The existence of chalcogen and other intermolecular interactions in 1 and 2 is further evaluated by QTAIM analysis.
Analysis of molecular electrostatic potential surface
The molecular electrostatic potential surface maps for X-ray and optimized structures of 1 and 2 were generated. The distribution of electrostatic potentials is very similar in both structures including their corresponding optimized structures (Fig. 4). However, the values of positive (Vs,max) and negative (Vs,min) electrostatic potentials are varied. In 1, the most positive electrostatic potential has occurred at the amine proton of the pyrimidine ring with the Vs,max value of 53.5 kcal mol−1 and at other important protons H72 and H12 have Vs,max values of 23.2 and 21.4 kcal mol−1, respectively. The most negative electrostatic potential observed for carbonyl oxygen (O6; Vs,min: −46.3 kcal mol−1) followed cyano nitrogen (N2; Vs,min: −41.1 kcal mol−1).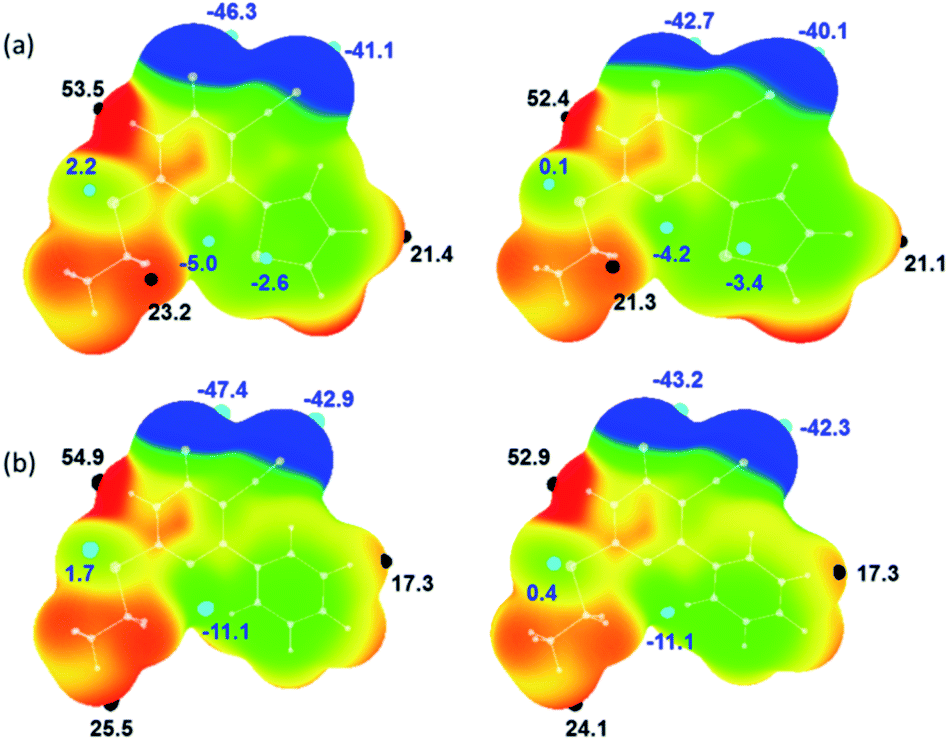 | ||
| Fig. 4 Molecular electrostatic potential of (a) X-ray structure of 1 (left) and its optimized structure (right) and (b) X-ray structure of major disordered component of 2 (left) and its optimized structure (right) mapped over the electron density isosurface at 0.001 au. Colour scale (in kcal mol−1): red: greater than 20; yellow: between 20 and 0; green: 0 to −20 and blue: greater than −20. The important most positive (Vs,max) and negative (Vs,min) electrostatic potentials are given. | ||
It is of interest to note that the pyrimidine N3 atom has a weak accepting tendency as evident from the Vs,min value of −5.0 kcal mol−1. The sulphur atoms in 1 show distinct features of electrostatic potentials and the thiophene S2 atom has relatively more negative electrostatic potential (Vs,min: −2.6 kcal mol−1) compared to S1 atom (Vs,min: 2.2 kcal mol−1). As mentioned above, compound 2 possesses similar electrostatic potentials and the values of Vs,max and Vs,min are comparable in the X-ray structures of 1 and 2. As shown in Fig. 4, the values of positive and negative electrostatic potentials vary between X-ray and optimized structure for some of the donor and acceptors atoms. This variation is indicating the effect of crystal packing.
Dimers in the crystal structure of 1
The intermolecular interactions energies calculated by the PIXEL method for dimers of 1 are summarized in Table 2 and these energies are ranging from −20.9 to −4.3 kcal mol−1. These dimers (Fig. 5 and 6) are stabilized by both classical and non-classical non-covalent interactions such as N–H⋯O, C–H⋯π, C–H⋯N, C⋯C, S⋯S, S⋯C and S⋯OC interactions. The latter two non-covalent interactions can be classified as chalcogen bonds.
| Dimer | CD | Symmetry | Important interactions | Geometry H⋯A (Å)/∠D–H⋯A (°) | ECoul | Epol | Edisp | Erep | Etot |
|---|---|---|---|---|---|---|---|---|---|
| a Neutron values are given for all D–H⋯A interactions. CD: centroid-to-centroid distance of the molecular pair. | |||||||||
| Compound 1 | |||||||||
| D1 | 8.353 | −x + 1, −y, −z + 1 | N1–H1⋯O6 | 1.75/179 | −29.3 | −13.9 | −7.1 | 29.4 | −20.9 |
| D2 | 4.991 | x, y − 1, z | C7–H71⋯Cg1 | 2.90/156 | −2.8 | −2.4 | −13.0 | 10.6 | −7.6 |
| S2⋯C9 | 3.520 (4) | ||||||||
| D3 | 9.058 | −x + 2, −y + 1, −z + 1 | C11⋯C12 | 3.487 (3) | −3.0 | −0.9 | −5.3 | 3.2 | −6.0 |
| D4 | 10.092 | −x + 2, −y, −z + 1 | C12–H12⋯N2 | 2.63/129 | −3.8 | −1.4 | −3.0 | 2.7 | −5.4 |
| D5 | 7.068 | −x + 1, −y + 1, −z + 1 | S1⋯O6C6 |
3.683 (4) | −2.7 | −1.5 | −5.5 | 4.9 | −4.9 |
| D6 | 6.827 | −x + 3/2, y − 1/2, −z + 1/2 | S2⋯S2 | 3.497 (4) | −2.2 | −2.0 | −9.0 | 8.5 | −4.6 |
| D7 | 9.017 | −x + 1/2, y + 1/2, −z + 1/2 | C7–H72⋯N2 | 2.57/132 | −3.2 | −0.9 | −2.5 | 2.3 | −4.3 |
|
|||||||||
| Compound 2 | |||||||||
| D1 | 8.266 | −x + 1, −y + 1, −z + 2 | N1–H1⋯O6 | 1.72/177 | −29.2 | −13.9 | −6.8 | 32.8 | −17.2 |
| D2 | 10.071 | −x + 2, −y + 1, −z + 1 | C12A–H121⋯N2 | 2.67/121 | −4.2 | −1.6 | −4.6 | 3.9 | −6.4 |
| D3 | 9.535 | x − 1, y − 1, z | C8–H83⋯O6 | 2.67/131 | −1.5 | −0.5 | −1.9 | 0.9 | −3.1 |
| D4 | 11.853 | x − 2, y − 1, z | C8–H81⋯N2 | 2.45/161 | −2.9 | −0.8 | −1.3 | 2.0 | −3.0 |
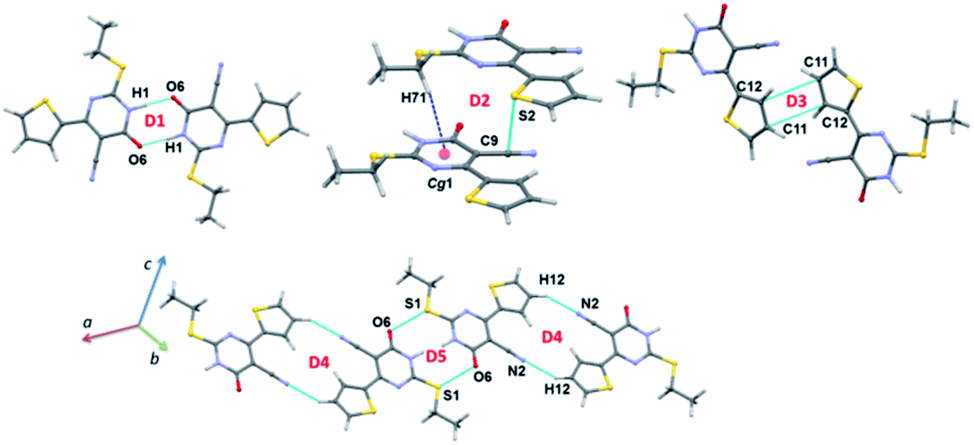 | ||
| Fig. 5 Different dimers observed in the crystal structure of 1. | ||
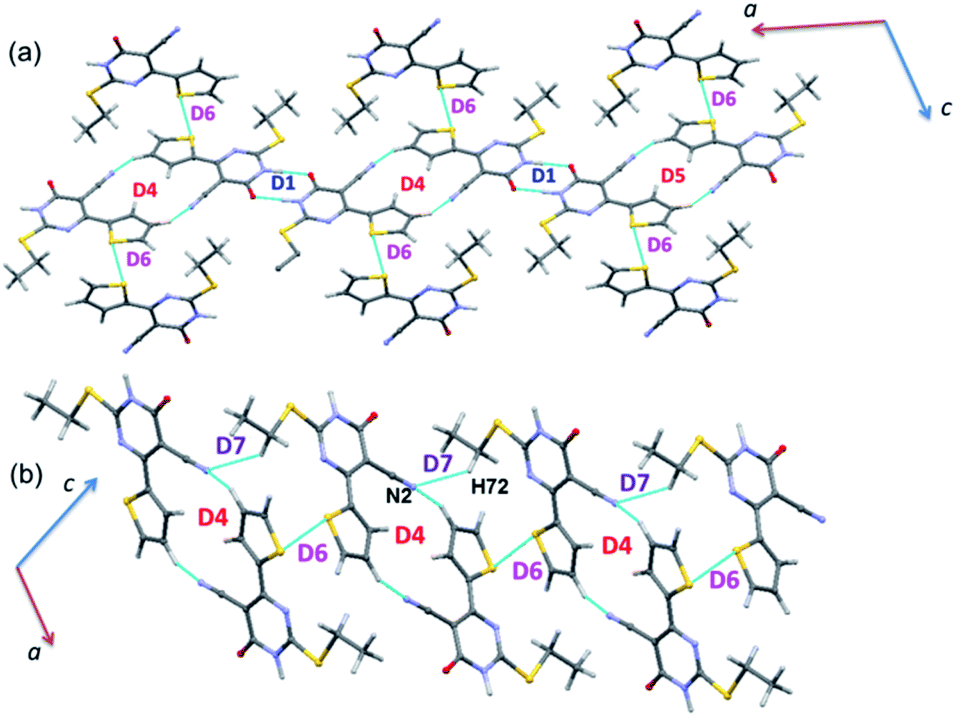 | ||
| Fig. 6 Molecular sheets formed by various combinations of motifs in the crystal structure of 1. | ||
The strong dimer (D1; Etot: −20.9 kcal mol−1) forms by a linear N–H⋯O hydrogen bond involving amine group (N1–H1) and carbonyl O6 atom and this hydrogen bond produces a cyclic R22(8) synthon. The contribution of electrostatic energy was calculated to be 86% towards the stabilization of this cyclic synthon. The dimer D2 (Etot: −7.6 kcal mol−1) is stabilized by intermolecular C–H⋯π (centroid of the pyrimidine ring) and stabilization is further supported by a weak chalcogen bond S⋯C(π) contact. This weak chalcogen bond is established by sum of vdW radii of S and C atoms +0.03 Å. It should be noted that this dimer is predominantly dispersive with a contribution of 76%. It is realized that this one of the rare chalcogen bonds formed between thiophene S and cyano C atom. To support this statement, a CSD search was performed using sum of the vdW radii of S and C +0.03 Å cut-off and we found that 46 hits contain this contact and the contact distance ranging from 3.359 to 3.529 Å.
It is to be noted that adjacent D1 dimers are interlinked by dimer D2 which is further extended along the crystallographic b axis. Dimer D3 (Etot: −6.0 kcal mol−1) supports by a weak and long intermolecular C⋯C (involving C11 and C12) contact. This contact only appears when the distance being sum of vdW radii of interacting atoms +0.09 Å. The electrostatic and dispersion energies contribute 42% and 58%, respectively towards the stabilization of this dimer. Moreover, each molecule in this dimer (D3) interacts with the neighbouring molecule via S⋯S (involving thiophene S2 atoms) contact (dimer D6). The intermolecular interaction energy for dimer D6 being −4.6 kcal mol−1 and the dispersion energy contributes 68% towards the stabilization of D6.
Intermolecular C–H⋯N interaction (involving H12 and N2) stabilizes the dimer D4 (Etot: −5.4 kcal mol−1) with 63% of electrostatic energy contribution towards the stabilization. This dimer generates a R22(16) motif. Interestingly, another chalcogen bond of the type S⋯OC (involving S1 and O6) stabilizes the dimer D5 with an intermolecular interaction energy value of −4.9 kcal mol−1. It should be noted that this chalcogen bond is established only when distance cutoff is relaxed i.e. sum of the vdW radii of S and O +0.23 Å. The electrostatic and dispersion energies contribute 43 and 57%, respectively, towards the stabilization of this dimer. It is also pointed out that dimer D5 is flanked by on both sides by dimer D4. A CSD search identifies this chalcogen bond in 993 structures.
The energetically least dimer D7 (Etot: −4.3 kcal mol−1 with 62% contribution of electrostatic energy towards stabilization) forms by an intermolecular C–H⋯N (involving H72 and N2) interaction. This non-covalent interaction links the molecules of 1 into a zigzag C(9) chain which runs parallel to the c axis. Further, dimers D4, D6 and D7 collectively generate two different closed synthons and these synthons are alternately arranged in a zigzag manner in the solid state (Fig. 6).
Dimers in the crystal structure of 2
PIXEL energy analysis was carried out for both major and minor disordered components. For the major component, four molecular dimers were identified and the intermolecular interactions for these dimers are ranging from −17.2 to −3.0 kcal mol−1 (Table 2). These dimers are stabilized by intermolecular N–H⋯O, C–H⋯O and C–H⋯N interactions (Fig. 7). No non-classical contacts including chalcogen bond exist in this structure.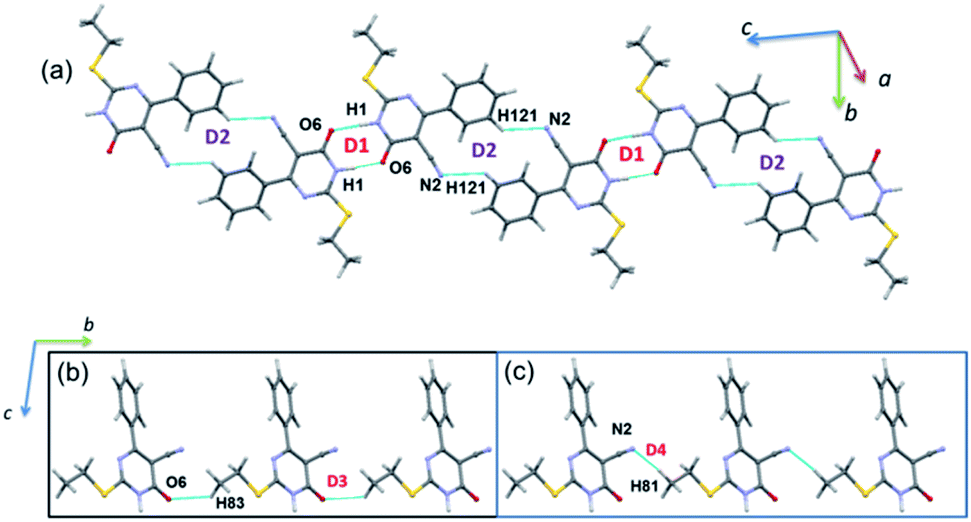 | ||
| Fig. 7 (a) Molecular ribbon formed by motifs D1 and D2 and (b) one-dimensional molecular chain formed by motifs D3 and D4 in the crystal structure of 2. | ||
As observed in 1, the strong dimer (D1; Etot: −17.2 kcal mol−1) stabilizes by an intermolecular N–H⋯O hydrogen bond with a cyclic R22(8) synthon. The contribution of electrostatic energy was calculated to be 86% towards the stabilization of this hydrogen-bonded dimer. Moreover, this dimer is relatively weaker than dimer D1 in 1 due to the repulsion contribution as compared to 1. The dimer D2 (Etot: −6.4 kcal mol−1) forms by an intermolecular C–H⋯N interaction in which cyano N2 atom functions as an acceptor, as observed in 1. Though the donor moiety is different in 1 (thiophene ring) and 2 (phenyl ring), a cyclic synthon (R22(16)) formed by this interaction is the same. Further, the electrostatic and dispersion energies contribute about 56 and 44% towards stabilizing D2 dimer in 2. The dimers D1 and D2 are arranged in an alternate fashion that runs parallel to the crystallographic c axis (Fig. 7(a)). Unlike in 1, the carbonyl group is also involved as an acceptor for the intermolecular C–H⋯O interaction (dimer D3, Etot: −3.1 kcal mol−1). This interaction links the molecules into a C(8) chain which runs parallel to the b axis (Fig. 7(b)). Further, the intermolecular C–H⋯N interaction stabilizes the dimer D4 (Etot: −3.0 kcal mol−1). The adjacent molecules of 2 interlinked via this interaction forms as one-dimensional chain with the graph-set motif of C(10) which extended along the b axis (Fig. 7(b)). It is important to emphasize that the contribution of electrostatic (51%) and dispersion (49%) energies are nearly the same towards the stabilization of dimer D3. In contrast, dimer D4 is predominantly electrostatic in nature with the contribution of 74% towards the stabilization.
As mentioned above, the phenyl ring has participated in an intermolecular C–H⋯N (involving C12A–H121 and N2) interaction in the major disordered component of 2. The equivalent atoms are also engaged in an intermolecular C–H⋯N (involving C12B–H122 and N2) interaction in the minor disordered part. The remaining interactions come from the non-disordered atoms of 2 which are identical with those of interactions observed in the major disordered component.
Crystal packing and energy frameworks
The crystal packing of 1 can be described as a typical herringbone (viewed down a axis) or columnar (viewed down b axis). Molecules of 2 also packed as a columnar fashion (viewed down a and b axes). Recently, the energy frameworks have received considerable attention to understanding the polymorphism, effect of substituents on the crystal packing and mechanical properties of the crystals. The 3D topology of the energy frameworks for crystals 1 and 2 are depicted in Fig. 8. Overall, both electrostatic (red) and total interaction energy (blue) show similar topology for crystals 1 and 2. The large and small cylinders are placed alternately in the horizontal layer. In 1, the large cylinders represent intermolecular N–H⋯O hydrogen bond and a weak chalcogen bond (S⋯OC). A small cylinders in each horizontal layer represent intermolecular C–H⋯N interaction. The large cylinders in two adjacent layers are further interconnected by a small cylinder which represents a weak S⋯S interaction.
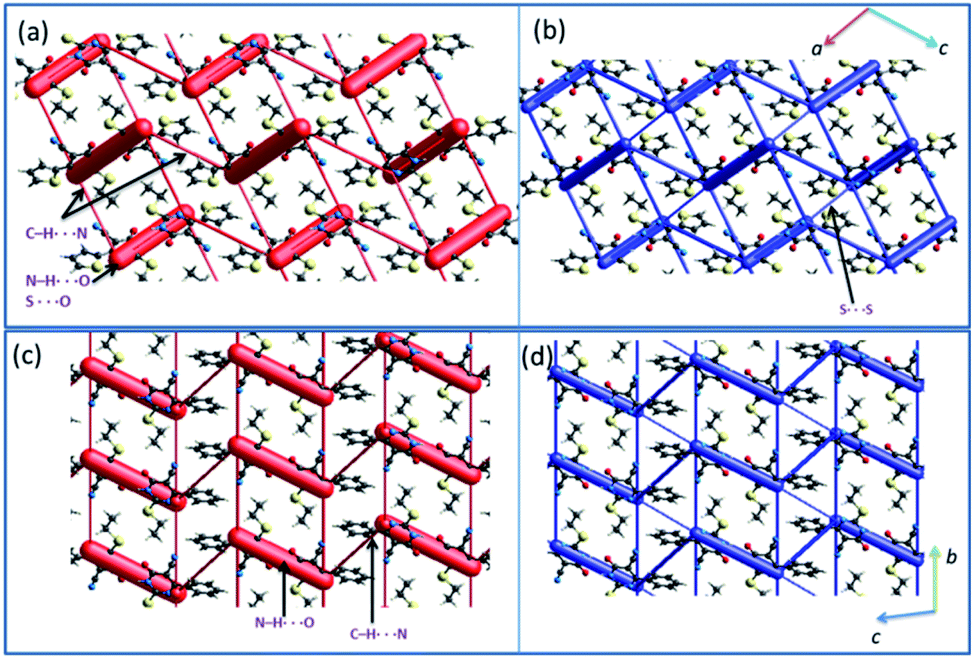 | ||
| Fig. 8 Energy frameworks for the crystal structures of (a and b) 1 and (c and d) 2 (major disordered component). The electrostatic (red) and total interaction energy (blue) components are shown as cylinders. | ||
In 2, the large cylinders belong to intermolecular N–H⋯O hydrogen bond and a small cylinder which links neighbouring large cylinders in the horizontal layer represent intermolecular C–H⋯N interaction. Although 3D topology of the energy frameworks looks similar for both 1 and 2, there is a difference in the large cylinders geometry. In 2, these cylinders are oriented in a parallel fashion in the same layer and as well as in the adjacent layers. These features are different in 1 i.e. the large cylinders are oriented in a parallel fashion in the same layers and not in the adjacent layers. Desiraju and co-workers investigated the bending behavior of a large number of structures. They found that the bending behavior was well correlated with the shortest crystallographic axis of ca. 4 Å.62 As shown in Fig. 8, the crystal packing of 1 (b = 4.99 Å) and 2 (a = 4.69 Å) viewed along the respective shortest crystallographic axis and these structures showed similar 3D topology of the energy frameworks. Based on the energy frameworks, one can conclude that the bending behavior could be similar to 1 and 2.
Analysis of isostructurality
The concept of supramolecular constructs (SC) which are subcomponents of a complete crystal structure was introduced by Gelbrich and Hursthouse.63,64 In this study, we used this concept to identify the similar packing arrangements in the title compounds and their closely related structures. The common packing motifs observed in the title compounds and their closely related structures are depicted in Fig. S2 and S3.† The structures 1 and 2 show 1D isostructurality (molecular chain) and the molecular chain has alternate N–H⋯O and C–H⋯N bonded synthons. The structures of 1 and ILEREY display 2D isostructurality and in both structures respective molecule forms as layers. These layers are primarily stabilized by N–H⋯O and C–H⋯N interactions. It is important to note that structures OKUWEY and ZAPQEO share 0D isostructurality (N–H⋯O mediated dimer) with the structure of 1. The cell parameters of 2 and ILEREY structures are comparable and these two structures share 3D isostructurality. This 3D isostructurality is further quantified using isostructurality index (Π) proposed by Fábián and Kálmán65 and the Π value is found to be 0.02 for this pair. Further, structures of 2 and OKUWEY show 0D SC (N–H⋯O mediated dimeric pair). However, no supramolecular construct observed between 2 and ZAPQEO.Evaluation of intermolecular interactions using QTAIM analysis
The topological parameters for the non-covalent interactions observed in structures of 1 and 2 (major disordered component) are summarized in Tables 3 and 4. The molecular graphs exhibiting the non-covalent interactions in these dimers are illustrated in Fig. S4 and S5.† In 1, there are three intermolecular interactions of the type N–H⋯O (dimer D1) and C–H⋯N (dimers D4 and D7) and all of these interactions showing hydrogen bonding character according to the KP-4 rule. As mentioned in the previous section, dimer D2 stabilizes with two interactions, namely C7–H71⋯Cg1(π) and a chalcogen bond S2⋯C9. The topological analysis suggests a bond path that established between H71 and N1 (pyrimidine ring) atoms resulting in the formation of C–H⋯N interaction. This interaction is found to be van der Waals in nature based on KP-4 rule.| Interaction | Rij (Å) | ρ(r) (e Å−3) | ∇2ρ(r) (e Å−5) | V(r) | G(r) | H(r) |
|
De |
|---|---|---|---|---|---|---|---|---|
| D1 | ||||||||
| N1–H1⋯O6 | 1.772 | 0.281 | 2.676 | −108.6 | 90.8 | −17.9 | 1.20 | 13.0 |
|
||||||||
| D2 | ||||||||
| C7–H71⋯N1 | 2.825 | 0.043 | 0.517 | −8.5 | 11.3 | 2.8 | 0.75 | 1.0 |
| S2⋯C9 | 3.614 | 0.041 | 0.482 | −7.5 | 10.3 | 2.8 | 0.73 | 0.9 |
|
||||||||
| D3 | ||||||||
| C11⋯C12 | 3.874 | 0.034 | 0.412 | −5.9 | 8.5 | 2.7 | 0.69 | 0.7 |
|
||||||||
| D4 | ||||||||
| C12–H12⋯N2 | 2.666 | 0.048 | 0.631 | −9.4 | 13.3 | 3.9 | 0.70 | 1.1 |
|
||||||||
| D5 | ||||||||
| S1⋯O6C6 |
3.571 | 0.038 | 0.419 | −7.1 | 9.3 | 2.1 | 0.77 | 0.9 |
|
||||||||
| D6 | ||||||||
| S2⋯S2 | 3.512 | 0.055 | 0.633 | −11.0 | 14.1 | 3.1 | 0.78 | 1.3 |
|
||||||||
| D7 | ||||||||
| C7–H72⋯N2 | 2.599 | 0.055 | 0.704 | −10.9 | 15.0 | 4.1 | 0.73 | 1.3 |
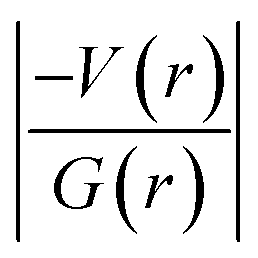
| Interaction | Rij (Å) | ρ(r) (e Å−3) | ∇2ρ(r) (e Å−5) | V(r) | G(r) | H(r) |
|
De |
|---|---|---|---|---|---|---|---|---|
| D1 | ||||||||
| N1–H1⋯O6 | 1.742 | 0.301 | 2.767 | −120.0 | 97.7 | −22.3 | 1.23 | 14.4 |
|
||||||||
| D2 | ||||||||
| C12A–H121⋯N2 | 2.736 | 0.046 | 0.596 | −9.1 | 12.7 | 3.6 | 0.72 | 1.1 |
|
||||||||
| D4 | ||||||||
| C8–H83⋯O4 | 2.710 | 0.041 | 0.513 | −8.5 | 11.2 | 2.7 | 0.76 | 1.0 |
|
||||||||
| D5 | ||||||||
| C8–H81⋯N2 | 2.476 | 0.064 | 0.841 | −13.3 | 18.1 | 4.8 | 0.74 | 1.6 |
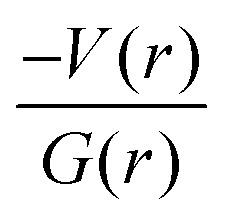
It is of interest to note that the N–H⋯O hydrogen bond exposes an intermediate bonding character between shared and closed-shell interaction as concluded using the following conditions: (i) ∇2ρ > 0; (ii) 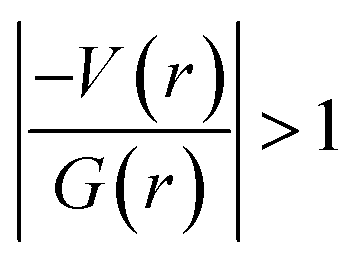 and (iii) H(r) < 0. Further, the remaining hydrogen bonds (C–H⋯N) belong to closed-shell interaction categories as evident from (i) ∇2ρ > 0; (ii)
and (iii) H(r) < 0. Further, the remaining hydrogen bonds (C–H⋯N) belong to closed-shell interaction categories as evident from (i) ∇2ρ > 0; (ii) 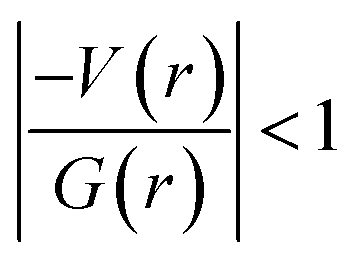 and (iii) H(r) > 0. The N–H⋯O hydrogen bond is calculated to be strong among other intermolecular interactions in 1 and the dissociation energy for this hydrogen bond is 13.0 kcal mol−1. The corresponding value for C–H⋯N hydrogen bonds is in the range of 1.0–1.3 kcal mol−1. As noted earlier, the chalcogen bonds of the type S⋯O and S⋯C interaction are weak in strength compared to hydrogen bonds and S⋯S contact and the dissociation energy for these interactions are the same. We also noted that the strength of S⋯S contact is comparable to that of one of the C–H⋯N (dimer D7) hydrogen bonds.
and (iii) H(r) > 0. The N–H⋯O hydrogen bond is calculated to be strong among other intermolecular interactions in 1 and the dissociation energy for this hydrogen bond is 13.0 kcal mol−1. The corresponding value for C–H⋯N hydrogen bonds is in the range of 1.0–1.3 kcal mol−1. As noted earlier, the chalcogen bonds of the type S⋯O and S⋯C interaction are weak in strength compared to hydrogen bonds and S⋯S contact and the dissociation energy for these interactions are the same. We also noted that the strength of S⋯S contact is comparable to that of one of the C–H⋯N (dimer D7) hydrogen bonds.
In 2 (major disordered component), though all four intermolecular (N–H⋯O, C–H⋯N and C–H⋯O type) interactions showing hydrogen bonding character based on KP-4 rule, one of the hydrogen bonds (N–H⋯O) demonstrate intermediate bonding character between shared and closed-shell interaction as observed in 1. This conclusion has been made based on the positive value of Laplacian of electron density (∇2ρ > 0), 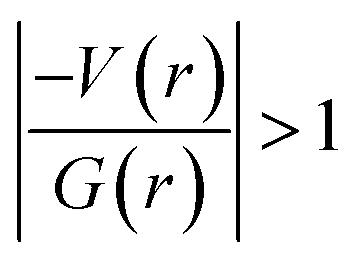 and H(r) < 0. The remaining three hydrogen bonds display closed-shell character of bonding (based on (∇2ρ > 0),
and H(r) < 0. The remaining three hydrogen bonds display closed-shell character of bonding (based on (∇2ρ > 0), 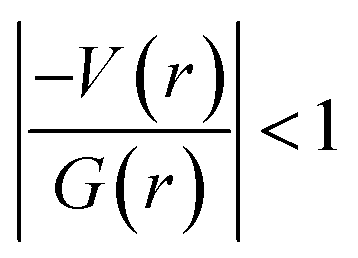 and H(r) > 0). The dissociation energy for the closed-shell interactions is in the range of 1.0–1.6 kcal mol−1. It should be important to note that the N–H⋯O hydrogen bond is found be to stronger (De = 14.4 kcal mol−1) compared to that of the corresponding hydrogen bond in 1. The distribution of Laplacian of electron density and the total electronic energy density (H(r)) for N–H⋯O hydrogen bond in 1 and 2 is depicted in Fig. 9. As can be seen from this figure that the total electronic energy density (H(r)) has uninterrupted regions of negative values represent the intermediate bonding character.
and H(r) > 0). The dissociation energy for the closed-shell interactions is in the range of 1.0–1.6 kcal mol−1. It should be important to note that the N–H⋯O hydrogen bond is found be to stronger (De = 14.4 kcal mol−1) compared to that of the corresponding hydrogen bond in 1. The distribution of Laplacian of electron density and the total electronic energy density (H(r)) for N–H⋯O hydrogen bond in 1 and 2 is depicted in Fig. 9. As can be seen from this figure that the total electronic energy density (H(r)) has uninterrupted regions of negative values represent the intermediate bonding character.
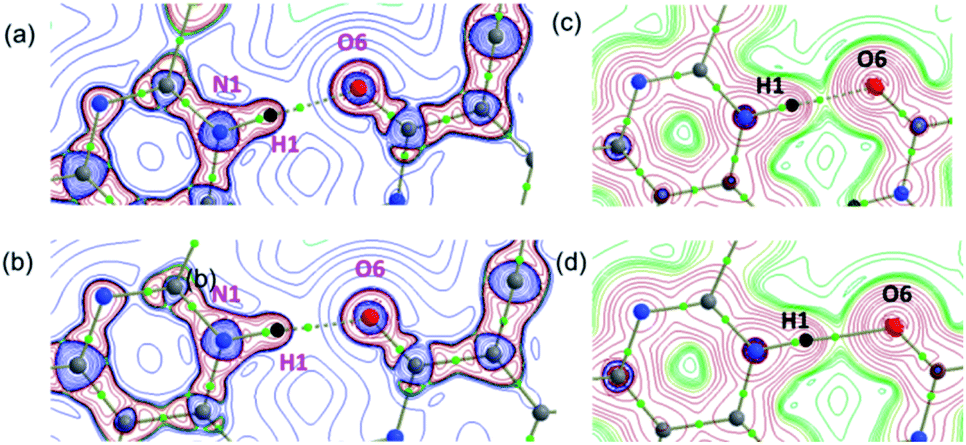 | ||
| Fig. 9 The distribution of the (a and b) Laplacian of the electron density and (c and d) the total energy density showing the formation of strong intermolecular N–H⋯O hydrogen bond in 1 and 2, respectively. These diagrams were plotted in the plane comprising the atoms involved in the hydrogen bond. Small green spheres indicate the bond critical points. | ||
Molecular docking
In the present study, we explored the anticancer and antibacterial activities of the title compounds against dihydrofolate reductase (DHFR) enzymes from two different species, namely Homo sapiens and Staphylococcus aureus, respectively employing in silico molecular docking approach. The glide XP docking scores of the title compounds are compared with the control inhibitors trimethoprim (Table 5). The result suggests that the glide XP scores for the title compounds are comparable with trimethoprim for both targets. Both title compounds make at least one hydrogen bond with the active residue of the DHFR enzymes. The predicted binding pose of the title compounds at the active site of the H.s DHFR and S.a DHFR and hydrogen bond interaction formed between ligand and protein is illustrated in Fig. 10. In all cases, either donor or acceptor group of the pyrimidine moiety has participated in the hydrogen bonding interaction with the active site residues. Specifically, the amine group in compound 1 is involved in a hydrogen bond with the backbone carbonyl group of the Val 115 residue of H.s DHFR. In contrast, the carbonyl group of 1 has participated in a hydrogen bond with the backbone amine group of Ala 7 of S.a DHFR enzyme. Similarly, the carbonyl group of 2 makes a hydrogen bond with the amine group of the conserved Ala 9 residue of H.s DHFR. In contrast, the carbonyl group of 2 involves a hydrogen bond with amine group of conserved Ala 7 of S.a DHFR.| Compound | Glide XP docking score | |
|---|---|---|
| DHFR | ||
| H. sapiens | S. aureus | |
| 1 | −6.5 | −6.9 |
| 2 | −5.4 | −7.5 |
| Trimethoprim (control inhibitor) | −6.0 | −7.3 |
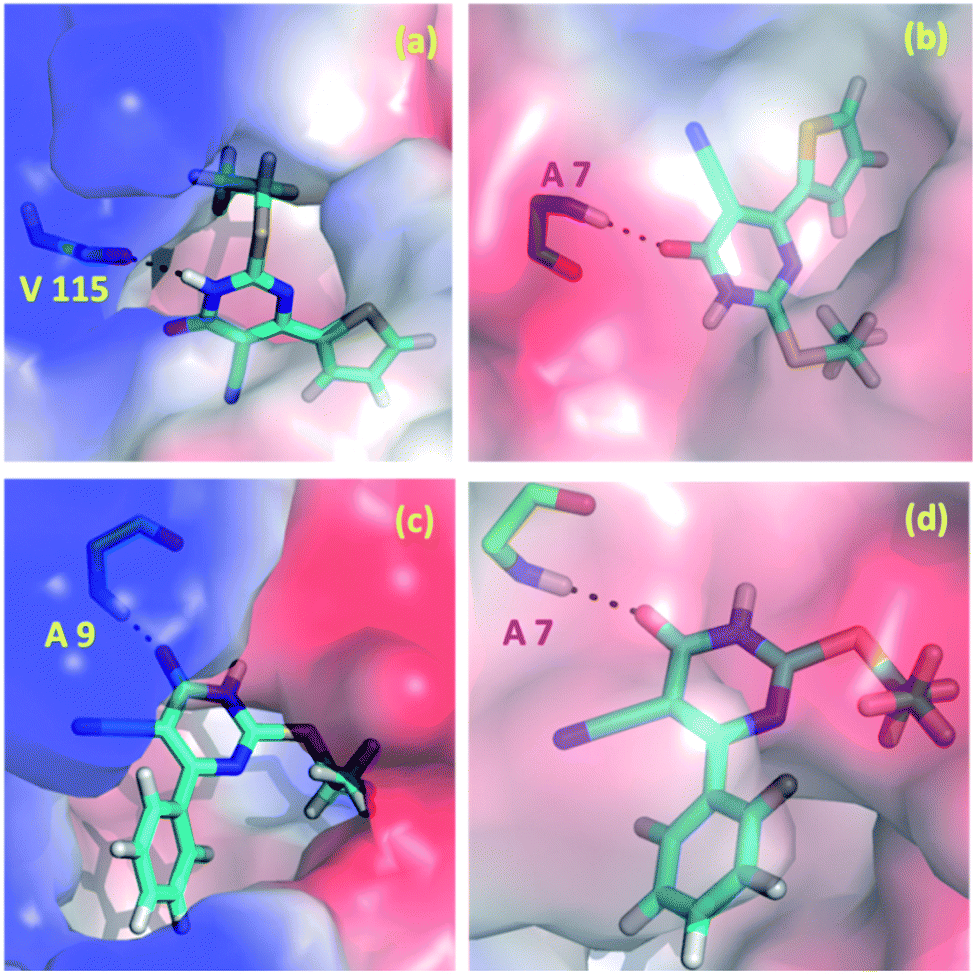 | ||
| Fig. 10 Predicted binding poses of the compounds 1 (a and b) and 2 (c and d) at the active site of (a and c) H.s DHFR (b and d) S.a DHFR. | ||
Conclusions
In the present investigation, we explored role of various types of non-covalent interactions present in the crystal structures of two pyrimidine-5-carbonitrile derivatives. The X-ray analysis revealed that the molecular conformation is locked by an intramolecular C–H⋯C interaction which is one of the common interaction observed in nitrile containing organic compounds. If aliphatic group is present at the 2nd position of the pyrimidine ring, this intramolecular interaction is not existing. Both structures 1 and 2 show common 3D topology of the energy frameworks and these features suggesting that they have similar bending properties. The molecular electrostatic potential surface revealed potential sites which are involved in non-covalent interactions and donating and accepting tendencies of different donor and acceptors sites present in these compounds. The Hirshfeld surface analysis suggested that the chalcogen bond is only preferred in thiophene containing derivative. This result is in good agreement with the PIXEL energy analysis. This chalcogen bond (formed between thiophene S and cyano carbon) is one of the rare non-covalent interactions as revealed by CSD analysis. The strong dimer in both compounds formed by highly directional N–H⋯O hydrogen bond. The topological analysis indicated that this hydrogen bond showed intermediate bonding character between shared and closed-shell interaction. All other interactions observed in both structures are of closed-shell type of interactions. The common packing motifs observed in the title compounds and their closely related structures are discussed. A detailed investigation of molecular docking analysis was performed to probe the inhibitory activities of the title compounds against DHFR enzyme from two different species (human and Staphylococcus aureus). The docking results are compared with known DHFR inhibitor trimethoprim. This in silico docking analysis suggested that the title compounds have comparable inhibitory potential against these targets.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was funded by the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University through the Research Groups Program (Grant No. RGP-1442-0010(4)).References
- S. Kumar and B. Narasimhan, Chem. Cent. J., 2018, 12, 38 CrossRef.
- K. Ghoshal and S. T. Jacob, Biochem. Pharmacol., 1997, 53, 1569–1575 CrossRef CAS.
- R. S. Klein, M. Lenzi, T. H. Lim, K. A. Hotchkiss, P. Wilson and E. L. Schwartz, Biochem. Pharmacol., 2001, 62, 1257–1263 CrossRef CAS.
- S. Matsushita, T. Nitanda, T. Furukawa, T. Sumizawa, A. Tani, K. Nishimoto, S. Akiba, K. Miyadera, M. Fukushima, Y. Yamada, H. Yoshida, T. Kanzaki and S.-i. Akiyama, Cancer Res., 1999, 59, 1911 CAS.
- M. Boisdron-Celle, G. Remaud, S. Traore, A. L. Poirier, L. Gamelin, A. Morel and E. Gamelin, Cancer Lett., 2007, 249, 271–282 CrossRef CAS.
- A. Y. Bedikian, J. Stroehlein, J. Korinek, D. Karlin and G. P. Bodey, Am. J. Clin. Oncol., 1983, 6, 181–186 CrossRef CAS.
- B. L. De Corte, J. Med. Chem., 2005, 48, 1689–1696 CrossRef CAS.
- K. Andries, H. Azijn, T. Thielemans, D. Ludovici, M. Kukla, J. Heeres, P. Janssen, B. De Corte, J. Vingerhoets, R. Pauwels and M.-P. de Béthune, Antimicrob. Agents Chemother., 2004, 48, 4680 CrossRef CAS.
- P. A. J. Janssen, P. J. Lewi, E. Arnold, F. Daeyaert, M. de Jonge, J. Heeres, L. Koymans, M. Vinkers, J. Guillemont, E. Pasquier, M. Kukla, D. Ludovici, K. Andries, M.-P. de Béthune, R. Pauwels, K. Das, A. D. Clark, Y. V. Frenkel, S. H. Hughes, B. Medaer, F. De Knaep, H. Bohets, F. De Clerck, A. Lampo, P. Williams and P. Stoffels, J. Med. Chem., 2005, 48, 1901–1909 CrossRef CAS.
- V. Summa, A. Petrocchi, F. Bonelli, B. Crescenzi, M. Donghi, M. Ferrara, F. Fiore, C. Gardelli, O. Gonzalez Paz, D. J. Hazuda, P. Jones, O. Kinzel, R. Laufer, E. Monteagudo, E. Muraglia, E. Nizi, F. Orvieto, P. Pace, G. Pescatore, R. Scarpelli, K. Stillmock, M. V. Witmer and M. Rowley, J. Med. Chem., 2008, 51, 5843–5855 CrossRef CAS.
- W. Semaine, M. Johar, D. L. J. Tyrrell, R. Kumar and B. Agrawal, J. Med. Chem., 2006, 49, 2049–2054 CrossRef CAS.
- R. Kumar, M. Nath and D. L. J. Tyrrell, J. Med. Chem., 2002, 45, 2032–2040 CrossRef CAS.
- K. K. Gauri and H. Kohlhage, Chemotherapy, 1969, 14, 158–169 CrossRef CAS.
- R. Ramajayam, K.-P. Tan, H.-G. Liu and P.-H. Liang, Bioorg. Med. Chem. Lett., 2010, 20, 3569–3572 CrossRef CAS.
- W. Brumfitt and J. M. T. Hamilton-Miller, J. Chemother., 1993, 5, 465–469 CrossRef CAS.
- S. G. B. Amyes, J. Chemother., 1993, 5, 417–421 CrossRef CAS.
- H. H. Locher, H. Schlunegger, P. G. Hartman, P. Angehrn and R. L. Then, Antimicrob. Agents Chemother., 1996, 40, 1376 CrossRef CAS.
- C. A. Sincak and J. M. Schmidt, Ann. Pharmacother., 2009, 43, 1107–1114 CrossRef CAS.
- B. I. Schweitzer, A. P. Dicker and J. R. Bertino, FASEB J., 1990, 4, 2441–2452 CrossRef CAS.
- I. M. Kompis, K. Islam and R. L. Then, Chem. Rev., 2005, 105, 593–620 CrossRef CAS.
- A. F. Cowman, M. J. Morry, B. A. Biggs, G. A. Cross and S. J. Foote, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 9109–9113 CrossRef CAS.
- S. Bunyarataphan, U. Leartsakulpanich, S. Taweechai, B. Tarnchompoo, S. Kamchonwongpaisan and Y. Yuthavong, Antimicrob. Agents Chemother., 2006, 50, 3631–3637 CrossRef CAS.
- O. K. Mc Carthy, A. Schipani, A. M. Buendía, L. M. Ruiz-Perez, M. Kaiser, R. Brun, D. G. Pacanowska and I. H. Gilbert, Bioorg. Med. Chem. Lett., 2006, 16, 3809–3812 CrossRef CAS.
- S. N. Suryawanshi, B. A. Bhat, S. Pandey, N. Chandra and S. Gupta, Eur. J. Med. Chem., 2007, 42, 1211–1217 CrossRef CAS.
- (a) J. Larsen, B. S. Rasmussen, R. G. Hazell and T. Skrydstrup, Chem. Commun., 2004, 202–203 RSC; (b) K. T. Mahmudov, M. N. Kopylovich, A. M. Maharramov, M. M. Kurbanova, A. V. Gurbanov and A. J. L. Pombeiro, Coord. Chem. Rev., 2014, 265, 1–37 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- L. Lihua, Y. Shan, X. Sheng, C. Peijun and R. Liangce, Chin. J. Org. Chem., 2012, 612–615 Search PubMed.
- A. S. Al-Tamimi, H. A. Ghabbour and A. A. El-Emam, Z. Kristallogr. - New Cryst. Struct., 2016, 231, 583–585 CAS.
- A. A. El-Emam, H. A. Ghabbour, O. A. Al-Deeb, M. S. M. Abdelbaky and S. García-Granda, Z. Kristallogr. - New Cryst. Struct., 2016, 231, 285–287 CAS.
- A. A. El-Emam, G. Demirtas, N. Dege, O. A. Al-Deeb and N. R. El-Brollosy, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o1379 CrossRef CAS.
- (a) C. B. Aakeroy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo and G. Resnati, Pure Appl. Chem., 2019, 91, 1889–1892 CAS; (b) P. Scilabra, G. Terraneo and G. resnati, Acc. Chem. Res., 2019, 52, 1313–1324 CAS; (c) K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2017, 345, 54–72 CrossRef CAS; (d) K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC; (e) L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS.
- R. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, USA, 1994 Search PubMed.
- U. Koch and P. L. A. Popelier, J. Phys. Chem. B, 1995, 99, 9747–9754 CrossRef CAS.
- A. A. El-Emam, E. Saveeth Kumar, K. Janani, L. H. Al-Wahaibi, O. Blacque, M. I. El-Awady, N. H. Al-Shaalan, J. M. Percino and S. Thamotharan, RSC Adv., 2020, 10, 9840–9853 RSC.
- A. A. Al-Mutairi, B. K. P. Katari, Y. Narasimhan, O. Blacque, L. H. Al-Wahaibi, M. A. Al-Alshaikh, A. A. El-Emam, M. J. Percino and S. Thamotharan, J. Mol. Struct., 2020, 1221, 128883 CrossRef CAS.
- S. A. Rizk, A. M. El-Naggar and A. A. El-Badawy, J. Mol. Struct., 2018, 1155, 720–733 CrossRef CAS.
- S. Kambe, K. Saito, H. Kishi, A. Sakurai and H. Midorikawa, Synthesis, 1979, 1979, 287–289 CrossRef.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887–897 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 CrossRef.
- A. Spek, Acta Crystallogr., 2009, D65, 148–155 CrossRef.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017 Search PubMed.
- A. Gavezzotti, J. Phys. Chem. B, 2002, 106, 4145–4154 CrossRef CAS.
- A. Gavezzotti, J. Phys. Chem. B, 2003, 107, 2344–2353 CrossRef CAS.
- A. Gavezzotti, Z. Kristallogr. - Cryst. Mater., 2005, 220, 499–510 CAS.
- A. Gavezzotti, New J. Chem., 2011, 35, 1360–1368 RSC.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2015, 51, 3735–3738 RSC.
- T. A. Keith, AIMAll (Version 19.10.12), TK Gristmill Software, Overland Park KS, USA, 2019 Search PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- F. A. Bulat, A. Toro-Labbé, T. Brinck, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 1679–1691 CrossRef CAS.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS.
- M. Udayakumar, M. Cerón, P. Ceballos, P. Venkatesan, M. J. Percino and S. Thamotharan, J. Chem. Sci., 2019, 131, 60 CrossRef.
- M. Udayakumar, M. Cerón, P. Ceballos, M. J. Percino and S. Thamotharan, J. Mol. Struct., 2019, 1195, 32–42 CrossRef CAS.
- P. Venkatesan, M. Cerón, S. Thamotharan, F. Robles and M. J. Percino, CrystEngComm, 2018, 20, 2681–2697 RSC.
- C. M. Reddy, R. C. Gundakaram, S. Basavoju, M. T. Kirchner, K. A. Padmanabhan and G. R. Desiraju, Chem. Commun., 2005, 3945–3947 RSC.
- T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2005, 7, 324–336 RSC.
- T. Gelbrich, T. L. Threlfall and M. B. Hursthouse, CrystEngComm, 2012, 14, 5454–5464 RSC.
- L. Fábián and A. Kálmán, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 1099–1108 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Relative contributions of different atom–atom contacts, selected torsion angles, 2D-fingerprint plots, molecular graphs for intermolecular interactions and experimental details of in vitro bioactivity. CCDC 2022218 and 2022219. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra07215j |
| This journal is © The Royal Society of Chemistry 2020 |