
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation on the composition and corrosion resistance of cerium-based conversion treatment by alkaline methods on aluminum alloy 6063
Cen Lu a,
Songlin Mu*a,
Jun Dua,
Kai Zhangb,
Mincheng Guoa and
Ling Chena
a,
Songlin Mu*a,
Jun Dua,
Kai Zhangb,
Mincheng Guoa and
Ling Chena
aSchool of Materials Science and Engineering, South China University of Technology, Wu Shan Road 381, Tian He District, Guangzhou 510640, China. E-mail: mslucen95@mail.scut.edu.cn; musonglin@scut.edu.cn; Fax: +86 20 8711 3747; Tel: +86 20 8711 3747
bGuangDong Provincial Academy of Building Research Group Co.,Ltd, Guangzhou 510500, China
First published on 6th October 2020
Abstract
Cerium conversion coating (CeCC) and Ce–Mo conversion coating (CeMCC) were prepared on aluminum alloy 6063 (AA6063) by immersion in alkaline conversion baths. Surface morphology and composition of the conversion coatings were characterized by scanning electron microscopy (SEM) and X-ray photoelectron spectroscopy (XPS). And electrochemical measurements were used to assess corrosion performance of the coatings. The SEM observations showed that CeMCC possessed a smoother and more uniform structure than CeCC, and the thickness of CeCC and CeMCC was about 0.8 and 1.2 μm respectively. The XPS depth analysis indicated that CeMCC contained a considerable amount of molybdenum and the cerium content was higher than that of CeCC at all coating depths. CeCC comprised of Al2O3, Ce2O3, CeO2, and cerium hydroxides, and the composition of CeMCC also included MoO2, MoO3, Al2(MoO4)3 and Na2MoO4 besides the above mentioned components. A potentiodynamic polarization (PDP) test revealed that the corrosion current density (icorr) values for bare alloy and CeCC were 13.36 and 4.38 μA cm−2 respectively in 3.5 wt% NaCl solution, while CeMCC exhibited the lowest icorr value of 0.24 μA cm−2, about two orders of magnitude lower than that of the substrate. Furthermore, the results obtained from both a cupric sulfate drop test and electrochemical impedance spectroscopy (EIS) characterization suggested that CeMCC possessed higher corrosion resistance in comparison with CeCC.
Introduction
Aluminum alloys are widely used in aerospace, shipping, architecture, and 3C electronic products because of their high strength/weight ratio, excellent electrical and thermal conductivity, and good machining performance.1–3 However, the high chemical activity of aluminum makes it particularly liable to corrosion. In a slightly harsh (such as humid) environment, especially in the presence of Cl−, aluminum and its alloys are prone to localized corrosion, including pitting corrosion, crevice corrosion, intergranular corrosion and stress corrosion cracking, which seriously shortens the service life of aluminum products.4 Hence, there is an urgent need to improve the corrosion resistance of aluminum alloys prior to its application. Chemical conversion treatment is known as a very important approach to achieve appropriate corrosion resistance for metals surface due to its simple processing equipment, easy operation, and low cost.5 While as the most common traditional anticorrosive treatment, chromate conversion treatment with remarkable corrosion performance and favorable adhesion, has been used in industrial applications for several decades.6 However, European Union has banned the use of hexavalent chromium through RoHS and WEEE regulations because of its highly toxic and carcinogenic nature, which brings about many serious environmental and health-related problems.7 Therefore, considerable effort has been devoted to developing viable alternatives to chromate, including permanganate,8 phosphate,9 titanate/zirconate,10 molybdate,7 and rare earth salts.11–15Extensive researches have shown that rare earth metal (REM) salts are effective inhibitors for corrosion protection of aluminum alloys,12,15–17 zinc,18 magnesium,11,14,19 steel,20 and metal matrix composites.13,21,22 Due to the non-toxicity and acceptable anticorrosive property of REM salts,23 rare earth conversion treatment of aluminum alloys is considered to be one of the most promising substitutes for chromate conversion treatment.11,15,17,19 In addition, most studies imply that cerium salts show better corrosion protection than other REM salts.24,25 K. A. Yasakau et al.26,27 synthesized amorphous cerium molybdate nanowires, which was used alone or as corrosion inhibitor in sol–gel coatings for protection of 2024 aluminum alloy, and the mechanism of active protection was based on the high solubility of cerium molybdate nanomaterials. Various addition of inorganic and/or organic additives have been used to modify the microstructure and corrosion resistance of cerium-based conversion coatings. Z. Mahidashti et al.28 studied the effect of CeCC modified by lanthanum as additive and post-treatment on the corrosion resistance and surface morphology of the mild steel. They found that the CeCC with 0.4 g L−1 La additive or post-treated by the same amount of La for 90 s exhibit the best anti-corrosion performance compared to the CeCC and untreated steel. Mohamed et al.29 examined the synergistic inhibition effect of Ce4+ and melamine on the corrosion of aluminum alloy 2024 (AA2024) in 3.5% NaCl solution. Results of this study showed that a combination of 50% Ce4+ and 50% melamine results in the lowest corrosion rates, and the mechanism of protection revealed that the reduction of Ce4+ to Ce3+ occurs during corrosion protection of AA2024. In another study, the synergistic effect of cerium acetate and sodium sulfate on corrosion inhibition of AA2024-T3 was studied by Peter et al.30 The results present a more effective corrosion inhibition of AA2024-T3 immersed in NaCl + Ce(OAc)3 solution with Na2SO4 owing to a synergistic effect leading to the formation of more compact and more durable film. Furthermore, the authors also investigated the synergistic or adverse effects of additives NaOAc, NaNO3 and Na2SO4 on the performance of Ce(OAc)3 as inhibitor of corrosion of aluminum alloy 7075-T6 in 0.1 M NaCl solution, and the most efficient corrosion inhibition was attained by synergism between Ce(OAc)3 and Na2SO4 and attributed to low solubility of the product formed.31 Maryam et al.32 have utilized H2O2 and NaCl to accelerate the formation of CeCC deposited on high pressure die cast (HPDC) Al–Si alloys. The results showed that NaCl promotes micro-galvanic coupling accelerating the corrosion reactions and thus the coating deposition, and H2O2 accelerates cerium hydroxide/oxide precipitation and modifies the coating morphology from localized deposited islands to a continuous layer with a crack-mud structure in some regions. Kanani et al.33 developed a cerium-based conversion treatment on aluminum 6063 with the addition of glycerin and H2O2. They found that immersion time in the conversion solution had an important influence on the corrosion performance and the optimum coating behavior was achieved by 600 s immersion. Hossein et al.34 studied the effect of chitosan on cracking and corrosion behavior of cerium oxide conversion coating on AA2024. The result showed that the cracking of coating was reduced noticeably by adding chitosan to the solution and the coating exhibited extremely high corrosion resistance when the content of chitosan was 0.01 wt%.
In order to improve the coating formation rate, oxidants (H2O2,22,35,36 KMnO4,37 etc.) and accelerators (for instance NaCl38 and NaF10) are often added to the rare earth conversion solution. It is worth noting that most researches were conducted in acidic conversion baths, which exhibited poor stability. As is known to all, H2O2 is easy to decompose in aqueous solution, accelerating the aging of conversion solution; KMnO4 itself is not stable enough, and it is apt to be reduced to low valence oxides (such as MnO2 precipitation) in solution containing accelerators and Ce3+, while in turn MnO2 will accelerate the decomposition of KMnO4 and make it lose its oxidation property.39 All of this leads to poor stability of acidic baths, fast aging of conversion solution and short service life. However, the use of liquid acid can be avoided in alkaline conversion solution, making the operation safer. In addition, there is no acid mist in the alkaline one, which will not cause damage to workers' health. In contrast with the abundant studies of acidic treatment, the research on alkaline conversion treatment, especially on the rare earth involved alkaline treatment, is considerably rare. In this paper, Ce(NO3)3 was used as the main salts of the conversion bath, EDTA-2Na was used as the complexing agent, and the rare earth conversion treatment was conducted under alkaline condition. Moreover, Na2MoO4 was added into the alkaline bath for comparison. The microstructure, composition and corrosion resistance of the coatings were investigated by means of scanning electron microscopy (SEM), energy dispersive X-ray spectroscopy (EDS), X-ray photoelectron spectroscopy (XPS), and electrochemical workstation, respectively.
Experimental
Materials and treatment process
The material used in this study was AA6063 aluminum alloy panels, bought from Dongguan GuanMei Metal Materials Co., Ltd, of size 30 mm × 30 mm × 3 mm, with the chemical composition (in wt%) of 0.95 Mg, 0.43 Si, 0.22 Cu, 0.10 Zn, 0.22 Cr, 0.20 Fe, 0.01 Mn, 0.02 Ti, and Al balance.The specimens were successively wet ground to 1000 grade by SiC abrasive papers. Then, the specimens were ultrasonically cleaned in ethanol for 5 min and rinsed in deionized water. In order to fully remove the native oxide layer from the surface of aluminum alloy, a chemical pretreatment was conducted by the following method: etched in industrial acid (containing H2SO4, H3PO4, HF, and emulsifier OP-10) at room temperature for 1 min, rinsed three times with deionized water, then etched in mixed acid solution which contained HNO3, H3PO4 and H2SO4 at room temperature for 2 min, rinsed three times with deionized water. The acid etching promoted the activation of the specimen surface favoring the formation of conversion coating on the substrate. When the pretreatment was finished, conversion treatment was carried out immediately to avoid the surface of the samples to be oxidized again. After the preliminary exploration experiments, the components of alkaline conversion bath were optimized as follows: urea 20 g L−1, Ce(NO3)3·6H2O 8 g L−1, EDTA-2Na 4 g L−1, Na2CO3 5 g L−1. The pH value of the solution was adjusted around 9 by 50 g L−1 Na2CO3 solution. Samples were prepared by immersion of the aluminum alloy panels in bath at 70 °C for 20 min. To study the effect of molybdate on the CeCC, 10 g L−1 Na2MoO4·2H2O was added to obtain CeMCC, and the conversion treatment was performed under the same conditions (temperature and treatment time) as aforementioned. Conversion treated samples were then dried and aged in ambient condition for about 24 h before further microstructural characterization and electrochemical measurements. All the reagents used were AR grade, bought from Guangzhou Chemical Reagent Factory.
Surface characterization
The surface morphology and microstructure of the conversion coatings were characterized by field emission scanning electron microscopy (FESEM, Merlin, Zeiss, Germany), under high vacuum condition with 5 kV beam energy. An EDS system attached to FESEM was employed to examine the chemical composition of the conversion coatings.The chemical state of elements presented in the conversion coatings were investigated by X-ray photoelectron spectroscopy (XPS, Thermo K-Alpha+). A monochromatic Al Kα (1486.8 eV) X-ray source (analysis area was c.a. 400 μm2) was used in the XPS, which was operated at 15 kV and 10 mA, along with pressure in the analysis chamber of 2 × 10−9 mbar. Overview spectra were recorded in the energy range of 1350 to 0 eV with pass energy of 150 eV. The elemental composition depth profiles of the coatings were analyzed by way of Ar+ sputtering. Firstly, the XPS data were collected on the outermost surface of the samples, then the surface was sputtered by Ar+ for 5 min, the XPS data were collected again. This sputtering-collection process was repeated for 9 times on each conversion coating. While as for the high-resolution spectra, the pass energy and energy step of the analyzer were 30 eV and 0.05 eV respectively. The binding energy (BE) of obtained data was calibrated by a linear shift so that the peak of the C 1s originated from adventitious carbon corresponded to 284.8 eV.40 The experimental data were curve fitted by XPSPEAK 4.1 software after background subtraction. The background shape was Shirley, and Lorentzian–Gaussion ratio was fixed at 20%. The ∑χ2 (the degree of deviation between actual value and theoretical value) values of fitted results were less than 10.
Cupric sulfate drop experiment and electrochemical measurements
The cupric sulfate drop test was carried out to quickly evaluate the corrosion resistance of the samples, with solution composed of 2 g CuSO4·5H2O, 2 mL hydrochloric acid and 98 mL deionized water. When the solution was dropped on the surface – start time; when the color of the dropped region changed from blue to dark red – end time. To guarantee the reliability of the data, four points were taken on the surface of each sample, and the mean time was taken as the anticorrosion time of the sample. The recorded time was used to evaluate the corrosion performance of the coatings.Electrochemical measurements including EIS and PDP were performed in 3.5 wt% NaCl solution at room temperature with a CHI604E electrochemical workstation. A conventional three-electrode cell was employed for electrochemical tests, where the working electrode was the samples with an exposure area of 1.0 cm2, the reference electrode was a saturated calomel electrode (SCE), and the counter electrode was a platinum sheet of 1.5 cm2. The ratio of corrosive solution to tested area is 250 mL to 1 cm2. The working electrode was immersed in testing electrolyte for 10 min to establish a steady open circuit potential (Eocp) before EIS experiment. The EIS measurements were conducted in the frequency range from 100 kHz to 10 mHz and the sinusoidal amplitude of 10 mV with respect to the Eocp. After the EIS tests, the PDP measurements were carried out at the same position of the samples. The purpose to do this is to avoid the error caused by different testing locations. The potential was scanned from −300 to +300 mV versus Eocp at a scanning rate of 2 mV s−1. And three different spots on the samples were tested to confirm the reproducibility of the electrochemical measurements.
Results and discussion
SEM studies
The digital photos of the samples before and after conversion treatment are displayed in Fig. 1. As shown in Fig. 1(b) and (c), the prepared CeCC is colorless and transparent, and the CeMCC exhibits a uniform brown appearance. Fig. 1(d)–(f) depict the microstructure of AA6063, CeCC and CeMCC under SEM observation. The scratches and abrasive dust originated from the abrading procedure are still clearly visible for the bare AA6063. From the Fig. 1(e), there are a few corrosion pits and a great many of small strip-shaped crevices on the surface of CeCC, which is mainly due to random dissolution of intermetallic particles (IMPs) during the acid etching step,35 and the layer is relatively loose. As is evident in Fig. 1(f), a smooth, continuous and uniform “dried-riverbed”-shaped coating with some bulge of spherical particles has completely covered the entire surface of aluminum alloy. However, the CeMCC surface also shows some micro-cracks, which were mainly created by internal stress release resulted from the coating shrinkage during the drying process.21 From the SEM images the CeMCC possesses more compact structure compared with the CeCC. | ||
| Fig. 1 Digital photos and SEM images of bare AA6063 (a and d), CeCC (b and e) and CeMCC (c and f). | ||
Table 1 exhibits the elemental contents of the four spots marked in Fig. 2. The EDS measurement shows the presence of Ce (3.55 wt%), Al (87.88 wt%) and O (7.24 wt%) on spot 1 for CeCC. It can be expected that the CeCC may be composed of oxides and/or hydroxides of aluminum and cerium. As shown in Fig. 2(a), there are some irregular white regions in the CeCC. According to the corresponding EDS spectrum on spot 2, in which Ce, Al and O account for 5.41 wt%, 58.41 wt% and 12.66 wt% respectively. And the content of Si, Fe and Cr in this area is significantly increased in comparison with the matrix composition due to the presence of Al(Fe, Cr)Si intermetallic compounds. The amount of Ce on the intermetallic compounds is higher than that on areas without intermetallics, indicating that the deposition of Ce occurred preferentially at the location of Fe-rich intermetallics thanks to their cathodic behavior with respect to the aluminum matrix.41 In the case of CeMCC (Fig. 2(b)), elements such as Al (74.83 wt%), Ce (2.19 wt%), Mo (8.59 wt%) and O (13.37 wt%) have been detected on its surface (spot 3), which may consist of oxides and/or hydroxides of aluminum, cerium and molybdenum. A white particle (spot 4) can be seen in the CeMCC, mainly containing elements of Al (72.65 wt%), Ce (3.57 wt%), Mo (11.40 wt%), and O (12.37 wt%). Obviously, the amount of Ce and Mo on spot 4 is higher than that on spot 3, while the content of Al and O is just the opposite. It is worthy to note that the amount of Ce is decreased in the CeMCC (2.19 wt% Ce) compared to the CeCC (3.55 wt% Ce), this decrease may be resulted from the co-deposition of Mo compounds, because the anions of Mo compounds should combine with Ce ions to form Ce insoluble compounds if there is no Mo ions. Besides, the Al content (74.83 wt%) in the CeMCC is lower than that (87.88 wt%) of CeCC, while the O (13.37 wt%) content in the CeMCC is nearly twice as much as that (7.24 wt%) of CeCC. The EDS results show that both CeCC and CeMCC may be composed of oxides, hydroxides of the inorganic elements (Al, Ce, Mo).
| Spots | Elemental contents | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Al | O | Ce | Mo | Mg | Si | Cr | Fe | ||
| 1 | wt% | 87.88 | 7.24 | 3.55 | — | 0.84 | 0.49 | — | — |
| at.% | 86.37 | 11.62 | 0.67 | — | 0.89 | 0.45 | — | — | |
| 2 | wt% | 58.41 | 12.66 | 5.41 | — | 0.45 | 5.34 | 2.35 | 15.38 |
| at.% | 61.43 | 22.45 | 1.10 | — | 0.53 | 5.40 | 1.28 | 7.81 | |
| 3 | wt% | 74.83 | 13.37 | 2.19 | 8.59 | 0.59 | 0.43 | — | — |
| at.% | 73.88 | 22.26 | 0.42 | 2.38 | 0.65 | 0.41 | — | — | |
| 4 | wt% | 72.65 | 12.37 | 3.57 | 11.40 | — | — | — | — |
| at.% | 74.58 | 21.42 | 0.71 | 3.29 | — | — | — | — | |
 | ||
| Fig. 2 EDS illustrations for CeCC (a) and CeMCC (b). | ||
Fig. 3 shows the cross-sectional SEM images and line-scanning EDS spectra of CeCC and CeMCC. In order to highlight the elements of cerium and molybdenum, aluminum with strong signal is not added in the EDS spectra. It can be observed from Fig. 3(a and b) that the O content of the CeCC increases evidently along with scanning line, while the Ce content presents a slight increase. The cross-sectional micrograph and line-scanning spectrum of CeCC exhibit that the thickness of the coating is about 0.8 μm. As shown in Fig. 3(c and d), the content of O and Mo in the CeMCC is obviously higher than that in the matrix, and the Ce content shows a slight increase. Thus it can be deduced that the main components of the CeMCC are oxides of Al, Mo and Ce. And according to the peak of element Mo, the coating thickness of the CeMCC is 1.2 μm or so.
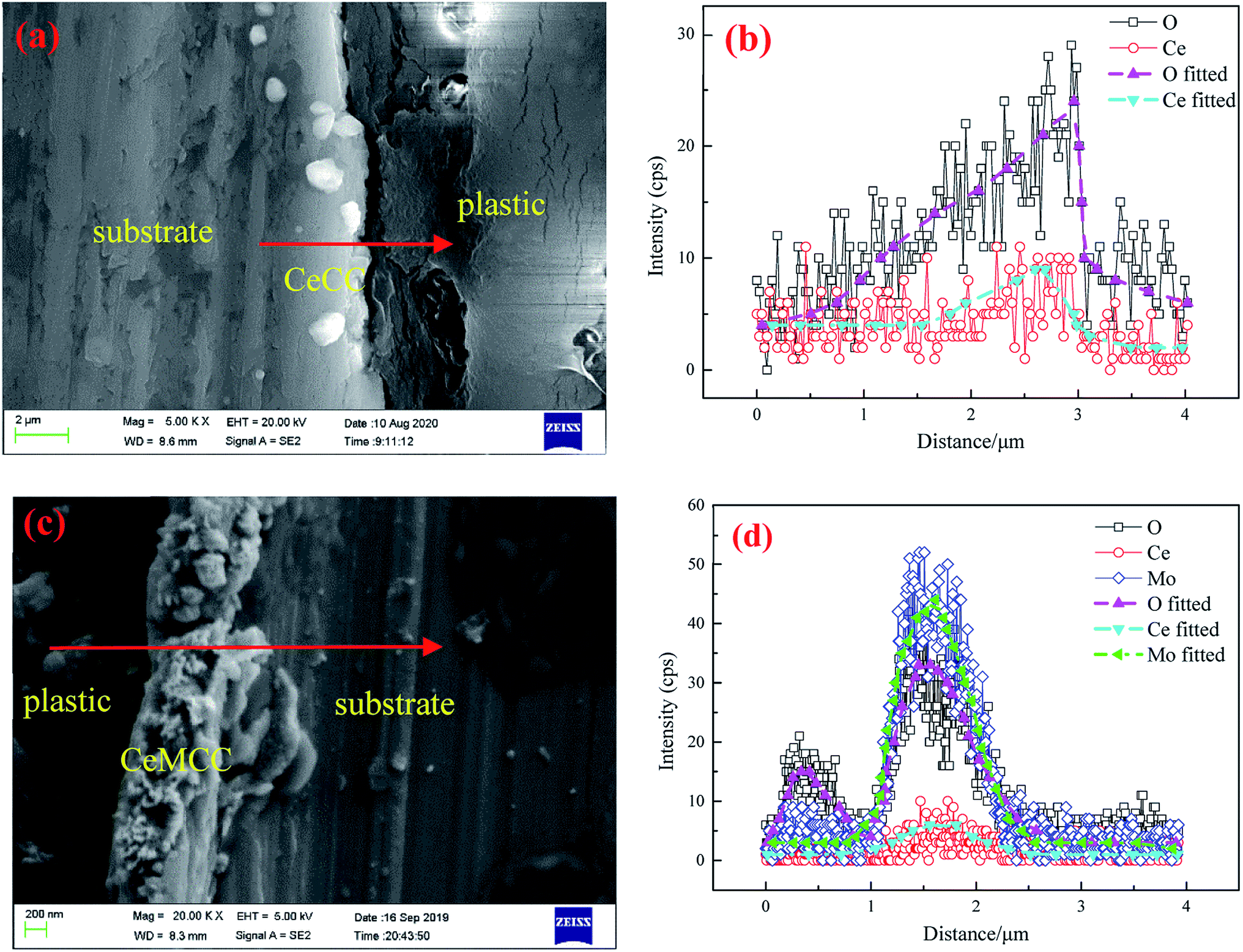 | ||
| Fig. 3 Cross-sectional images and line-scanning EDS spectra of CeCC (a and b) and CeMCC (c and d). | ||
XPS investigation
XPS was utilized to further analyze the element composition and chemical valence of the conversion coatings. Fig. 4 presents the depth profile spectra for CeCC and CeMCC. Both the CeCC and CeMCC were sputtered 9 layers and each layer was sputtered for 5 min. As can be seen in Fig. 4(a), as the Ar+ etch time increases from 0 to 15 min, the Al content increases sharply from 6.6 at% to 70.6 at%, while the C content decreases from 67.9 at% to 8.9 at%. Then the amount of Al slowly increases and C slowly decreases as a function of the etch time. A considerable amount of O is detected in the CeCC. After sputtering for 5 min, the maximum value of O element reaches about 50 at% and then it decreases slowly. The amount of O decreases to its initial value after about 12 min of sputtering. As for the element Ce, the low concentration (1.4 at%) indicates that the deposited cerium oxides only covered a small fraction of the whole surface area, mainly located at the Al(Fe, Cr)Si intermetallics.42 Similar to the CeCC, the C curve of CeMCC varies significantly after 5 min of sputtering because of the removal of surface carbon. The content of C decreases sharply from 70.9 at% in the outermost surface to 17.1 at% in the 5 min-sputtering layer. It is worth noting that after 5 min of sputtering the contents of Ce, Mo and O reach their maximum, 10.8 at%, 21.3 at% and 47.9 at% respectively. It suggests that the outer layer of CeMCC may be mainly composed of oxides and/or hydroxides of Ce and Mo. In addition, the O curve exhibits an elongated peak between 5 and 10 min of sputtering, while the O curve for CeCC appears a peak at 5 min-sputtering position. This may imply that the coating thickness of CeMCC is larger than that of CeCC, which is in accordance with the result of line-scanning (Fig. 3). After 20 min of sputtering, the contents of O, C, Mo and Ce gradually decrease with the sputtering time, while the Al curve shows the opposite behavior. By the XPS analysis of the element content with the coating depth, it can be concluded that the content of Ce in the CeMCC is higher than that in the CeCC at all coating depths, which indicates that the addition of molybdate may promote the deposition of cerium insoluble compound(s).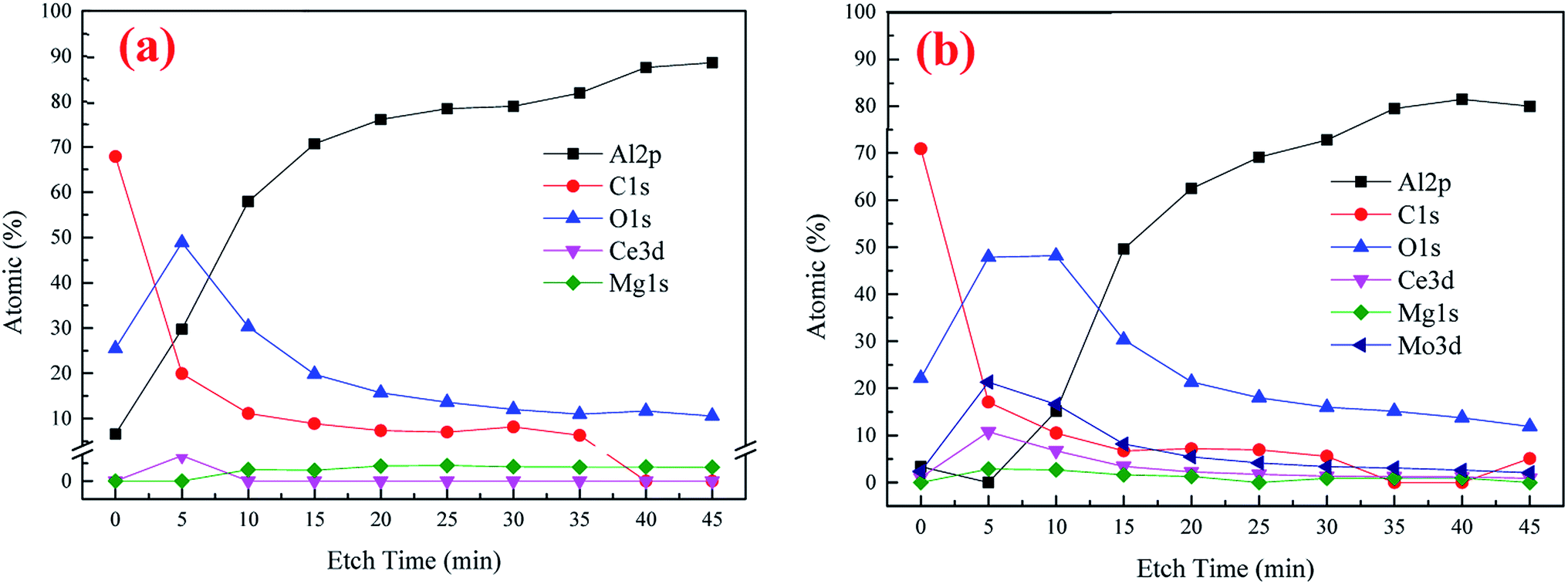 | ||
| Fig. 4 The XPS depth profile spectra of conversion coatings: (a) CeCC, (b) CeMCC. | ||
The high-resolution spectra of Al 2p, C 1s, O 1s and Ce 3d for CeCC are presented in Fig. 5. The peak of Al 2p observed at BE of 74.58 eV suggests the presence of Al2O3.43
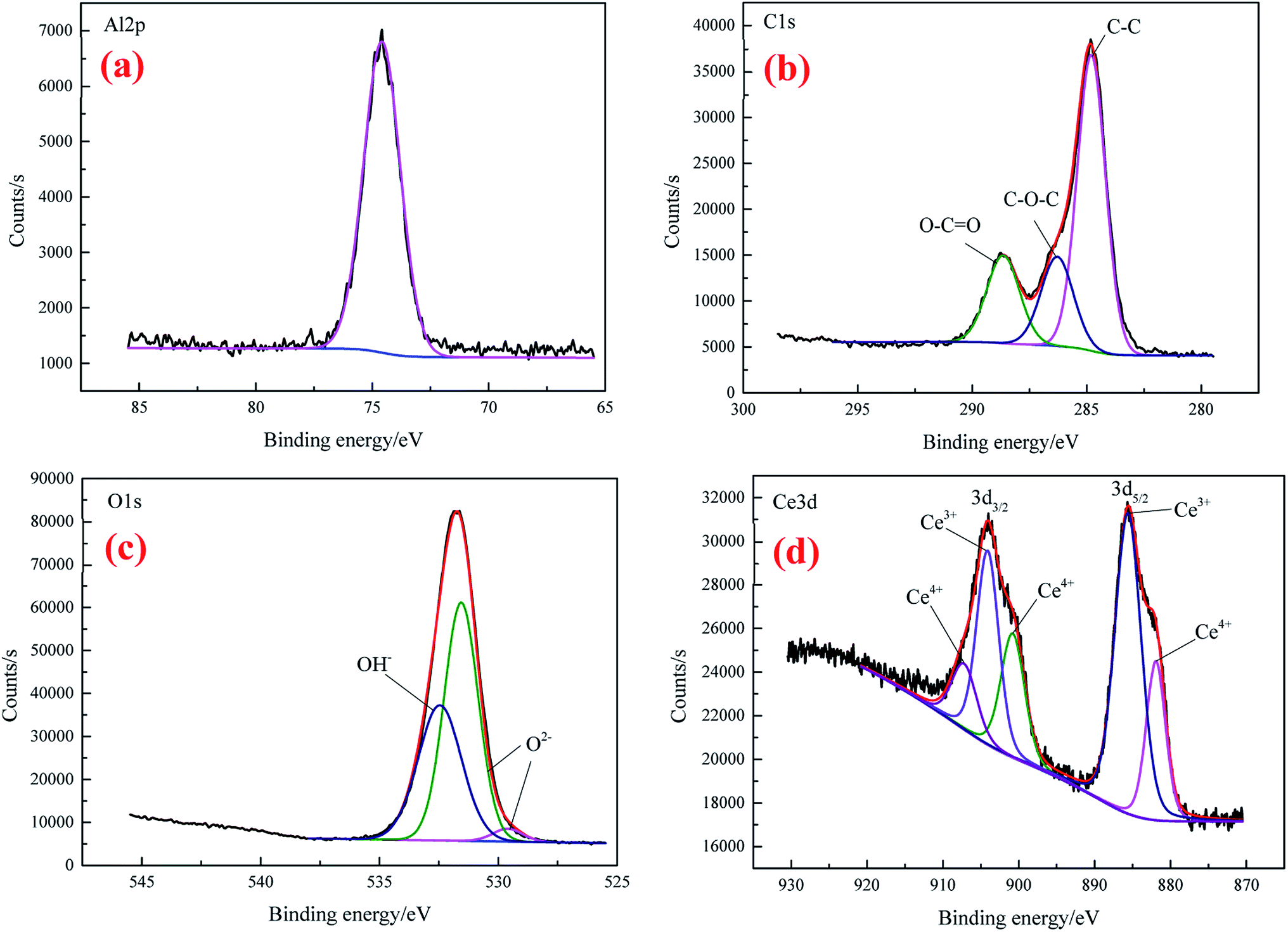 | ||
| Fig. 5 The high-resolution XPS spectra of Al 2p (a), C 1s (b), O 1s (c), and Ce 3d (d) for CeCC. | ||
The C 1s (Fig. 5(b)) can be fitted with three peaks at 284.8 eV, 286.29 eV and 288.65 eV, which are assigned to the peak of C–C, the amorphous carbon C–O–C on the natural aluminum oxide and the carboxyl carbon O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (derived from EDTA-2Na), respectively.40,44 As shown in the XPS spectrum of O 1s (Fig. 5(c)), the peak with BE of 529.6 eV is consistent with the characteristic peak of CeO2,45 the O2− component located at 531.46 eV is attributed to the coexistence of Ce2O3 (BE O 1s = 530.2 eV) and Al2O3 (BE O 1s = 531.9 eV),36 while the peak centered at 532.46 eV matches closely to the BE of Ce–OH group.44 Thus the element O in the CeCC may come from Al2O3, CeO2, Ce2O3, and cerium hydroxides. It is reasonable that the presence of partial C and O may be due to the accumulation of contaminants during exposure to air. The physical and chemical perturbation of the surface influenced the Ce 3d electron emission which displays a noisy signal.17 From the Fig. 5(d), it is evident that Ce 3d exhibits a broad and complex spectrum which reveals the presence of multiple oxidation states for Ce. The Ce 3d spectrum is fitted in spin–orbit doublets of Ce 3d5/2 and Ce 3d3/2. The peaks at 881.93 eV and 900.8 eV are corresponding to the satellite peaks of Ce4+ 3d5/2 and Ce4+ 3d3/2 respectively, which should be originated from Ce–O bonding in CeO2.46–48 The signals of Ce3+ 3d5/2 and Ce3+ 3d3/2 spin–orbit doublets are observed at BE of 885.6 eV and 904.1 eV, respectively, which can be assigned to Ce2O3.46 While the shoulder at 907.2 eV of Ce4+ 3d3/2 can be attributed to the presence of CeO2.46 Accordingly, both the O 1s and Ce 3d spectra indicate that cerium in the CeCC existed in trivalent and tetravalent states, and the peak area of Ce(III) is about 59% higher than that of Ce(IV). It can be inferred that a considerable part of Ce3+ was oxidized to Ce4+ during the conversion process (O2 + 2H2O + 2e → H2O2 + 2OH−, Ce3+ + H2O2 + 2OH− → Ce(OH)4 (CeO2·2H2O)↓),49 since the main salt of conversion bath was cerium(III) nitrate. The results above reveal that CeCC is comprised of Al2O3, Ce2O3, CeO2, and cerium hydroxides.
O (derived from EDTA-2Na), respectively.40,44 As shown in the XPS spectrum of O 1s (Fig. 5(c)), the peak with BE of 529.6 eV is consistent with the characteristic peak of CeO2,45 the O2− component located at 531.46 eV is attributed to the coexistence of Ce2O3 (BE O 1s = 530.2 eV) and Al2O3 (BE O 1s = 531.9 eV),36 while the peak centered at 532.46 eV matches closely to the BE of Ce–OH group.44 Thus the element O in the CeCC may come from Al2O3, CeO2, Ce2O3, and cerium hydroxides. It is reasonable that the presence of partial C and O may be due to the accumulation of contaminants during exposure to air. The physical and chemical perturbation of the surface influenced the Ce 3d electron emission which displays a noisy signal.17 From the Fig. 5(d), it is evident that Ce 3d exhibits a broad and complex spectrum which reveals the presence of multiple oxidation states for Ce. The Ce 3d spectrum is fitted in spin–orbit doublets of Ce 3d5/2 and Ce 3d3/2. The peaks at 881.93 eV and 900.8 eV are corresponding to the satellite peaks of Ce4+ 3d5/2 and Ce4+ 3d3/2 respectively, which should be originated from Ce–O bonding in CeO2.46–48 The signals of Ce3+ 3d5/2 and Ce3+ 3d3/2 spin–orbit doublets are observed at BE of 885.6 eV and 904.1 eV, respectively, which can be assigned to Ce2O3.46 While the shoulder at 907.2 eV of Ce4+ 3d3/2 can be attributed to the presence of CeO2.46 Accordingly, both the O 1s and Ce 3d spectra indicate that cerium in the CeCC existed in trivalent and tetravalent states, and the peak area of Ce(III) is about 59% higher than that of Ce(IV). It can be inferred that a considerable part of Ce3+ was oxidized to Ce4+ during the conversion process (O2 + 2H2O + 2e → H2O2 + 2OH−, Ce3+ + H2O2 + 2OH− → Ce(OH)4 (CeO2·2H2O)↓),49 since the main salt of conversion bath was cerium(III) nitrate. The results above reveal that CeCC is comprised of Al2O3, Ce2O3, CeO2, and cerium hydroxides.
The high-resolution spectra of Al 2p, C 1s, O 1s, Ce 3d, and Mo 3d for CeMCC are shown in Fig. 6(a)–(e), respectively. It can be seen that the noise in Al 2p spectrum (Fig. 6(a)) is extensive, and the intensity is lower than that of CeCC, implying less proportion of aluminum in the coating. The Al 2p peak that appeared at 74.5 eV is possibly attributed to the compounds Al2O3 and Al2(MoO4)3.43,50 The C 1s spectrum (Fig. 6(b)) clearly exhibits that peak components centered at BE of 284.8 eV, 286.1 eV and 288.32 eV are ascribed to C–C bonding, C–O bonding and the carboxyl carbon O–CO group (originated from EDTA-2Na) respectively.40,44 As can be seen in Fig. 6(c), the O 1s core-level spectrum was curve-fitted into three peak components. The peak observed at 530.38 eV can be ascribed to the oxygen in Ce2O3, MoO2 or/and Al2(MoO4)3,36,51,52 while the peaks at BE of 531.52 eV and 532.44 eV are respectively assigned to the presence of Al2O3 or/and MoO3, and cerium hydroxides.44,53 The BE of 885.8 eV and 904.1 eV in Ce 3d spectrum (Fig. 6(d)) respectively represent the Ce 3d5/2 and Ce 3d3/2 of Ce2O3,46 while the peaks for CeO2 are observed at 881.93 eV, 900.5 eV, and 907.1 eV, associating with Ce 3d5/2 and Ce 3d3/2 photoelectron line of cerium.46,48 The area ratio of Ce(III) to Ce(IV) is about 1.42, which is lower than that of CeCC, indicating more Ce3+ are oxidized to Ce4+ owing to the addition of molybdate (MoO2−4 + 2e + 2H2O → MoO2·+ 4OH−, O2 + 2H2O + 2e → H2O2 + 2OH−, Ce3+ + H2O2 + 2OH− → Ce(OH)4 (CeO2·2H2O)↓).27,49 Fig. 6(e) displays the narrow-scan spectrum of the Mo 3d which were fitted into five peak components. The peaks located at BE of 229.25 eV, 230.41 eV and 233.71 eV are attributed to Mo 3d5/2 and Mo 3d3/2 in MoO2,54 while MoO3 is identified in the Mo 3d5/2 region centered at BE of 232.32 eV and Mo 3d3/2 at 235.30 eV.50,55 Besides, the peak at 232.32 eV may also be assigned to Mo 3d5/2 in Al2(MoO4)3 and Na2MoO4,53 which possibly co-deposited with insoluble compounds. Moreover, the content of Mo(IV) is almost equal to that of Mo(VI) according to the calculated area under relative peaks. In conclusion, the CeMCC is mainly composed of Al2O3, CeO2, Ce2O3, MoO2, MoO3, Al2(MoO4)3 and Na2MoO4, and cerium hydroxides.
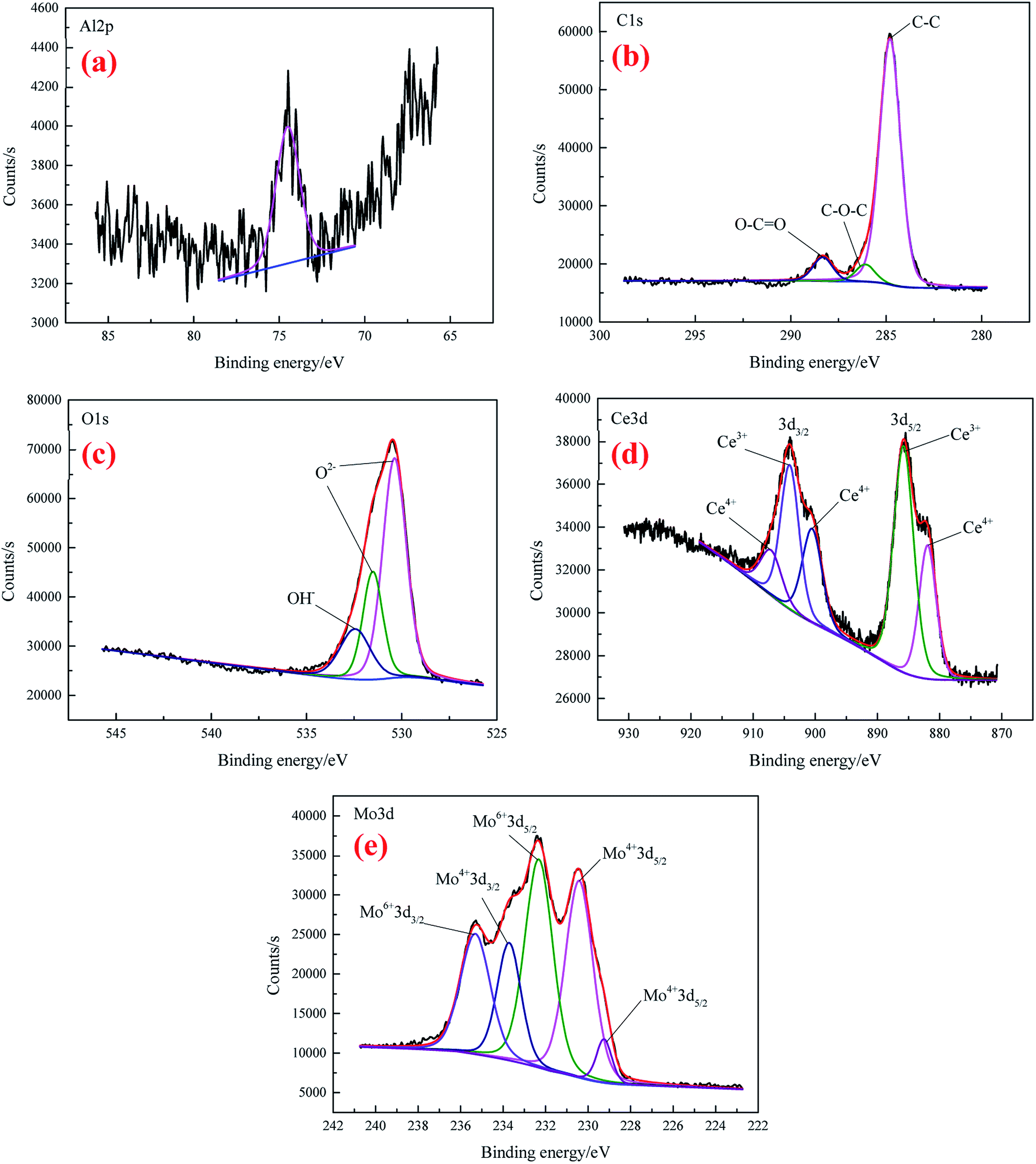 | ||
| Fig. 6 The high-resolution XPS spectra of Al 2p (a), C 1s (b), O 1s (c), Ce 3d (d), and Mo 3d (e) for CeMCC. | ||
Corrosion resistance analysis
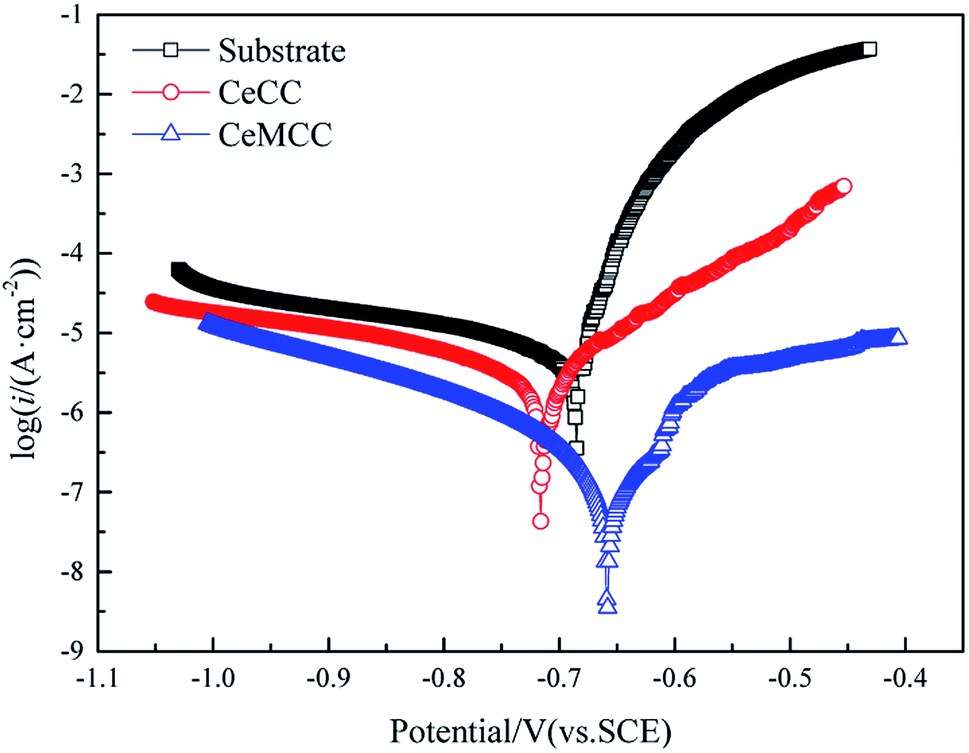 | ||
| Fig. 7 Polarization curves of substrate and coated samples. | ||
| Samples | Ecorr (V vs. SCE) | βa (mV dec−1) | −βc (mV dec−1) | Rp (kΩ cm2) | icorr (μA cm−2) | Std dev. (μA cm−2) |
|---|---|---|---|---|---|---|
| Substrate | −0.69 | 64 | 297 | 1.7 | 13.36 | 1.27 |
| CeCC | −0.72 | 102 | 256 | 7.2 | 4.38 | 0.15 |
| CeMCC | −0.66 | 122 | 150 | 120.7 | 0.24 | 0.03 |
It is well established that icorr is one of the most important parameters to evaluate the corrosion performance of material, the smaller the icorr value is, the better the corrosion resistance is.10 Reproducibility of the results was confirmed with two other spots on the sample in each case. In this study, the icorr for bare sample is 13.36 μA cm−2, while the icorr for CeMCC is 0.24 μA cm−2, about two orders of magnitude lower than the former. The icorr of CeMCC is lower than that of Mohamed et al.,29 but worse than that of Kanani's research,33 in which the icorr lowered from 5.1 μA cm−2 to 0.075 μA cm−2. The strong current reduction observed for the CeMCC can be attributed to the large amount of cerium/molybdenum compounds precipitation on the substrate. Because these deposits are hydroxides or hydrated oxides with high resistance, and can also provide good barrier effect, which can insulate the substrate from the corrosive species. As a consequence, the electrochemical reactions are greatly hindered and the corrosion current density is accordingly reduced. The CeCC coated sample also shows a lower icorr (4.38 μA cm−2) as compared with the bare one. However, the CeCC cannot offer the same protection as the CeMCC does, which is mainly due to the fact that the aggressive ions can migrate to the metal substrate through the crevices or pores within the CeCC. The standard deviation values of icorr for substrate, CeCC and CeMCC calculated by the three repeated tests were 1.27 μA cm−2, 0.15 μA cm−2 and 0.03 μA cm−2, respectively. The Rp of the CeCC is 7.2 kΩ cm2, while the Rp of the CeMCC obtained in our alkaline Ce–Mo conversion bath is 120.7 kΩ cm2, nearly 20 times higher than the CeCC. The Rp of CeCC is higher than the result obtained by Peter et al.,31 and the Rp of CeMCC is also significantly higher that of Kanani et al.33 These results demonstrate that the CeMCC possesses higher corrosion resistance than the CeCC in the corrosive electrolyte of 3.5 wt% NaCl solution.
For the sake of further investigating the anticorrosion property of the conversion coatings, EIS measurements were conducted in 3.5 wt% NaCl solution. The spectra corresponding to uncoated samples have been included for reference. As can be seen in the Nyquist plots (Fig. 8(a)), the arc for each coated sample presents a larger diameter in comparison with that of the uncoated sample, which implies a higher degree of corrosion protection. While the semicircle diameter of the CeMCC coated sample is significantly larger than that of the CeCC one, which suggests the presence of CeMCC could offer a better level of protection than CeCC. This result is in good agreement with the polarization test results above. The figure also shows an inductive behavior for both CeCC and CeMCC coated samples. The physical explanation of low-frequency inductive loop is not sufficiently clear; it has been attributed to the relaxation of adsorbed intermediate species57 and/or formation of a passive layer and/or redox activity.29 In this work, the inductive loop of the two coated samples may be related to redox activity, which may indicate that Ce(IV) undergoes a redox process in the low frequency region.
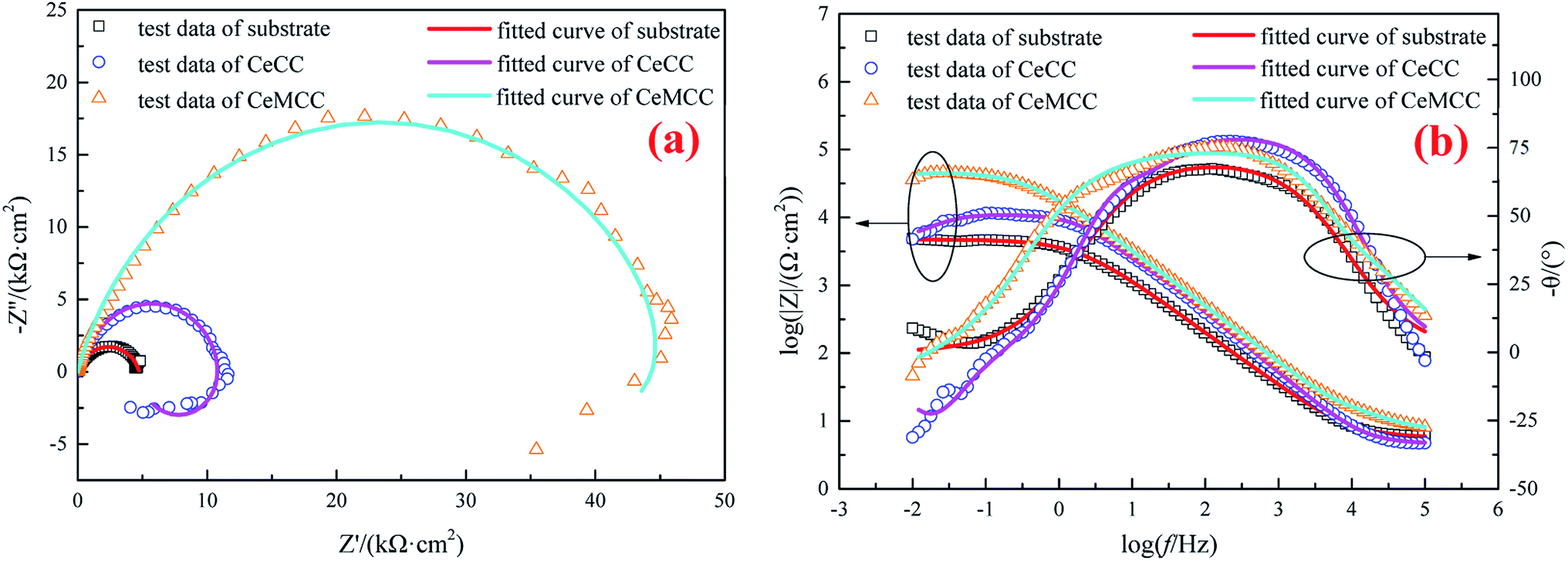 | ||
| Fig. 8 EIS spectra for substrate and conversion coatings: (a) Nyquist plots and (b) Bode plots. | ||
The Bode plots are presented in Fig. 8(b). It is well known that the impedance modulus (|Z|) and phase angle (θ) values extracted from Bode plots are often used to investigate the corrosion resistance of coated samples. According to the literature,58 the |Z| values imply the total resistance (solution, charge transfer and coating/oxide film resistance) and the θ values indicate the coating barrier performance. From the Bode modulus plots (the plots of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) f versus log|Z|), it can be seen that both the CeCC and the CeMCC possess higher |Z| values at low frequencies compared to the bare aluminum alloy. The impedance modulus |Z| value (log|Z| is nearly 4.0) of the CeCC coated sample is about three times greater than the uncoated sample, while the CeMCC coated sample presents the |Z| value (log|Z| is nearly a constant about 4.7) more than one order of magnitude higher than the bare one. The increase in |Z| value in the case of CeCC coated sample may be thanks to the active inhibition role of Ce(III)/Ce(IV) species in the coating which could heal the corroded areas and thus inhibit the corrosion reactions.28 Different from CeCC, the increment of |Z| value for CeMCC may be attributed to the insoluble molybdenum oxides precipitation on the metal surface. These compounds act as a barrier layer to impede the access of Cl− to the substrate and reduce the number of anodic sites on the alloy surface. As a result, the rate of electron generation can be reduced, leading to the decrease of corrosion rate. In the Bode phase plots (the curves of logf versus −θ), the first relaxation process at the frequency region ranged from 1000 to 1 Hz of the two conversion coatings has a θ value very close to 80°, indicating the electrochemical process on the surface shows a capacitive behavior with good dielectric properties.59 In other words, the interface of solution/conversion coatings has the ability to charge and hence avoid the ionic flow of corrosive medium.59 What is more, the CeMCC coated sample exhibits a greater flattening of the maximum in the diagram. This is a further indication that these compounds could provide a better protection level owing to their barrier effect. In contrast, the substrate shows a θ value slightly higher than 65°, since its capacitive properties are poorer than those of the two conversion coatings. In addition, with the decrease of the frequency, the phase angle tends to drop and results in a second relaxation process, related to the permeation of the electrolyte through the pores or cracks reaching the underlying metal surface.60
f versus log|Z|), it can be seen that both the CeCC and the CeMCC possess higher |Z| values at low frequencies compared to the bare aluminum alloy. The impedance modulus |Z| value (log|Z| is nearly 4.0) of the CeCC coated sample is about three times greater than the uncoated sample, while the CeMCC coated sample presents the |Z| value (log|Z| is nearly a constant about 4.7) more than one order of magnitude higher than the bare one. The increase in |Z| value in the case of CeCC coated sample may be thanks to the active inhibition role of Ce(III)/Ce(IV) species in the coating which could heal the corroded areas and thus inhibit the corrosion reactions.28 Different from CeCC, the increment of |Z| value for CeMCC may be attributed to the insoluble molybdenum oxides precipitation on the metal surface. These compounds act as a barrier layer to impede the access of Cl− to the substrate and reduce the number of anodic sites on the alloy surface. As a result, the rate of electron generation can be reduced, leading to the decrease of corrosion rate. In the Bode phase plots (the curves of logf versus −θ), the first relaxation process at the frequency region ranged from 1000 to 1 Hz of the two conversion coatings has a θ value very close to 80°, indicating the electrochemical process on the surface shows a capacitive behavior with good dielectric properties.59 In other words, the interface of solution/conversion coatings has the ability to charge and hence avoid the ionic flow of corrosive medium.59 What is more, the CeMCC coated sample exhibits a greater flattening of the maximum in the diagram. This is a further indication that these compounds could provide a better protection level owing to their barrier effect. In contrast, the substrate shows a θ value slightly higher than 65°, since its capacitive properties are poorer than those of the two conversion coatings. In addition, with the decrease of the frequency, the phase angle tends to drop and results in a second relaxation process, related to the permeation of the electrolyte through the pores or cracks reaching the underlying metal surface.60
The EIS data for the substrate and the coated samples were fitted using equivalent circuits (ECs) illustrated in Fig. 9, where Rs represents the resistance of the electrolyte between the reference electrode and the sample surface. For the bare 6063, the EC (Fig. 9(a)) is formed by Rs series with a constant phase element (CPE) in parallel with a second resistance (Rc) representing the aluminum oxide film properties. The impedance of the CPE is defined as:61
| ZCPE = Q/(jω)n |
 | ||
| Fig. 9 Equivalent circuits used to fit the impedance data for (a) bare AA6063 and (b) coated samples. | ||
While in the EC for coated samples (see in Fig. 9(b)), the Rc accounts for the resistance of cracks or pores when immersion in NaCl solution, and in series with a circuit consisting of Cdl, Rct and RL series with L in parallel, representing the double layer capacitance on the interface of coating/substrate, the charge transfer resistance associated with corrosion process, the inductance resistance, and the inductance element, respectively. Since a conversion coating is generally not simplified as a pure capacitance, the two conversion coatings were simulated as CPE components in the electrochemical model. It is noteworthy that these models were choice due to displayed highest goodness to fit experimental data of the as-prepared samples.
The EIS spectra of the samples were fitted according to the ECs in Fig. 9, and the fitted results are in good agreement with the experimental data (see Fig. 8(a) and (b)). The chi-squared (χ2) error value was of the order of 10−3 for all cases, indicating that the ECs used to model sample/NaCl solution system were appropriate. The simulated parameters extracted from the models are given in Table 3. The table shows that the Rs values are between 4 and 7 Ω cm2, indicating high conductivity of the 3.5 wt% NaCl solution. The lower values of Y for CeCC and CeMCC may confirm the excellent insulating property of the coatings, which results in high resistance for electrochemical reactions taking place on the surface. And the Cdl for CeMCC is 0.5 μF cm−2, nearly 10 times lower than the CeCC one, indicating that the number of cracks or pores developed on the CeMCC is less than those on CeCC surface. The value of Rct for CeMCC is almost 8 times higher than the CeCC one, corroborating the enhanced barrier property of the conversion coating and the fact that the compact layer of CeMCC offers better corrosion resistance than the CeCC does. In addition, the RL and L values of CeMCC are about 40 times and 130 times respectively higher than that of CeCC. The results above correspond well to the PDP results.
| Samples | Rs (Ω cm2) | CPE | Rc (Ω cm2) | Cdl (μF cm−2) | Rct (kΩ cm2) | RL (kΩ cm2) | L (kH cm−2) | |
|---|---|---|---|---|---|---|---|---|
| Y (μS sn·cm−2) | n | |||||||
| Substrate | 5.6 | 29.7 | 0.79 | 4751 | — | — | — | — |
| CeCC | 4.6 | 7.1 | 0.90 | 4820 | 4.5 | 6.1 | 5.6 | 35.8 |
| CeMCC | 6.9 | 10.2 | 0.79 | 38.1 | 0.5 | 47.1 | 229.2 | 4610 |
According to the literature,62,64,65 the inhibition mechanism of cerium treatment consists of two steps: (1) insoluble oxides/hydroxides of cerium are formed owing to interaction with hydroxyl ions on the cathodic sites and (2) the formed insoluble cerium compounds precipitate on the intermetallics, creating a barrier to the supply of oxygen or electrons to the oxygen reduction reaction. Thus the cathodic reaction is inhibited and the corresponding anodic reaction significantly reduces. And Yasakau et al. also reported that the inhibiting mechanism of cerium molybdate nanowires is related to the formation of thin protective layer on the top of intermetallics and surrounding alloy matrix.27,66
While in the present study, the mechanism of inhibition of CeMCC is most probably related to the synergistic inhibiting action of cerium and molybdate ions present in the conversion solution. The formation of cerium oxides/hydroxides deposits is expected to occur near cathodic areas, while molybdenum oxides/hydroxides may precipitate on the entire alloy surface. Considering the criteria for corrosion inhibitors to be used in active protective treatments: soluble in water combined with fast, effective and irreversible inhibition,67 the inhibitive activity of CeMCC treatment makes it functionally attractive for inclusion in active protective treatments.
Influenced by the inertia thinking that the metallic materials could only be dissolved in acid solution, nearly all researches on chrome-free conversion treatment have been concentrated in acid range for several decades. However, for aluminum alloys, researchers in the field may have overlooked the fact that aluminum can also be dissolved in alkaline solutions. In this study, a Ce–Mo based conversion coating was prepared with an alkaline conversion bath, and the studies on the coating indicated that the conversion treatment was a cathodic protection treatment according to the results of EDS, PDP, EIS and βc. And compared with the poor stability of acidic bath, the stability of our alkaline conversion bath can be maintained for at least three months. However, there are some problems that need to be further studied. The treatment time is too long, the treatment temperature is too high, and the coating formation mechanism under alkaline condition is still unknown. The results of these studies will be reported in our next article.
Conclusion
Cerium-based conversion treatment (without and with molybdate) by alkaline methods was investigated on aluminum alloy 6063. The CeMCC possesses a smoother and more uniform morphology compared to the CeCC. The thickness of CeCC and CeMCC are about 0.8 and 1.2 μm respectively. The XPS depth analysis indicates that CeMCC possesses considerable amount of molybdenum and its cerium content is higher than that of CeCC at all coating depths. CeCC is comprised of Al2O3, Ce2O3, CeO2, and cerium hydroxides; and CeMCC is made up of Al2O3, CeO2, Ce2O3, MoO2, MoO3, Al2(MoO4)3, Na2MoO4 and cerium hydroxides. The anticorrosion time of cupric sulfate drop test for CeCC and CeMCC are 62 and 95 s, respectively. The CeMCC exhibits better corrosion protection, with a lower icorr of 0.24 μA cm−2 compared to 4.38 μA cm−2 for the CeCC in 3.5 wt% NaCl solution. And the CeMCC also presents a higher Rp value (120.7 kΩ cm2), nearly 20 times higher than the CeCC. In the EIS analysis, the Rct of 47.1 kΩ cm2, Cdl of 0.5 μF cm−2, RL of 229.2 kΩ cm2 and L of 4610 kH cm−2 for CeMCC well explained the reason why CeMCC presents better barrier property than the CeCC one.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Science and Technology Program of Guangzhou, China (grant no. 201804010348) and by the Guangdong Basic and Applied Basic Research Foundation (grant no. 2020A1515010006).References
- M. Witkowska, G. E. Thompson, T. Hashimoto and E. Koroleva, Surf. Interface Anal., 2013, 45, 1585–1589 CrossRef CAS.
- L. Xu, R. Guo, C. Tang and J. ZHANG, Surf. Technol., 2011, 40, 78–80 CAS.
- J. C. Williams and E. A. Starke Jr, Acta Mater., 2003, 51, 5775–5799 CrossRef CAS.
- Z. Feng, G. S. Frankel and C. A. Matzdorf, J. Electrochem. Soc., 2014, 161, C42–C49 CrossRef CAS.
- O. Lunder, F. Lapique, B. Johnsen and K. Nisancioglu, Int. J. Adhes. Adhes., 2004, 24, 107–117 CrossRef CAS.
- R. Grilli, M. A. Baker, J. E. Castle, B. Dunn and J. F. Watts, Corros. Sci., 2011, 53, 1214–1223 CrossRef CAS.
- C. Liang, Z. Lv, Y. Zhu, S. Xu and H. Wang, Appl. Surf. Sci., 2014, 288, 497–502 CrossRef CAS.
- R. Uma Rani, A. K. Sharma, S. M. Mayanna, H. Bhojraj and D. R. Bhandari, Surf. Eng., 2005, 21, 198–203 CrossRef.
- B. Ramezanzadeh, H. Vakili and R. Amini, Appl. Surf. Sci., 2015, 327, 174–181 CrossRef CAS.
- A. Yi, W. Li, J. Du and S. Mu, Appl. Surf. Sci., 2012, 258, 5960–5964 CrossRef CAS.
- L. Lei, J. Shi, X. Wang, D. Liu and H. Xu, Appl. Surf. Sci., 2016, 376, 161–171 CrossRef CAS.
- A. K. Mishra and R. Balasubramaniam, Corros. Sci., 2007, 49, 1027–1044 CrossRef CAS.
- J. Hu, X. H. Zhao, S. W. Tang, W. C. Ren and Z. Y. Zhang, Appl. Surf. Sci., 2007, 253, 8879–8884 CrossRef CAS.
- M. Dabala, K. Brunelli, E. Napolitani and M. Magrini, Surf. Coat. Technol., 2003, 172, 227–232 CrossRef CAS.
- M. Bethencourt, F. J. Botana, J. J. Calvino, M. Marcos and M. A. Rodriguez-Chacon, Corros. Sci., 1998, 40, 1803–1819 CrossRef CAS.
- A. De Nicolò, L. Paussa, A. Gobessi, A. Lanzutti, C. Cepek, F. Andreatta and L. Fedrizzi, Surf. Coat. Technol., 2016, 287, 33–43 CrossRef.
- A. Uhart, J. B. Ledeuil, D. Gonbeau, J. Dupin, J. Bonino, F. Ansart and J. Esteban, Appl. Surf. Sci., 2016, 390, 751–759 CrossRef CAS.
- B. Hinton and L. Wilson, Corros. Sci., 1989, 29, 967–985 CrossRef CAS.
- A. P. Loperena, I. L. Lehr and S. B. Saidman, J. Magnesium Alloys, 2016, 4, 278–285 CrossRef CAS.
- C. Wang, F. Jiang and F. Wang, Corros. Sci., 2004, 46, 75–89 CrossRef CAS.
- C. Wang, G. Wu, Q. Zhang and L. Jiang, J. Mater. Sci., 2008, 43, 3327–3332 CrossRef CAS.
- W. Chunyu, Z. Qiang, Z. Ji and W. Gaohui, J. Rare Earths, 2006, 24, 64–67 CrossRef.
- F. Mansfeld, S. Lin, S. Kim and H. Shih, Electrochim. Acta, 1989, 34, 1123–1132 CrossRef.
- M. A. Arenas, M. Bethencourt, F. J. Botana, J. De Damborenea and M. Marcos, Corros. Sci., 2001, 43, 157–170 CrossRef CAS.
- D. R. Arnott, B. Hinton and N. E. Ryan, Corrosion, 1989, 45, 12–18 CrossRef CAS.
- K. A. Yasakau, S. Kallip, M. L. Zheludkevich and M. G. S. Ferreira, Electrochim. Acta, 2013, 112, 236–246 CrossRef CAS.
- K. A. Yasakau, J. Tedim, M. L. Zheludkevich, R. Drumm, M. Shem, M. Wittmar, M. Veith and M. G. S. Ferreira, Corros. Sci., 2012, 58, 41–51 CrossRef CAS.
- Z. Mahidashti and B. Ramezanzadeh, J. Rare Earths, 2018, 36, 1112–1120 CrossRef CAS.
- M. Gobara, A. Baraka, R. Akidb and M. Zorainy, RSC Adv., 2020, 10, 2227–2240 RSC.
- P. Rodič, I. Milošev, M. Lekka, F. Andreatta and L. Fedrizzi, Electrochim. Acta, 2019, 308, 337–349 CrossRef.
- P. Rodič and I. Milošev, Corros. Sci., 2019, 149, 108–122 CrossRef.
- M. Eslami, M. Fedel, G. Speranza, F. Deflorian and C. Zanella, J. Electrochem. Soc., 2017, 164, C581–C590 CrossRef CAS.
- M. Kanani, I. Danaee and M. H. Maddahy, Mater. Corros., 2014, 65, 1073–1079 CrossRef CAS.
- H. Hassannejad, M. Moghaddasi, E. Saebnoori and A. R. Baboukani, J. Alloys Compd., 2017, 725, 968–975 CrossRef CAS.
- P. Campestrini, H. Terryn, A. Hovestad and J. De Wit, Surf. Coat. Technol., 2004, 176, 365–381 CrossRef CAS.
- M. Dabalà, L. Armelao, A. Buchberger and I. Calliari, Appl. Surf. Sci., 2001, 172, 312–322 CrossRef.
- J. Zhang, W. Li, J. Du, D. Han and X. Zheng, Chin. Sci. Bull., 2010, 55, 3345–3349 CrossRef CAS.
- M. Eslami, M. Fedel, G. Speranza, F. Deflorian, N. Andersson and C. Zanella, Electrochim. Acta, 2017, 255, 449–462 CrossRef CAS.
- S. A. Kulinich, A. S. Akhtar, P. C. Wong, K. C. Wong and K. A. R. Mitchell, Thin Solid Films, 2007, 515, 8386–8392 CrossRef CAS.
- T. Lostak, A. Maljusch, B. Klink, S. Krebs, M. Kimpel, J. Flock, S. Schulz and W. Schuhmann, Electrochim. Acta, 2014, 137, 65–74 CrossRef CAS.
- F. Andreatta, A. Turco, I. De Graeve, H. Terryn, J. De Wit and L. Fedrizzi, Surf. Coat. Technol., 2007, 201, 7668–7685 CrossRef CAS.
- O. Lunder, F. Lapique, B. Johnsen and K. Nisancioglu, Int. J. Adhes. Adhes., 2004, 24, 107–117 CrossRef CAS.
- D. Chidambaram, C. R. Clayton and G. P. Halada, Electrochim. Acta, 2006, 51, 2862–2871 CrossRef CAS.
- X. Yu and G. Li, J. Alloys Compd., 2004, 364, 193–198 CrossRef CAS.
- J. A. DeRose, T. Suter, A. Bałkowiec, J. Michalski, K. J. Kurzydlowski and P. Schmutz, Corros. Sci., 2012, 55, 313–325 CrossRef CAS.
- H. Ardelean, I. Frateur and P. Marcus, Corros. Sci., 2008, 50, 1907–1918 CrossRef CAS.
- M. Romeo, K. Bak, J. El Fallah, F. Le Normand and L. Hilaire, Surf. Interface Anal., 1993, 20, 508–512 CrossRef CAS.
- R. C. Furneaux, G. E. Thompson and G. C. Wood, Corros. Sci., 1978, 18, 853–881 CrossRef CAS.
- K. A. Yasakau, M. L. Zheludkevich, S. V. Lamaka and M. G. Ferreira, J. Phys. Chem. B, 2006, 110, 5515–5528 CrossRef CAS.
- C. D. Wanger, W. M. Riggs, L. E. Davis, J. F. Moulder and G. E. Muilenberg, Handbook of X-ray photoelectron spectroscopy, Perkin-Elmer Corporation, Eden Prairie, Minnesota, 1979 Search PubMed.
- B. Brox and I. Olefjord, Surf. Interface Anal., 1988, 13, 3–6 CrossRef CAS.
- D. S. Zingg, L. E. Makovsky, R. E. Tischer, F. R. Brown and D. M. Hercules, J. Phys. Chem., 1980, 84, 2898–2906 CrossRef CAS.
- T. A. Patterson, J. C. Carver, D. E. Leyden and D. M. Hercules, J. Phys. Chem., 1976, 80, 1700–1708 CrossRef CAS.
- T. Schroeder, J. Zegenhagen, N. Magg, B. Immaraporn and H. Freund, Surf. Sci., 2004, 552, 85–97 CrossRef CAS.
- J. Choi and L. T. Thompson, Appl. Surf. Sci., 1996, 93, 143–149 CrossRef CAS.
- G. Yoganandan, K. Pradeep Premkumar and J. N. Balaraju, Surf. Coat. Technol., 2015, 270, 249–258 CrossRef CAS.
- U. Donatus, J. V. de Sousa Araujo, C. de Souza Carvalho Machado, N. V. V. Mogili, R. A. Antunes and I. Costa, Corros. Eng., Sci. Technol., 2019, 54, 205–215 CrossRef CAS.
- B. Ramezanzadeh and M. Rostami, Appl. Surf. Sci., 2017, 392, 1004–1016 CrossRef CAS.
- E. Onofre-Bustamante, M. A. Dominguez-Crespo, A. M. Torres-Huerta, A. Olvera-Martínez, J. Genescá-Llongueras and F. J. Rodríguez-Gómez, J. Solid State Electrochem., 2009, 13, 1785–1799 CrossRef CAS.
- E. Cano, D. M. Bastidas, V. Argyropoulos, S. Fajardo, A. Siatou, J. M. Bastidas and C. Degrigny, J. Solid State Electrochem., 2010, 14, 453–463 CrossRef CAS.
- G. J. Brug, A. L. G. van den Eeden, M. Sluyters-Rehbach and J. H. Sluyters, J. Solid State Electrochem., 1984, 176, 275–295 CAS.
- K. A. Yasakau, M. L. Zheludkevich, S. V. Lamaka and M. G. S. Ferreira, J. Phys. Chem. B, 2006, 110, 5515–5528 CrossRef CAS.
- A. E. Hughes, J. D. Gorman, P. R. Miller, B. A. Sexton, P. J. K. Paterson and R. J. Taylor, Surf. Interface Anal., 2004, 36, 290–303 CrossRef CAS.
- B. R. W. Hinton, J. Alloys Compd., 1992, 180, 15–25 CrossRef CAS.
- M. A. Jakab, F. Presuel-Moreno and J. R. Scully, J. Electrochem. Soc., 2006, 153, B244–B252 CrossRef CAS.
- K. A. Yasakau, J. Tedim, M. F. Montemor, A. N. Salak, M. L. Zheludkevich and M. G. S. Ferreira, J. Phys. Chem. C, 2013, 117, 5811–5823 CrossRef CAS.
- M. W. Kendig and R. G. Buchheit, Corrosion, 2003, 59, 379–400 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |