
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-induced self-catalysis of nano-Bi2MoO6 for solar energy harvesting and charge storage†
Jiangju Si ab,
Changmeng Guoa,
Haojie Liua,
Weiwei Liab,
Xiaowei Guoab,
Peidong Baia,
Yanghong Liua,
Gairong Chen*ab and
Ningbo Sun*ab
ab,
Changmeng Guoa,
Haojie Liua,
Weiwei Liab,
Xiaowei Guoab,
Peidong Baia,
Yanghong Liua,
Gairong Chen*ab and
Ningbo Sun*ab
aSchool of Chemistry and Materials Engineering, Xinxiang University, Xinxiang, Henan 453003, China. E-mail: sunningbo682@163.com
bHenan Photoelectrocatalytic Material and Micro-Nano Application Technology Academician Workstation, Xinxiang, Henan 453003, China
First published on 15th October 2020
Abstract
Efficient, sustainable, and integrated energy systems require the development of novel multifunctional materials to simultaneously achieve solar energy harvesting and charge storage. Bi-based oxysalt aurivillius phase materials are potential candidates due to their typical photovoltaic effect and their pseudo-capacitance charge storage behavior. Herein, we synthesized nano-Bi2MoO6 as a material for both solar energy harvesting and charge storage due to its suitable band gap for absorption of visible light and its well-defined faradaic redox reaction from Bi metal to Bi3+. The irradiation of visible light significantly affected the electrochemical processes and the dynamics of the Bi2MoO6 electrode. The photo-induced self-catalytic redox mechanism was carefully explored by adding sacrificial agents in photocatalysis reaction. In accordance with the rule of energy matching, the photo-generated holes oxidized the Bi metal to Bi3+, and the corresponding peak current increased by 79.5% at a scanning rate of 50 mV s−1. More importantly, the peak current retention rate remained higher than 92.5% during the entire 200 cycles. The photo-generated electrons facilitated a decrease of 184 mV in the overpotential of the reduction process. Furthermore, the irradiation of visible light also accelerated the ionic diffusion of the electrolyte. These investigations provide a unique perspective for the design and development of new multifunctional materials to synergistically realize solar energy harvesting and charge storage.
Highly efficient and sustainable green energy conversion and storage are available solutions to the intensifying energy and environmental conservation issues.1–17 New integrated energy systems with simplified processes, that realize the complementarity between multiple clean energy technologies, are attracting increasing amounts of attention.1,4–6,9,12–14,17 Among the numerous energy conversion and storage technologies available, solar energy is one of the most promising as an abundant, low-cost, eco-friendly, and renewable energy source. Electrochemical rechargeable devices can achieve highly efficient energy conversion and storage. Therefore, it is necessary to develop an integrated system that synchronously achieves solar energy harvesting and charge storage within one device.1,11,18 Some photo-rechargeable electrochemical energy storage devices, such as photo-rechargeable batteries7,8,12,13,16,19–22 and photo-rechargeable capacitors,2–6,9–11,14,15,18,23–27 have been emerged. These new devices usually contain an independent solar energy harvesting unit (such as photovoltaic solar cells that include a dye-sensitized solar cell,4 organometal halide perovskite cells,23 polymer solar cell,9 and Si solar cells13), as well as separate rechargeable energy storage units (such as lithium-ion batteries,5,7,12,13,21 lithium oxygen batteries,22 zinc-ion batteries8 and flow batteries,19 supercapacitors,6,14,23 and hybrid capacitors25), which are expensive and lead to the energy-level mismatch and ohmic transport losses at the interface between the two units.3,17 Thus, it is highly desirable to explore new materials that are capable of harvesting and storing energy simultaneously.8,25 Bi-based oxysalt aurivillius phase materials are the potential candidates due to their typical photovoltaic effect28 and electrochemical energy storage behavior.29–40 However, there are few reports on the integrated photo-rechargeable devices that use Bi-based oxysalt,41 and the synergy mechanism between the photovoltaic effect and the electrochemical process has not yet been revealed. Here, we selected nano-Bi2MoO6 as the material for both solar energy harvesting and charge storage because it has a suitable band gap for the absorption in visible light,28 a layered structure that favors a high electron transfer rate, and a well-defined faradaic redox from Bi metal to Bi(III).36 The irradiation of visible light significantly influenced the electrochemical processes and the dynamics of the Bi2MoO6 electrode, and the photo-induced self-catalytic redox mechanism was carefully explored.
Nano Bi2MoO6 was prepared by a simple hydrothermal reaction, and the specific method was shown in the ESI.† The X-ray diffraction (XRD) test was conducted to confirm the phase of the as-prepared Bi2MoO6 sample. As shown in Fig. 1a, the sharp and intense diffraction peaks demonstrated that the as-prepared Bi2MoO6 powder had high crystallinity, and the characteristic diffraction peaks at 10.9°, 23.6°, 28.4°, 32.6°, 33.3°, 36.0°, 39.8°, 47.2°, 55.7°, 56.3°, 58.6°, and 76.2° were well indexed to (020), (111), (131), (200), (002), (151), (062), (331), (133), (262), and (400) planes of the pure orthorhombic phase of the aurivillius Bi2MoO6 (JCPDS card no. 21-0102), respectively.35 The morphology of the Bi2MoO6 was investigated by scanning electronic microscopy (SEM), transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) (Fig. 1b–h). The SEM and TEM images of the Bi2MoO6 showed a mixed morphology consisting of nanorods with diameters in 50–100 nm and nanosheets of 20–30 nm thickness; the elements of Bi, Mo, and O had a uniform distribution on Bi2MoO6's surface by STEM coupled with energy dispersive X-ray spectroscopy (EDX) (Fig. 1d–g). Further, the lattice interplanar spacing of 0.24 nm in the high-resolution (HR)-TEM image corresponded to the (022) crystal plane of the orthorhombic Bi2MoO6 (Fig. 1h).36 The nanostructures of the aurivillius Bi2MoO6 facilitate fast transportation of electrons and ions during the electrochemical and photo reactions. In addition, Fig. 1i showed the photo absorption behavior of the nano-Bi2MoO6, its visible-light response could extend to 600 nm, and the band gap was 2.6 eV, thus confirming that the as-prepared nano-Bi2MoO6 was able to harvest the solar energy.
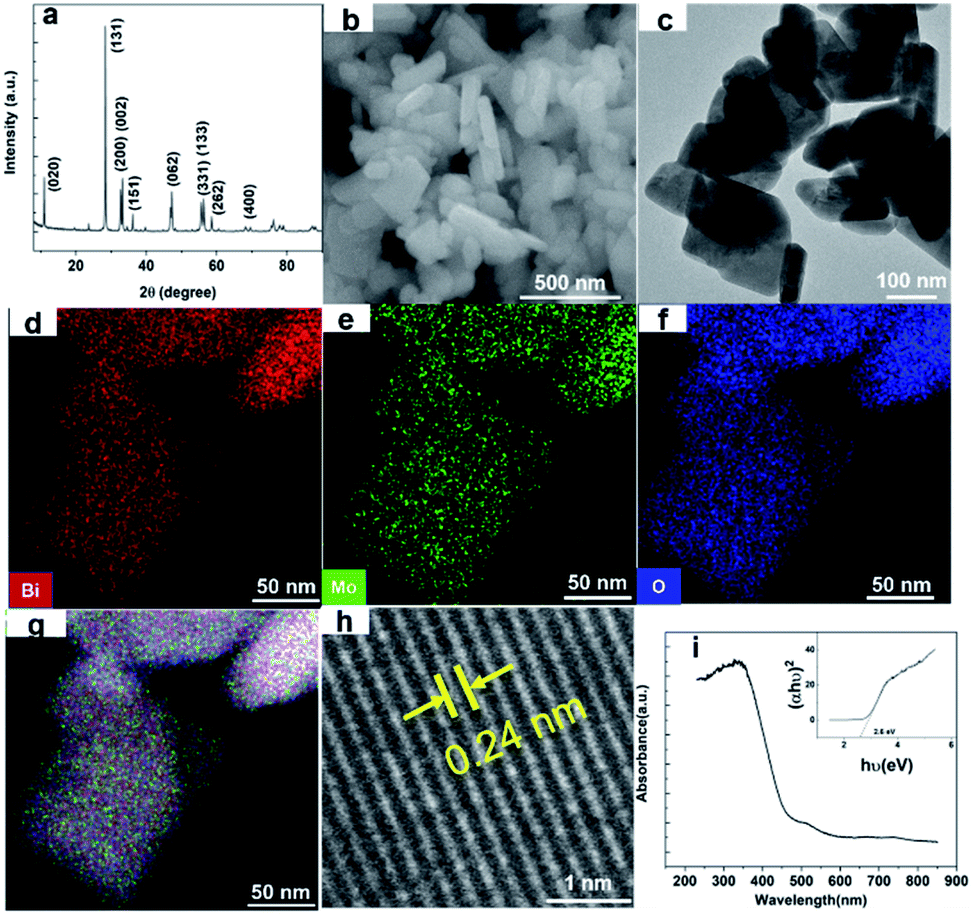 | ||
| Fig. 1 (a) XRD pattern, (b) FESEM images, (c) TEM graphics, (d–g) STEM images, (h) high-resolution TEM graphic, and (i) optical absorption spectrum of the as-synthesized Bi2MoO6. | ||
The electrochemical processes of the Bi2MoO6 electrodes were measured using a standard three-electrode system, which contained the working electrode of Bi2MoO6, the platinum counter electrode, and the Hg/HgO reference electrode in a 1 M KOH electrolyte at room temperature and atmospheric pressure. The experimental measurements of the irradiation of visible light were carried out in a 100 ml quartz electrochemical cell. A 300 W xenon arc lamp was utilized as a visible-light source, and the light intensity is 8 mW cm−2. In the dark, the cyclic voltammetry (CV) curves of the Bi2MoO6 electrodes exhibited well-defined faradaic redox peaks at −0.35 V, −0.50 V, and −0.75 V, corresponding to the conversion of the Bi metal to Bi(III) being mediated by OH− from the electrolyte, which clearly indicated the typical pseudo-capacitance charge storage behavior of the Bi2MoO6 electrode (Fig. S1†).31,42 Moreover, the cathodic and anodic peak currents depended linearly on the square roots of the scan rates (Fig. S2†), revealing that the diffusion of the electrolyte was a rate-controlling step during the pseudo-capacitance charge storage process of the Bi2MoO6 electrodes.
Under illumination of visible light, the Bi2MoO6 electrode showed similar pseudo-capacitance charge storage behavior to that exhibited in the dark from the CV results (Fig. S3†), but the intensity and position of anodic and cathodic peaks showed significant changes (Fig. 2a–f). Specifically, the anodic peak at −0.35 V (Pa2) became much sharper and its intensity increased significantly. Correspondingly, the charging platform at −0.46 V appeared much more obvious from galvanostatic charge–discharge (GCD) result (Fig. 2h). These results clearly manifested that the irradiation of visible light improved the reversibility of the oxidation reaction from Bi0 to Bi3+ and enhanced the peak current of Pa2 dramatically.41 Furthermore, we quantitively compared the peak current of Pa2 at different scan rates, and found that the maximum gain reached 79.5% at the scanning rate of 50 mV s−1 under the irradiation of visible light (Fig. 2g). In addition, the anodic peak at −0.5 V (Pa1) shifted negatively, demonstrating that the oxidation from metal bismuth located near the electrode–electrolyte interface to Bi3+ occurred more easily under the irradiation of visible light.43 For the reduction process, the cathodic peak at −0.74 V (Pc) had a positive shift, demonstrating that the reduction of Bi3+ had a smaller overpotential. Meanwhile, Pc split into a shoulder peak (Pc1) and a main peak (Pc2), also, Pc1 became increasingly strong as the sweep rate increased. Pc2 is attributed to the reduction of Bi3+ the dissolved species Bi3+ to Bi0, and the shoulder peak Pc1 may correspond to water splitting to produce hydrogen. Simultaneously, two charging platforms arose during the discharging (Fig. 2h). These results highlighted that the irradiation of visible light had a great influence on the electrochemical behavior of the Bi2MoO6 electrode, and that it can improve the reversibility, decrease the overpotential and increase the peak current during the anodic and cathodic processes. Furthermore, the irradiation of visible light also affected the dynamics of the processes at the Bi2MoO6 electrode (Fig. 2i). As that in the dark, the transformation between Bi0 and Bi3+ is still a diffusion of the electrolyte dominant process under illumination, because IPa2 and IPc2 depend linearly on the square roots of the scan rates. However, the oxidation of metal bismuth switched to a capacitive process, because IPa1 was linearly dependent on the scan rates. This result illustrated the irradiation of visible light also accelerated the mobility of ions, which enabled the diffusion of the electrolyte opposed to the rate-controlling step. Further, the electrochemical impedance spectroscopy (EIS) of the Bi2MoO6 electrode is performed in the dark and under the irradiation of visible light, and the results are shown in Fig. S5.† Compared with the quasi-semicircle plot in the dark condition, the Bi2MoO6 electrode shows a sharper plot in the low frequency region under the irradiation of visible light, verifying that the irradiation of visible light accelerated the ionic diffusion of the electrolyte again.
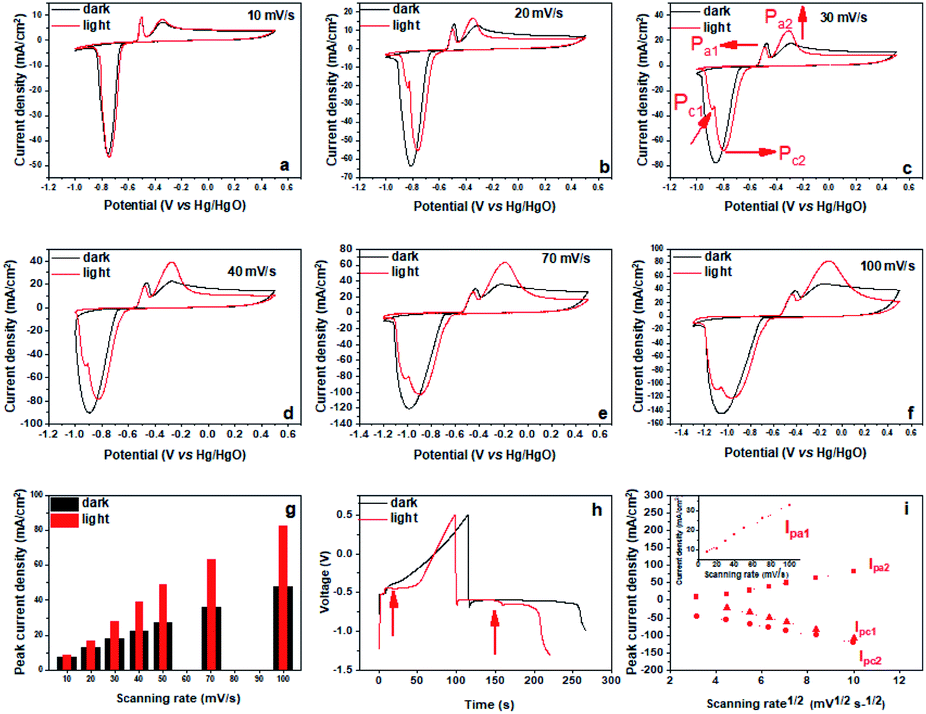 | ||
| Fig. 2 (a–f) The CV curves of the Bi2MoO6 electrode and (g) the Pa2 current density at different scan rates in the dark and under the irradiation of visible light; (h) the GCD profiles at 1 A g−1 of the Bi2MoO6 electrode in the dark and under the irradiation of visible light with a light intensity of 8 mW cm−2; (i) the relationship between the peak currents of the anodic and cathodic peaks and the sweep rate of the Bi2MoO6 electrode (the currents of the anode and cathode are abbreviated as IPa1, IPa2, IPc1, and IPc2, respectively). | ||
The above results demonstrated that the irradiation of visible light influenced the electrochemical processes and dynamics of the Bi2MoO6 electrode remarkably, but this influence mechanism has rarely been reported because of the complexity of the photo processes. As a typical n-type semiconductor, Bi2MoO6 can generate excitons, and electron–hole pairs after absorbing incident photons. Then the holes and electrons, separating in the electric field, play important roles as oxidants and reducing agents in the following process, respectively. The same photo process of the Bi2MoO6 also occurs in the photocatalysis reactions, and the catalytic mechanism of photo-generated carriers has been studied in depth by adding appropriate sacrificial agents.28,44
Inspired by the sacrificial agents in photocatalysis, we added triethanolamine (TEA) to the electrolyte as the hole sacrificial agent to explore the influence mechanism of the photo-generated holes and electrons on the electrochemical behavior of Bi2MoO6 electrode. Under the irradiation of visible light, the photo-generated holes on the Bi2MoO6 electrode were exhausted by the TEA, avoiding the recombination of photo-generated electrons at the same time. The valence band (VB) edge of Bi2MoO6 located at −0.32 + 2.6 V (vs. NHE, pH 7), which is much lower than the oxidation potential of Bi0, therefore, the photo-generated holes on the Bi2MoO6 electrode can oxidize Bi0. However, there is no obvious change for the anodic peak Pa1 when compared with the CV curves without the TEA (Fig. 3), and only the Pa2 declined dramatically. This result clearly confirmed that the photo-generated holes, preferring to accumulate in the bulk electrode, selectively enhanced the oxidation from Bi0 in the electrode bulk to Bi3+, but the Bi-metal in the interface cannot be oxidized. Summarizing the above oxidation process, we can find that it is the holes induced by photo on the Bi2MoO6 electrode, catalyze the oxidation of Bi2MoO6, which means that the above oxidation process is a typical photo-induced self-catalytic oxidation reaction of Bi2MoO6 (Fig. 4).45 The conduction band (CB) edge of the Bi2MoO6 is located at −0.32 V (vs. NHE, pH 7), which is much higher than the reduction potential of Bi3+, thus, the photo-generated electrons on the Bi2MoO6 electrode are unable to reduce the Bi3+. Meanwhile, the cathodic peak of the Bi2MoO6 electrode had a further positive shift, signifying that the overpotential of the reduction reaction decreased continuously, and this phenomenon became much more obvious as the scan rate increased. When the scan rate increased to 100 mV s−1, the decrement in the reduction overpotential reached 184 mV (Fig. 3h), verifying the photo-induced self-catalytic reduction reaction of Bi2MoO6 (Fig. 4). In other words, the photo-generated electrons on the electrode can transfer rapidly to an external circuit and accelerate the reduction of the Bi2MoO6 electrode.
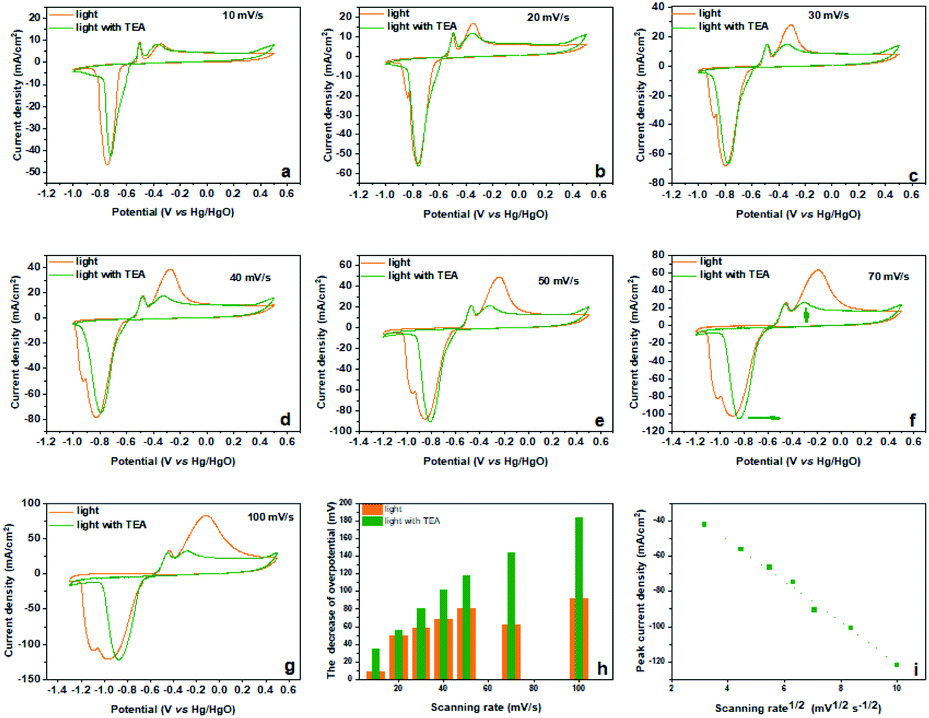 | ||
| Fig. 3 (a–g) The CV curves of the Bi2MoO6 electrode at different scan rates and (h) the decrement in the reduction overpotential of the reduction reaction under the irradiation of visible light before and after the addition of the TEA, and the visible light intensity is 8 mW cm−2; (i) the relationship between the peak current of the cathodic peak and the sweep rate of the Bi2MoO6 electrode. | ||
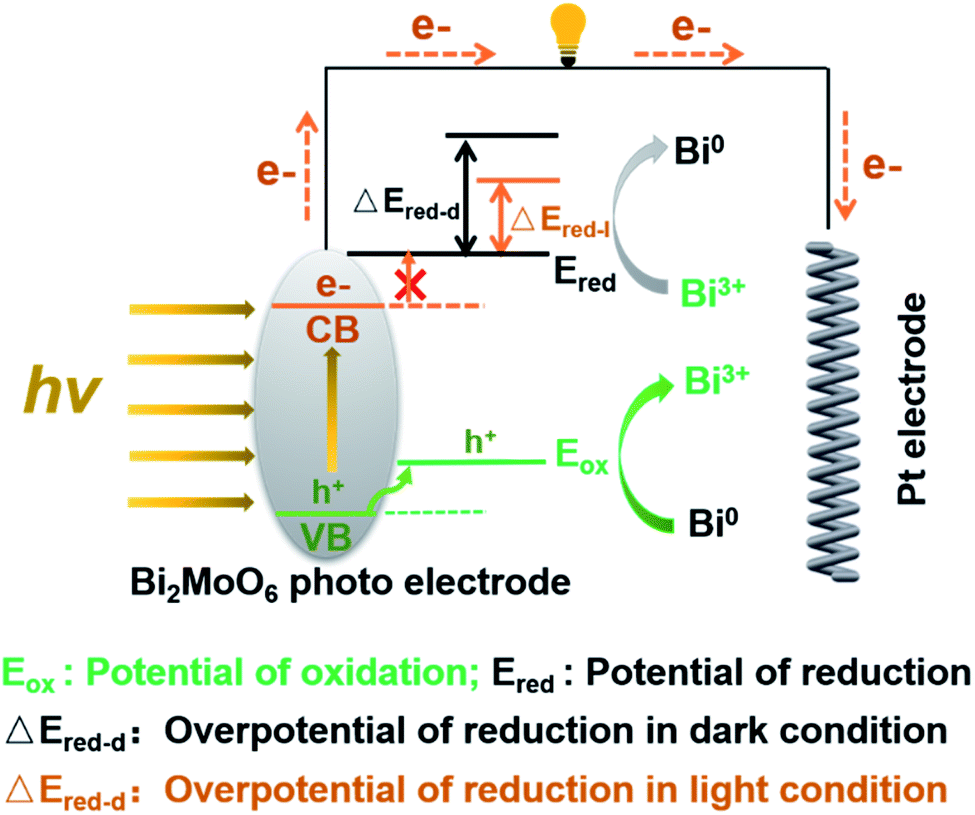 | ||
| Fig. 4 The proposed photo-induced self-catalytic redox mechanism of the Bi2MoO6 electrode under the irradiation of visible light. | ||
The stability of the Bi2MoO6 electrode under the irradiation of visible light was further investigated by cycling at a scan rate of 50 mV s−1, and the results were shown in Fig. 5 and S4.† For the entire 200 cycles, the peak current retention rate of Pa2 maintained higher than 92.5%, suggesting that photo-generated holes on the Bi2MoO6 electrode had a good stability. For the reduction process, the potential of Pc2 continued to shift slightly to positive direction, but the Pc2 had an obvious decay (Fig. S4†), the possible reason was the reduction of water.
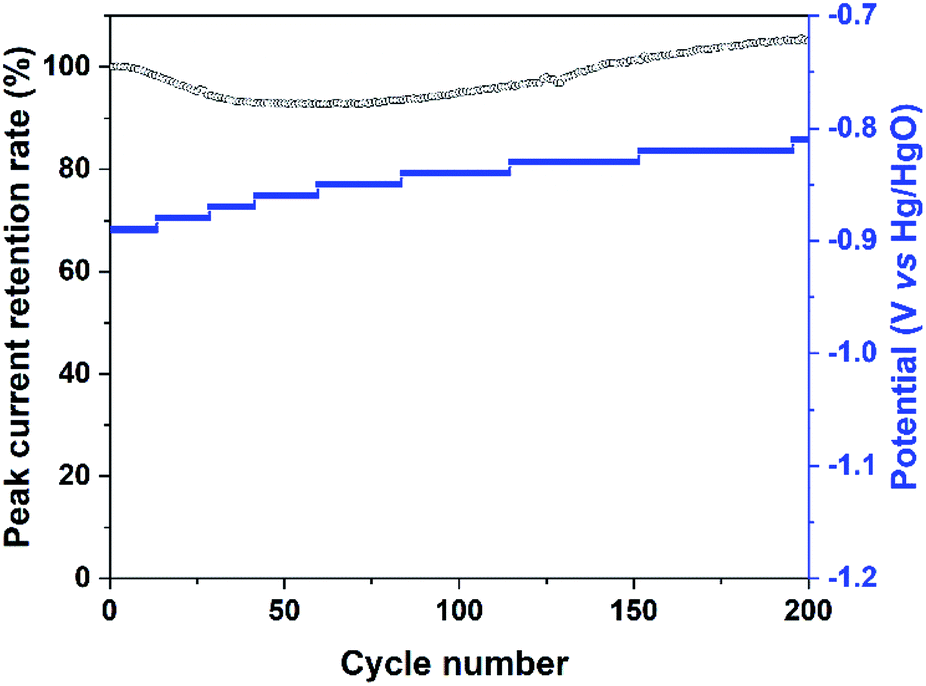 | ||
| Fig. 5 The peak current retention rate of Pa2 and the potential of Pc2 in the CV curves of the Bi2MoO6 electrode cycling at a scan rate of 50 mV s−1 under the irradiation of visible light. | ||
In summary, we aimed to develop novel multifunctional materials to achieve solar energy harvesting and charge storage synchronously. Nano-Bi2MoO6 was synthesized as a material for both solar energy harvesting and charge storage due to its suitable band gap for the absorption in visible light and its well-defined faradaic redox from Bi metal to Bi3+. The irradiation of visible light had a considerable influence on the electrochemical processes and the dynamics of the Bi2MoO6 electrode. In accordance with the rule of energy matching, the photo-generated holes could oxidize the Bi metal to Bi3+, and the photo-generated electrons could decrease the overpotential of the reduction process. Furthermore, the irradiation of visible light also accelerated the ionic diffusion of the electrolyte. This work provides a unique perspective for the development of new multifunctional materials to simultaneously achieve solar energy harvesting and electrical energy storage, and thereby open pathways towards the efficient integrated energy systems with simplified processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by grants from the National Natural Science Foundation of China (No. 51901200, U1904189, 51801171), Key Scientific Research Project Plans of Higher Education Institutions in Henan Province (No. 20B430015), and the Regional Joint Fund of Basic and Applied Basic Research of Guangdong Province (No. 2019A1515110725).References
- T. Chen, L. Qiu, Z. Yang, Z. Cai, J. Ren, H. Li, H. Lin, X. Sun and H. Peng, Angew. Chem., Int. Ed., 2012, 51, 11977–11980 CrossRef CAS.
- Y. Jin, Z. Li, L. Qin, X. Liu, L. Mao, Y. Wang, F. Qin, Y. Liu, Y. Zhou and F. Zhang, Adv. Mater. Interfaces, 2017, 4, 1700704 CrossRef.
- E. Navarrete-Astorga, D. Solis-Cortes, J. Rodriguez-Moreno, E. A. Dalchiele, R. Schrebler, F. Martin and J. R. Ramos-Barrado, Chem. Commun., 2018, 54, 10762–10765 RSC.
- T. Song and B. Sun, ChemSusChem, 2013, 6, 408–410 CrossRef CAS.
- Y. Fu, H. Wu, S. Ye, X. Cai, X. Yu, S. Hou, H. Kafafy and D. Zou, Energy Environ. Sci., 2013, 6, 805–812 RSC.
- X. Chen, H. Sun, Z. Yang, G. Guan, Z. Zhang, L. Qiu and H. Peng, J. Mater. Chem. A, 2014, 2, 1897–1902 RSC.
- A. Lee, M. Vörös, W. M. Dose, J. Niklas, O. Poluektov, R. D. Schaller, H. Iddir, V. A. Maroni, E. Lee, B. Ingram, L. A. Curtiss and C. S. Johnson, Nat. Commun., 2019, 10, 4946 CrossRef.
- B. D. Boruah, A. Mathieson, B. Wen, S. Feldmann, W. M. Dose and M. De Volder, Energy Environ. Sci., 2020, 13, 2414–2421 RSC.
- Z. Zhang, X. Chen, P. Chen, G. Guan, L. Qiu, H. Lin, Z. Yang, W. Bai, Y. Luo and H. Peng, Adv. Mater., 2014, 26, 466–470 CrossRef CAS.
- F. Zhou, Z. Ren, Y. Zhao, X. Shen, A. Wang, Y. Y. Li, C. Surya and Y. Chai, ACS Nano, 2016, 10, 5900–5908 CrossRef CAS.
- S. Safshekan, I. Herraiz-Cardona, D. Cardenas-Morcoso, R. Ojani, M. Haro and S. Gimenez, ACS Energy Lett., 2017, 2, 469–475 CrossRef CAS.
- W. Guo, X. Xue, S. Wang, C. Lin and Z. L. Wang, Nano Lett., 2012, 12, 2520–2523 CrossRef CAS.
- H.-D. Um, K.-H. Choi, I. Hwang, S.-H. Kim, K. Seo and S.-Y. Lee, Energy Environ. Sci., 2017, 10, 931–940 RSC.
- R. Liu, Y. Liu, H. Zou, T. Song and B. Sun, Nano Res., 2017, 10, 1545–1559 CrossRef CAS.
- H. Meng, S. Pang and G. Cui, ChemSusChem, 2019, 12, 3431–3447 CrossRef CAS.
- A. Gurung and Q. Qiao, Joule, 2018, 2, 1217–1230 CrossRef CAS.
- B. Luo, D. Ye and L. Wang, Adv. Sci., 2017, 4, 1700104 CrossRef.
- A. Das, S. Deshagani, R. Kumar and M. Deepa, ACS Appl. Mater. Interfaces, 2018, 10, 35932–35945 CrossRef CAS.
- N. F. Yan, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2013, 1, 7012 RSC.
- P. Liu, H. X. Yang, X. P. Ai, G. R. Li and X. P. Gao, Electrochem. Commun., 2012, 16, 69–72 CrossRef CAS.
- A. Paolella, C. Faure, G. Bertoni, S. Marras, A. Guerfi, A. Darwiche, P. Hovington, B. Commarieu, Z. Wang, M. Prato, M. Colombo, S. Monaco, W. Zhu, Z. Feng, A. Vijh, C. George, G. P. Demopoulos, M. Armand and K. Zaghib, Nat. Commun., 2017, 8, 14643 CrossRef CAS.
- H. Gong, T. Wang, H. Xue, X. Fan, B. Gao, H. Zhang, L. Shi, J. He and J. Ye, Energy Storage Materials, 2018, 13, 49–56 CrossRef.
- R. Liu, C. Liu and S. Fan, J. Mater. Chem. A, 2017, 5, 23078–23084 RSC.
- B. J. Trześniewski and W. A. Smith, J. Mater. Chem. A, 2016, 4, 2919–2926 RSC.
- D. B. D. Boruah, A. Mathieson, B. Wen, C. Jo, F. Deschler and M. De Volder, Nano Lett., 2020, 20(8), 5967–5974 CrossRef.
- B. D. Boruah and A. Misra, ACS Appl. Energy Mater., 2019, 2, 278–286 CrossRef CAS.
- T. N. Murakami, N. Kawashima and T. Miyasaka, Chem. Commun., 2005, 26, 3346–3348 RSC.
- H. Yu, L. Jiang, H. Wang, B. Huang, X. Yuan, J. Huang, J. Zhang and G. Zeng, Small, 2019, 15, e1901008 CrossRef.
- X. Hu, W. Zhang, X. Liu, Y. Mei and Y. Huang, Chem. Soc. Rev., 2015, 44, 2376–2404 RSC.
- J. Wen, S. Sun, B. Zhang, N. Shi, X. Liao, G. Yin, Z. Huang, X. Chen and X. Pu, RSC Adv., 2019, 9, 4693–4699 RSC.
- B. Senthilkumar, R. K. Selvan, L. Vasylechko and M. Minakshi, Solid State Sci., 2014, 35, 18–27 CrossRef CAS.
- Z.-Q. Liu, L.-Y. Tang, N. Li, K. Xiao, J. Wang, J.-H. Zhang, Y.-Z. Su and Y.-X. Tong, J. Electrochem. Soc., 2012, 159, D582–D586 CrossRef CAS.
- D. Zhu, W. Wang, J. Zhu, S. Chen and X. Liu, J. Solid State Electrochem., 2016, 21, 403–408 CrossRef.
- F. Wu, X. Wang, W. Zheng, H. Gao, C. Hao and C. Ge, Electrochim. Acta, 2017, 245, 685–695 CrossRef CAS.
- T. Yu, Z. Li, S. Chen, Y. Ding, W. Chen, X. Liu, Y. Huang and F. Kong, ACS Sustainable Chem. Eng., 2018, 6, 7355–7361 CrossRef CAS.
- K. J. Samdani, J. H. Park, D. W. Joh and K. T. Lee, ACS Sustainable Chem. Eng., 2018, 6, 16702–16712 CrossRef CAS.
- P. V. Shinde, N. M. Shinde, J. M. Yun, R. S. Mane and K. H. Kim, ACS Omega, 2019, 4, 11093–11102 CrossRef CAS.
- S. S. Patil, D. P. Dubal, V. G. Deonikar, M. S. Tamboli, J. D. Ambekar, P. Gomez-Romero, S. S. Kolekar, B. B. Kale and D. R. Patil, ACS Appl. Mater. Interfaces, 2016, 8, 31602–31610 CrossRef CAS.
- A. Martínez-de la Cruz, S. Obregón Alfaro, E. López Cuéllar and U. Ortiz Méndez, Catal. Today, 2007, 129, 194–199 CrossRef.
- J. Hu, Y. Xie, J. Zheng, Y. Lai and Z. Zhang, Nano Res., 2020, 13, 2650–2657 CrossRef CAS.
- M. Zargazi and M. H. Entezari, Ultrason. Sonochem., 2020, 67, 105145 CrossRef CAS.
- V. D. Nithya, R. Kalai Selvan, D. Kalpana, L. Vasylechko and C. Sanjeeviraja, Electrochim. Acta, 2013, 109, 720–731 CrossRef CAS.
- V. Vivier, A. Régis, G. Sagon, J. Y. Nedelec, L. T. Yu and C. Cachet-Vivier, Electrochim. Acta, 2001, 46, 907–914 CrossRef CAS.
- Z. Wang, H.-C. Chiu, A. Paolella, R. Gauvin, K. Zaghib and G. P. Demopoulos, Sustainable Energy Fuels, 2020, 4, 4789–4799 RSC.
- S. H. DuVall and R. L. McCreery, J. Am. Chem. Soc., 2000, 122, 6759–6764 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07020c |
| This journal is © The Royal Society of Chemistry 2020 |