
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFuro[3,2-c]coumarins carrying carbon substituents at C-2 and/or C-3. Isolation, biological activity, synthesis and reaction mechanisms
Iván Cortés
 ,
L. Javier Cala
,
Andrea B. J. Bracca
and
Teodoro S. Kaufman
*
,
L. Javier Cala
,
Andrea B. J. Bracca
and
Teodoro S. Kaufman
*
Instituto de Química Rosario (IQUIR, CONICET-UNR), Facultad de Ciencias Bioquímicas y Farmacéuticas – Universidad Nacional de Rosario, Suipacha 531, S2002LRK Rosario, Argentina. E-mail: kaufman@iquir-conicet.gov.ar
First published on 10th September 2020
Abstract
The isolation, biological activity and synthesis of natural furo[3,2-c]coumarins are presented, covering mainly the developments in the last 35 years. The most relevant approaches toward the synthesis of 2-substituted, 3-substituted and 2,3-disubstituted heterocycles are also discussed, with emphasis on the efficiency of the processes and their mechanisms.
 Iván Cortés | Iván Cortés was born in Pergamino (Province of Buenos Aires, Argentina). He received his Bachelor degree in Chemistry in 2017 from the National University of Rosario (UNR), where he is a teaching assistant in General and Inorganic Chemistry. Currently, he is a PhD student in Chemistry under the supervision of Dr Kaufman at the Institute of Chemistry of Rosario (IQUIR-UNR) with a fellowship from the Argentine National Research Council (CONICET). His research interests include the total synthesis of heterocyclic natural products and their analogs, as well as the development of new synthetic methodologies. |
 L. Javier Cala | L. Javier Cala was born in Bucaramanga, Colombia. He obtained his Bachelor degree in 2015 from the School of Chemistry of the Industrial University of Santander (UIS, Bucaramanga, Colombia), from where he also obtained his Master degree in 2019. He is currently a PhD Student at the National University of Rosario (UNR), working at IQUIR with a Fellowship from CONICET, under the supervision of Drs Kaufman and Bracca. His current research focuses on the total synthesis of heterocyclic natural products of fungal origin. |
 Andrea B. J. Bracca | Andrea B. J. Bracca was born in Rosario (Santa Fe, Argentina) and graduated in 2001 with a BS in Biotechnology from the National University of Rosario. She received her PhD in 2009 under the guidance of Prof. Kaufman. After a two years period of post-doctoral training, she returned to work in Dr Kaufman's group as an Assistant Research Scientist of the Argentine National Research Council (CONICET) at the Institute of Chemistry of Rosario (IQUIR). Currently, Dr Bracca is an Adjunct Researcher and develops research work in the area of total synthesis of heterocyclic natural products and their most relevant analogues. |
 Teodoro S. Kaufman | Teodoro S. Kaufman was born near Moisés Ville (Santa Fe, Argentina). He graduated as Biochemist (1982) and Pharmacist (1985) at the National University of Rosario (UNR), and received his PhD (1987) under the guidance of Prof. Edmundo A. Rúveda. After a two-year post-doctoral training at the University of Mississippi (USA), he returned to Rosario. Currently, he is full Professor of the UNR, Research Head of the Argentine National Research Council (CONICET) and Head of the Institute of Chemistry of Rosario (IQUIR). His research interests include heterocyclic chemistry and the synthesis and evaluation of bioactive natural products and analogues. |
1 Introduction and scope of the review
Furocoumarins are a relevant family of natural and synthetic heterocycles, endowed in many cases with properties of biological and/or technological interest.1 These compounds are characterized by a furan ring unit fused onto a coumarin motif. The fusion can be in either a 2,3- or 3,2- fashion and may take place at the faces c (lactone ring), f, g or h (benzene ring) of the coumarin skeleton, resulting in linear and angular compounds (Fig. 1).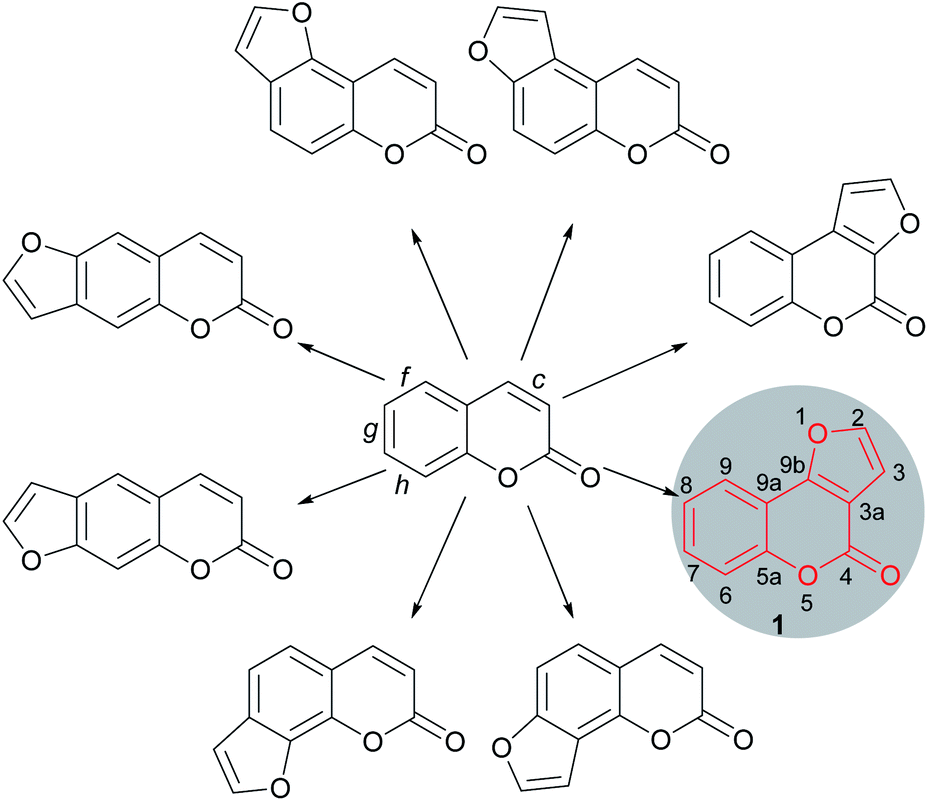 | ||
| Fig. 1 Part of the furocoumarin family of oxygen heterocycles. | ||
Related heterocycles are also obtained when the ring fusion is with the face b of the coumarin moiety, and still other heterocycles are possible, where the fusion to the furan is of the 3,4- type and the oxygen of the furan moiety is not bonded to the coumarin core. However, such kinds of compounds are relatively less common.2
The furo[3,2-c]coumarins (4H-furo[3,2-c]chromen-4-ones) are prominent members within the furocoumarin family. Their representatives are very widespread in nature, being produced by a variety of plants.3 The elegant architecture of the angular tricyclic skeleton of these compounds (1) and their importance in medicinal and natural products chemistry has motivated sustained interest in the development of synthetic tools and methodologies towards this heterocyclic ring system.
The furo[3,2-c]coumarins are a complex family, with a high number of members. Therefore, in order to gain focus, this review has been compiled aiming specifically at those heterocycles carrying substituents on positions 2 and/or 3 of the furan ring, and with the objective of providing ordered and systematized information on the main advances which took place in the last 35 years in the fields of isolation, synthesis and biological activity of these heterocycles.
Hence, a high number of representatives carrying aromatic rings fused to the 2,3-position of the furan moiety have been omitted from the discussion. These comprise important compounds, like the coumestans (Fig. 2) exemplified by wedelolactone (2), an anti-hepatotoxic from Eclipta prostrata,4 medicagol (3), isolated from alfalfa,5 plicadin (4) found in Psoralea plicata6 and coumestrol (5), a heterocycle found in a variety of legumes,7 which exhibits high antitumor power and selectivity against breast cancer cell lines.8
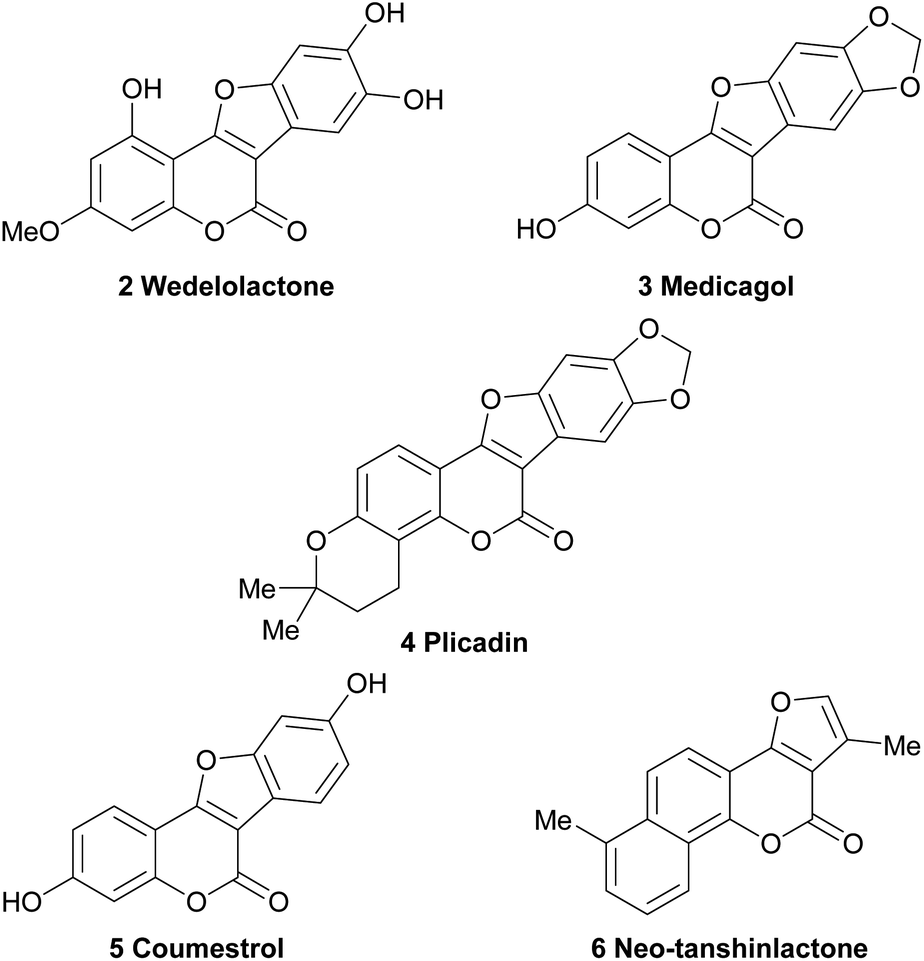 | ||
| Fig. 2 Chemical structures of some representative polycyclic natural products bearing the furo[3,2-c]coumarin core. | ||
On the other hand, however, furo[3,2-c]coumarins bearing additional rings attached to the coumarin framework, such as neo-tanshinlactone (6), have been included.
2 Naturally-occurring furo[3,2-c]coumarins
The naturally occurring furo[3,2-c]coumarins comprise, among others, fully aromatic and dihydro-compounds, carrying 1–3 substituents on the (dihydro)furan moiety, with different patterns and stereochemical features. They may also display a 9-methyl group (C-5 of the coumarin ring) and oxygen functionalities (OH, OMe), mainly on C-7 and/or C-9. Most of them have been isolated after 1980.9 The least functionalized members include those which carry alkyl (methyl, prenyl, farnesyl, etc.) residues attached to the furan ring; the presence of diastereomers and rearranged products has also been detected (Fig. 3).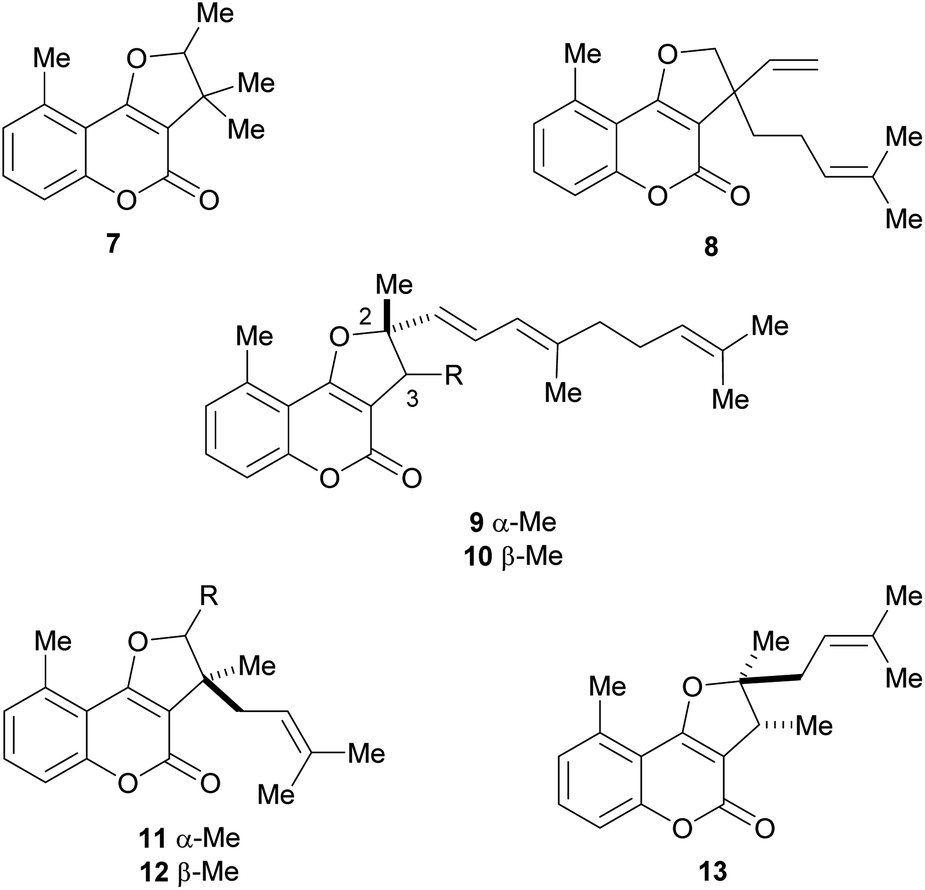 | ||
| Fig. 3 Representative natural products bearing hydrocarbon (methyl and prenyl)-type C-2 and/or C-3 side chains. | ||
Mutisia (Compositae, tribe Mutisieae) is a South American genus with about 60 species of shrubs and vines distributed along the Andes from Colombia to South Argentina and Chile, being also present in South East Brazil, Paraguay, Uruguay and North East Argentina.10
Bohlmann et al. found that the extract of the aerial parts of Trichocline caulescens Phil. (tribe Mutisieae, subtribe Nassauvimae) grown in Chile afforded the 5-methyl coumarin derivative 7.11 The same group studied the Bolivian species, M. orbignyana Wedd, isolating compound 8,12 the structure of which was confirmed by synthesis.13 The authors conjectured that the presence of 5-methylcoumarin derivatives in this species could have chemotaxonomic relevance.
On the other hand, isotriptiliocoumarin (9) and its C-3 isomer (10) were isolated in 1988 from Triptilion benaventei,14 whereas cycloisobrachycoumarin (11), 2′-epi-cycloisobrachycoumarin (12) and cyclobrachycoumarin (13) were isolated in 1986 from Brachyclades megalanthus.13,15
Some natural products bear tertiary alcohol moieties in their side chains, as shown in Fig. 4. The study of Chilean plants of the tribe Mutisieae uncovered the tertiary alcohols mutisicoumarin (14), which was isolated from Mutisia spinosa,15 whereas isoerlangeafusciol (15) was obtained from Erlangea fusca (Compositae, tribe Vernonieae) in 1980.16
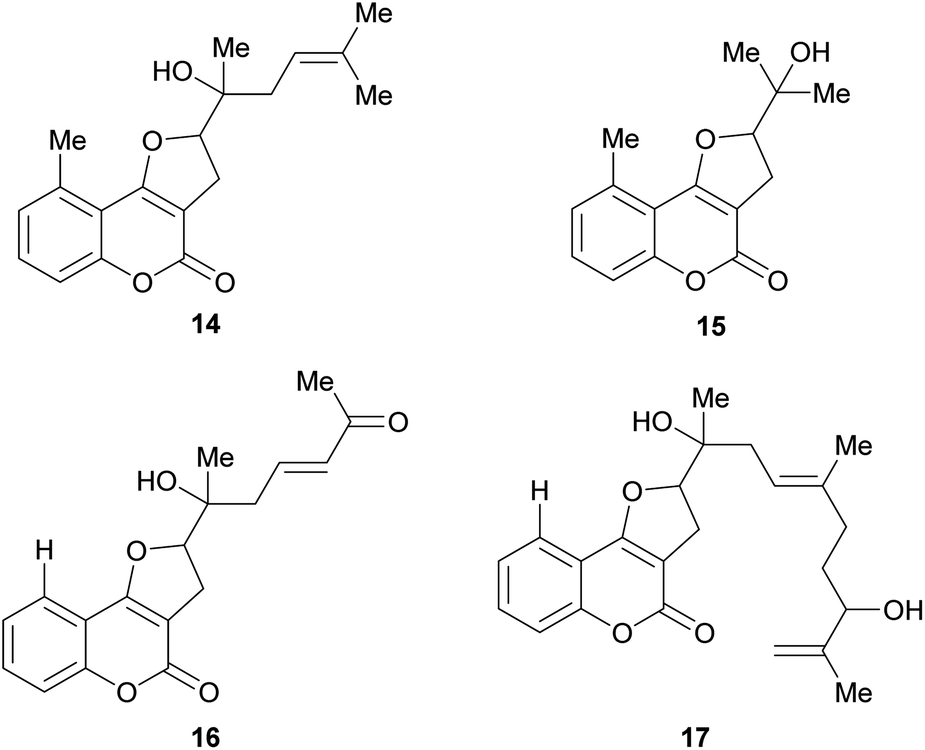 | ||
| Fig. 4 Naturally occurring furo[3,2-c]coumarins carrying a tertiary alcohol motif. | ||
Several studies on various Sardinian, Moroccan and Spanish populations17,18 of Ferula communis revealed that the chemical variability of this species depends on its geographical location and that the toxic principles of the poisonous varieties were farnesylated cyclic 4-oxygenated coumarins. Based on these studies, Misky and Jakupovic isolated the farnesylcoumarin derivatives fercoprenol (16) and fercoprolone (17), from the polar fractions of the benzene extract of the roots of F. communis subsp. communis, grown in the Marmara region of Turkey.19 These heterocycles lack the 9-methyl group Ferula communis is a medicinal plant of the Eastern Mediterranean areas since ancient times.20 However, in the Western Mediterranean regions, cases of livestock and human poisoning (ferulosys) from ingestion of this species have been occasionally reported.
In addition, a series of related compounds were obtained from Ferula feruloides grown in Mongolia, where it is used for the treatment of spasm.21a,b The isolated heterocycles 18–24 comprise different tricyclic phenols, phenolic ethers and isomers bearing rearranged farnesyl side chains (Fig. 5). Compounds 18 and 21 were also isolated from Ferula caspica M. Bieb.21c
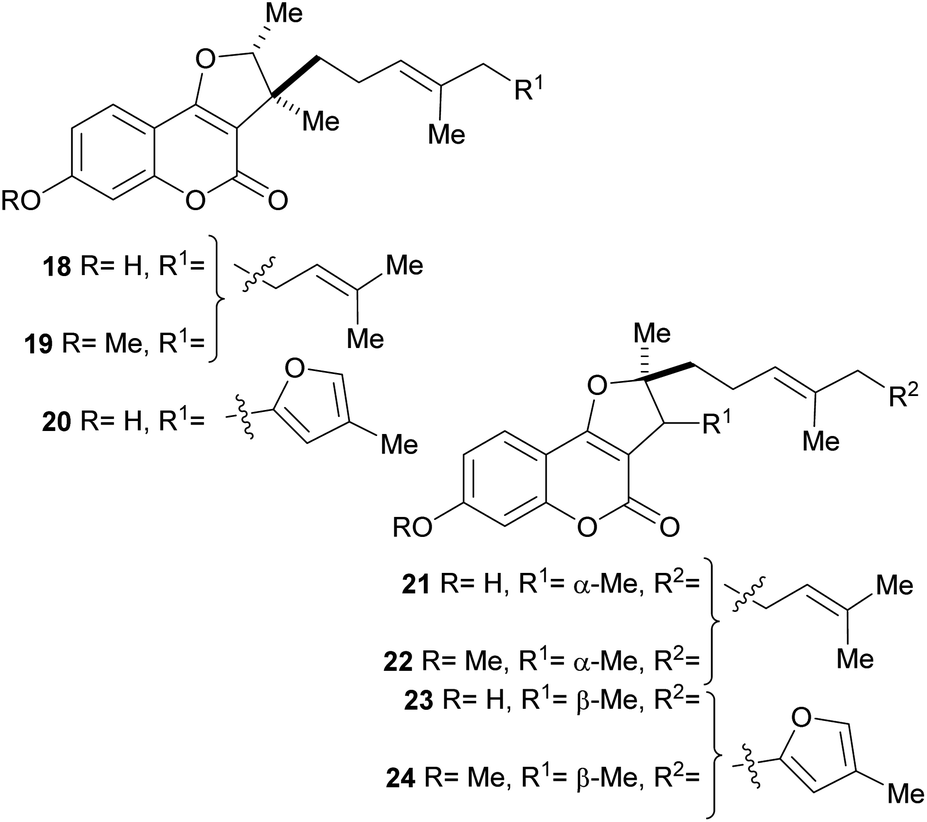 | ||
| Fig. 5 Naturally occurring furo[3,2-c]coumarins with phenolic functions and rearranged farnesyl side chains. | ||
It was proposed that biogenetically, some of these compounds might be derived from the enzymatic cyclization of 2′,3′-epoxidized ferulenol (3-farnesyl-4-hydroxycoumarin) 26 (Scheme 1).
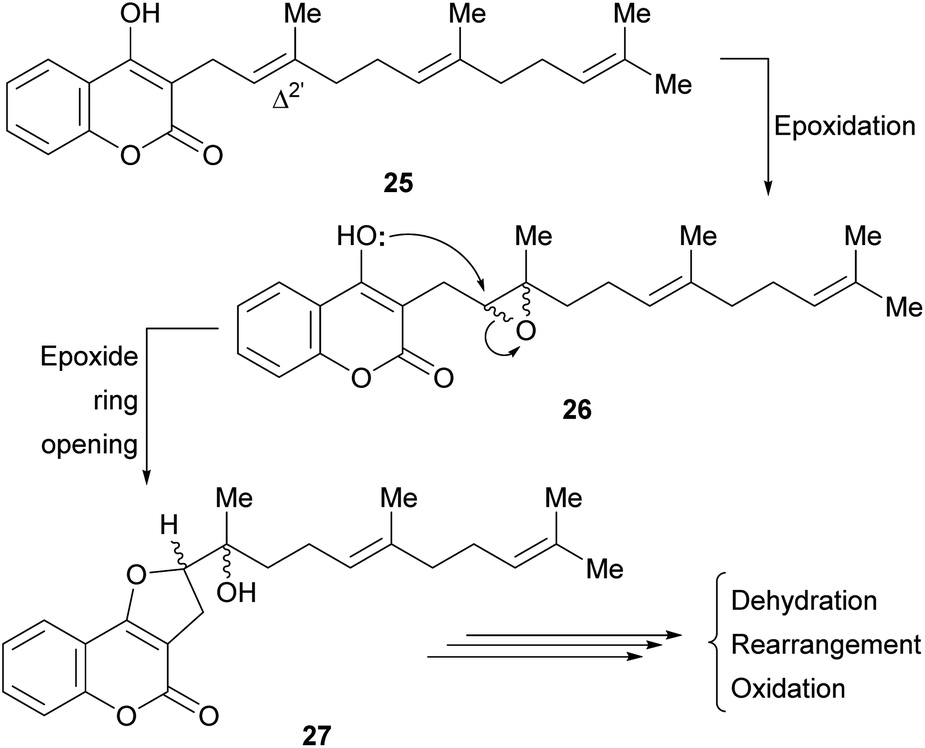 | ||
| Scheme 1 Proposed biogenetic path for some furo[3,2-c]coumarins. | ||
Tertiary alcohols, presumably arising from an epoxide ring opening during the biogenesis are also found among the members of this family. Their racemic nature (27) could be an outcome of the non-enantioselective epoxidation of the Δ2′ double bond of ferulenol (25). However, the regioselectivity of this epoxidation process clearly indicates the involvement of an enzymatic system.
A biosynthetic pathway has been proposed for these compounds (Scheme 2), where the hypothetical precursor 28 undergoes hydration, affording intermediate 29. In turn, the latter suffers lactonization to give intermediate 30 and further dehydration to furnish the tricyclic derivative 31. Additional oxidation and cyclization steps provide the furan derivative 32.
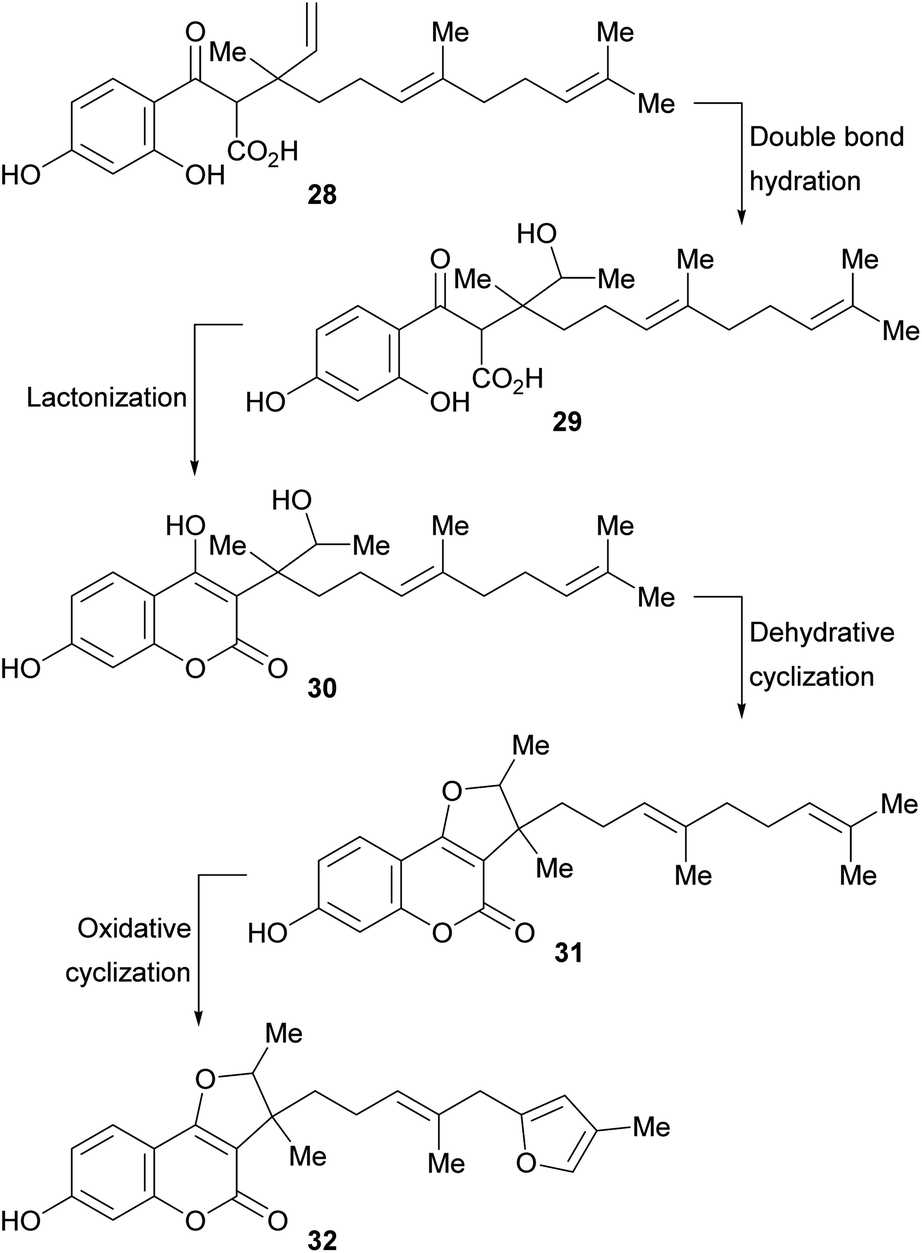 | ||
| Scheme 2 Biosynthetic pathway toward 3-substituted furo[3,2-c]coumarins carrying rearranged farnesyl side chains. | ||
The isolation of compounds bearing other oxygenated functions, such as ketones, lactones and epoxides have also been recorded (Fig. 6). Compounds 33 and 34 were obtained from Ferula feruloides, whereas the heterocycles 35 and 36 were isolated from Vernonia brachycalyx.1c,22
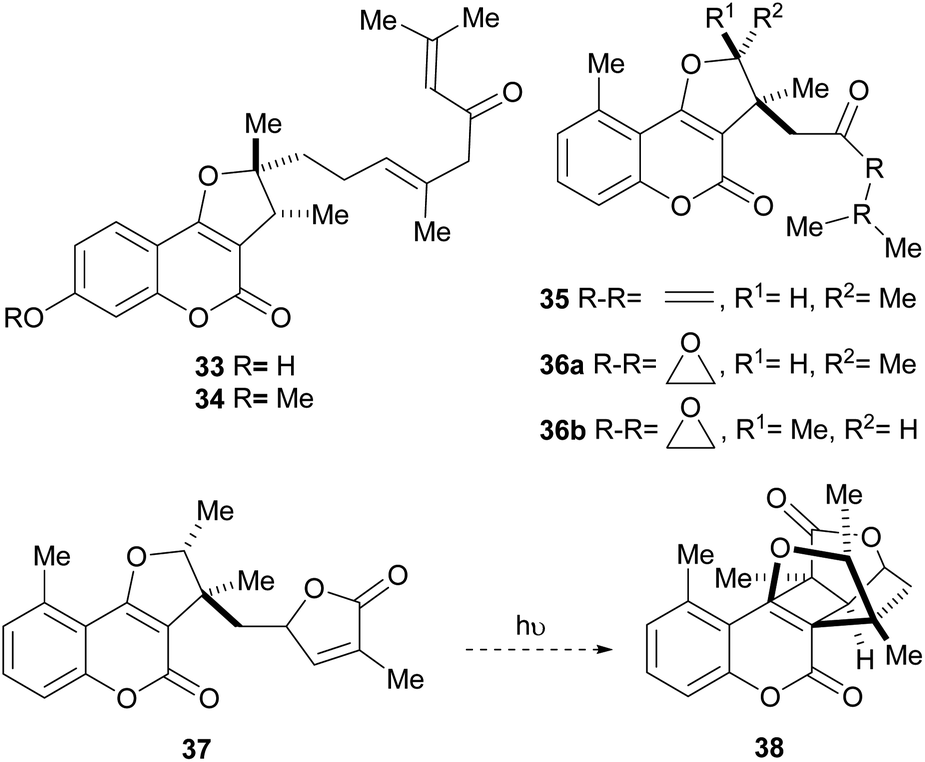 | ||
| Fig. 6 Naturally occurring furo[3,2-c]coumarins bearing different oxygenated functions. | ||
The leaves and roots of the plant are used in Kenia by various indigenous tribes for treatment of parasitic diseases, stomachache and as a purgative. On the other hand, the lactone 37 (hoehmelia coumarin) was isolated from the Kenyan shrub Ethulia vernonioides, together with the unique compound 38 (cyclohoehmelia coumarin). This related structure is a [2 + 2] adduct, presumably resulting from natural irradiation of 37.3a In fact, 37 spontaneously cyclized during optical rotation measurements and irradiation of 37 in benzene for 1 h gave 38 quantitatively.
The pterophyllins 1, 2, 4 and 5 (39–42) are 5-methyl coumarin derivatives (Fig. 7), isolated from the bark and wood of Ekebergia pterophylla (C.D.C.) Hofmeyr (Meliaceae), a small evergreen tree known as Rock Ash, which grows on the Natal Group Sandstone outcrops, in South Africa.23
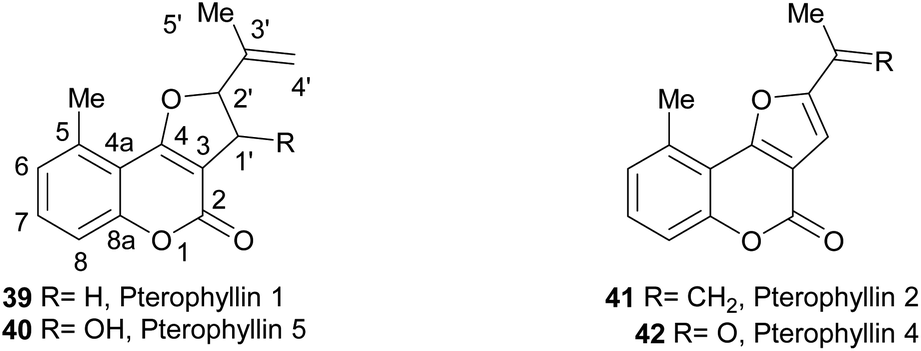 | ||
| Fig. 7 The pterophyllins as naturally occurring furo[3,2-c]coumarins. Original ring numbering. | ||
It has been proposed that the pterophyllins are biogenetically related (Scheme 3). Pterophyllin 4 (42) may be the last metabolite of a chain of events resulting from 43, the product of prenylation at C-3 of 5-methyl-4-hydroxycoumarin (44).16b,24
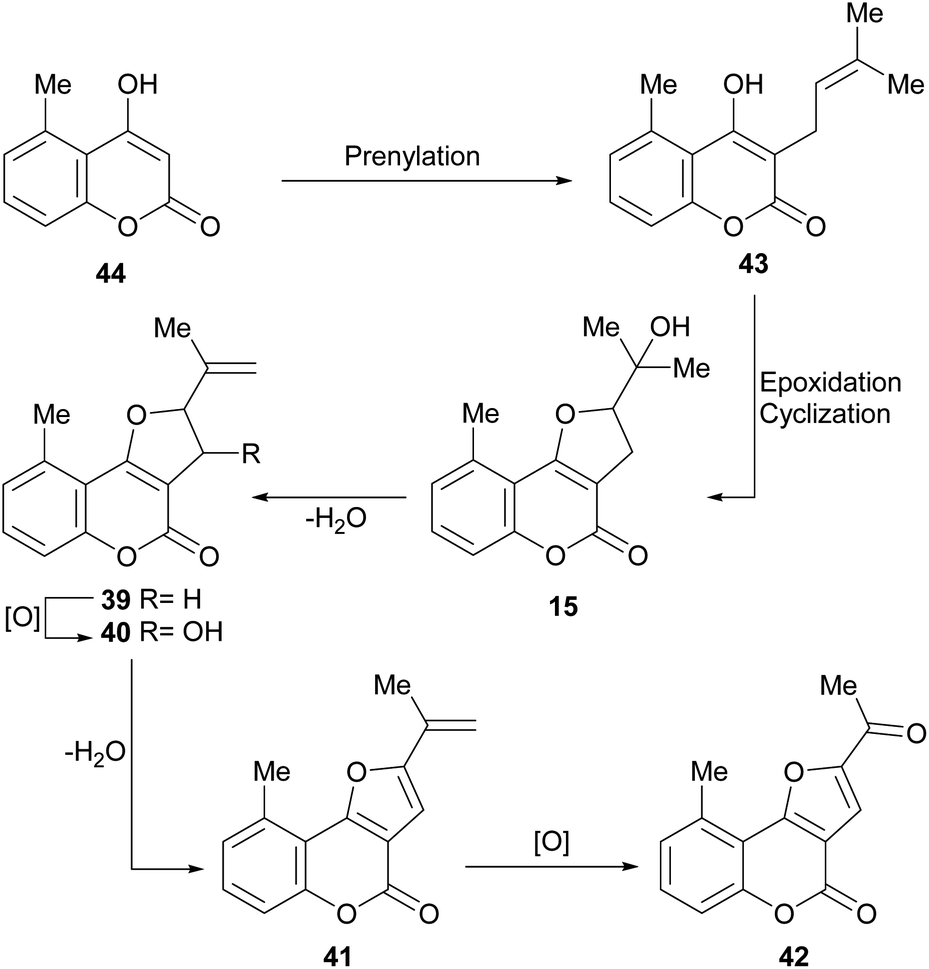 | ||
| Scheme 3 Possible biogenetical path toward the natural products 39–42 from 4-hydroxy coumarin 44. | ||
Compound 43 could undergo epoxidation and a subsequent cyclization to give a tertiary alcohol intermediate (15).25a In turn, the latter could suffer sequential dehydration of the tertiary alcohol moiety to pterophyllin 1 (39), followed by further allylic oxidation to pterophyllin 5 (40) and a final aromatization driven dehydration, resulting in pterophyllin 2 (41).25 Pterophyllin 2 may experience a final oxidative fission of the exocyclic double bond, being transformed into pterophyllin 4 (42).
3 Bioactivities of some furo[3,2-c]coumarins
Numerous members of the furo[3,2-c]coumarin family are bioactive,1c,3a,15,16a,19,26 and possess significant biological, pharmacological and therapeutic activities, such as fungicidal, insecticidal, insect anti-feedant, anti-HIV and anticancer.27 They are also anticoagulants, anthelmintics, hypnotics, and HIV protease inhibitors.28Some of this type of heterocycles, derived from osthole, demonstrated ability to form covalent bonds with DNA and other biological macromolecules.1c In addition, other analogues of this coumarin derivative were evaluated in vitro for their antifungal activity against six phytopathogenic fungi. The compounds 45–48 shown in Fig. 8 were the most active ones.29
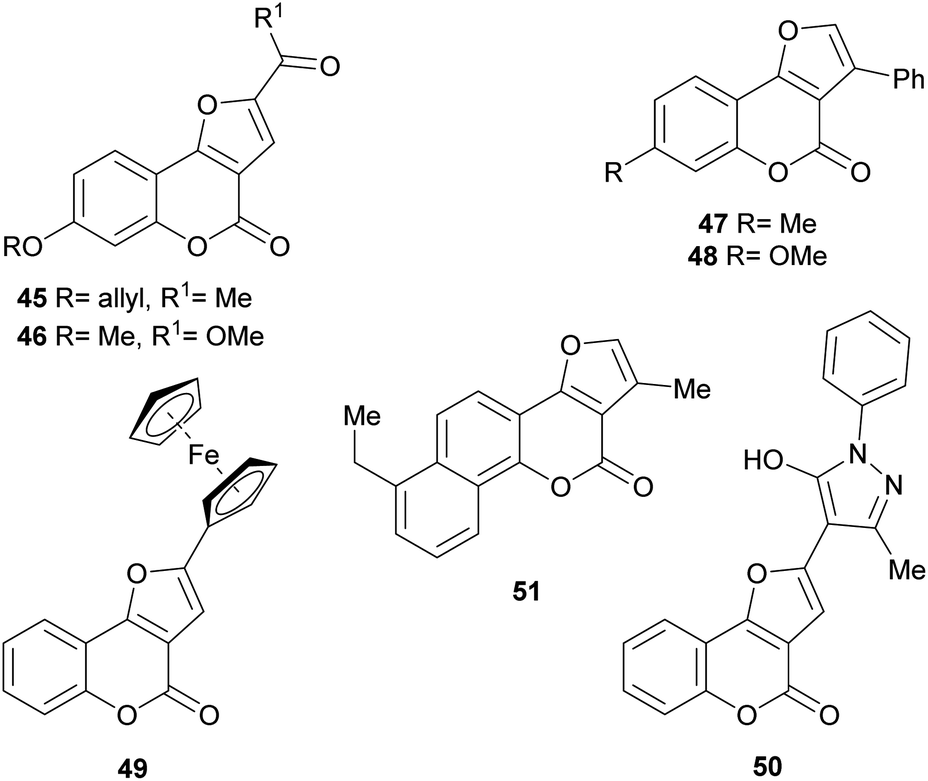 | ||
| Fig. 8 Selected furo[3,2-c] coumarin derivatives with activity against phytopathogenic fungi. | ||
Also, as part of the effort to find a new class of antimicrobial agents, Bondock et al. synthesized pyrazoles attached to furo[3,2-c]coumarins (50). Some of these compounds showed potent antifungal activity in vitro with MIC (minimum inhibitory concentration) of 6.25 μg mL−1 against Fusarium oxysporum and Botrytis fabae.30 Other 2-substituted and 2,3-disubstituted furo[3,2-c]coumarins were shown to be active against Botrytis cinerea, Collecterichum capsica, Alternaria solani, Gibberella zeae, and Rhizoctorzia solani, with EC50 values in the range 2–8 μg mL−1.31
On the other hand, the ferrocenyl-substituted furo[3,2-c]coumarin 49 was prepared by a one-pot aerobic reaction from 3-bromochromone and an alkynyl ferrocene, under palladium and copper(I) catalysis.32 The heterocycle was tested for its anticancer activity against several cell lines, showing IC50 (half maximal inhibitory concentration) values in the range 19–43 μM.
In addition, some members of this family also display interesting photochemical, photophysical and photobiological activities,33 being photosensitizers that increase the sensitivity of biological objects to UVA radiation. Hence, they have potential pharmaceutical applications for skin and autoimmune diseases.
Neo-tanshinlactone (6) is a polycyclic compound isolated from Salvia miltiorrhiza, known in Traditional Chinese Medicine as “Tanshen”,34 and found in many other plants such as S. przewalskii Maxim35 and Pholidota cantonensis.36 Neo-tanshinlactone showed selective cytotoxicity against two estrogen receptor-positive (ER+) breast cancer cell lines, MCF-7 and ZR-75-1,37 being 10-fold more potent than tamoxifen.34a
It was observed that certain tanshinones, bearing ortho-quinone C rings, inhibited telomerase. On the contrary, neo-tanshinlactone, was a significantly less effective inhibitor (25% inhibition). Since neo-tanshinlactone has a lactone C ring, it was suggested that the ortho-quinone motif is a critical requirement for telomerase inhibition.38
In initial SAR (structure–activity relationship) studies, the first generation neo-tanshinlactone analog 4-ethyl-neo-tanshinlactone (51) displayed a median effective dose (ED50) of 0.45 μg mL−1, being more potent than neo-tanshinlactone (ED50 = 0.18 μg mL−1) against these two cell lines. In addition, it potently inhibited the SK-BR-3 breast cancer cell (ED50 0.1 μg mL−1), which is estrogen receptor negative (ER−), but overexpresses HER2 (HER2+).39
Neo-tanshinlactone was also tested in mice against cancer cell xenografts. The natural product delayed tumor growth compared to the control at the 10 mg kg−1 level, and showed significant and selective antitumor activity against the human ZR-75-1 breast ductal carcinoma xenograft.40
We have shown that pterophyllin 2 (41) and pterophyllin 4 (42) have some antifungal activity against phytopatogenic fungi41 (Fig. 9). It has also been proposed that the natural role of the pterophyllins is in the plant defense against phytopathogenic fungi.42 The progressive increase in selectivity and potency with metabolite maturation (39 → 40 → 41 → 42),43 has also been noticed.
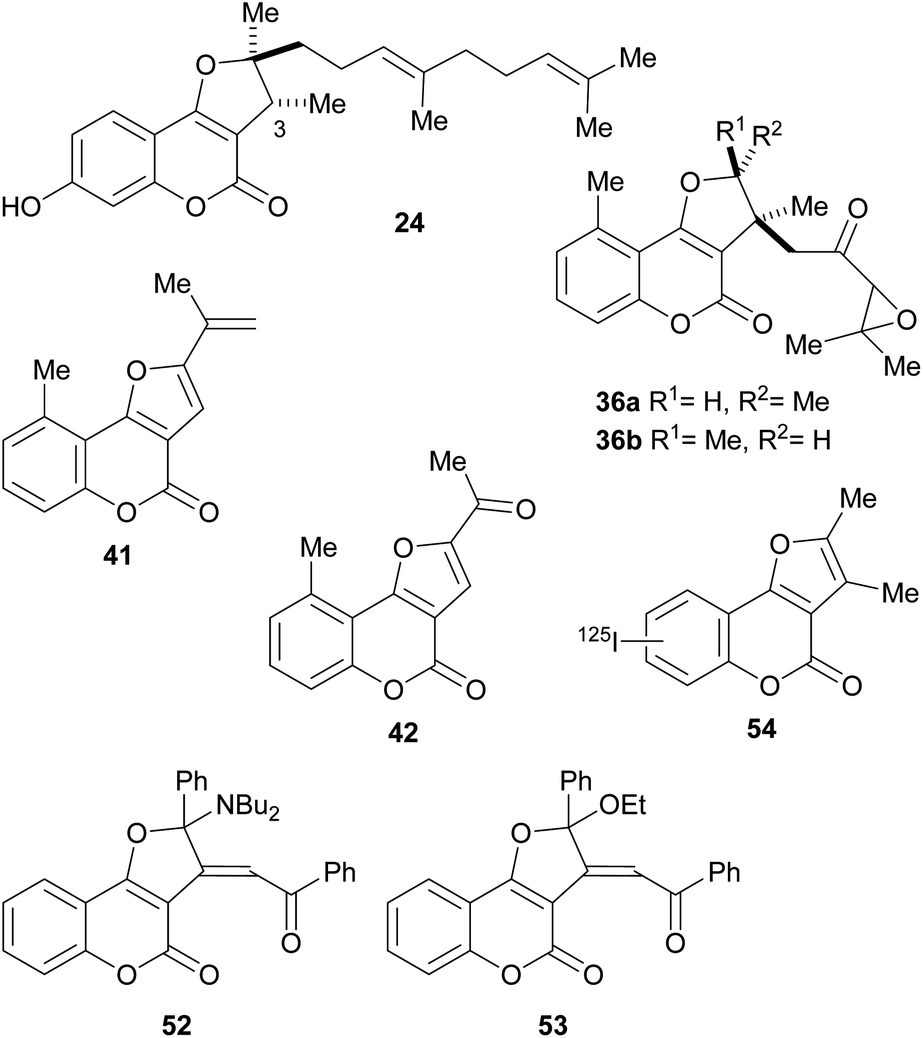 | ||
| Fig. 9 Some selected bioactive furo[3,2-c] coumarins. | ||
In addition, compound 24 and its C-3 epimer were patented as antimicrobials against methicillin-resistant strains of Staphylococcus aureus,44 and the heterocycles 36a and 36b isolated from Vernonia brachycalyx proved to have weak antiprotozoal activity against Leishmania major promastigotes and schizonts of Plasmodium falciparum (approximately three orders of magnitude less than chloroquine). They also exhibited weak inhibition of the proliferation of human lymphocytes.22b
The group of Rajabi demonstrated the antiproliferative activity of the furo[3,2-c]coumarin derivatives 52 and 53 against MCF-7 breast and HCT-15 colon cancer cell lines using the sulforhodamine B (SRB) assay. They also determined their DNA binding affinity K1b = 8.1 × 103 M−1, K1d = 1.1 × 104 M−1.45
In an experiment carried out by the group of Ibrahim, 2,3-dimethyl-4H-furo[3,2-c]coumarin was iodinated with 125I and the uptake of the resulting radiolabelled heterocycle 54 was studied in albino mice with Ehrlich ascites carcinoma cells. The results (5% uptake) supported the potential use of this heterocycle as a probe for imaging of tumor sites.46
4 Chemical synthesis of 2-substituted furo[3,2-c] coumarins
Given the high level of interest conceited by these heterocycles, a number of procedures have been developed for their synthesis. Most of them use the corresponding and easily available 4-hydroxycoumarins as starting materials, becoming in fact strategies for building a properly substituted furan ring attached to the enolether motif of the starting heterocycle. The main approaches are classified and briefly discussed below, bearing in mind that the boundaries between them are not always well defined.4.1. Reaction of 4-hydroxycoumarins with terminal alkynes
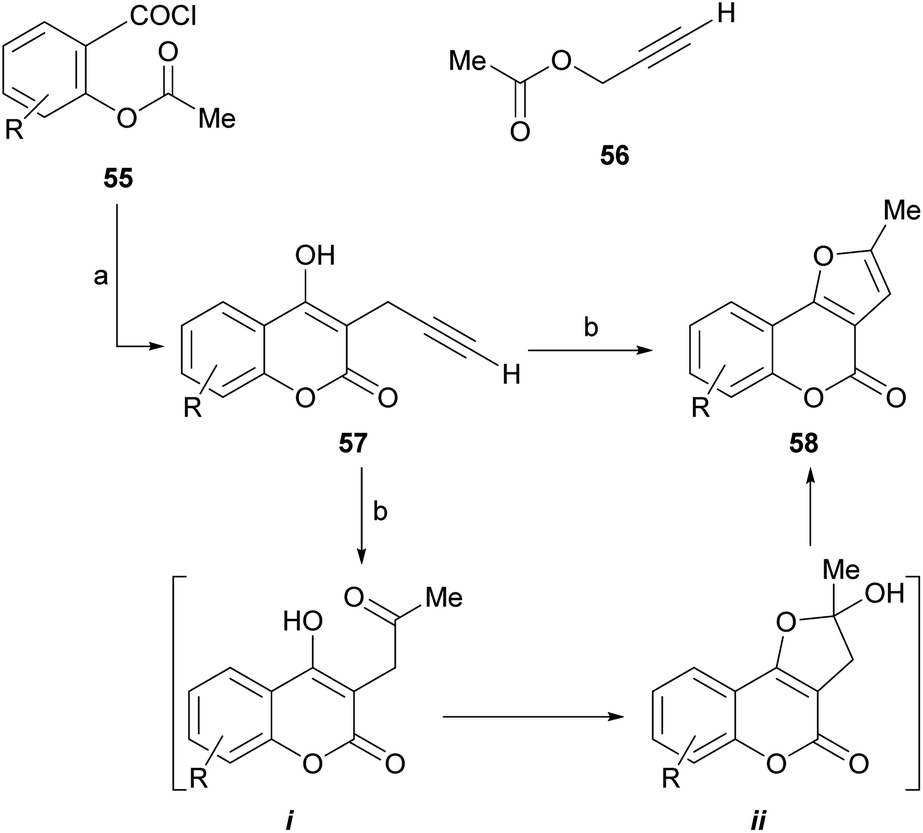 | ||
| Scheme 4 Reagents and conditions: (a) 56, NaH, Et2O (65%); (b) H2SO4, H2O (70–90%, overall). | ||
Mechanistically, it appears that, in the presence of aqueous acid, the double hydration of the terminal alkyne would take place to give an intermediate methyl ketone (i). This one would condense with the 4-OH and the resulting hemiacetal ii would dehydrate to conclude the methylfuran motif of the final product.
The group of Saidi group perfected the process (Scheme 5), informing that the propargyl ethers 60 derived from 4-hydroxycoumarins (59) can undergo rearrangements to 61 and finally give furo[3,2-c] coumarin 62.48 The unequivocal assignement of the 13C NMR data of 62 has been disclosed.49
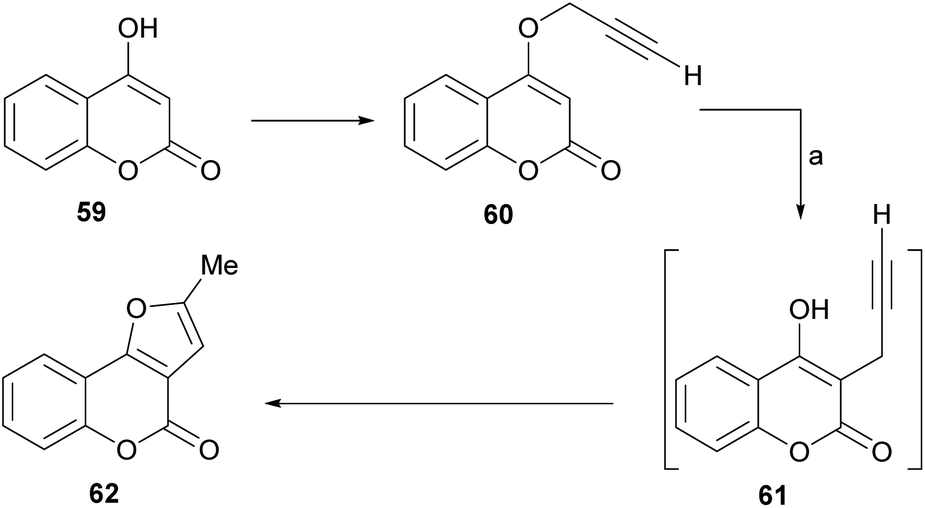 | ||
| Scheme 5 Reagents and conditions: (a) HCONHMe, MW, 18 min (65%), or HCONHEt, 14 h, reflux (52%). | ||
These rearrangements take place when the compounds are exposed to conventional microwave or thermal heating in amide-type solvents [N-ethylformamide (NEF) or N-methylformamide (NMF)], and pyranocoumarins can also be obtained. NEF and NMF are advantageous, since unlike DMF or DMA, they are soluble in water and can be removed very easily from the product.
The authors observed that the rearrangement is more efficient under microwave promotion. Mechanistically, the transformation proceeds through a thermal Claisen rearrangement of the O-propargyl ether 60 followed by cyclization of the resulting 3-propargyl intermediate 61, which is not isolated. The yields of product are moderate to good (52–65%).
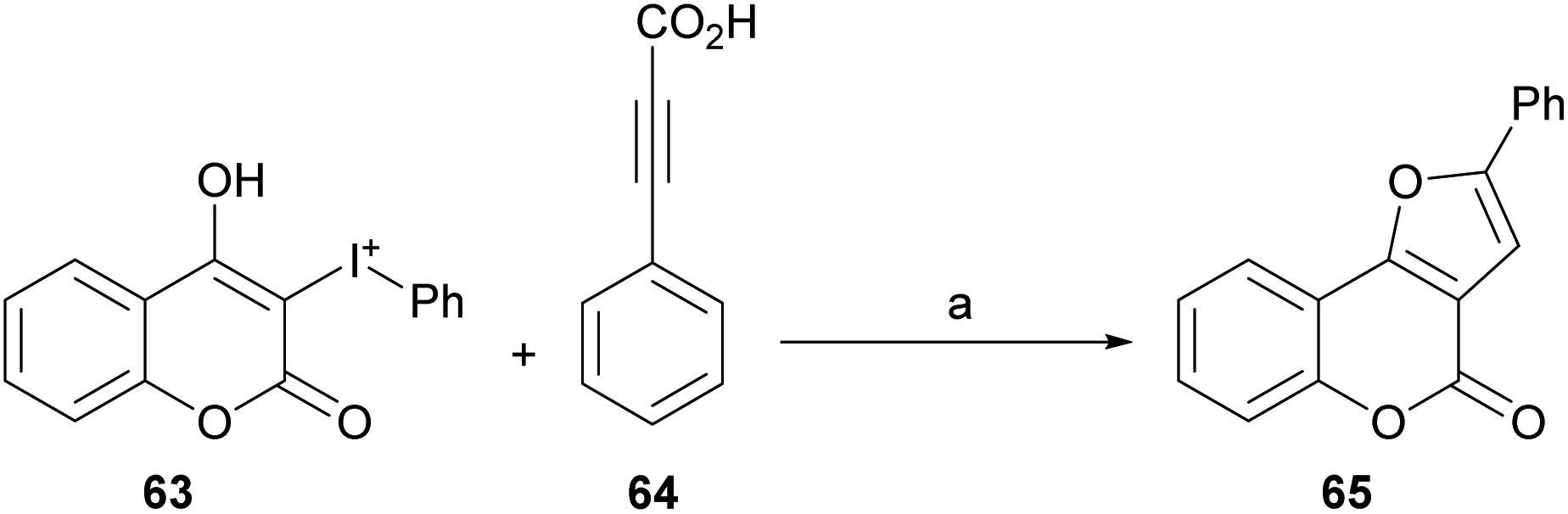 | ||
| Scheme 6 Reagents and conditions: (a) [Cp*RhCl2]2, Ag2O, MeCN, 60 °C, 12 h (79%). | ||
The reaction does not require directing groups and the mild decarboxylation undergone by the carboxylic acid is compatible with a wide range of functional groups and generates the derivatives with good yields (∼80%) of the isolated products.51
Based on previous reports,52 a possible reaction mechanism was proposed (Scheme 7). Heating the hypervalent iodine starting material 63 could generate a carbene,53 which could then interact with [Cp*RhCl2]2 and release iodobenzene, providing intermediate i. In turn, this intermediate should undergo transmetalation with alkynylsilver,54 generated in situ from the reaction of the phenylpropiolic acid 64 and Ag2O to give intermediate ii.
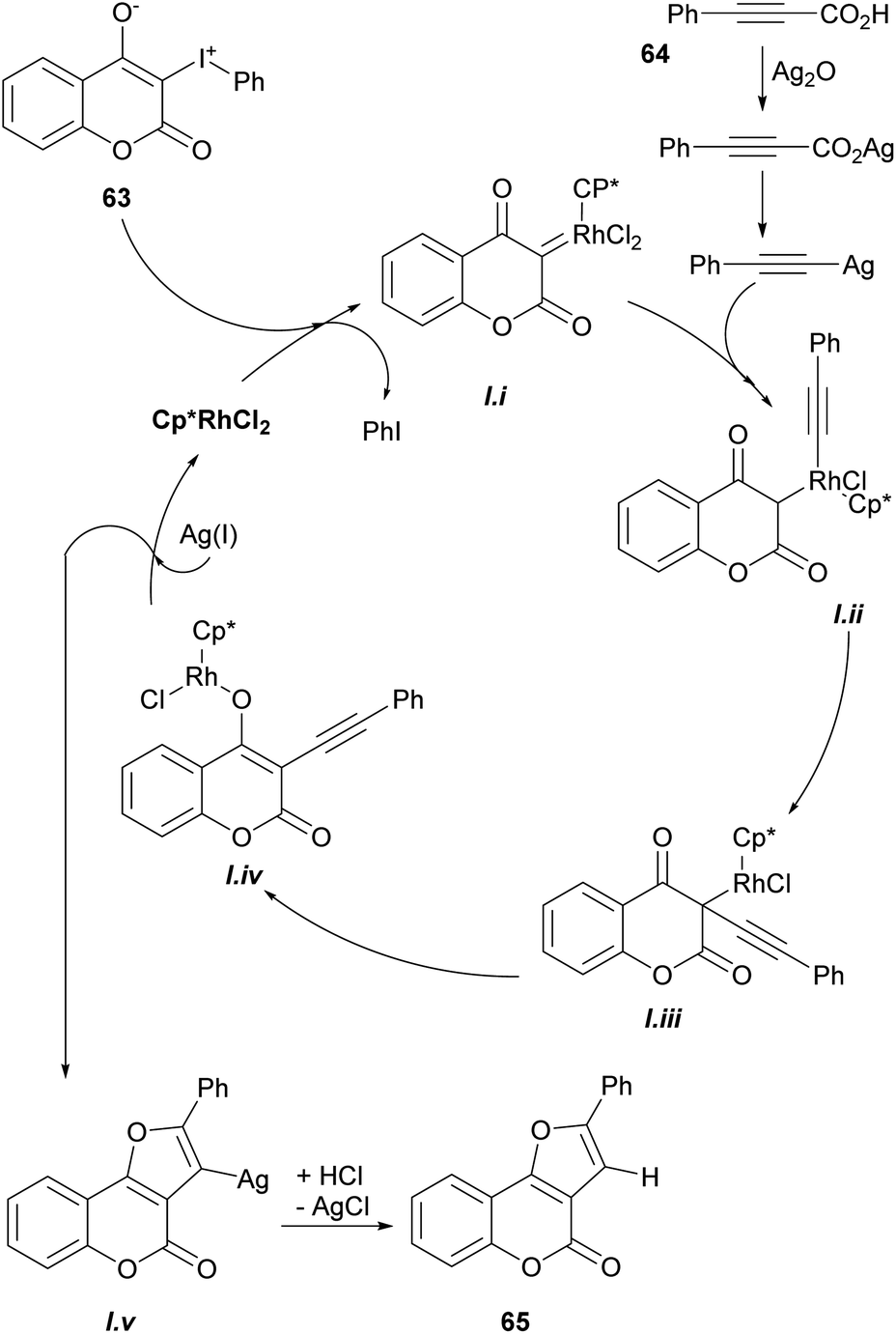 | ||
| Scheme 7 Mechanism of the Rh-catalyzed decarboxylative cyclization. | ||
Then, intermediate iii could be formed by direct insertion of the alkyne into the Rh-carbene, which could migrate to the oxygen (iv) and further react with Ag(I) to give intermediate v and regenerate the catalyst. Finally, removal of the silver by protonation with HCl or H2O would afford the tricyclic product 65.
Inspired in their previous success with the preparation of pyrrolocoumarins,55 the group of Chen achieved a fast option for the construction of furo[3,2-c]coumarins by means of a one-pot sequential coupling/cyclization strategy, from 3-bromo-4-acetoxycoumarins and terminal alkynes (Scheme 8).56
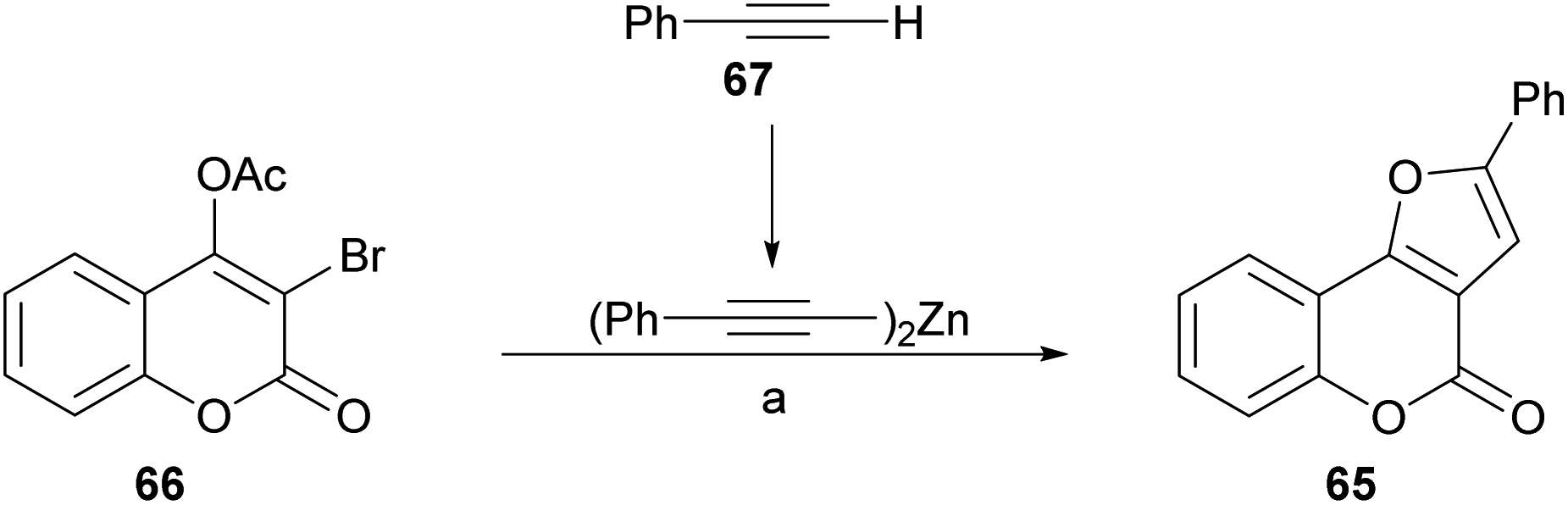 | ||
| Scheme 8 Reagents and conditions: (a) (1) 67, ZnMe2, PhMe, 60 °C, 4 h; (2) Pd(PPh3)4, CuI, dppf, Et3N, THF, 60 °C, 6 h; (3) K2CO3, H2O, overnight. | ||
The required 3-bromo-4-acetoxycoumarin 66 was prepared by treatment of 4-hydroxycoumarin (59) with N-bromosuccinimide (NBS) and Mg(ClO4)2 as a Lewis acid.57 A safer, practical and mild alternative employed ZnCl2 combined with NBS in EtOAc,56 which gave the 3-bromo-4-hydroxy derivative in 81% yield. In both cases, this stage was followed by conventional acetylation (Ac2O, pyridine) toward 66.
Sonogashira58 conditions [Pd(PPh3)4, CuI, Et3N, THF, 60 °C] proved to be inefficient (8% yield) due to extensive debromination of 66. However, this inconvenience was partly alleviated upon addition of dppf [1,1′-bis(diphenylphosphino)ferrocene] as ligand and K2CO3 as base to promote hydrolysis of the acetate at a second stage (29% yield). Similar results (25% yield of 65) were obtained in the Negishi cross-coupling attempt with alkynyl zinc derivatives.59
However, the use of dialkynylzinc reagents prepared in situ from alkynes and Et2Zn, furnished 38% product yield, which was increased to 81% upon addition of dppf.60 The last stage of the sequence involved an intramolecular hydroalkoxylation.61 The yields of the 3-phenyl furo[3,2-c]coumarin products 65 with different substitution patterns were moderate to excellent (51–96%).
The group of Conreaux noticed that various coumarin derivatives proved to be convenient precursors of furocoumarins and developed a sequence complementary to Chen's work (Scheme 9). Their approach includes the Sonogashira coupling of 68 with the copper acetylides formed in situ from the corresponding acetylenes 69.
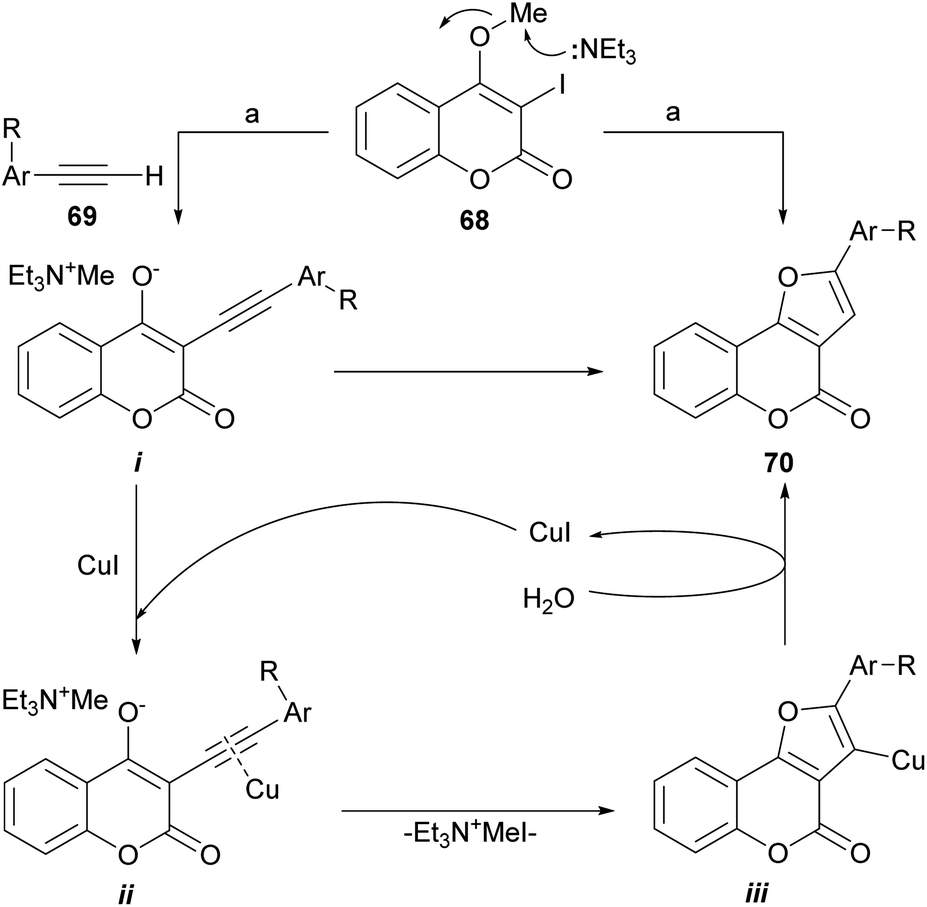 | ||
| Scheme 9 Reagents and conditions: (a) 69, Pd(PPh3)2Cl2 (cat.), CuI (cat.), Et3N, MeCN, 60 °C, 15 h (70%). | ||
According to the proposed reaction mechanism, the SN2-type nucleophilic displacement of the methyl group by Et3N62 would generate a delocalized triethylmethyl ammonium enolate i and the latter would then undergo a regioselective anionic 5-endo-dig cyclization to form the tricyclic furan intermediate iii through the intermediacy of the copper-coordinated acetylene ii.63 Upon capture of a proton from water,64 iii would furnish the desired furo[3,2-c]coumarin 70.
Dey and Hajra developed an aerobic synthesis of angularly fused furans catalyzed by FeCl3/ZnI2 (Scheme 10), by intermolecular coupling between enols and alkynes as 69.65 This methodology was applied to 4-hydroxycoumarin (59) providing regioselectively good to very good yields of the furan derivatives 70. Other active methylene compounds were also successfully tested.
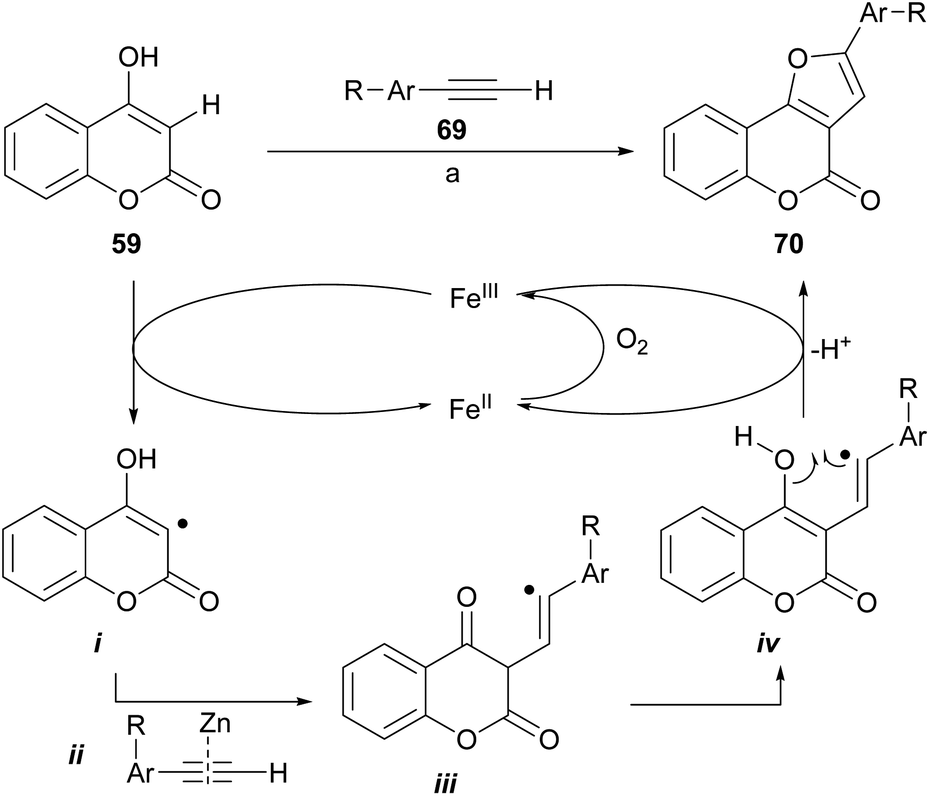 | ||
| Scheme 10 Reagents and conditions: (a) FeCl3, ZnI2, DMSO, O2 (air), 130 °C, 8 h (59–80%). | ||
The authors suggested a radical-type mechanism, where the 4-hydroxycoumarin is converted by FeIII into a carbon-centered radical (i). In turn, this radical reacts with the zinc-activated phenylacetylene (ii)66 to form a vinyl radical intermediate iii. Subsequent tautomerization into iv and further cyclization under FeIII assistance results in furan 70. In the presence of air, the FeIII promoter is regenerated from FeII.
Recently, the group of Ma reported the synthesis of furo[3,2-c] coumarins by a I2/TBHP-mediated reaction of 4-hydroxycoumarins with terminal alkynes, in the presence of KOAc as a mild base, under aerobic conditions.67 The transformation accepts aryl as well as alkyl substituted alkynes; however, in the case of the latter the yields are rather low.
The authors also proposed a reaction mechanism (Scheme 11), where the starting coumarin 59 is first iodinated by I+, produced by exposure to I2 to TBHP, to afford 3-iodocoumarin (59a), which originates the radical intermediate i. Next, the addition of the alkyne generates the vinyl radical intermediate ii, which cyclizes with the carbonyl group to give iv through the intermediacy of iii. The former is finally oxidized by TBHP to form the target molecule 70.
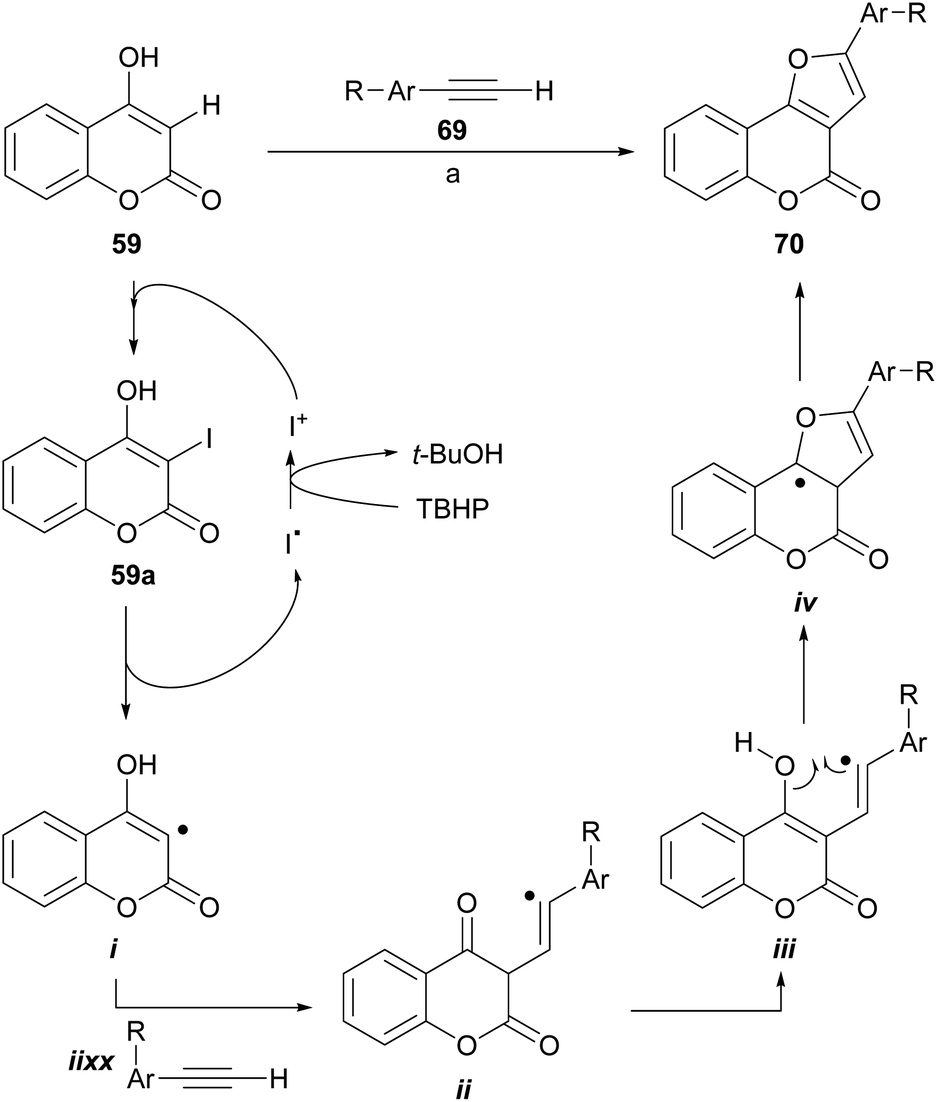 | ||
| Scheme 11 Reagents and conditions: (a) I2, TBHP, KAcO, air, dioxane, 90 °C, 8 h (30–88%). | ||
Visible-light photoredox catalysis has been utilized as a powerful tool to build C–C and C-heteroatom bonds through a single-electron transfer (SET) pathway. The construction of furo[3,2-c]coumarins 70 using a photo-redox process (Scheme 12) promoted by visible light and under iridium catalysis has been described, through the neutral coupling between 3-bromo-4-hydroxycoumarins (71) and functionalized aromatic alkynes (69). This is a special case of the synthesis of polysubstituted furans using visible-light-promoted radical alkyne insertion with 2-bromo-1,3-dicarbonyl compounds.68
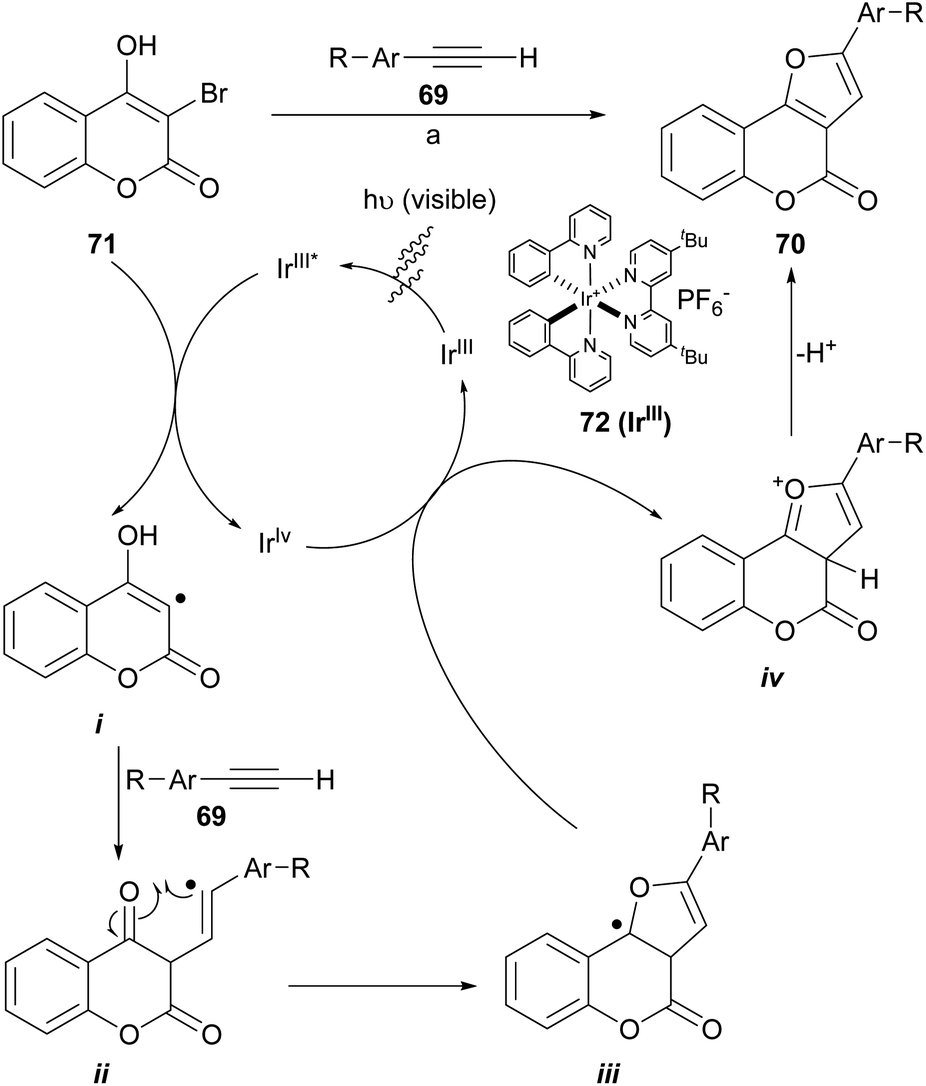 | ||
| Scheme 12 Reagents and conditions: (a) NaHCO3, Ir(ppy)2(dtbbpy)PF6 (72), DMSO, white LEDs (13 W), r.t. (41–93%). | ||
The transformations take place at room temperature under irradiation with visible light. They accepted alkyl, fluoro and methyl ether substituents on the alkyne side and methyl ethers as well as halogens on the coumarin component and provided moderate to excellent product yields (41–93%) when 72 was used as catalyst. Some of the so synthesized heterocycles were moderate inhibitors of the enzyme acetylcholinesterase (IC50 = 2.16 ± 0.13 μM).69
A similar photo-catalytic process was reported involving methylene blue as sensitizer and K2S2O8 as oxidant in the photo-redox cycle. In the only example provided, the transformation was performed in MeCN at room temperature and under blue LED light irradiation, furnishing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the linear and angularly fused furocoumarin products in 71% overall yield.70
:1 mixture of the linear and angularly fused furocoumarin products in 71% overall yield.70
A catalytic cycle was proposed based on literature precedents.71,72 There, the IrIII complex 72 is irradiated to its excited state IrIII* and oxidatively quenched by the 3-bromocoumarin 71 with the generation of an IrIV complex and the radical species i. Then, the radical adds onto the alkyne (69) to generate the vinyl radical intermediate ii, which can perform an intramolecular attack to the carbonyl group and give radical intermediate iii. The oxidation of this intermediate by the IrIV species forms the cationic species iv and regenerates the catalyst. Final base-assisted deprotonation of iv gives the furocoumarin product 70.
4.2. Oxidative cyclization of 3-alkynyl chromones
Cheng and Hu developed a novel metal-free73 one-pot cyclization/oxidation two-stage cascade which uses water as nucleophile (Scheme 13), for the regioselective synthesis of furo[3,2-c]coumarins as 74 from 3-alkynyl chromones (73). In the initial version,74 the transformation took place through reaction with MeSO3H and HCO2H as acidic promoters in DMSO–1H2O, to which a catalytic amount of HBr was added as an oxidant, at 105–120 °C.75 However, the yields were rather low (5–25%).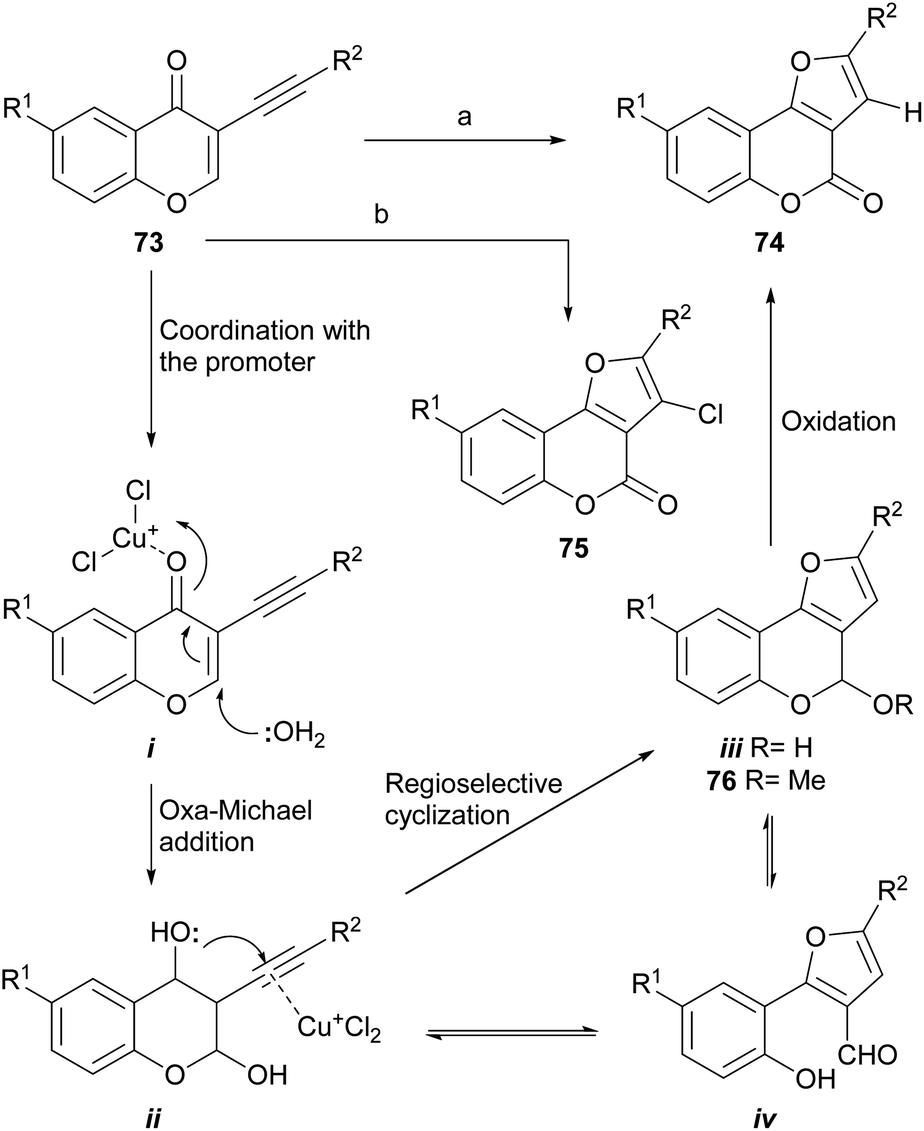 | ||
| Scheme 13 Reagents and conditions: (a) CuCl (cat.), O2, DMF, H2O, 90 °C; (b) CuBr (cat.), CuCl (10 equiv.), DMF, H2O, 75 °C. R1 = H, Me, Cl, OMe, NO2; R2 = alkyl, aryl (37–89%). | ||
In an improved version, the authors modified the conditions to use CuCl2 as the oxidant, employing a DMF–1H2O solvent mixture, at 90 °C for 20 h.7a In this way, the reaction proceeded at a lower temperature, with good yields, and evolved easily without the need of anhydrous solvents and an inert atmosphere. Furthermore, the yields ranged from satisfactory to very good (37–89%). It was conjectured that a single electron transfer mechanism might be operative in this sequence.76
In a still more refined version (Scheme 13), where the addition of reagents is done in stages,74b the authors removed the methanesulfonic acid as reagent and developed two efficient one-pot conditions to generate different furo[3,2-c]coumarins, by an addition/cyclization/oxidation sequence with a final chlorination step.
The use of MeOH as the nucleophile resulted in 97% yield of a mixed acetal (76), which under the reaction conditions cannot be oxidized to afford the coumarin. Under an aerobic atmosphere and promotion by CuCl as Lewis acid, compound 74 was obtained, while using CuBr as a catalyst and excess CuCl as oxidant, the 3-chloro derivative 75 could also be obtained. The latter was used as a scaffold to introduce aryl moieties through a Suzuki reaction, which enabled an easy access to 2,3-disubstituted furo[3,2-c]coumarins.74b
Mechanistically, it was conjectured that the transformation would take place by coordination of the acid promoter with the carbonyl (i), to trigger an oxa-Michael reaction with water on the chromone and generate the intermediate ii. Subsequent activation of the alkyne with a Lewis acid would induce the regioselective cyclization to form the new oxygenated ring (iii). The final oxidation of the lactol ii or its open form (iv), would provide the product 74.
A variation of this sequence was also developed. It entails using α-enaminones (77)77 as replacement nucleophiles instead of water, for the Michael addition to 73 and a subsequent 5-endo-dig cyclization (Scheme 14). This process does not require contaminating transition metals, nor inert atmosphere or anhydrous solvents.78
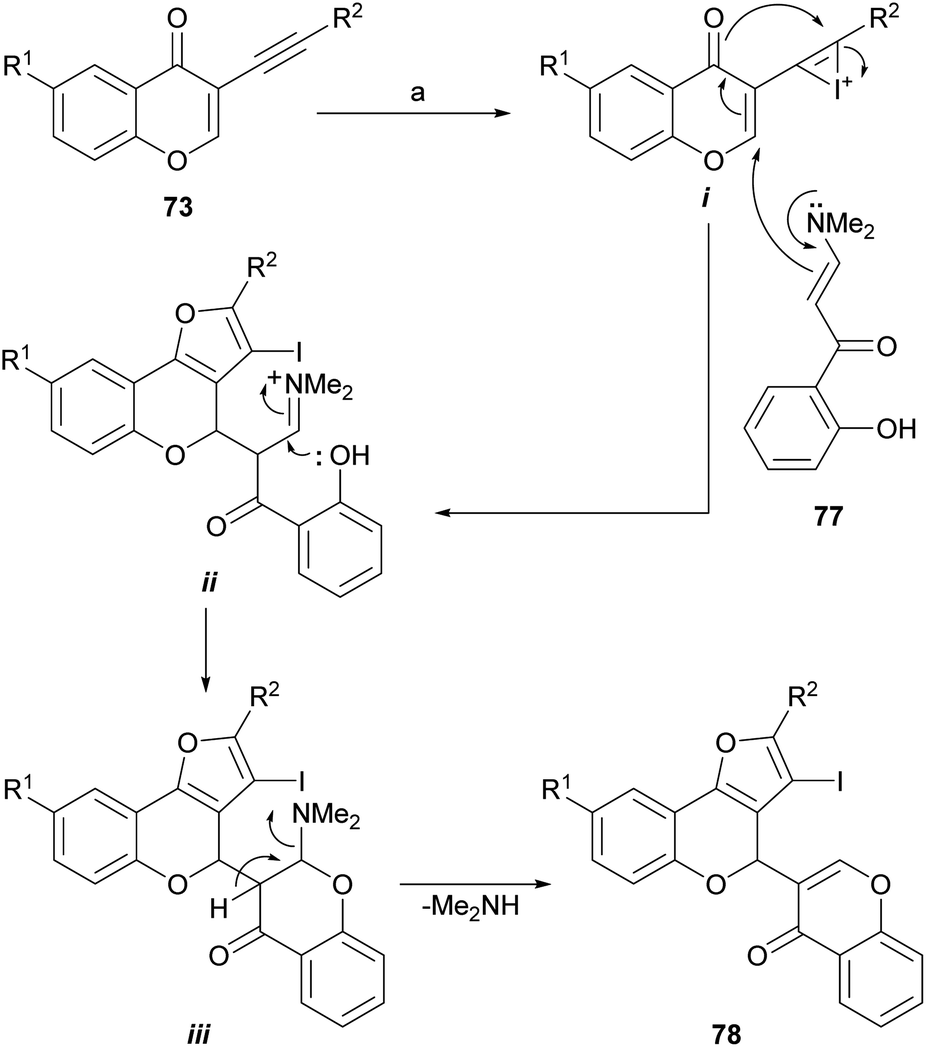 | ||
| Scheme 14 Reagents and conditions: (a) CuBr, DMF, 89–90 °C, 4 h (70%) or I2, CH2Cl2, r.t., 15 h (67%). | ||
When CuBr or molecular iodine were used as promoters, it was possible to construct more complex heterocyclic derivatives, such as 4-(3-chromonyl)furo[3,2-c]-coumarins (78) with good yields; however, the use of ICl in place of I2 resulted in diminished product yield.
The formation of 78 under both conditions can be rationalized similarly, as exemplified with iodine (Scheme 14), considering the initial activation of the alkyne moiety by the promoter to form the intermediate i. This one undergoes a vinylogous aza-Michael addition with the enamine 77 followed by a dehydrative cyclization between the enolic intermediate and the alkyne to form the furan ring in the first intermediate ii.
The appropriately placed phenolic hydroxyl group can then attack the iminium ion moiety and cause the second ring closure, giving in this way the intermediate iii, which would eliminate dimethylamine and finally produce 78. The iodo derivative can be either deiodinated or employed as a scaffold toward more complex structures.
4.3. Diazocarbonyl heterocyclization catalyzed by rhodium salts
The rhodium-mediated decomposition of diazocarbonyl compounds has become an important method in the synthesis of heterocyclic structures such as furans.79The group of Tollari reported (Scheme 15) that the decomposition of 3-diazobenzopyran-2,4(3H)-diones (79) catalyzed by rhodium(II) acetate, in the presence of terminal alkynes (80) and in reflux of halogenated solvents, produces intermediate rhodium carbenoids that give rise to a mixture of isomeric derivatives furo[3,2-c]coumarin (81) and the linear furo[3,2-b]coumarin (82), resulting from a formal [3 + 2] cycloaddition.80
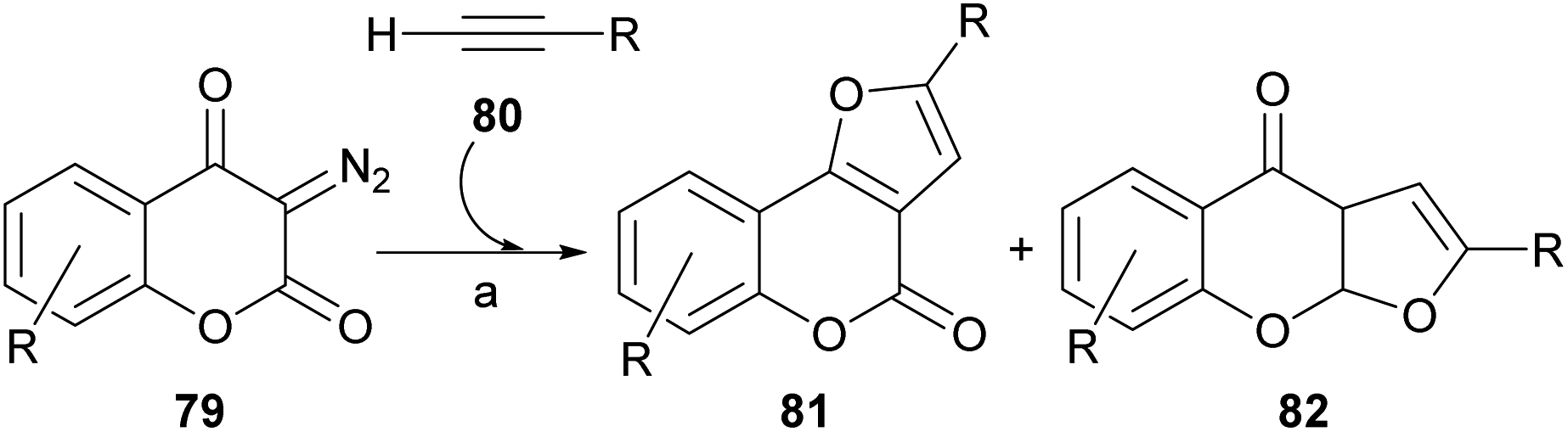 | ||
| Scheme 15 Reagents and conditions: Rh2(OAc)4, C6H5F, reflux (81, 4–45%; 82, 25–41%). | ||
Although the mass balance of the reaction was reasonable, mixtures of two compounds were always produced, and the yield in the studied cases never exceeded 45% of each of them. It was shown that the reaction accepts aryl-, alkyl- and even silyl-substituted terminal alkynes as the alkyne component.
From the mechanistic perspective (Scheme 16), the metal-carbene i is generated in situ by the Rh(II)-catalyzed decomposition of the starting diazocoumarin (79). This intermediate then adds the alkyne to give the highly strained rhodacyclobutene ii, in equilibrium with the strained cyclopropene iii, which could undergo a reversible [2 + 2] electrocyclic [π2σ + σ2σ] ring opening as a means of releasing strain.
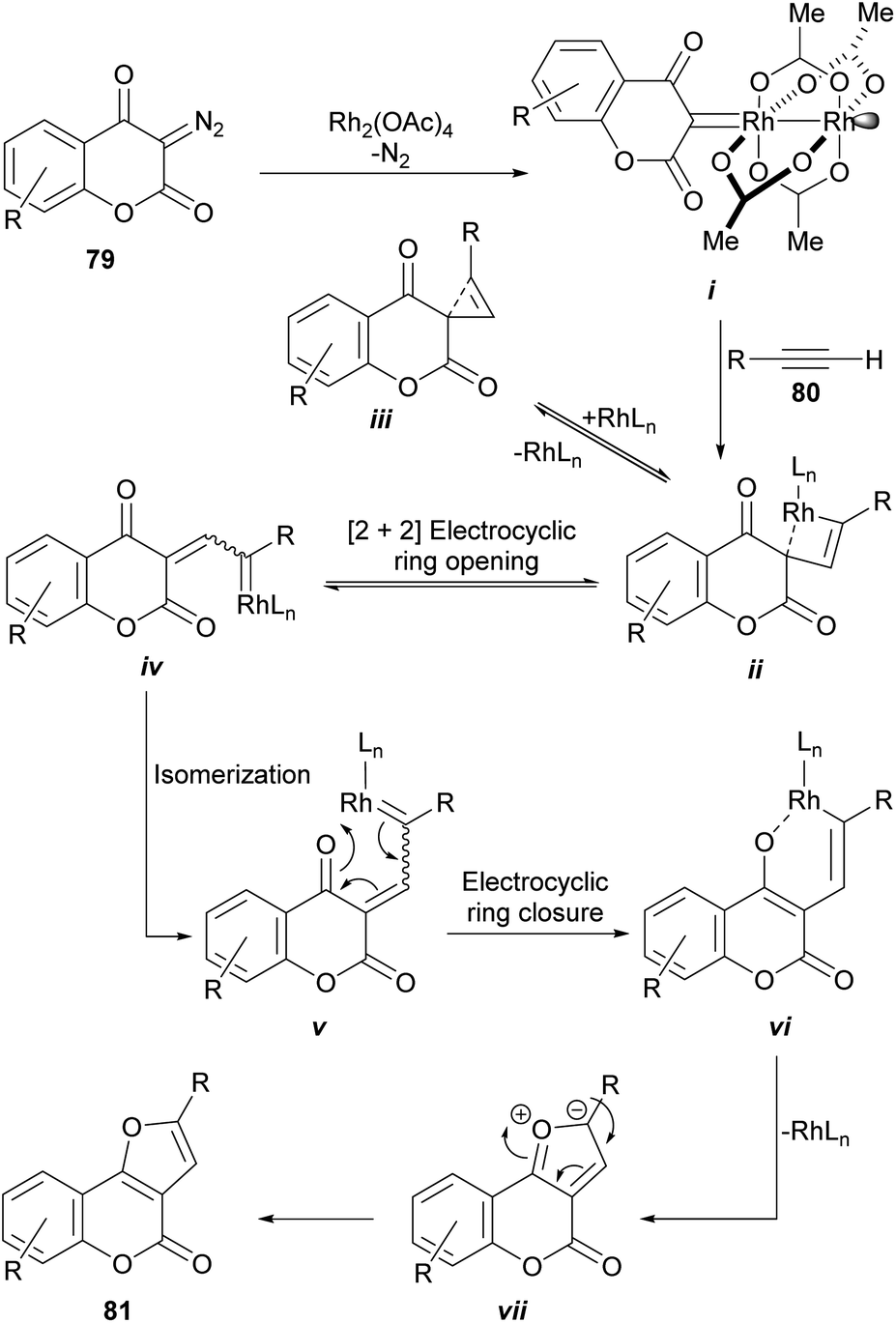 | ||
| Scheme 16 Proposed mechanism for the rhodium-catalyzed synthesis of furo[3,2-c]coumarins from diazocoumarins. | ||
In turn, this should give the corresponding Rh-coordinated vinylcarbene iv, which can exist as an E/Z pair of equilibrating diastereomers with v. The following electrocyclic ring closure of v would then give the corresponding oxarhodacycle vi.81 The collapse of which, possibly through the intermediacy of the ylide vii, would afford the furan motif 81 via reductive elimination and further double bond rearrangement and aromatization.
Lee and Suk studied the synthesis of dihydrofurans and furans by cyclic diazodicarbonyl reactions, catalyzed by rhodium(II) salts.82 The diazodicarbonyl compounds (79) were prepared by a diazotransfer reaction of the corresponding 1,3-dicarbonyl type compounds (4-hydroxycoumarins, 82) with mesylazide (MsN3) according to the method of Taber.83
Their reactions with allyl iodide were carried out at room temperature for 5 h in the presence of 1 mol% of Rh2(OAc)4, obtaining the iodides 83, as a result of the cycloaddition, with moderate performance (Scheme 17).
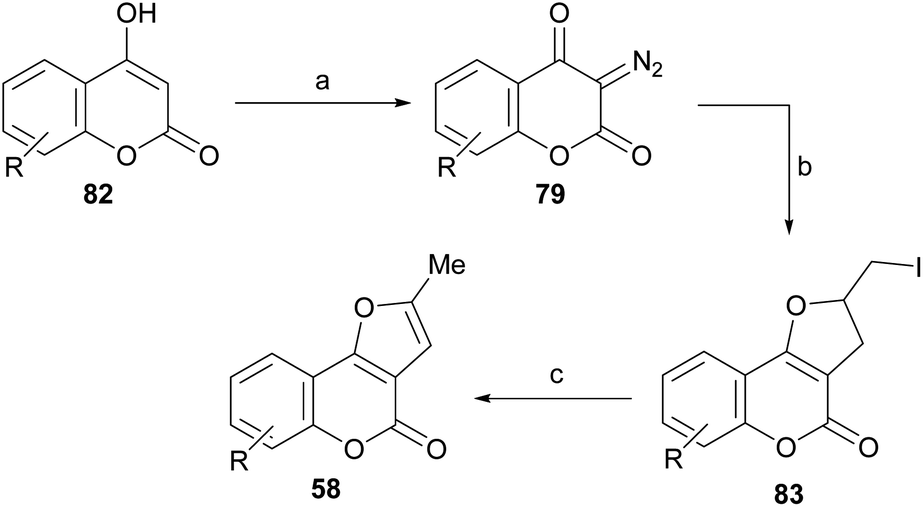 | ||
Scheme 17 Reagents and conditions: (a) MsN3; (b) Rh2(OAc)4, ICH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, r.t., 5 h (R = H, 5-Me, 7-Me and 8-Me, 58–78%); (c) DBU, PhH, r.t., 20 h (80–98%). CH2, r.t., 5 h (R = H, 5-Me, 7-Me and 8-Me, 58–78%); (c) DBU, PhH, r.t., 20 h (80–98%). | ||
The subsequent reaction of iodides 83 with DBU in benzene, at room temperature for 20 h caused dehydroiodination and resulted in the furocoumarins 58 in 80–98% yield. The strategy allows control of the regiochemistry of the transformation and ensures access to the fully aromatized heterocycle, free from its terminal methylene isomer.82
4.4. Cyclization of 4-hydroxycoumarin derivatives with alkenes
A non-oxidative intramolecular cyclization of 3-allyl-4-hydroxycoumarin derivatives (88) was reported by the group of Majumdar (Scheme 18).84 These researchers performed the successive Williamson etherification of 4-hydroxycoumarins 84 with 2,3-dichloropropene and rearrangement of the resulting allyl ethers 85 to obtain compounds 86.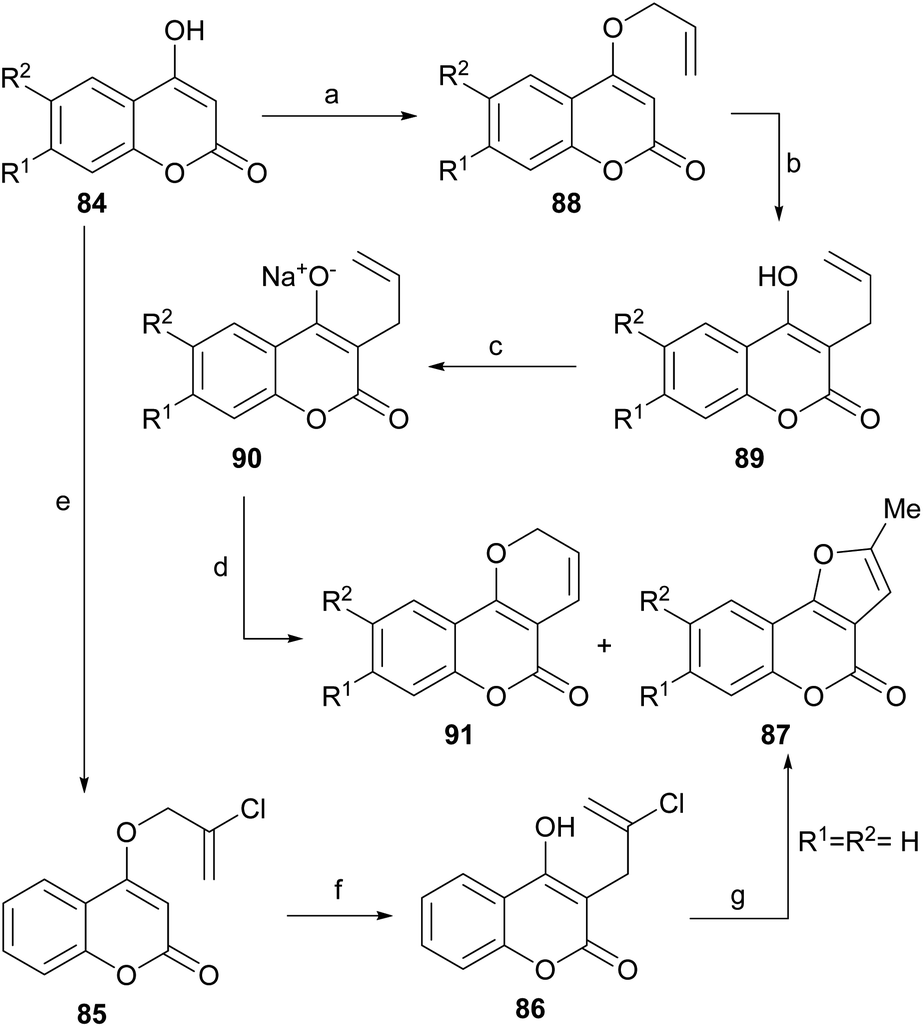 | ||
| Scheme 18 Reagents and conditions: (a) BrCH2CHCH2, K2CO3, Me2CO, reflux, 6 h (96–98%); (b) 180 °C, 0.1 torr, 2 h (90–92%); (c) NaOH, H2O (100%); (d) (1) Pd (PhCN)2Cl2, PhH, r.t., 30 min; (2) reflux, 2 h (40–46%); (e) ClCH2C(Cl)CH2, K2CO3, Me2CO, reflux, 20 h (50%); (f) PhCl, reflux, 8 h (90%); (g) H2SO4 (conc.), 0 °C, 2 h (85%) or DMA, reflux, 0.5 h (75%). | ||
The latter were subjected to a cationic cyclization in concentrated H2SO4 or under thermal conditions with DMA, to afford 87 in 75–85% yield. The overall yield (∼35%) of the sequence was moderate, but only one example was studied.
The group of Jagdish Kumar reported a complementary palladium-catalyzed oxidative intramolecular cyclization approach toward these compounds (Scheme 18). They observed that the reaction of different 4-hydroxycoumarins (84) with allyl bromide under Williamson etherification conditions produced the corresponding 4-allyloxy derivatives (88), which upon Claisen rearrangement gave the expected 3-allyl-4-hydroxy-2H-coumarins (89).
Their corresponding sodium salts were obtained (90) by treatment with aqueous NaOH and then they were exposed to an oxidative cyclization with equimolar amounts of PdCl2(PhCN)2,85 to give a 1:1 mixture of furo[3,2-c]coumarins (87) and pyrano[3,2-c] coumarin (91) products.86
The proposed mechanism (Scheme 19) for the generation of the furo[3,2-c] coumarins (87) entails the formation of the dimeric-π-allyl complex i, which was converted into the isomeric monomer iii, through the participation of intermediate ii, the cyclization of which resulted in the tricyclic compound v through the intermediacy of iv. Final aromatization released the catalyst and afforded the expected coumarin derivative 87.
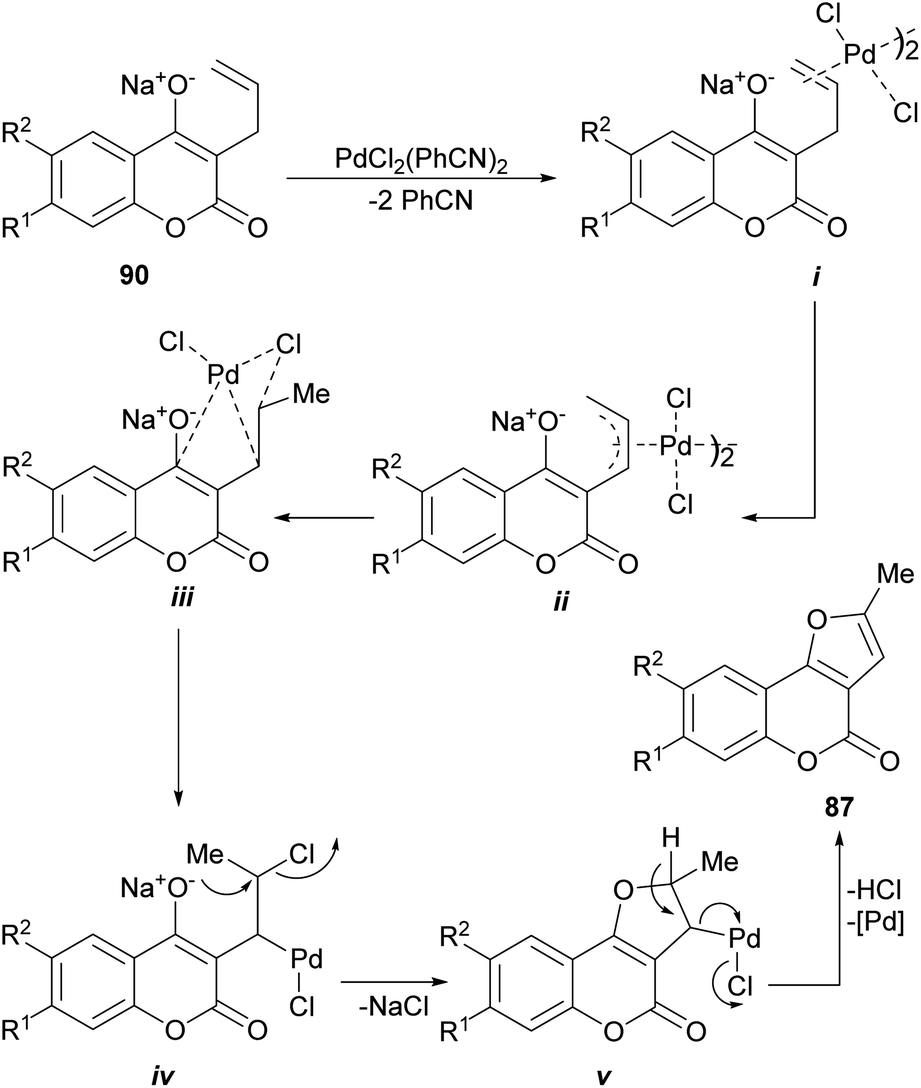 | ||
| Scheme 19 Proposed mechanism for the Pd-mediated oxidative cyclization of 3-allyl-4-hydroxycoumarins (90). | ||
4.5. Rearrangements of coumarin and chromone derivatives
The 3-halochromones are attractive starting materials for synthesis, because of their interesting structural characteristics,87 since they contain an α,β-unsaturated carbonyl residue linked to two leaving groups (halogen and aryloxy) and are prone to react with nucleophiles.The reactions of 3-halochromones with amines have been studied in detail and it was found that addition of primary amines produce cumarin-3-one derivatives.88 On the other hand, it was observed that compounds with active methylenes such as β-diketones or β-ketoesters react with 3-halochromones in the presence of a base to afford functionalized furanic derivatives.89
Considering the observed reactivity, the Bandyopadhyay group postulated that furo[3,2-c]coumarin derivatives 94 could be accessed (Scheme 20) by reaction of 2-aminochromones (92)90 with 3-halochromones like 3-iodochromone (93).91 With that intention, a mixture of equimolar amounts of both reactants was heated in AcOH in the presence of five equivalents of Cs2CO3 and the predicted compound 94 was obtained with good yield.89
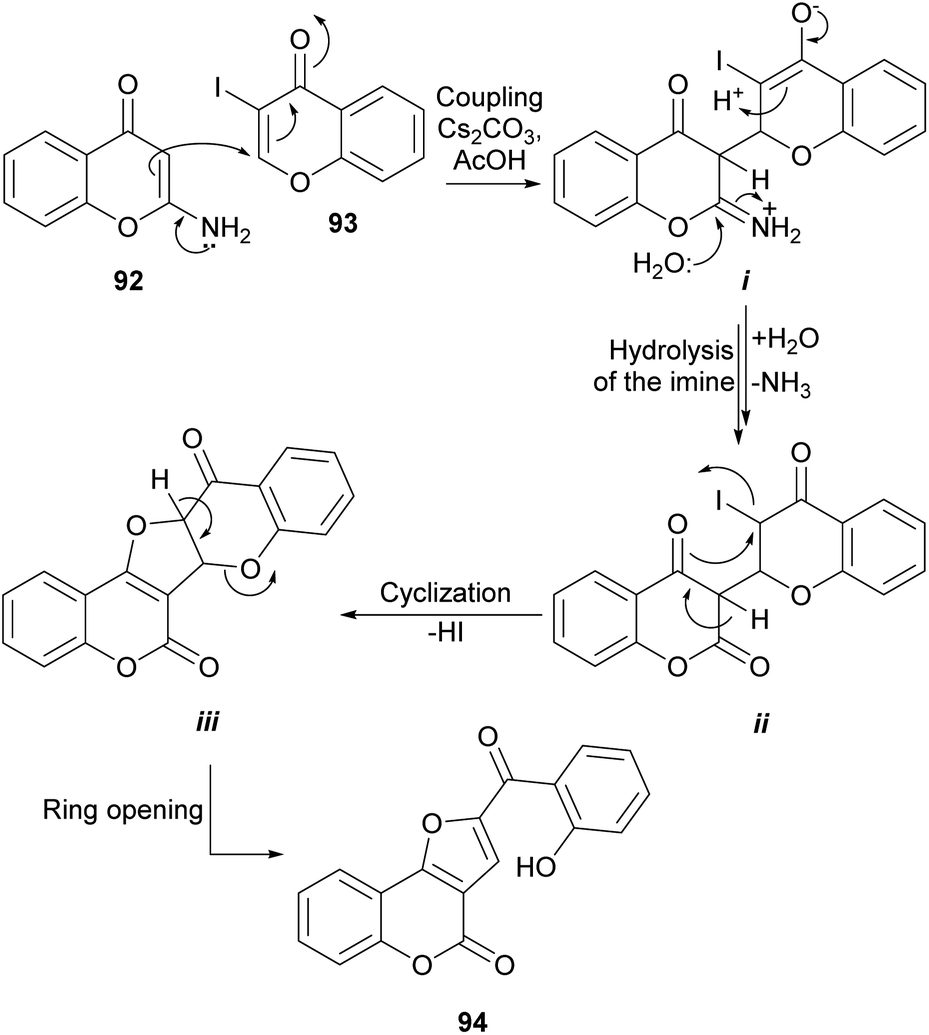 | ||
| Scheme 20 Mechanistic proposal for the formation of 94. | ||
DBU in CH3CN also demonstrated to be a suitable reaction condition. The formation of 94 was rationalized by assuming that the enamine moiety of the chromone 92 effects a 1,4 addition to the α,β-unsaturated carbonyl of 93 to form the imine intermediate i that once hydrolyzed affords the lactone intermediate ii. The cyclization of ii through its enol form to intermediate iii and the subsequent pyran ring opening reactions drive the reaction to the final product 94.
Analogously, the same group reported that DABCO is able to catalyze the one-pot synthesis of furo[3,2-c]coumarins 94 by reaction between 3-halochromones and coumarins.92 Other amines (Et3N, pyridine) are less efficient.
On the other hand, Khan et al. had previously demonstrated (Scheme 21) that heating of 3,3′-methylene dicoumarin (95) with the DMSO/acetic anhydride reagent system at 160 °C produces the rearrangement and functionalization of the starting 4-hydroxycoumarin (59) to afford the furocoumarin 94.93 These authors proposed an oxidative mechanism based on sulfur oxide intermediates, similar to those postulated for the Swern oxidation.
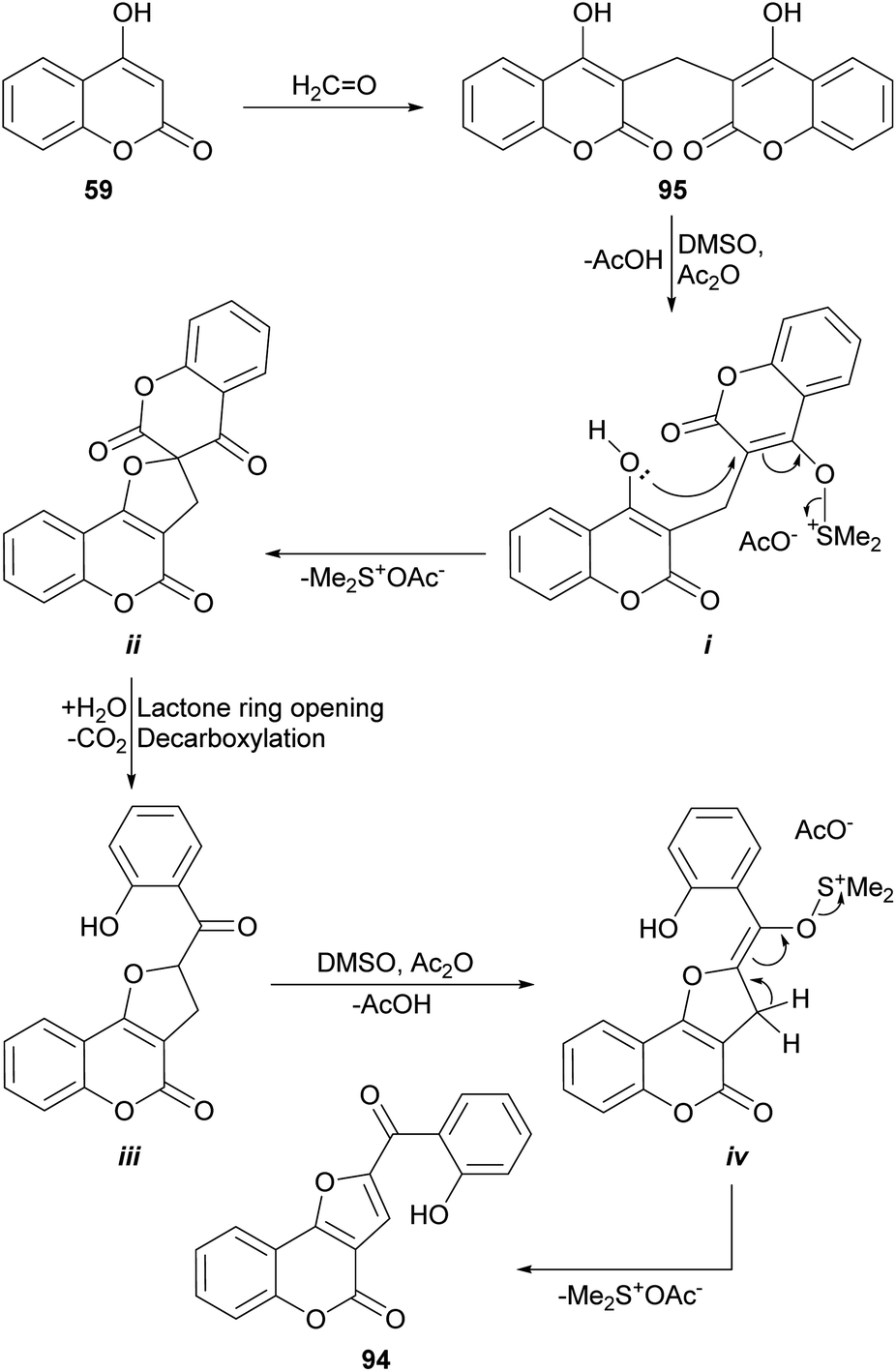 | ||
| Scheme 21 Proposed mechanism of the oxidative rearrangement of 3,3′-methylene dicoumarins to furo[3,2-c]coumarin derivatives. | ||
The process would proceed through the formation of the sulfur ylide (i) by reaction of one of the 4-hydroxycoumarin moieties with the DMSO/Ac2O reagent, which would give rise to the spirocyclic intermediate ii. In turn, this would be opened to give place to intermediate iii, capable of generating a second sulfur ylide (iv), that would trigger the formation of the furan ring of 94. An analogous sequence of events was used to explain the reactions with CuBr2 and iodine (Scheme 49).
4.6. Cyclization reactions with aldehydes
In the presence of a base, 4-hydroxycoumarins may exist as ambident anions, that can undergo O-alkylation84,94 under classical conditions (K2CO3, acetone) and partial C-alkylation in phase-transfer-catalyzed reactions with hydroxides.95 When 4-hydroxycoumarin (59) was treated with chloroacetaldehyde (96) in water, in the presence of K2CO3, 3-hydroxy-2,3-dihydro furo[3,2-c]coumarin (97) was obtained in 73% yield.96 A series of other 4-hydroxycoumarins behaved similarly. The products were then exposed to 1 M HCl, giving almost quantitative yields of the corresponding furo[3,2-c]coumarins (98).The reaction takes place by reversible nucleophilic addition of the enolate of the 4-hydroxycoumarin to the carbonyl group of the haloaldehyde to give the intermediate i. This is followed by base-catalyzed intramolecular cyclisation leading to the cyclized product 97 (Scheme 22). The nucleophilic addition of the 4-hydroxycoumarin to the carbonyl group of the aldehyde, which in water exists mainly as the hydrate, is facilitated by the electron-withdrawing inductive effect of the neighboring chlorine.
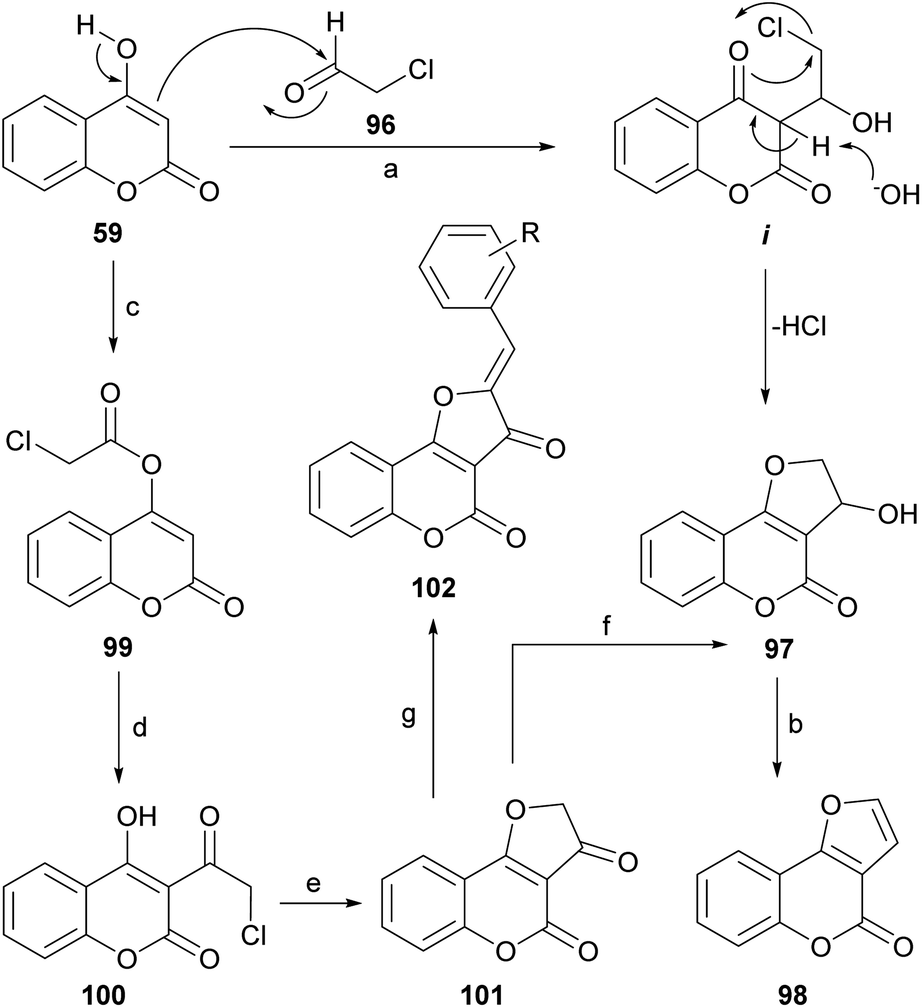 | ||
| Scheme 22 Reagents and conditions: (a) K2CO3, H2O, r.t., 1.5 h (73%); (b) HCl, H2O (∼100%) or 30% H2SO4, EtOH, reflux, 30 min (80–85%); (c) ClCH2COCl, pyridine, reflux (80%); (d) AlCl3, 140–150 °C; (e) K2CO3, Me2CO, r.t., 10 min (50% overall); (f) NaBH4, dioxane, MeOH, 30 °C, 1 h (80–90%); (g) ArCHO, AcOH, H2SO4 (cat.), reflux, 1 h (19–66%). | ||
Therefore, it was not surprising to observe that this methodology failed when the reaction was attempted with substrates such as 3-hydroxycoumarin, 3-hydroxy-2-quinolone and 7-hydroxycoumarin.
Perhaps, the reversible nucleophilic addition step from 59 to i is important. For example, in the case of 3-hydroxycoumarin and 3-hydroxy-2-quinolone, the equilibrium does not favor the intermediate; thus, the cyclisation is precluded.
An alternate and related sequence toward 98 which does not resort to the use of aldehydes has been reported by Traven et al.97 This group prepared the chloroacetyl ester of 4-hydroxycoumarin (99), effected its Fries rearrangement to 100 and then cyclized the chloromethyl ketone to the furanone 101.98 Reduction of the ketone moiety followed by dehydration of the resulting alcohol 97 furnished the tricyclic final product 98.
On the other hand, the Knoevenagel reaction of the furanone 101 with different aromatic aldehydes provided the corresponding crotonization products 102, which exhibited strong fluorescence, whereas reaction with aromatic amines afforded the corresponding imines, in tautomeric equilibrium with the related enamines.99
In an analogous fashion, the group of Traven prepared the 4-hydroxycoumarin 103 by the Pechmann condensation of 3-(N,N-diethylamino)phenol with diphenyl malonate in refluxing toluene (56% yield) and exposed the heterocycle to chloroacetaldehyde, to access the tricycle 104 (Scheme 23).100 The latter was then subjected to a Vilsmeier–Haack formylation, to access the formyl derivative 105 which, in turn, was condensed with different active methylene compounds or under modified Wittig conditions to afford the 2-substituted furo[3,2-c]coumarins 106a–g.
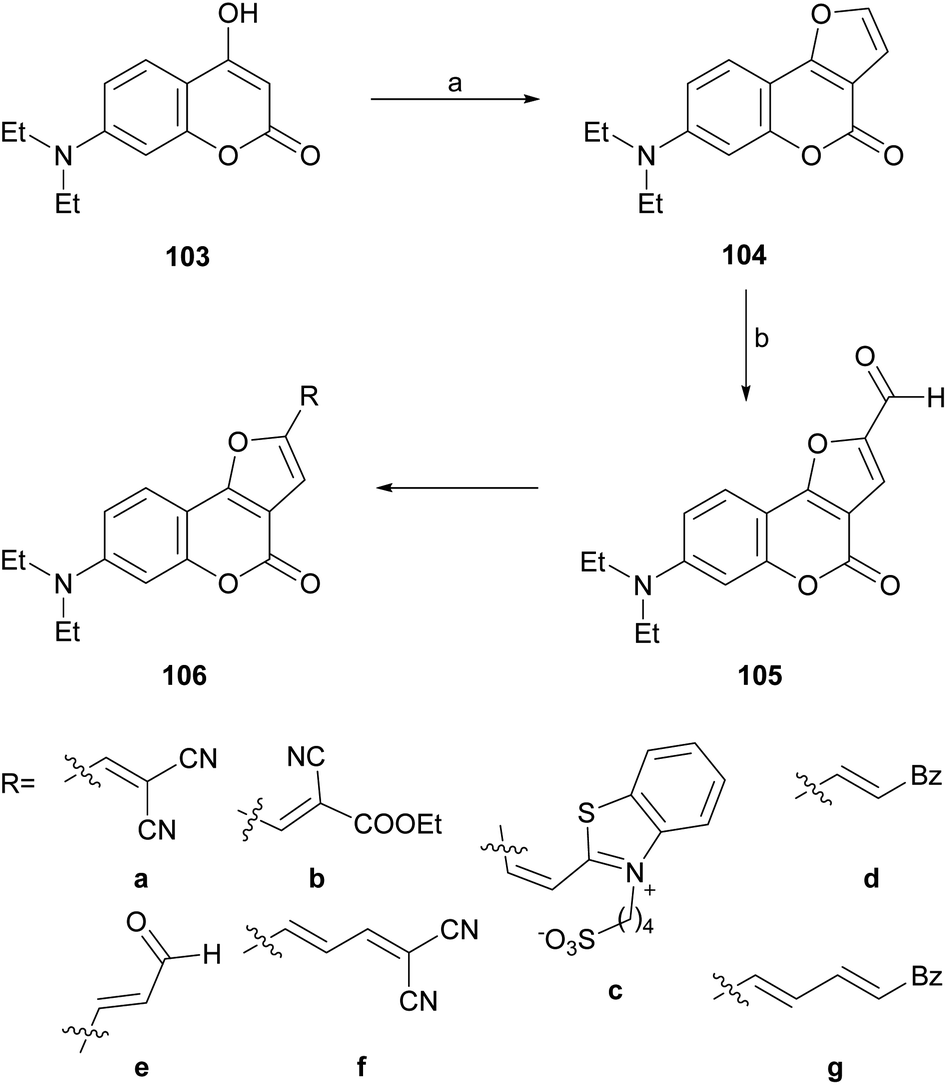 | ||
| Scheme 23 Reagents and conditions: (a) (1) ClCH2CHO, K2CO3, H2O, 0 °C; (2) 40% HCl, (b) DMF, POCl3, 70 °C (45% overall). | ||
The heterocycles displayed interesting electronic excitation and fluorescent spectra. It was concluded that the fusion of the five-membered heterocycle to the coumarin provides a definite increase of the Stokes shifts in all solvents and results in higher quantum yields of fluorescence.
Some of the studied compounds were submitted to TD-DFT calculations, which revealed that use of hybrid DFT functionals and an adequate representation of the molecular environment are essential for obtaining accurate predictions of the UV-Vis absorption spectra of these dyes with extended π-systems. The longest-wave electron transitions in the studied compounds were computationally shown to be of a push–pull nature.
A simple method for the synthesis of trisubstituted furans through the oxidative cyclization of 1,3-dicarbonyl compounds with aliphatic aldehydes under promotion by TsOH or AlCl3 was developed by Huang et al. using NBS as an oxidizing agent.101 This method was effective for the synthesis of several polysubstituted furan derivatives, allowing the preparation of previously practically inaccessible compounds.
Mechanistically (Scheme 24), this reaction involves a sequence of self-catalyzed and tandem events. Tandem auto-catalysis refers to the presence of very different processes, which occur sequentially and are promoted by the same type of catalyst. In this case, the sequence is initiated by a Knoevenagel type condensation between the 4-hydroxycoumarin 59, as the 1,3-dicarbonyl derivative, and the aliphatic aldehyde (107).
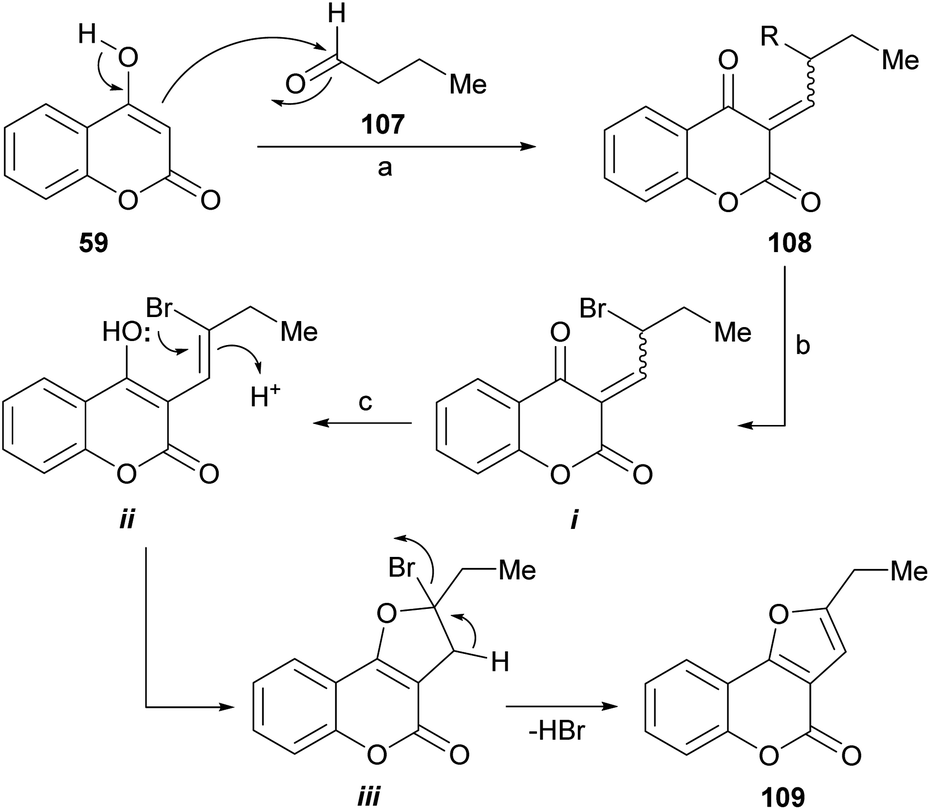 | ||
| Scheme 24 Reagents and conditions: (a) TsOH (5 mol%), MeCN; (b) NBS, MeCN; (c) TsOH, MeCN, 80 °C, 2 h (33%). | ||
Then, the resulting α,β-unsaturated dicarbonyl product (108) would undergo an acid-catalyzed allylic bromination mediated by NBS102 to give an intermediate (i) capable of enolization toward ii, which would suffer an intramolecular oxa-Michael type addition to form the new heterocyclic ring (iii). The final dehydrobromination stage would result in the tricyclic product 109.
A 33% overall yield was attained in the only furo[3,2-c]coumarin example provided. In this process, TsOH acts as a catalyst to promote the Knoevenagel condensation, the intramolecular oxo-Michael and the dehydrobromination reactions.
An unusual but easy approach to the synthesis of 2-benzofuranyl-3-hydroxyacetones (110) from “phenolics” (59) is the use of masked α,β-unsaturated aldehydes such as 6-acetoxy-β-pyrones (111), as developed by Bankar et al.103 From the point of view of the mechanism, the synthetic sequence (Scheme 25) involves a cascade which implies a transacetalization between the starting acetal 111 and the 4-hydroxycoumarin 59 to give i.
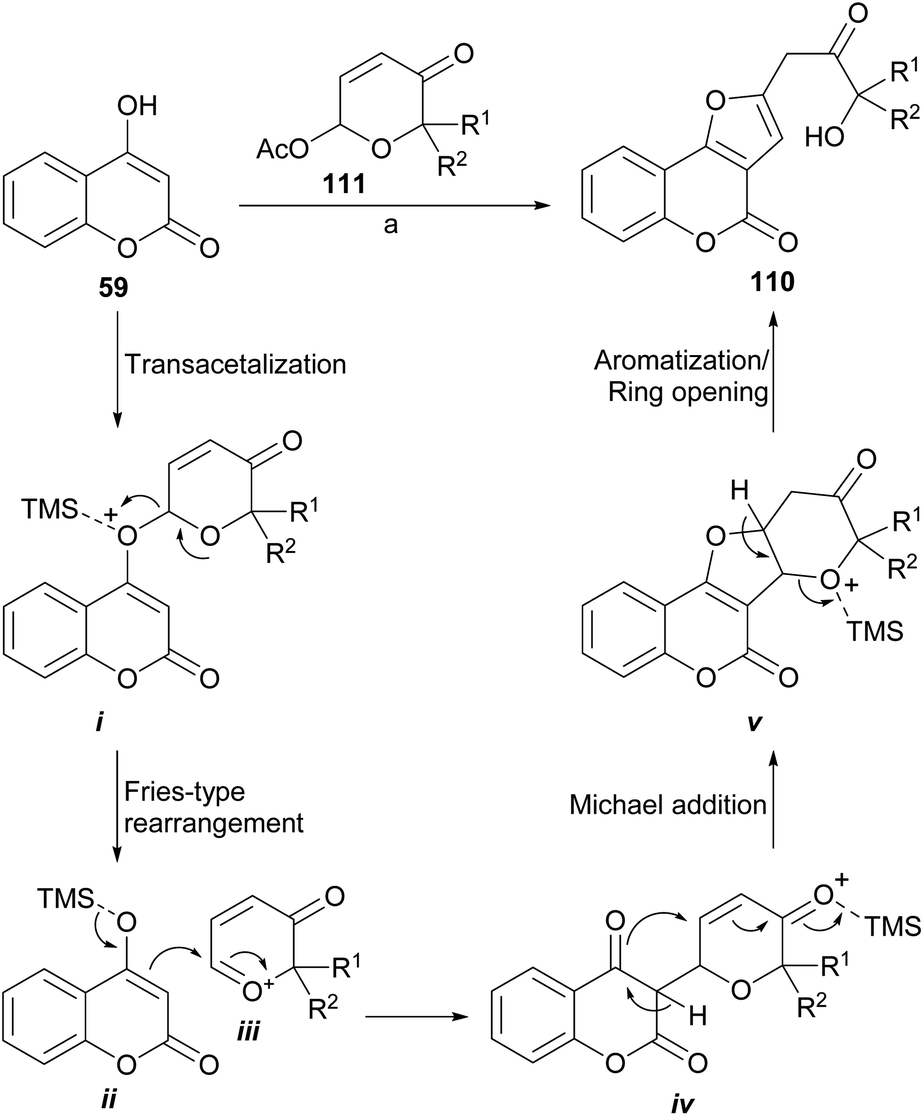 | ||
| Scheme 25 Reagents and conditions: TMSOTf (10 mol%), ClCH2CH2Cl, 0 °C → r.t. | ||
In turn, this one may undergo a Fries-like O→C rearrangement104 toward iv, through the intermediacy of the tight ion pair between the activated heterocycle ii and the oxocarbenium ion iii. This reaction should be followed by Michael addition to form the tetracyclic intermediate v and final acid-induced aromatization with concomitant ring opening toward 110. The method appears to be versatile and would allow access to complex structures in a simple way.
4.7. Oxidative rearrangement of 2,2′-diphenic acid
The group of Meyer observed (Scheme 26) that the exposure of methyl gallate (112) to peroxidase or FeCl3 produces a biaryl condensation to afford a 2,2′-diphenic acid derivative 113 which upon dehydration experiences a double lactonization, affording ellagic acid (114).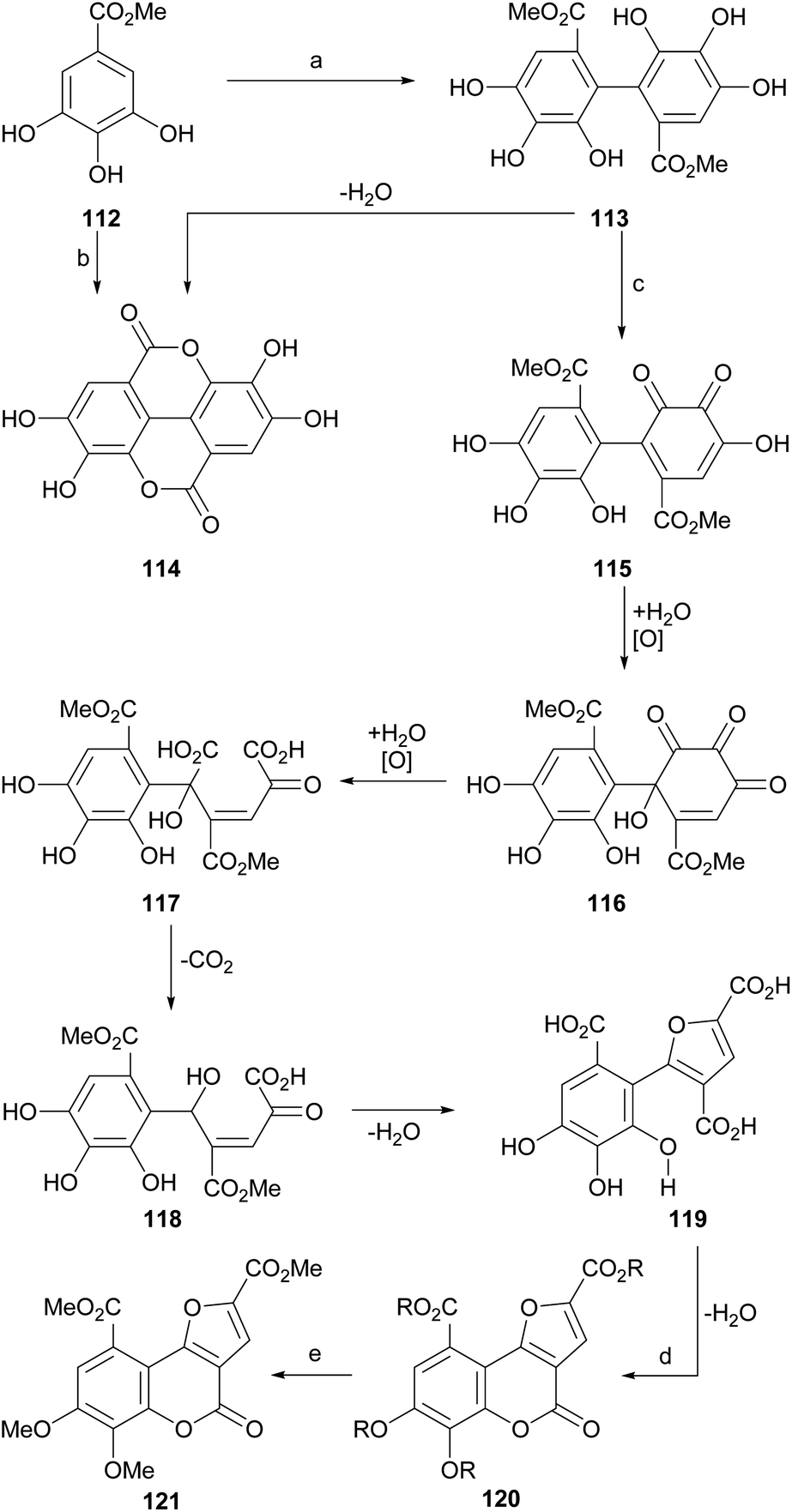 | ||
| Scheme 26 Reagents and conditions: (a) peroxidase, H2O (24%); (b) FeCl3, Me2CO, H2O, 24 h (19%); (c) FeCl3, Me2CO, H2O, 24 h (30%, from 112); (d) HCl, 100 °C, 6 h (81%); (e) CH2N2, Et2O, r.t. (66%). | ||
In addition, they detected that in the presence of FeCl3, the intermediate 113 suffers several oxidation stages on one of the aryl rings, passing through several polyoxygenated intermediates, which include oxidation to 115, hydroxylation to 116, oxidative ring opening to 117, decarboxylation (118) and dehydrative cyclization, to give rise to the trisubstituted furan derivative 119.
The dehydrative cyclization of 119 with aqueous HCl or H2SO4 gave rise to the polyfunctionalized furo[3,2-c]coumarin 120, whose etherification and esterification were carried out simultaneously by reaction with ethereal diazomethane, to afford the heterocycle 121.105
5 Chemical synthesis of 3-substituted furo[3,2-c] coumarins
5.1. Reaction of 4-hydroxycoumarins with allenyl sulfides
The group of Kanematsu reported a two-step synthesis of fused 3-methylfurans by the addition of the enolate anions of cyclic 1,3-dicarbonyl compounds to an allenic sulfonium salt.106 The sulfur reagent is easily obtained by the reaction of propargyl bromide and SMe2 in MeCN, followed by isomerization in EtOH. The yield of the single example disclosed is 50%.5.2. Reaction 4-hydroxycoumarins with β-nitrostyrenes
The group of Brahmbhatt disclosed the use of β-nitrostyrenes (123) as partners in the reaction with 4-hydroxycoumarins (123), to provide these heterocycles.107 This one-pot reaction (Scheme 27) takes place under piperidine promotion in refluxing MeOH and the yields of 124 are generally moderate (30–65%).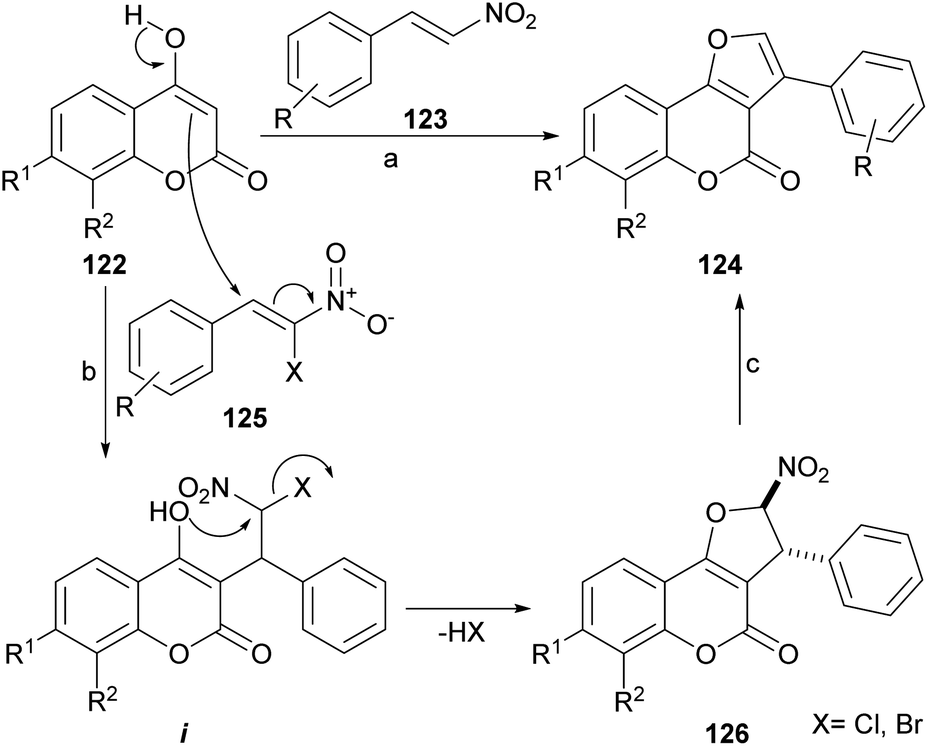 | ||
| Scheme 27 Reagents and conditions: (a) piperidine, MeOH, reflux, 5 h (35–65%); (b) KF, DME, reflux (X = Cl) or NaOAc, H2O, r.t., 72 h (X = Br); (c) Et3N, THF, reflux. | ||
The formation of the furan ring involves a base-catalyzed Nef reaction,108 where the 1,3-dicarbonyl starting heterocycle reacts with the β-nitrostyrene to afford the substituted furan derivative. It has been shown that these heterocycles can be further selenylated with (PhSe)2 and K2S2O8 in MeCN, in very good yields (82–85%).109
Alternately, β-chloro-β-nitrostyrenes (125) could be used and condensed under KF promotion to afford the nitro derivatives 126 in good to very good yields (70–90%), through the intermediacy of i.110 Upon exposure to Et3N in refluxing THF, the latter aromatized in good yield to afford 124. Using Et3N for the initial condensation resulted in an efficient one-pot procedure (82–92% yield). Interestingly, a 3-aryl furo[3,2-c]coumarin has been reported as a side product of the treatment of a 3-benzoyl-4hydroxycoumarin with ethereal diazomethane.111
In addition, employing β-bromo-β-nitrostyrenes, Xie and co-workers devised both, a racemic domino reaction in water and a high yielding (up to 98%) asymmetric organocatalyzed domino Michael-SN2 reaction toward 2-nitro derivatives 126, employing a bifunctional thiourea prepared from quinine as organocatalyst.112 The transformation was highly diastereo (de > 99%) and enantioselective (ee > 92%). A quite similar reaction was also disclosed by the group of Rueping.113
In another report, the use of aromatic aldehydes, ammonium acetate and β-nitrostyrenes resulted in a multicomponent reaction which furnished substituted furo[3,2-c]coumarins 127.114 The reaction sequence in the proposed mechanism (Scheme 28) is initiated by the Michael addition of 4-hydroxycoumarin 59 to the β-nitrostyrene to give the corresponding adduct i.
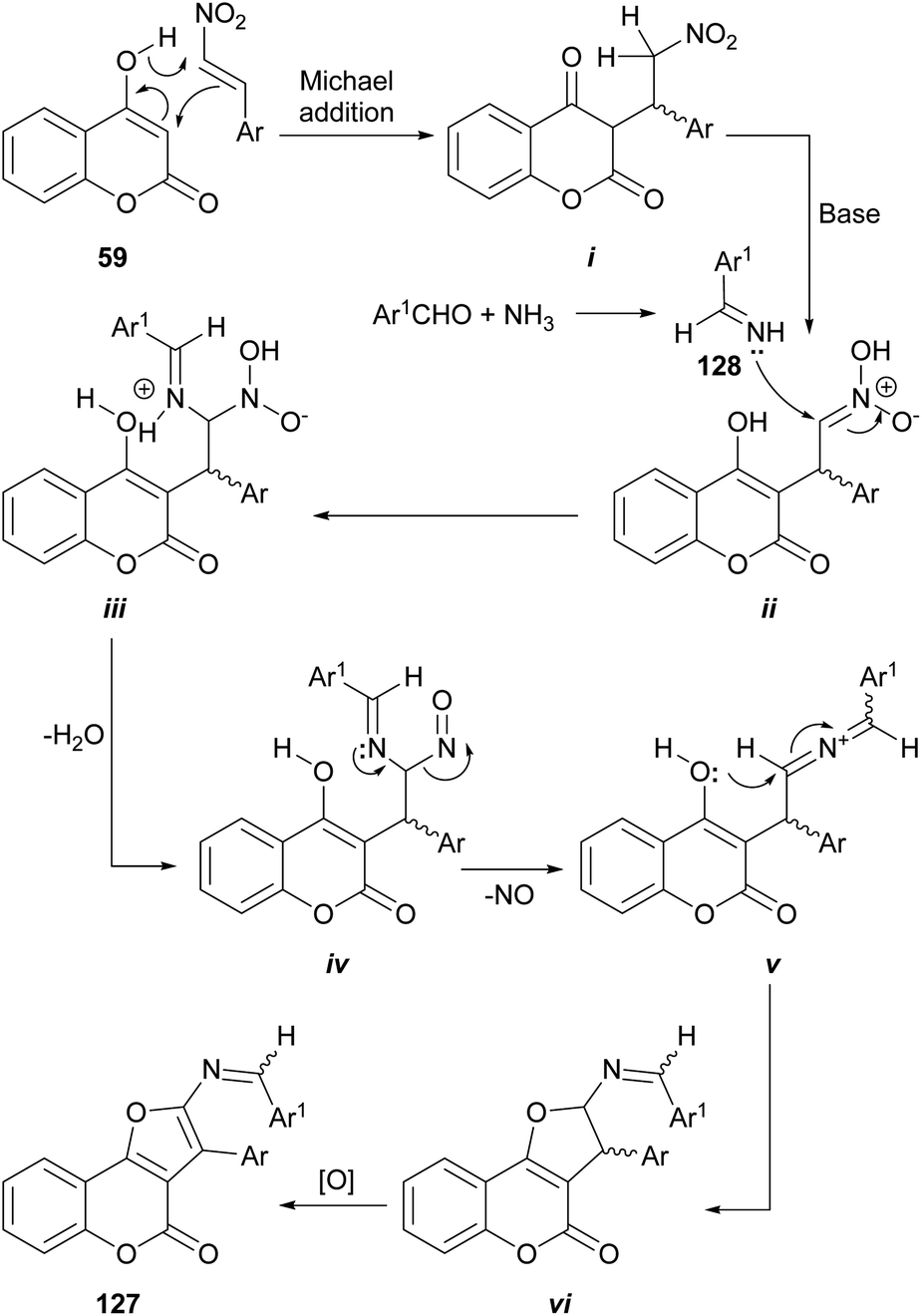 | ||
| Scheme 28 Reagents and conditions: (a) NH4AcO, piperidine, EtOH, r.t. → reflux, 27 h (53–73%). | ||
In the presence of piperidine as base, the nitro moiety of the resulting ii can suffer an aza-nucleophilic addition of the imine 128 (formed by imination of the benzaldehyde component and the NH4AcO) to the double bond, to give iii. Subsequent dehydration (iv), loss of NO (v) and cyclization should provide the dihydro furo[3,2-c]coumarin vi which upon oxidation furnishes the corresponding furo[3,2-c]coumarin 127.
On the other hand, the reaction of 4-hydroxycoumarins with isocyanides and benzaldehydes in refluxing benzene also resulted in good yields (61–93%) of 2-amino-3-aryl substituted furo[3,2-c] coumarins.115
5.3. Condensation with aldehydes
Another example was provided by the group of Malnar,116 who condensed 4-hydroxycoumarins (82) with glyoxal and 2,2-dimethoxyacetaldehyde, to provide methylene-3,3′-dicoumarin derivatives functionalized with a formyl group or its equivalent on the methylene bridge. Further condensation between the formyl moiety and one of the neighbor hydroxy groups gave different hemiacetalic or acetalic coumarin-substituted furo[3,2-c]coumarins (i–ii), which upon submission to reaction with AcOH or TFA lost water or MeOH to afford 129 (Scheme 29).117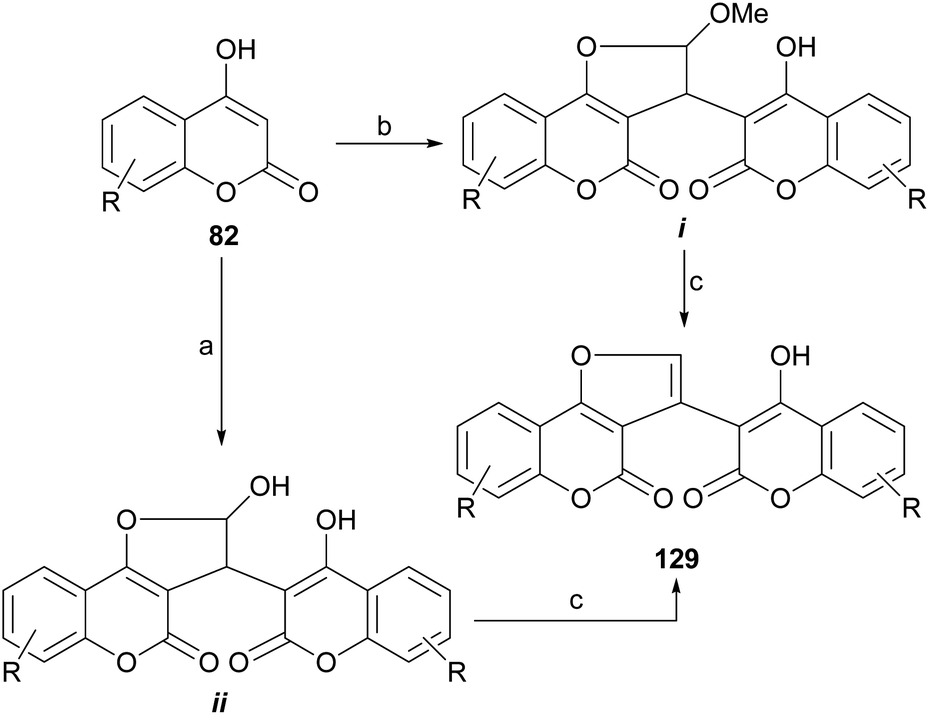 | ||
| Scheme 29 Reagents and conditions: (a) glyoxal, MeCN, reflux; (b) glyoxal, MeOH, reflux or dimethoxyacetaldehyde, MeOH, reflux; (c) AcOH or TFA, reflux. | ||
The compounds were tested as anti-inflammatories, and the most active derivatives showed significant inhibition of leukotriene B4 and mast cell degranulation induced by the activation of Fcε receptor type I or calcium ionophore. They also displayed significant local anti-inflammatory activity in the phorbol 12-myristate 13-acetate-induced ear edema test in mice, with a potency equal to zileuton, the reference drug used in the tests.
5.4. The Feist–Bénary reaction
The group of Risitano devised a simple and efficient synthesis of furo[3,2-c]coumarin derivatives from 4-hydroxycoumarin and α-haloketones.118 The authors proposed two alternate reaction mechanisms, involving initial either O- or C-alkylation; however, since they were unable to find linear furocoumarin products, they concluded that the transformation should be initiated by an O-alkylation and followed by a cyclization and further aromatization.3-Phenyl furo[3,2-c]coumarin was synthesized by the group of Risitano employing the Feist–Bénari reaction with phenacyl bromide, and subjected to X-ray diffraction.119 It was observed that the furo[3,2-c]coumarin fragment and the phenyl ring bonded to C-3 are both planar within the experimental error.
However, the phenyl ring is rotated by almost 40° with respect to the furocoumarin fragment and the C3–CPh bond distance of 1.482 Å indicates the absence of conjugation between both π-delocalized systems. The bond distances of the endocyclic furan ring indicate the existence of π-electron delocalization over the whole tricyclic system.
The group of Durrani devised a one-pot oxidative pseudo three component reaction which includes the in situ generation of phenacyl bromides, as a strategy toward 3-aryl furo[3,2-c] coumarins (132),120 thus avoiding to work with these lachrymator intermediates. In their synthetic embodiment (Scheme 30), mixtures of NBS, an acetophenone derivative (131) and a 4-hydroxycoumarin (130) were dissolved in water-PEG-400, treated with AcOH/NH4AcO and heated at 80–85 °C.
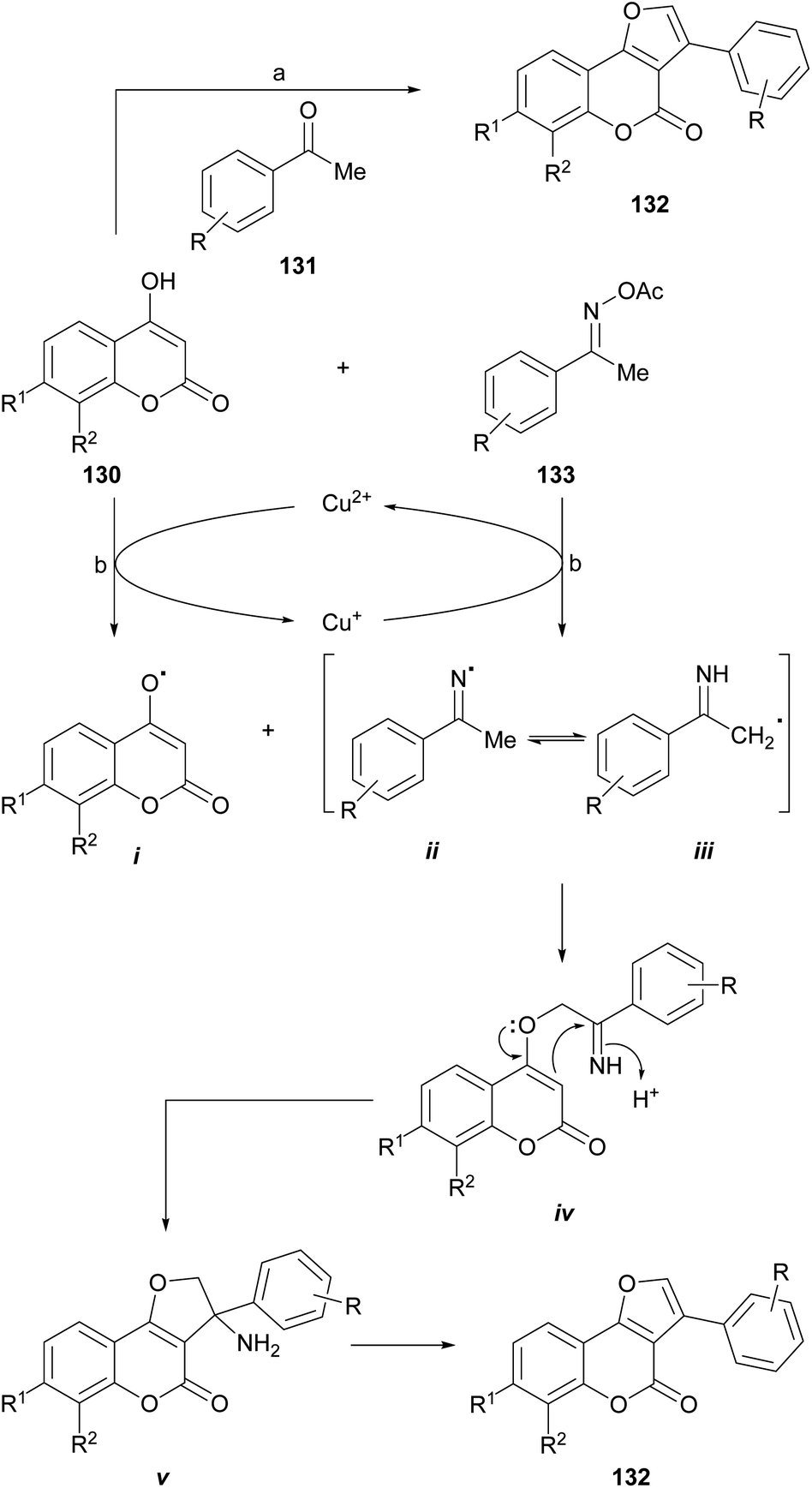 | ||
| Scheme 30 Reagents and conditions: (a) (1) PEG-400, H2O, 80–85 °C, 30 min; (2) NBS, AcOH, NH4AcO, 80–85 °C, 30 min (79–83%); (b) CuCl2, dioxane, 100 °C, 1 h (54–85%). | ||
Another interesting synthetic route is that proposed by the group of Lin, who developed a novel and efficient strategy for furo[3,2-c]coumarins via a copper-catalyzed radical/radical cross-coupling of ketoxime carboxylates (133) with 4-hydroxycoumarins. This process involved both, C–N/N–O/O–H bond cleavages and C–O/C–C bond formations. This approach has some advantages, such as the use of environmentally friendly copper salts, and the avoidance of additives or ligands and an external oxidant.
The authors proposed a mechanism for this reaction (Scheme 30), where the first step is the oxidation of 4-hydroxycoumarin by Cu2+ to generate the enolic oxygen radical i and Cu+. The latter reduces the oxime acetate to generate the iminium radical ii, which immediately isomerizes to a α-C(sp3) radical (iii). This transformation takes place with concomitant regeneration the oxidizing copper species.
In turn, the selective cross-coupling of the enolic oxygen radical i and radical iii takes place to build a new C–O bond and provide the intermediate iv. Subsequently, cyclization of intermediate iv generates the amino intermediate v, which affords the product 132 after losing NH3.121
The use of α-tosyloxyketones in place of α-haloketones has also been described.122 These reactants can easily be prepared by treating the ketones with [hydroxy(tosyloxy)iodo]benzene (Koser's reagent).123 The authors initially proposed that both, O-alkylation and C-alkylation routes are possible toward the final product. However, they isolated the O-alkylated intermediate, which did not undergo cyclization under similar conditions (3 h reflux in PhMe–EtOH, in the presence of AcOH and NH4OAc). Therefore, it was concluded that the reaction should proceed through the C-alkylation path, followed by a 5-exo-tet cyclization.
Following Risitano's approach, 3-(ω-bromoacetyl)coumarin (134) was condensed with various 4-hydroxycoumarins (82), in the presence of NH4AcO and AcOH to afford structurally similar heterocycles (135).124 These compounds (Scheme 31) were employed as neutral bidentate ligands for the preparation of copper(II) heterochelates (136) with clioquinol (137).
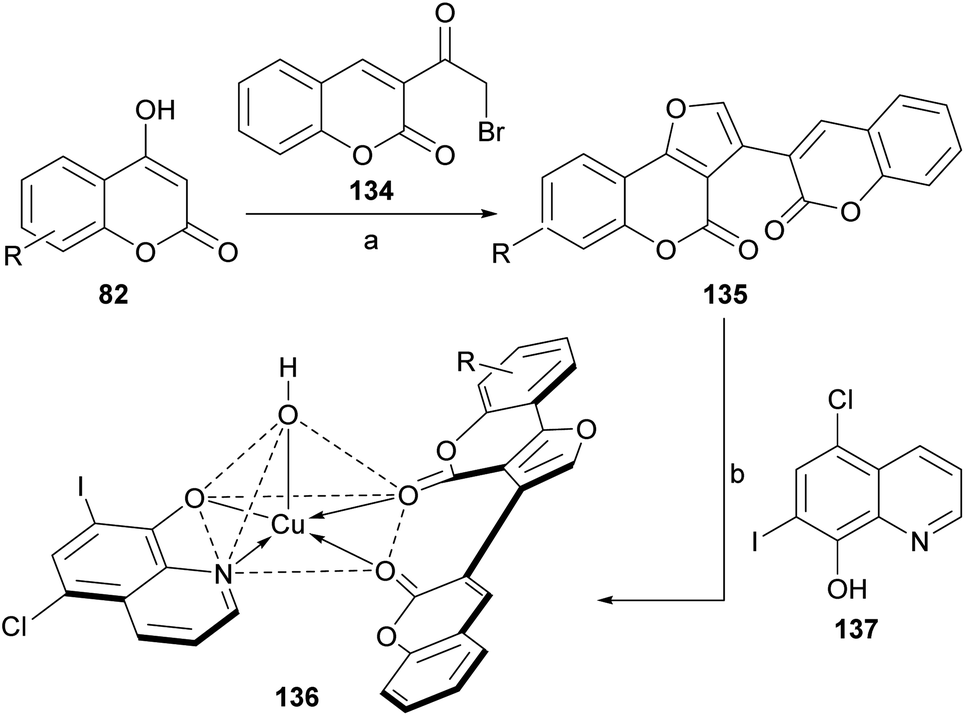 | ||
| Scheme 31 Reagents and conditions: (a) NH4AcO, AcOH, reflux, 16 h (72%); (b) Cu(NO3)2·3H2O, 6-chloro-7-iodo-8-hydroxyquinoline (clioquinol), DMF–EtOH (pH = 4.5–6.0), reflux, 7 h (61%). | ||
These high spin, square pyramidal compounds exhibited strong activities against Gram-negative and Gram-positive microorganisms. Their fluorescence spectra showed a red shift, which may be due to the chelating of the ligands to the metal ion.
Neo-tanshinlactone (Scheme 32) and some of its analogs are potent and selective anticancer agents in vitro. The Dong group optimized the synthetic route towards these compounds so that they can be obtained in five steps and in better overall performance and prepared the ethyl analog. The improved synthesis includes the addition of ethyl Grignard to the starting methoxytetralone 138 and dehydration with concomitant aromatization of the resulting product 139 to afford 140.
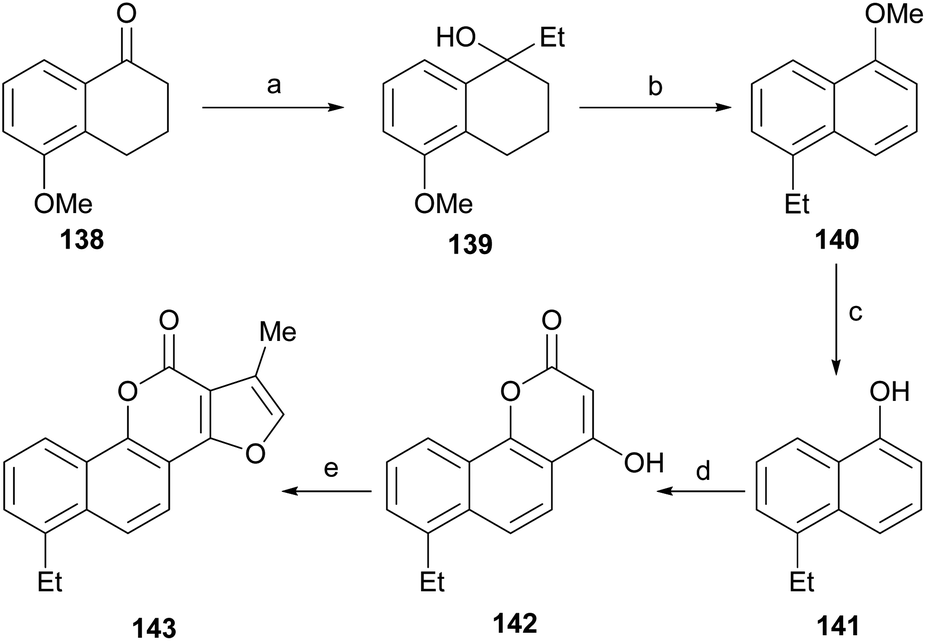 | ||
| Scheme 32 Reagents and conditions: (a) EtMgBr, ZnCl2, THF, r.t. (85%); (b) Pd/C, triglyme, reflux (60%); (c) BBr3, CH2Cl2 (60%); (d) malonic acid, PPA (85% P2O5), 75 °C, 3 h (95%); (e) ClCH2COMe, HOAc/NH4OAc, PhMe/EtOH, reflux, 24 h. | ||
Also, as part of the effort to find a new class of antimicrobial agents, Bondock et al. synthesized furo[3,2-c]pyrazoles (144) by the reaction of α-haloketone (146) and 4-hydroxycoumarins (59) under promotion by sodium ethoxide (Scheme 33). The authors claimed that this outcome, and not 145, is the result of employing a base stronger than usual.30
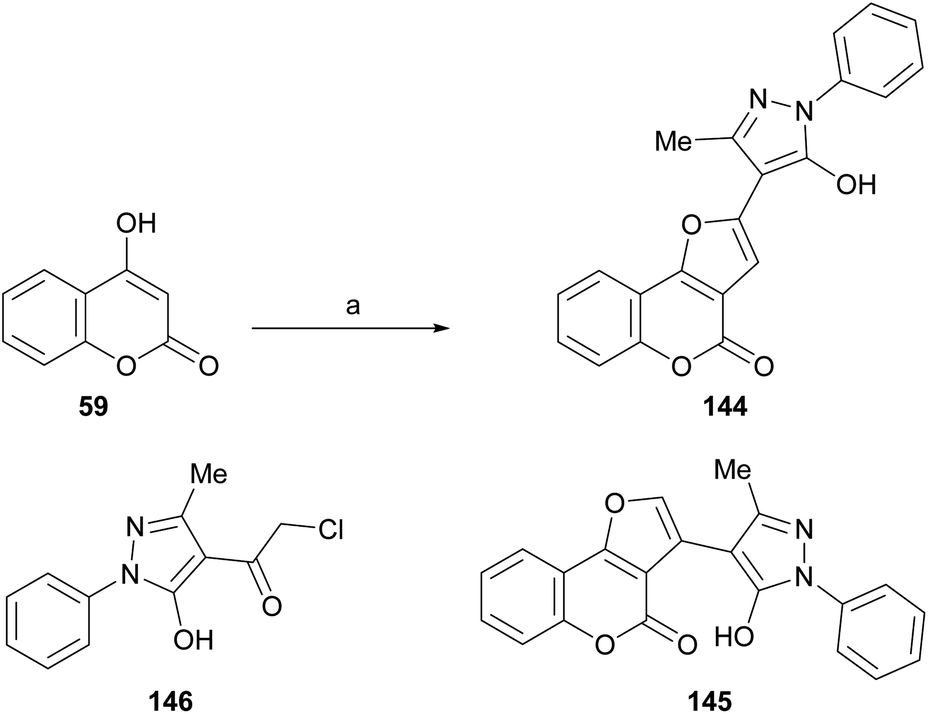 | ||
| Scheme 33 Reagents and conditions: (a) NaOEt, EtOH, r.t., 24 h (80%). | ||
Subsequent demethoxylation of the naphthalene derivative 140 to naphthol 141 and condensation with malonic acid gave rise to the 4-hydroxycoumarin derivative 142, which was treated with chloroacetone and HOAc/NH4OAc to give the final tetracycle 143 with yields globally close to 20%.40 Several derivatives were selective and potent against graft models and against glyphosate cancer cell lines with IC50 values of 0.3, 0.2, 0.1 and 0.1 μg mL−1.
6 Chemical synthesis of 2,3-disubstituted furo [3,2-c]coumarins
Coumestrol (3,9-dihydroxybenzofuro[3,2-c]coumarin) is a naturally occurring furocoumarin with estrogenic properties.125 Because of their structural resemblance with coumestrol, the synthesis of 2,3-disubstituted furo[3,2-c]coumarins have remained a subject of an active interest. However, the constant search for new alternative routes toward these heterocycles has resulted in the development of many imaginative procedures, as follows.6.1. Furanylation variations of the Feist–Bénary reaction
The Williamson-type reaction of 4-hydroxycoumarin (59) with 3-chloro-2-butanone and analogous α-halocarbonyl compounds has been employed by the groups of Al-Sehemi127 and Zhang31 to prepare a furo[3,2-c]coumarin derivatives (148) after basic (3% KOH in MeOH) or acid (PPA)-mediated cyclization (Scheme 34).
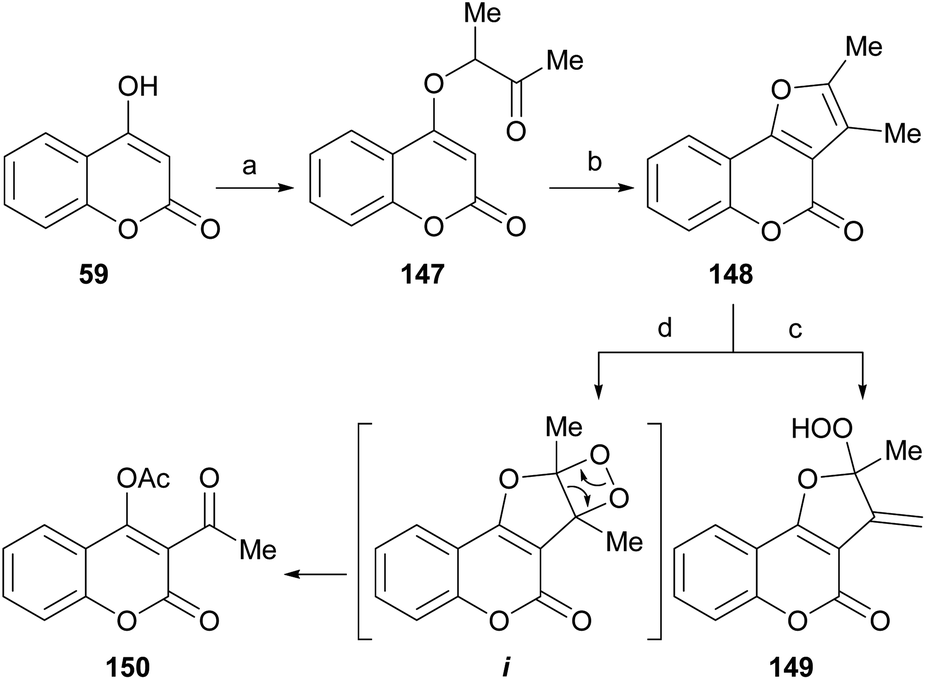 | ||
| Scheme 34 Reagents and conditions: (a) MeCHClCOMe, K2CO3, Me2CO, reflux, 24 h (75–80%); (b) PPA or 3% KOH in EtOH, reflux, 2 h (77%); (c) Na-lamp (500 W), TPP, CHCl3, r.t., 3 h (1O2, ene reaction, 18%); (d) Na-lamp (500 W), TPP, CHCl3, r.t., 3 h (O2, [2 + 2] cycloaddition, 73%). | ||
The room temperature photooxygenation of 148 in the presence of tetraphenylporphine (TPP) as singlet oxygen sensitizer, in a chloroformic solution gave a mixture of the stable allylic hydroperoxide 149 resulting from a Schenk-type ene reaction in 18% yield,128 and the unstable dioxetane i. The latter further rearranged to afford 150 in 73% yield.
The furocoumarin 150 is slightly antibacterial against Escherichia coli; on the other hand, according to differential pulse voltammetry studies, it is almost 40 times more potent than 8-methoxypsoralen in its ability to intercalate into DNA.
Zhang et al. reported the synthesis of furo[3,2-c]coumarins like 154 and 155 from 4-hydroxycoumarins 153, by their reaction under microwaves with α-halocarbonyl derivatives 151 and 152, derived from 1,3-dicarbonyl compounds.129 The authors proposed a mechanism for the transformations (Scheme 35), which involves nucleophilic attack of the 4-hydroxycoumarin on the ketone carbonyl of the other 1,3-dicarbonyl reactant to form an intermediate i.
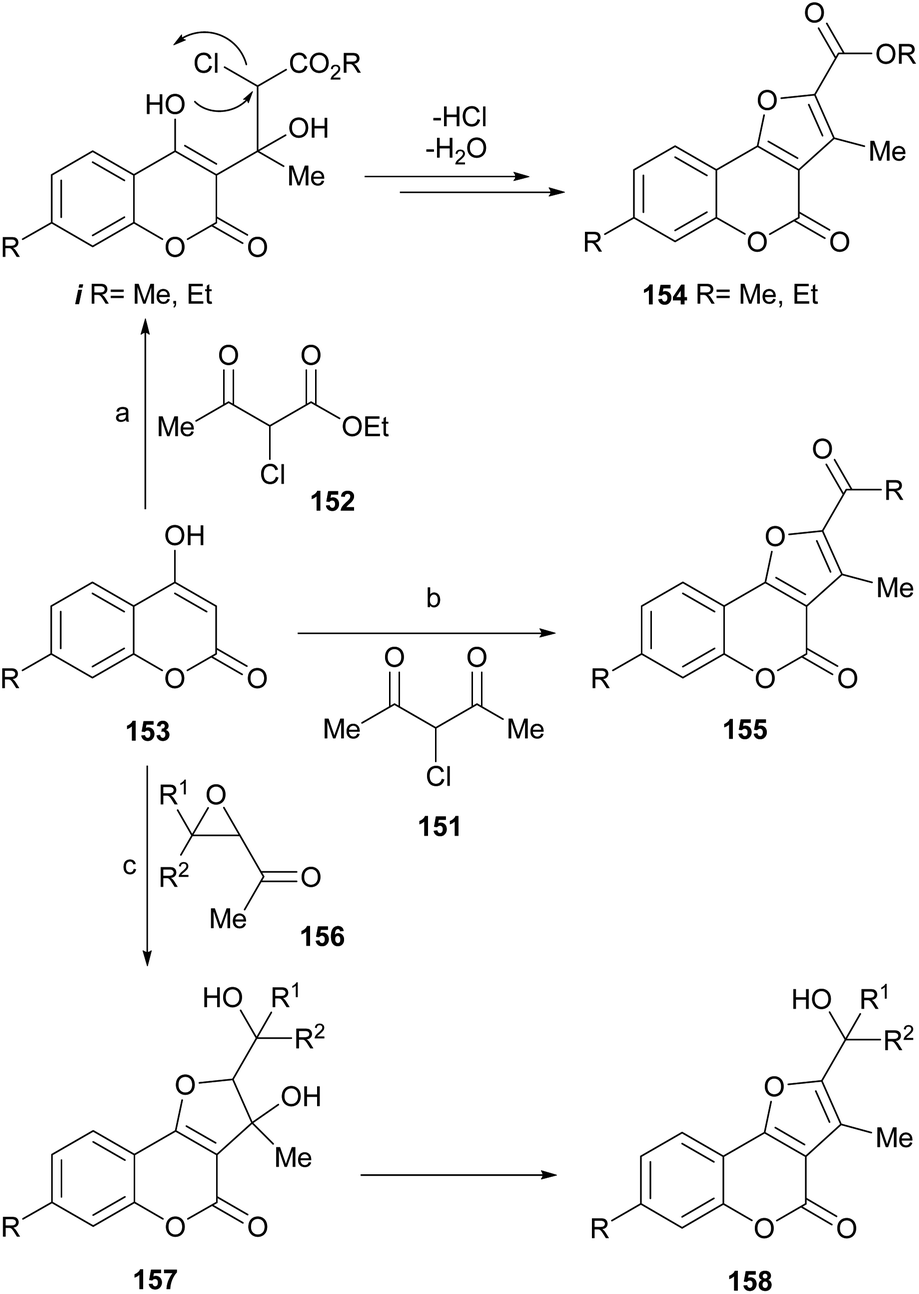 | ||
| Scheme 35 Reagents and conditions: (a) PhMe, MW (10–15 min), DMAP (63–77%); (b) PhMe, MW (10–15 min), DMAP (62–73%); (c) Et3N, DMF, r.t. | ||
Due to the good leaving ability of the halogen, it is displaced by an intramolecular nucleophilic substitution, which occurs from the oxygen of the 4-hydroxycoumarin. Subsequent dehydration afforded the final tricyclic product. Some of the synthesized 2,3-disubstituted furo[3,2-c]coumarins and others prepared from α-haloketones were employed as intermediates toward lactone-ring opened compounds, which were tested as antifungals.130
Acetyl oxiranes (156) can act as α-halocarbonyl surrogates. The group of Oskina demonstrated that 4-hydroxycoumarin (153) reacts with 2-acetyl-3-phenyloxirane in DMF in the presence of triethylamine at room temperature to give 3-hydroxy-2,3-dihydrofuro[3,2-c]coumarins 157, which affords compounds 158 after dehydrative aromatization.131
The acetyloxiranes were conveniently accessed in moderate yields by the Darzens reaction between aromatic aldehydes and acetone, or by alkaline H2O2-mediated epoxidation of α,β-unsaturated ketones. Similarly, the condensation with benzoin in refluxing xylene and under TsOH promotion afforded 50% yield of 2,3-diphenyl substituted heterocycles.132
An efficient and straightforward one-pot three component synthesis of 2,3-disubstituted furo[3,2-c]coumarins was reported.133 The synthesis takes place by condensation of aromatic aldehydes, 4-hydroxycoumarin and α-chloroketones in refluxing n-propanol. Pyridine or a mixture of AcOH and AcONH4 were used as a basic catalyst, to yield 2,3-disubstituted dihydrofuro[3,2-c]coumarins.
The yields of the products ranged from good to excellent. On the other hand, the synthesis of some analogs of coumestrol has been achieved by acid or base catalyzed condensation of 4-hydroxycoumarin with benzoin, acting as a surrogate of an α-halocarbonyl compound.134 The proposed raction mechanism was quite similar to that of the Feist–Bénary reaction.
In the proposed mechanism, the enol 59 effects a Michael addition on the nitroallylic acetate 159 (ref. 136) to give adduct i by an SN2′ process (Scheme 36). This is followed by a second 5-exo-trig Michael addition from the oxygen atom of the enol, to yield the five-membered ring (ii), as commanded by the Baldwin's rules.137 Final deprotonation with elimination of HNO2 completes the process, affording the aromatized ring compound 160.
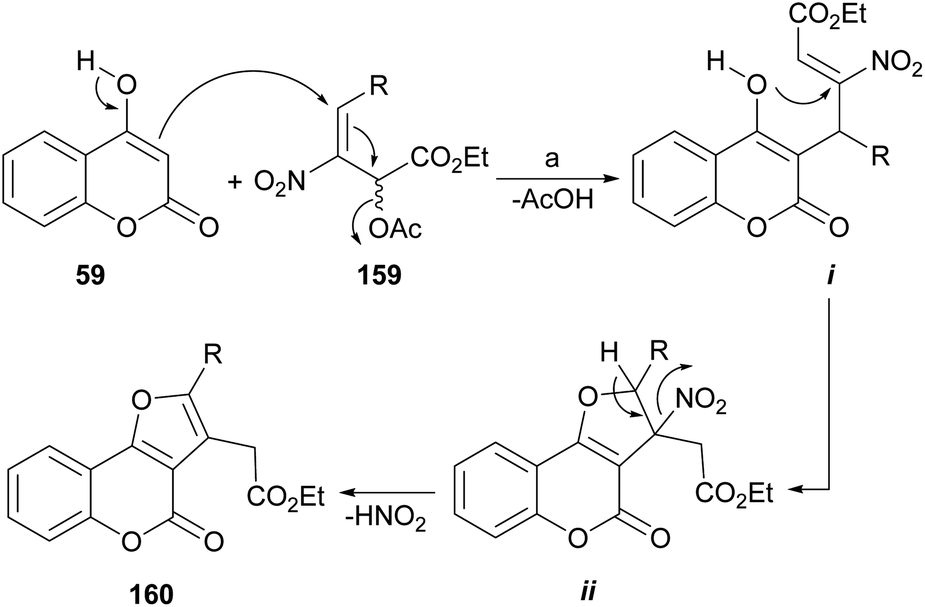 | ||
| Scheme 36 Reagents and conditions: (a) Cs2CO3, MeCN, r.t. (69–99%). | ||
6.2. Furanylation of polysubstituted olefins
On the other hand, Tan and coworkers (Scheme 37) developed a novel and efficient intermolecular aerobic catalytic procedure for the synthesis of furo[3,2-c]coumarins as 161 from 4-hydroxy coumarins (59) and alkenes (162), using Pd(CF3COO)2.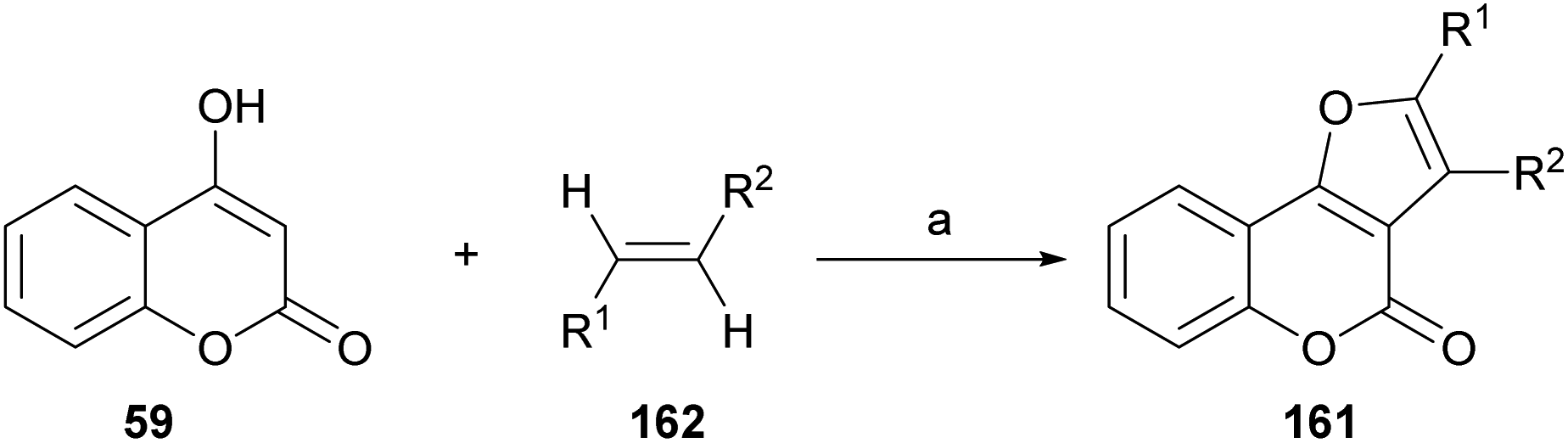 | ||
| Scheme 37 Reagents and conditions: Pd (CF3COO)2, PhCl, air, 90 °C, 4 h (70–85%). | ||
The method is mild and atom-economic, according to the concept of modern green chemistry.138 The yields are in the range of 70–85% if at least one of the ends of the olefin bears an aromatic substituent, although it also admits acrylates.
Based on literature precedents,139 a mechanism was proposed (Scheme 38) and exemplified with styrene (163), where initial palladation of the keto form of 59 provides the intermediate i.
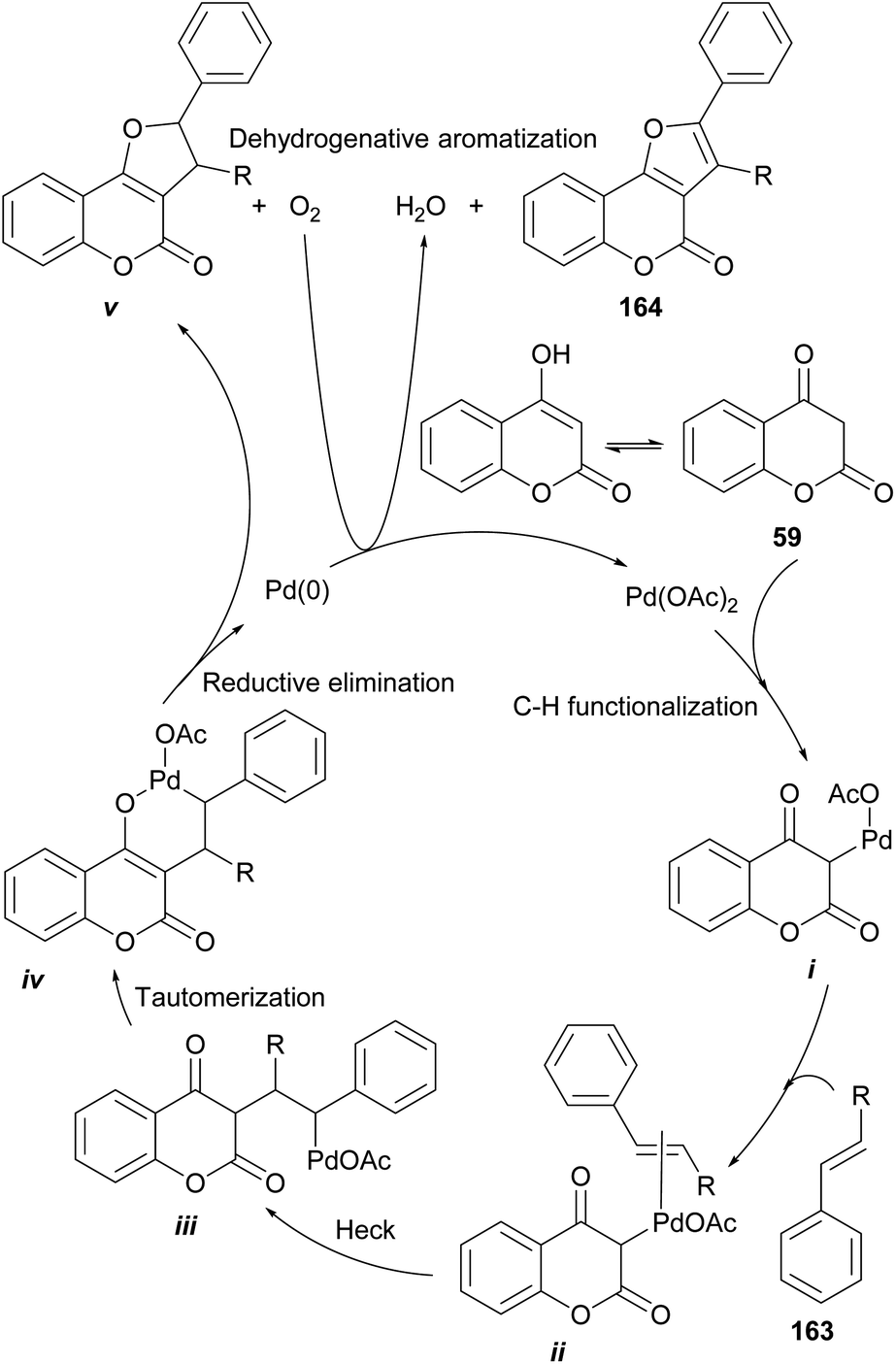 | ||
| Scheme 38 Proposed mechanism for the Pd-catalyzed oxidative alkoxylation of 4-hydroxycoumarins and alkenes. | ||
This is followed by coordination of the olefin (ii) and a Heck insertion to afford intermediate iii. Enolization of the latter toward the coordinated intermediate iv precedes a reductive elimination to produce the dihydro-furocoumarin v, which was detected and isolated. A final aerobic aromatization affords the desired product 164.
The groups of Appendino and Yilmaz reported that in the presence of metallic oxidants that are one-electron transfer agents [CAN, Mn(OAc)3·nH2O], 4-hydroxycoumarin (59) can undergo cycloaddition with alkenes (165) to give 3,2-dihydro-4H-furo[3,2-c]coumarins.140
As an example (Scheme 39), the reaction of the 4-hydroxycoumarin derivatives with a range of alkenes in the presence of ceric ammonium nitrate (CAN) gave the corresponding dihydro-furo[3,2-c]coumarins of type 166 or their respective linear tricyclic isomers, the dihydro-furo[3,2-b]coumarins 167. Alkenes bearing ester, alkyl and aryl groups were tested, and the yields were between low and moderate (15–62%).
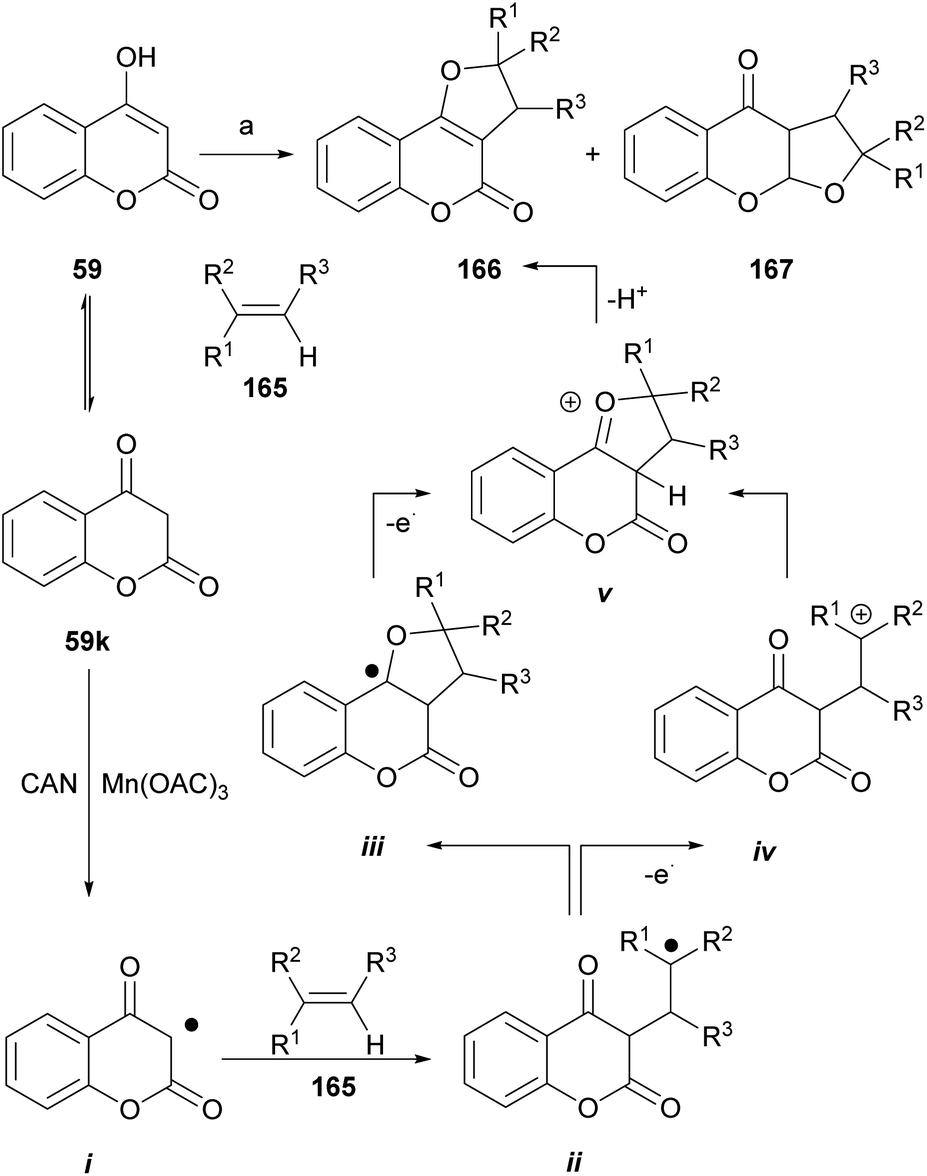 | ||
| Scheme 39 Reagents and conditions: (a) CAN (2 equiv.), MeCN, 0 °C, 40–90 min. R1 = H, Me, CH2OAc, OAc, OBz, Ph; R2 = H, Me; R3 = H, Me (21–62%). | ||
It was shown that the reaction can be extended to alkynes such as phenylacetylene, in which case it provides the corresponding angular and linear furocoumarins. On the other hand, the use of 4-hydroxy-1-methylquinolin-2(1H)-one as starting material afforded the corresponding nitrogen derivatives.141
The mechanism of the transformation was proposed on the basis of a [3 + 2] type cycloaddition of carbonyls,142,143 where the sequence is triggered by the oxidation of the carbonyl substrate (59k) to an electrophilic α-oxo alkyl radical intermediate i. This species adds the olefin to generate the corresponding radical ii, which by different routes (iii or iv) arrives at the cyclized cationic intermediate v, whose rearrangement and deprotonation finally generates the dihydro furo[3,2-c]coumarin product 166.
It was observed that 1,2-disubstituted alkenes gave the trans-substituted adducts, whereas trisubstituted olefins afforded a mixture of cis- and trans-substituted adducts. The yields of the transformation ranged from low to moderate and employing CAN gave better results than the use of Mn(OAc)3·nH2O.
The concomitant formation of the linear isomers was surprising since ketone carbonyls are better radical- and cation sinks than ester carbonyls.144 Dihydro furo[3,2-c]coumarins have been used as starting materials to obtain the corresponding furo[3,2-c]coumarins by means of a catalytic dehydrogenation (10% Pd/C, Ph2O, reflux, 2 h).84 On the other hand, under similar conditions, the group of Lee reported the reaction of phenylacetylene with 4-hydroxycoumarin, to give 2-phenylsubstituted linear and angular furocoumarins.145
In model systems, CAN also outperformed Mn(OAc)3·nH2O and Ag2CO3/Celite (Fétizon's reagent). However, it has been reported that the use of cyclic vinylsulfides146 as the olefin partner, in the presence of Ag2CO3/Celite147 affords medium- and large-sized ring substituted dihydro furo[3,2-c]coumarins in a one-pot procedure.148 On the other hand, the use of alkyl enol ethers in refluxing MeCN afforded the expected 3-alkyl substituted dihydro furo[3,2-c]coumarin acetals; Ag2O can also be used in this reaction.149
The mechanism of this transformation is similar to that involving CAN as oxidant. The dicarbonyl compound is first oxidized by silver(I) to generate an α-oxoalkyl radical, which then attacks vinyl sulfide to give a second radical species. In turn, the latter undergoes a new silverI-mediated oxidation to a cation, which suffers cyclization to the dihydrofuran and final elimination of the sulfide moiety to give the substituted furan.
6.3. Photochemical reactions with olefins
The group of Suginome reported the synthesis of linear (169) and angular (168) dihydro-furocoumarins by photolysis of photochemically generated radicals i from cyclobutyl hypoiodite derivatives (Scheme 40).150 The latter were formed by the [2 + 2] reaction between 4-hydroxycoumarin (59) and olefins 170 (cyclic and acyclic) in the presence of HgO and I2.151 The reaction also accepts enol-ethers and enol-esters as substrates, affording acetals as products. The yields strongly depend on the structure of the olefin, varying from rather low to very good (15–75%).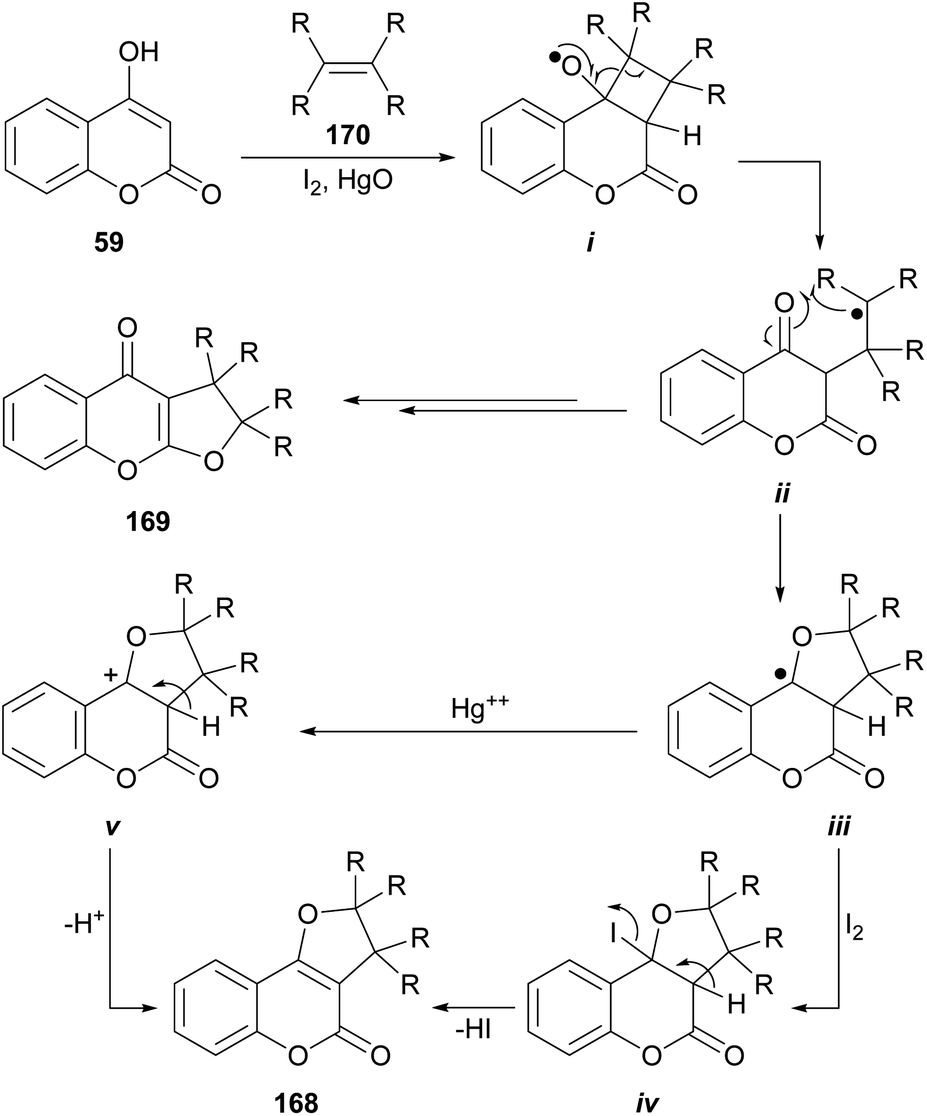 | ||
| Scheme 40 Reagents and conditions: (a) I2, HgO, RRCCRR, PhH, hυ (100 W high-pressure mercury arc), 2 h. | ||
The proposed mechanism entails the formation of radical i, followed by intramolecular combinations of the carbonyl oxygens with the carbon-centered radical ii resulting from β-scission, to afford linear furocoumarins (169). Alternatively, cyclization to iii and further reaction with HgO or I2 (the latter species could form the tertiary iodide iv or the cation v, respectively), could finally afford the tricycle 168.
6.4. Cyclization of 3-alkynyl-4-methoxycoumarins
The group of Monteiro reported a palladium-catalyzed cascade process initiated by alkyl or aryl iodides, which affords good yields of differentially 2,3-disubstituted furo[3,2-c]coumarins (172) from 3-alkynyl-4-methoxycoumarins (171) (Scheme 41).152 The alkynes were readily prepared by the Pd-catalyzed cross-coupling reaction of the 3-iodo-4-methoxy coumarins with terminal acetylenes under amine-free conditions to avoid the undesired amine-induced demethylation of the resulting acetylenic coumarins.153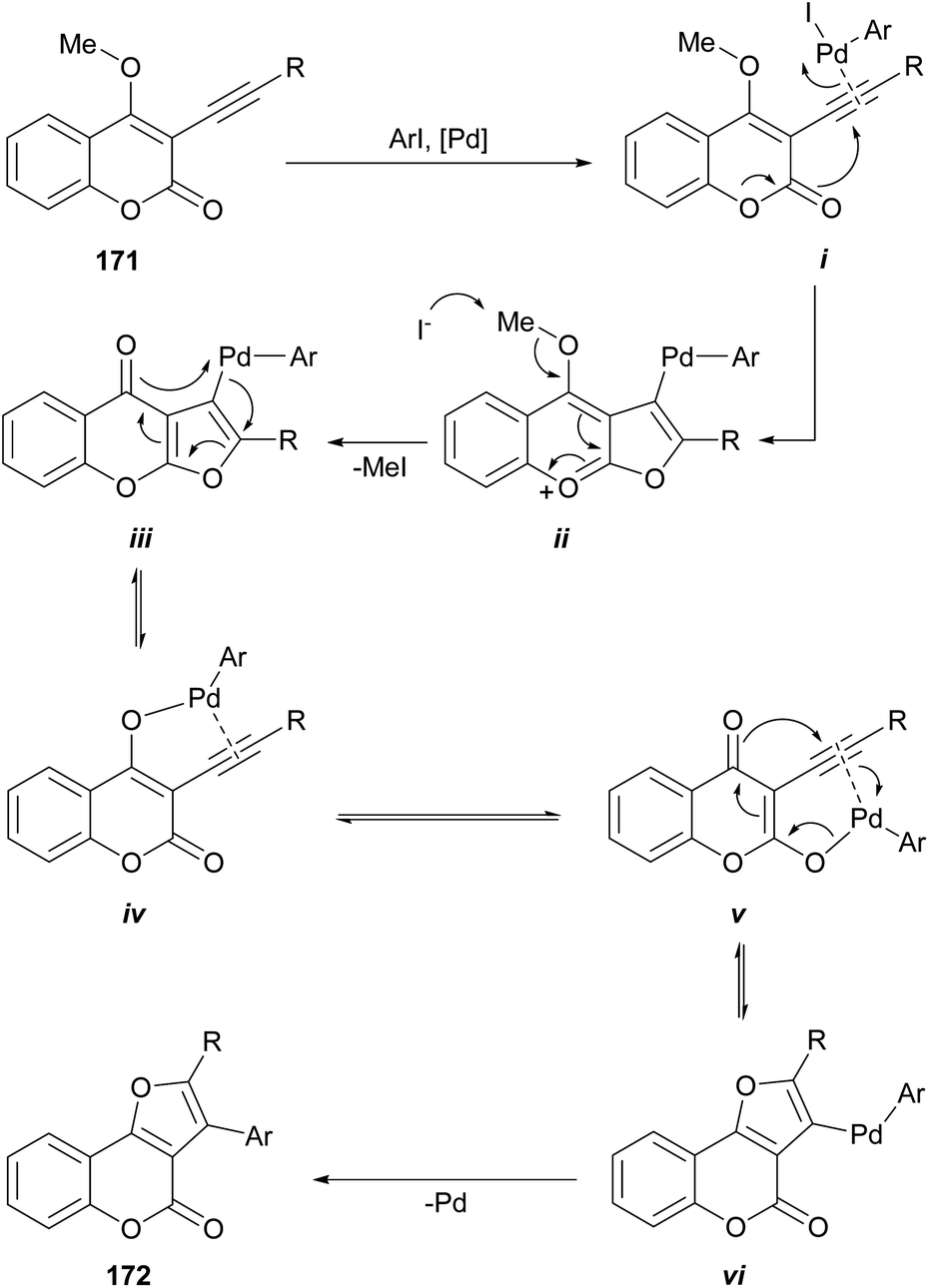 | ||
| Scheme 41 Reagents and conditions: (a) ArI, CuI, PdCl2(PPh3)2, K2CO3, THF, 50 °C. | ||
Mechanistically, the σ-aryl PdII iodide complex generated by oxidative addition of the organic iodide to the Pd0 catalyst would activate the alkyne triple bond of i toward intramolecular nucleophilic attack of the oxygen of the lactone to give the furochromenylium intermediate ii. Iodide-assisted demethylation of the latter is followed by reductive elimination to release MeI and give the corresponding Pd-containing furochromone intermediate iii (ref. 154) (which can also be considered a Miyaura cross-coupling product).
In addition to afford linearly fused heterocycles, this intermediate can rearrange and undergo furan ring-opening/ring-closure155 via palladium enolate intermediates iv and v to the isomeric, angularly fused Pd-containing furocoumarin vi.156 Then, a final reductive elimination affords the furo[3,2-c]coumarin 172.
The transformation furnishes moderate to good yields of product with aryl halides bearing electron withdrawing groups, probably reflecting a decreased electrophilicity of the organo-PdII complex cycloisomerization.
6.5. Furanylation of secondary propargyl alcohols
The group of Zhou157 devised in 2007 a simple synthesis of furo[3,2-c]coumarins by the Yb(OTf)3 catalyzed reaction between 4-hydroxycoumarins and secondary propargylic alcohols. The reaction uses only 5 mol% of the promoter, in nitromethane–dioxane (1:1, v/v) at 50 °C and the only example recorded was obtained in 37% yield.
In addition, the group of Cadierno reported a straightforward and more efficient one-pot approach to tetrasubstituted furans from secondary propargylic alcohols (173) and acyclic 1,3-dicarbonyl compounds (Scheme 42),158 and extended its scope to the synthesis of 2,3-disubstituted furo[3,2-c]coumarins (174), using 4-hydroxycoumarins such as 59, as starting materials.159
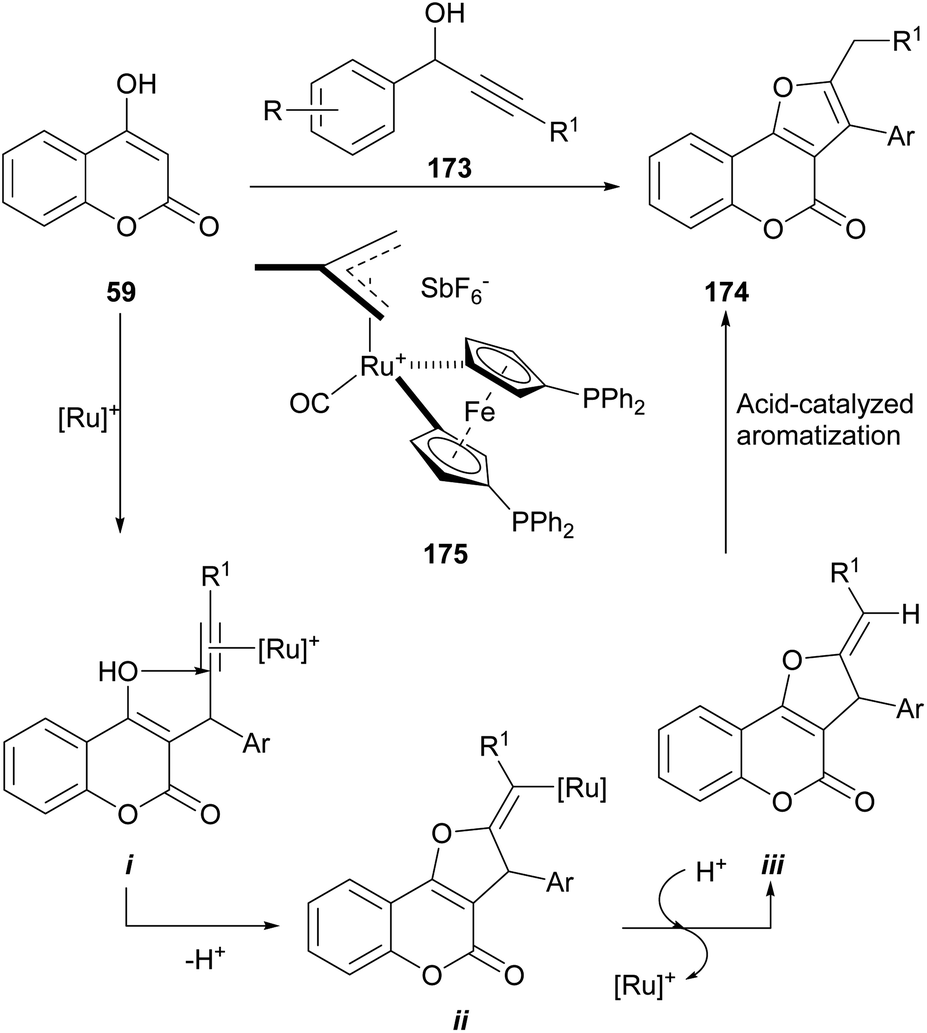 | ||
| Scheme 42 Reagents and conditions: (a) 175 (5 mol%); TFA (50 mol%), 75 °C. | ||
The reaction is catalyzed by 175, the 16-electron allyl-ruthenium(II) complex [Ru(η3-2-C3H4Me)-(CO)(dppf)][SbF6]. The reaction mechanism involves an initial TFA-promoted propargylation of the 4-hydroxy coumarin 59 with the alkynol 173 (ref. 160) via σ-coordination with the Ru-catalyst and intramolecular nucleophilic attack of the enol at the C-2 position of the coordinated alkyne161 to generate an alkenyl-ruthenium derivative (i) and promote an exo cyclization of the γ-ketoalkyne to intermediate ii.162
Protonolysis of the cyclized intermediate ii liberates the heterocycles iii, regenerating the catalytically active ruthenium species.163 Upon acid-catalyzed isomerization, iii gives the final product 174, in yields ranging from moderate to excellent (61–92%).
On the other side, Xie, Cui and coworkers demonstrated that, under CuBr promotion, the outcome of the propargylation-oxacyclization/isomerization reaction between 4-hydroxycoumarin (59) and certain secondary terminal propargylic acetates (176) depends on the reaction solvent (Scheme 43).164
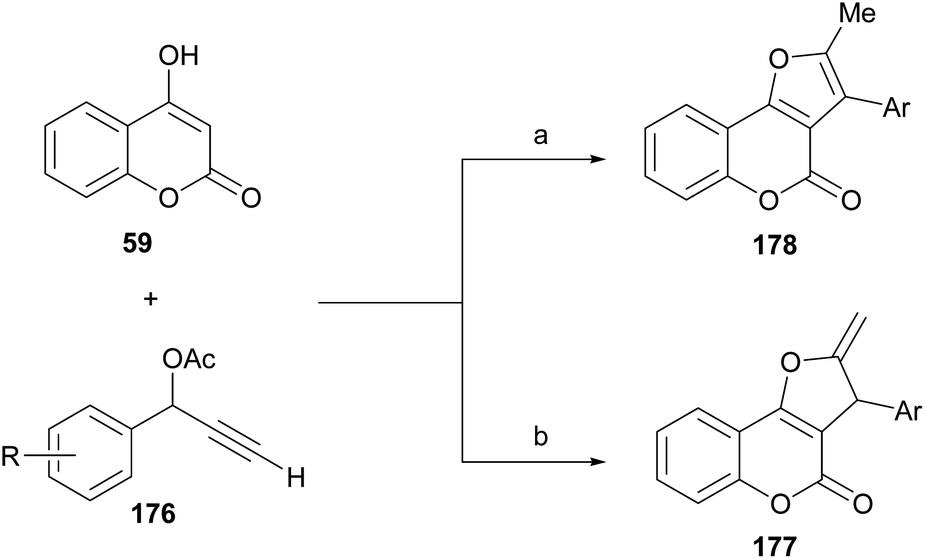 | ||
| Scheme 43 Reagents and conditions: (a) CuBr, DIPEA, DMSO, MW, 100 °C, 20 min (48–82%); (b) CuBr, DIPEA, ClCH2CH2Cl, MW, 100 °C, 20 min (36–85%). | ||
The transformation provides mainly the exocyclic olefin 177 when carried out in 1,2-dichloroethane, whereas the fully aromatized furan ring motif (178) is predominantly observed in transformations performed in DMSO. Control experiments suggested that the formation of a copper–allenylidene complex is essential for catalysis to occur. On the other hand, a similar transformation was reported under palladium catalysis, with Ag2CO3 as oxidant, which results in 2,3-disubstituted compound 178 through the Et3N-mediated isomerization of the related exo-olefins 177.165
6.6. Cyclization with dibenzoyl acetylene
Khalilzadeh et al. reported an efficient synthesis of furo[3,2-c] coumarin derivatives 180 by the NaH-promoted reaction between dibenzoylacetylene (179) and 4-hydroxycoumarin (59).166 The transformation (Scheme 44) took place in nearly quantitative yield.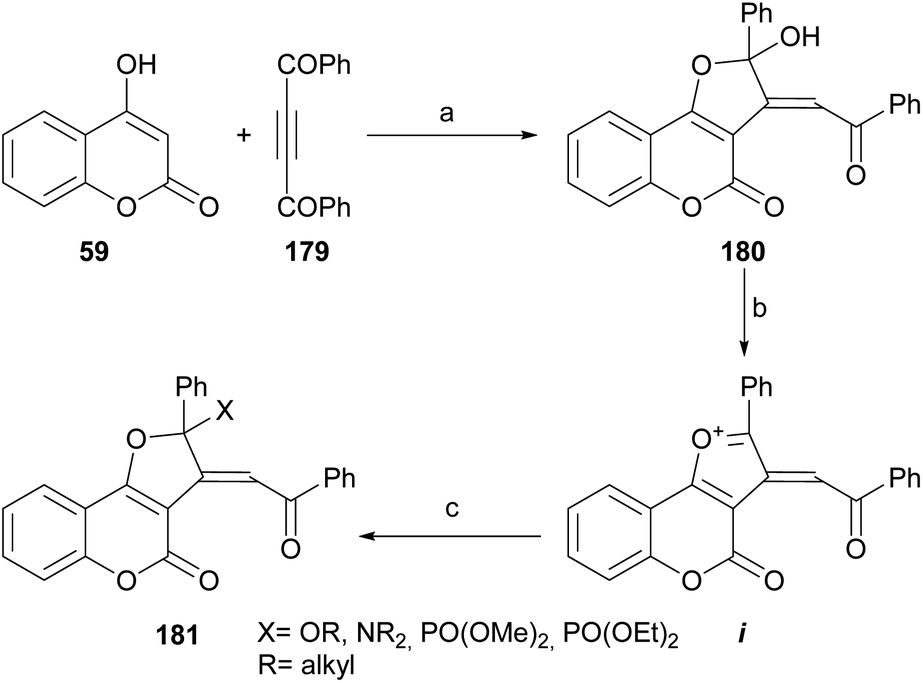 | ||
| Scheme 44 Reagents and conditions: (a) NaH, Et2O, 5 h (95%); (b) TMSCl, CHCl3, r.t., 8 h (90%); (c) ROH, Et3N, H2O, reflux, 40 min (78–87%) or R2NH, MeOH, H2O, reflux, 40 min (75–85%) or P(OMe)3/P(OEt)3, MeOH, Et3N, H2O, reflux, 40 min (85–90%). | ||
Treatment of the heterocycle with TMSCl followed by addition of nucleophiles, such as alcohols, amines or trialkyl phosphites in water gave excellent yields of the functionalized 2-phenyl-4H-furo[3,2-c]coumarin derivatives 181, through the intermediacy of i.
6.7. Furanylation through functional phosphorous zwitterions
The group of Lin demonstrated that multifunctionalized furan derivatives can be accessed through an intramolecular Wittig reaction by treating 4-hydroxycoumarins (59) as Michael acceptors, with Bu3P in the presence of acyl chlorides (Scheme 45).167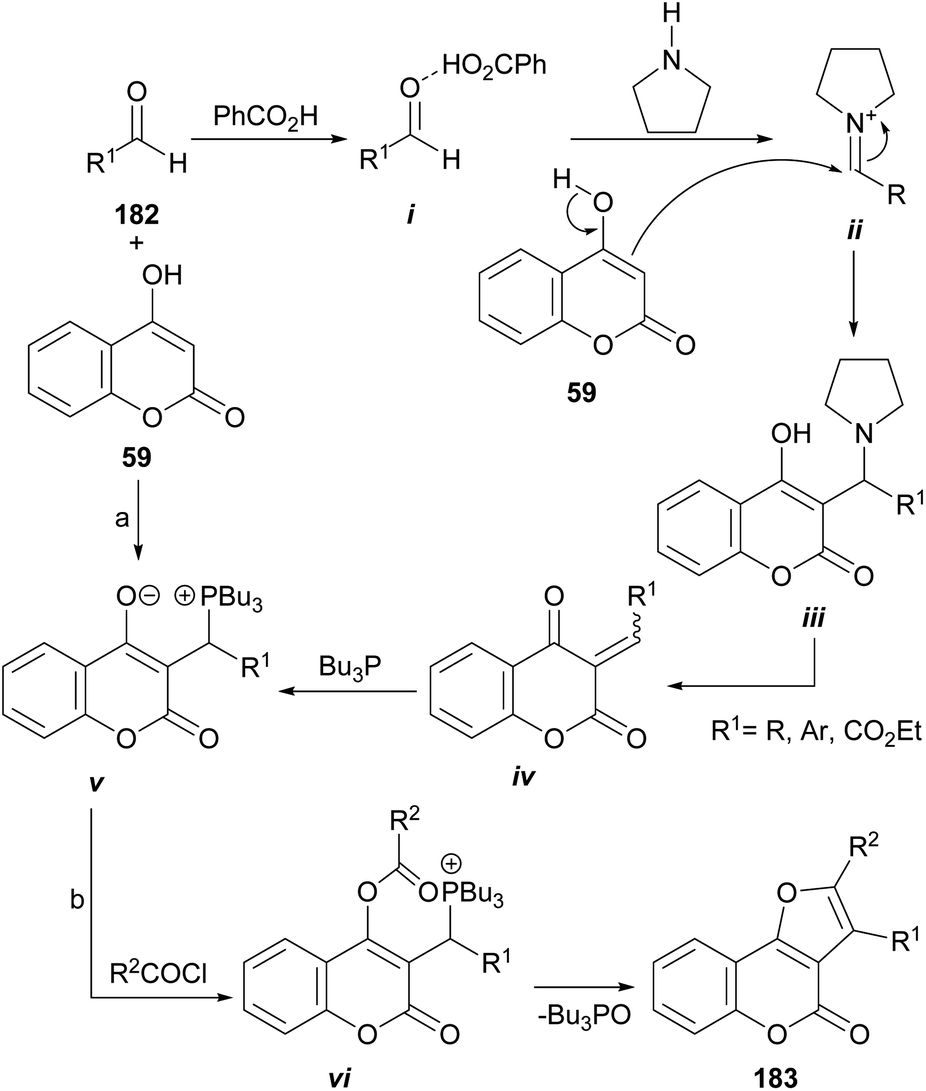 | ||
| Scheme 45 Reagents and conditions: (a) PhCO2H (cat.), pyrrolidine (cat.), Bu3P, r.t., THF (84–99%); (b) R2COCl, Et3N, THF, r.t. (92–95%). | ||
They disclosed a general preparation of highly functional phosphorus zwitterions by means of tandem three-component reactions between the corresponding 4-hydroxycoumarins, aldehydes (182), and Bu3P under the assistance of pyrrolidine and benzoic acid in catalytic amounts, to activate the aldehyde carbonyl moiety. In this way, the formation of intermediates i and ii was successively favored, before their reaction with 59 to form the corresponding adduct iii.
In the reaction mechanism, elimination of the pyrrolidine catalyst is followed by the Michael addition of Bu3P to the so formed Michael acceptor iv, to give v. Finally, upon reaction with acid chlorides, the resulting zwitterions vi undergo an intramolecular Wittig reaction to afford furo[3,2-c]coumarins 183 in a one-step procedure.168 The reaction conditions are mild, and yields are high to excellent.
The same group also informed that the reaction of 3-cinnamoyl-4-hydroxycoumarins (184) with Bu3P and an acid chloride also provides 2,3-disubstituted furo[3,2-c]coumarins (185 or 186) under base promotion. The outcome depends on the treatment given to the reaction and the workup conditions.169
The proposed reaction mechanism (Scheme 46) involves initial formation of phosphorus zwitterion (i) by Michael addition of Bu3P to the starting cinnamoylcoumarin 184. Then, this zwitterionic intermediate undergoes successive acylation with the acid chloride to provide the intermediate ii.
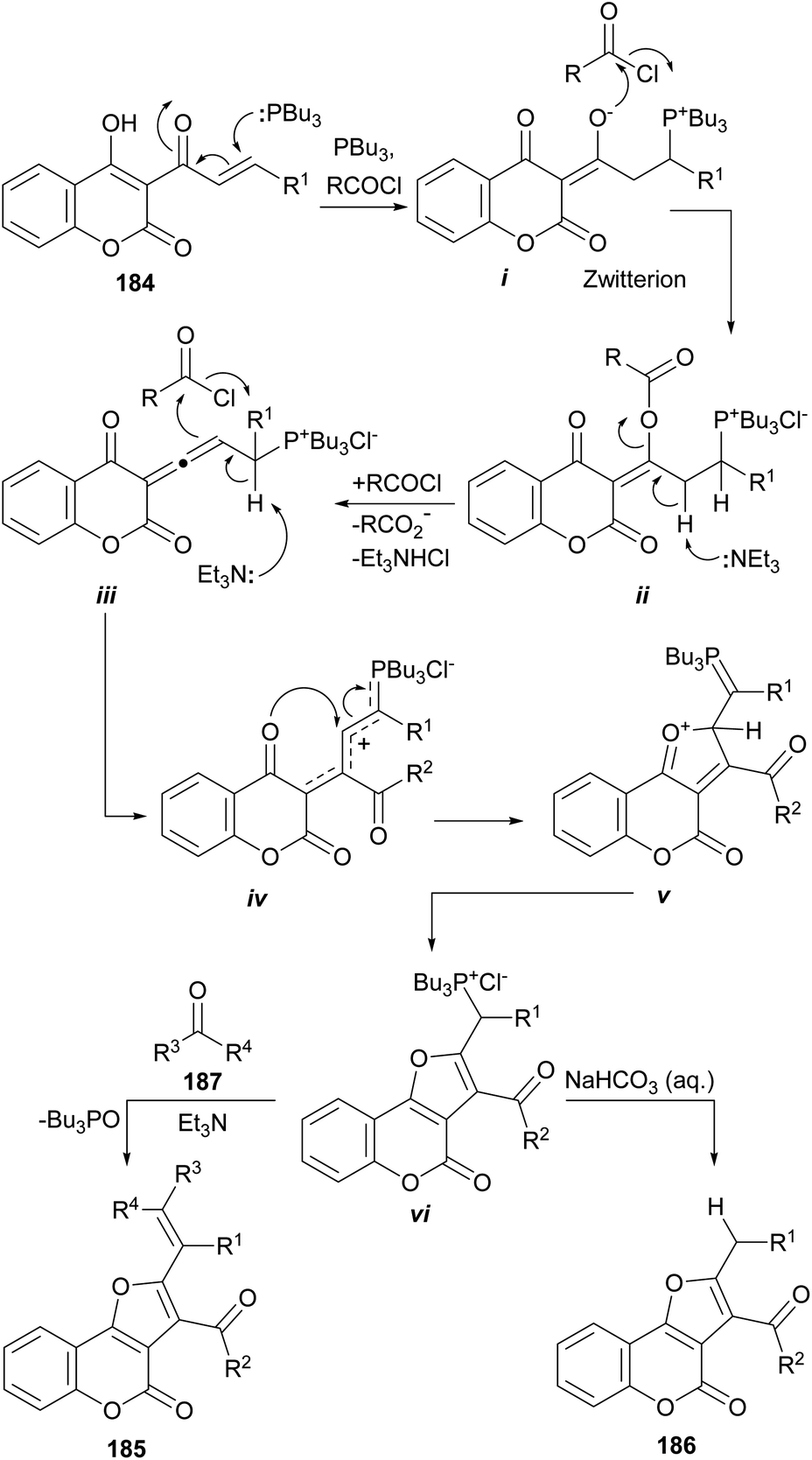 | ||
| Scheme 46 Reagents and conditions: (a) Bu3P, RCOCl, Et3N, MeCN, 30 °C, 1 h (75–90%). | ||
Under basic conditions, an elimination takes place, resulting in the formation of the allene intermediate iii.170 In turn, this intermediate undergoes a C-acylation with the acid chloride, furnishing the phosphonium chloride iv, able to undergo intramolecular cyclization to afford v. Isomerization of v takes place to give the ylide precursor vi, which can be activated with Et3N to undergo a Wittig reaction with a carbonyl electrophile (187), giving 185 or became hydrolyzed to afford 186.
6.8. Reaction with aldehydes and active methylenes
The group of Perumal recently reported a ionic liquid-mediated, three-component sequential approach toward the regio- and diastereo-selective synthesis of furo[3,2-c]coumarins under eco-conscious conditions (Scheme 47).171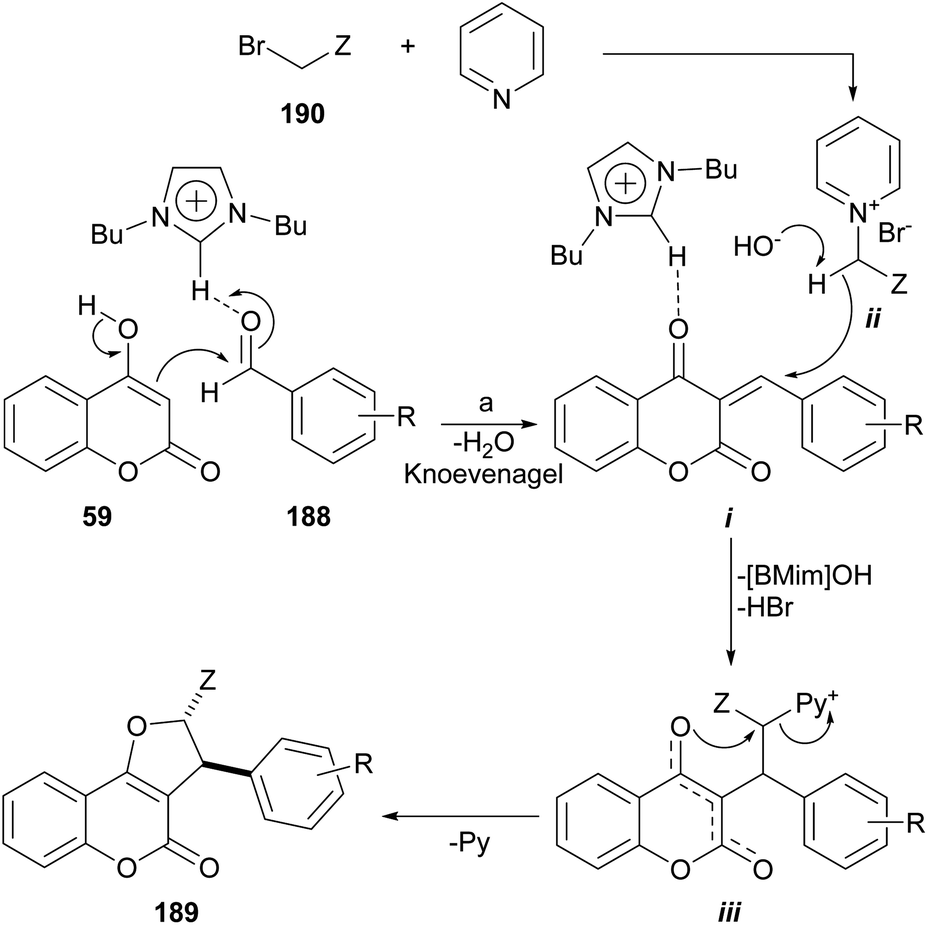 | ||
| Scheme 47 Reagents and conditions: (a) ArCHO, [BMim]OH, pyridine, BrCH2Z (ZCN, COPh, CO2Et), 80–90 °C, 1–4 h (81–93%). | ||
The method is based on the reaction of 4-hydroxycoumarin with aromatic aldehydes and in situ generated cyanomethyl pyridinium, phenacylpyridinium or (2-ethoxy-2-oxoethyl) pyridinium ylides. The transformation is run in [BMIm]OH, a ionic liquid which acts both, as a catalyst and the reaction medium. The yields are very good to excellent, provided the aromatic aldehyde does not bear strong electron withdrawing groups.
The mechanism possibly involves a [BMIM]OH-promoted abstraction of the hydroxyl proton from the 4-hydroxycoumarin 59, and activation of the aromatic aldehyde (188)172 to catalyse the initial aldol reaction, thus promoting the dehydration step during the Knoevenagel condensation with the aldehyde to give intermediate i.
The ionic liquid may also be involved in the abstraction of a proton from the pyridinium bromide derivative formed by reaction between pyridine and 190 to furnish an intermediate pyridinium ylide (ii) which could attack the Michael acceptor i, activated by the ionic liquid and afford the pyridinium intermediate iii. Final annulation through the displacement of pyridine furnishes the furo[3,2-c]coumarins 189 in a regio- and stereoselective way.
6.9. Multicomponent, domino approaches
The use of multicomponent reactions for the synthesis of different 2,3-disubstituted furo[3,2-c]coumarins derivatives from 4-hydroxycoumarins has been explored by various research teams (Scheme 48).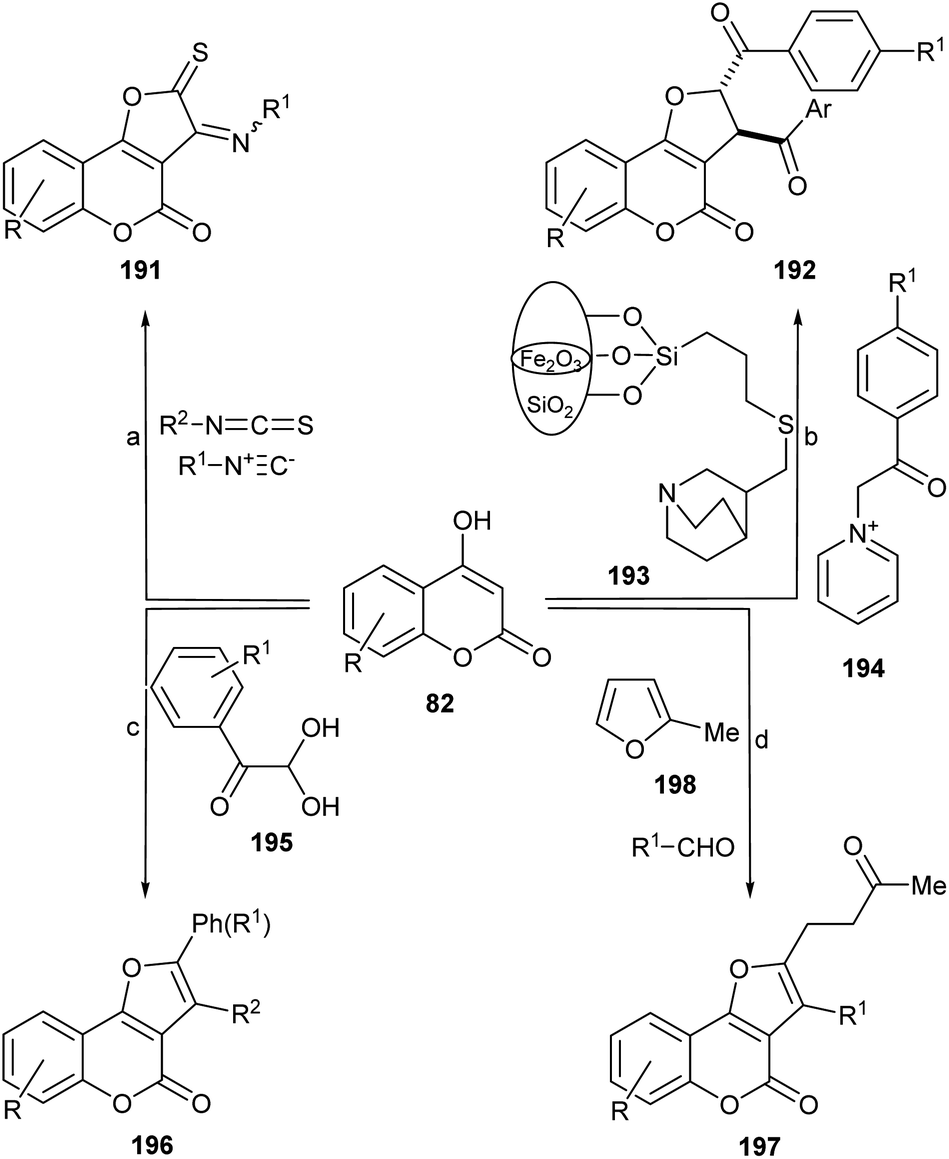 | ||
Scheme 48 Reagents and conditions: (a) R2–N+![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C−; R1–NCS, Fe3O4-MNPs (10 mol%), H2O, r.t., 4 h (alkyl, aryl, 85–92%); (b) 193, 194, ArC(O)CH(OH)2, H2O, reflux, 2 h (85–95%); (c) MsOH, PhMe, reflux [R1 = 3-(4-hydroxyxoumarin)] or ZnCl2, allylTMS, PhMe, reflux (R1 = allyl, 50–72%) or FeCl3, PhMe, 90 °C (R1 = 4Me–C6H4, 48–84%); (d) 198, R1CHO, PhMe, reflux, 6 h (R1 = alkyl, aryl, 68–87%). C−; R1–NCS, Fe3O4-MNPs (10 mol%), H2O, r.t., 4 h (alkyl, aryl, 85–92%); (b) 193, 194, ArC(O)CH(OH)2, H2O, reflux, 2 h (85–95%); (c) MsOH, PhMe, reflux [R1 = 3-(4-hydroxyxoumarin)] or ZnCl2, allylTMS, PhMe, reflux (R1 = allyl, 50–72%) or FeCl3, PhMe, 90 °C (R1 = 4Me–C6H4, 48–84%); (d) 198, R1CHO, PhMe, reflux, 6 h (R1 = alkyl, aryl, 68–87%). | ||
The group of Hossaini succeeded in developing a one-pot three components coupling of 4-hydroxycumarin (82), isothiocyanates, and isocyanides in water under promotion by magnetically recoverable Fe3O4 nanoparticles (Fe3O4-MNPs) in water at room temperature.173 This approach afforded the heterocycles 191 in good to excellent yields (85–92%).
On the other hand, Chen et al. disclosed the synthesis of these heterocycles with phenylglyoxal and phenol or allylTMS, under protic (MsOH) and Lewis acid (ZnCl2, FeCl3) promotion, in good yields and chemoselectivities.174 The use of heteroarylamines in refluxing MeCN gave heteroarylamino derivatives. It was proposed that this tandem process involves sequentially an aldol condensation, Michael addition, a ring closure, and a final dehydration reaction.175
Employing N-(2-(4-halobenzoyl)-2-oxoethyl)pyridinium bromide and a nano γ-Fe2O3-quinuclidine-based catalyst, the group of Mosslemin was able to prepare trans-2,3-disubstituted derivatives.176 The group of Nolland reported quite a similar transformation, employing aromatic aldehydes and 2-methylfuran under Fe2(SO4)3·xH2O catalysis in refluxing toluene.177
The group of Kolita (Scheme 49) developed a simple and efficient method for the synthesis of furo[3,2-c]coumarins.178 Their approach is based on a coupled oxidative process which involves the one-pot pseudo-condensation of three components (59, an aldehyde and I2), in the presence of DMSO as stoichiometric oxidant (15 mol% I2 was used). Similar conditions were more recently reported by the group of Fattah, employing only 7 mol% I2. 179
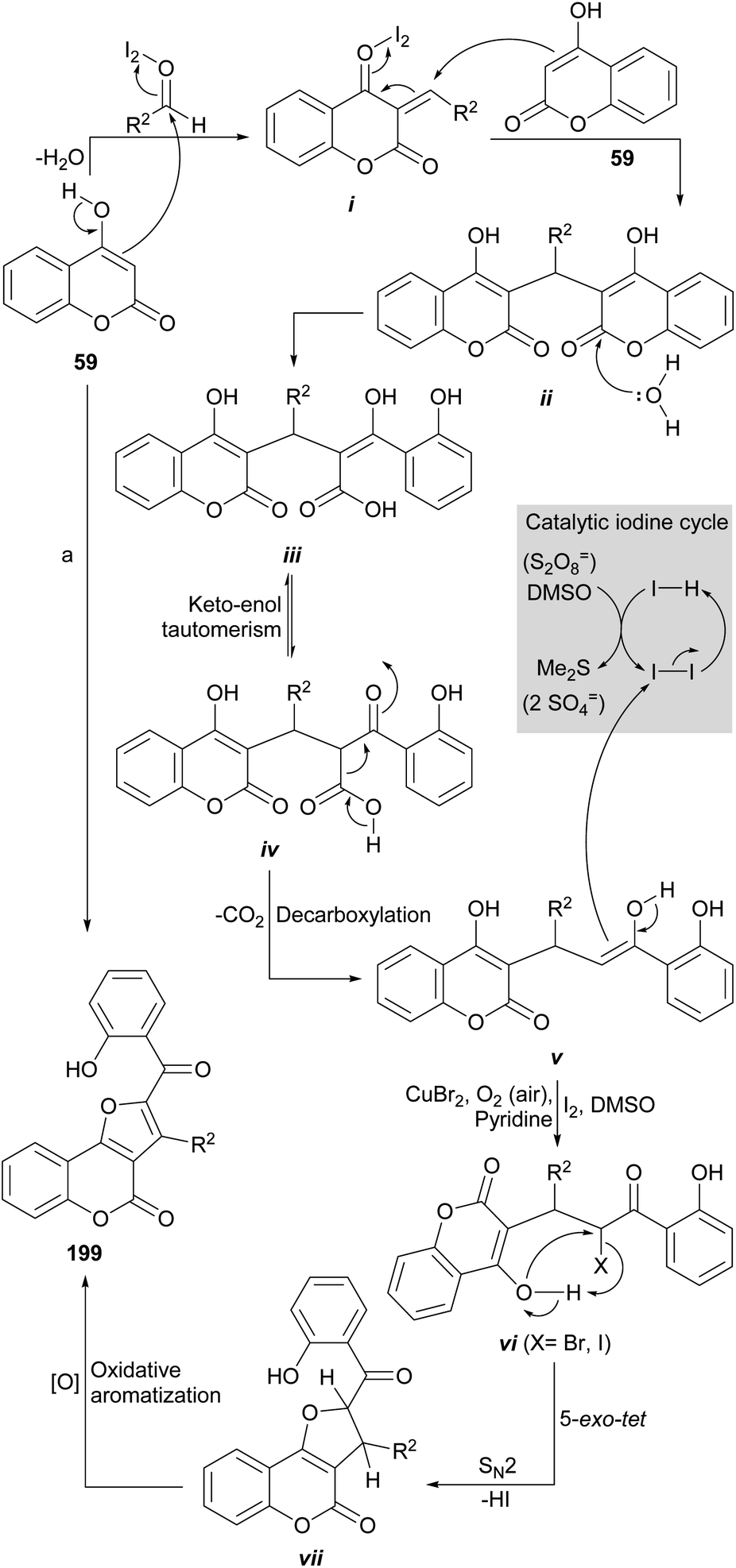 | ||
| Scheme 49 Reagents and conditions: (a) RCHO, I2 (cat.), DMSO, 80 °C (1–5 h, Kolita; 24 h, Fattah et al.); or (1) RCHO, I2, PEG, 80 °C; (2) I2, K2S2O8, Na2CO3, PEG, 120 °C (Shaffie et al., 42–75%; R = H, alkyl, aryl, thiophenyl, furyl); or RCHO, pyridine, CuBr2, O2, MeCN, 80 °C, 12 h (Zhang, 91%). | ||
The reaction of 4-hydroxycoumarins with aldehydes produces dicoumarins in the presence of Bronsted or Lewis acid catalysts.180 Iodine has been used for this purpose.181 In the reaction mechanism, it is assumed that the aldehyde is activated by iodine towards the attack of 59, which leads to the formation of intermediate i.
In turn, the latter, being a powerful Michael acceptor, may undergo conjugate addition of a second molecule of 59 to afford a 3,3′-methylene dicoumarin intermediate (ii), which is hydrolyzed to carboxylic acid iii, the enolic form of the 1,3-dicarbonyl system iv.
Decarboxylation182 of iv to afford intermediate v is followed by α-carbonyl halogenation to give vi,183 able to undergo a 5-exo-tet type cyclization via a SN2 attack of the free phenol to the halogen moiety (X), with concomitant loss of the elements of the corresponding hydracid (HX) to yield intermediate vii. Final oxidative aromatization of the five-membered ring would furnish the final product (199).184
The yields of this rather complex process exceeded 80% and different alkyl, aryl and hetaryl aldehydes (thiophene, furan derivatives) are accepted. Control experiments with TEMPO as radical scavenger excluded significant involvement of any radical intermediate and the access to the products from pure dicoumarins confirmed their intermediacy in the synthetic sequence.
Prompted by the observation that furo[3,2-c]coumarins form along with dicoumarins in the iodine-promoted reaction, Shafiee et al. developed a synthesis of furo[3,2-c]coumarins by first preparing dicoumarins in polyethyleneglycol (PEG) from 4-hydroxycoumarins with the aid of catalytic amounts of I2 and 2 equiv. of K2S2O8 as stoichiometric oxidizing agent, in the presence of Na2CO3.185
PEG is an inexpensive, recoverable, thermostable, and non-toxic hydrophilic polymer, which can replace less desirable organic solvents.186 The reaction conditions are mild, the procedure is simple and the products were obtained in moderate to high yield. In these transformations, the roles of DMSO and K2S2O8 are to oxidize the iodide ion to iodine, recycling this reagent along the process.187 The yields of the synthesis were between moderate and very good (42–75%).
The group of Zhang (Scheme 49) also developed a similar intramolecular decarboxylative functionalization of 4-hydroxy coumarin 59 to form 2,3-disubstituted furo[3,2-c]coumarins (199), which is a complement to the previous ones. This efficient and ecological approach employs CuBr2 catalysis in pyridine under aerobic conditions; the latter are useful to oxidize the CuI by-product, in the presence of pyridine.
7 Total synthesis of natural products
One of the best alternatives to test or extend the utility of the different approaches toward complex frameworks is to use them for the synthesis of natural products and their analogs. In the case of the furo[3,2-c]coumarins, this paradigm can be exemplified by the synthesis of the pterophyllins and neo-tanshinlactone.7.1. Pterophyllins and related compounds
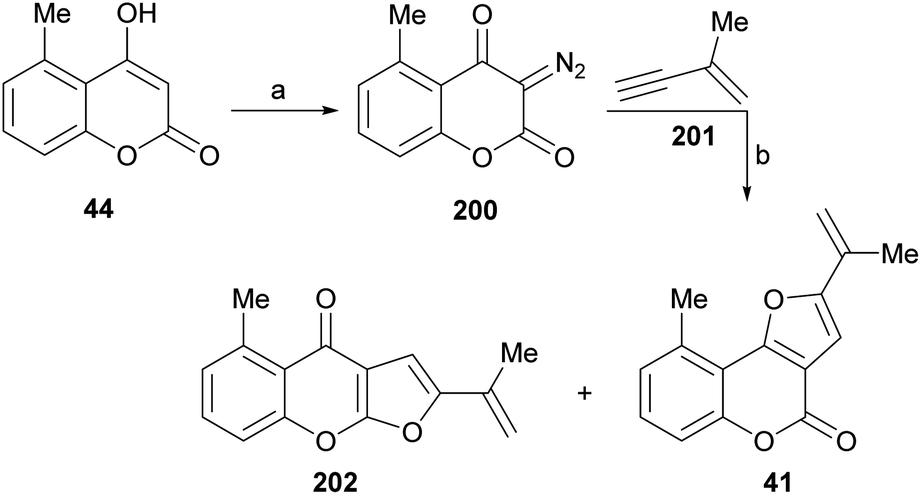 | ||
| Scheme 50 Reagents and conditions: (a) MsN3, Et3N, MeCN, 3 h; (b) 201 (15 equiv.), Rh2(OAc)4, reflux, 3 h (41, 33%; 202, 35%). | ||
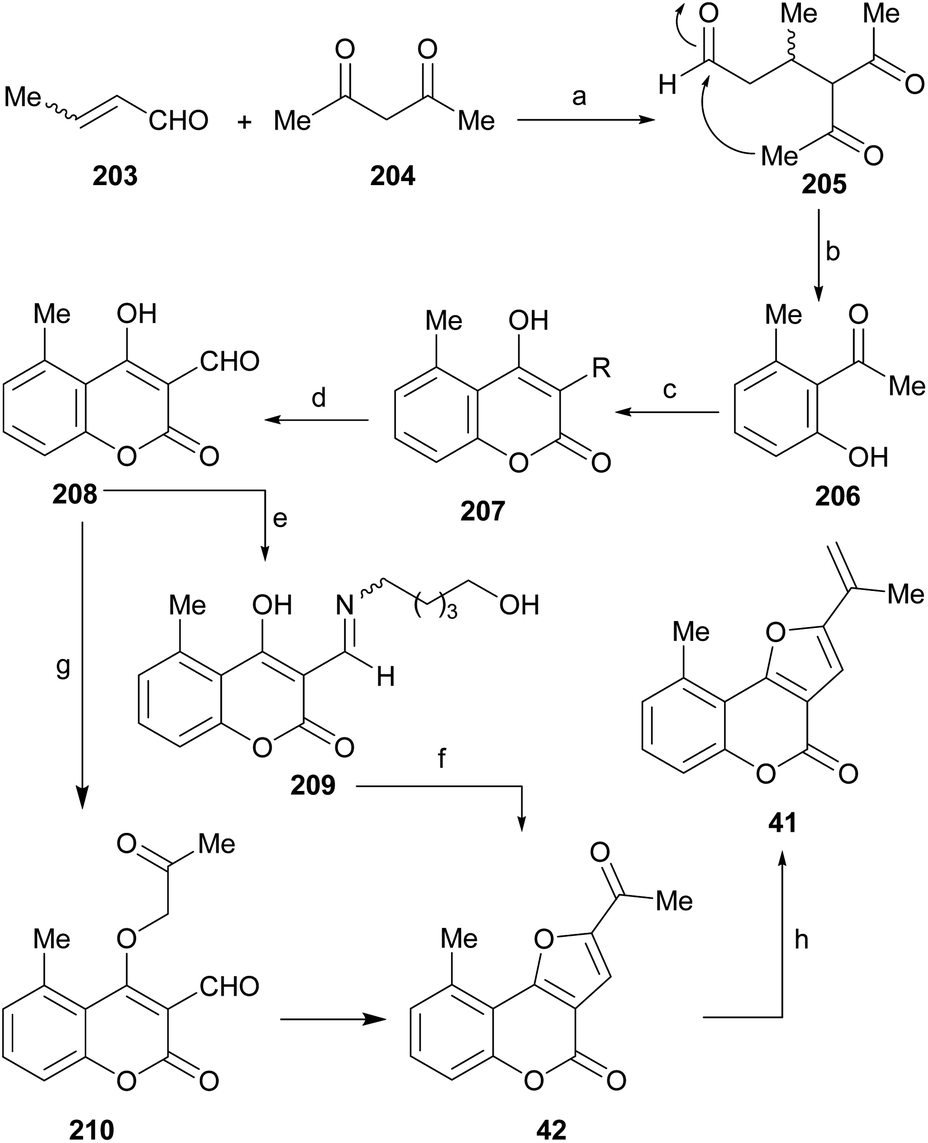 | ||
| Scheme 51 Reagents and conditions: (a) L-proline (0.1 equiv.), 0 °C, 20 h (58%) or 1-phenylbutane-1,4-diol (0.14 equiv.), 0 °C, 20 h (91%); (b) LiCl, CuCl2, DMF, 90 °C, 3 h (54%); (c) Et2CO3, KtBuO, r.t., 12 h (79%); (d) MgCl2 (anh.), Et3N, (CH2O)n, THF, 50 °C, 1 h (87%); (e) H2N(CH2)4CH2OH, PhMe, 90 °C, 1 h (85%); (f) ClCH2COCH3, K2CO3, DMF, 65 °C, overnight (15%); (g) ClCH2COCH3, Al2O3, CH2Cl2, 40 °C, 72 h (42, 88%); (h) Ph3PMe+I−, KtBuO, LiCl, THF, r.t., 1 h (41, 58%; 78%, based on recovered starting material). | ||
The sequence was continued with the acquisition of 207 in 79% yield,16b,191 by the Claisen reaction of 206 with (EtO)2CO in DMF, using KtBuO (3 equiv.) or sodium (5 equiv.) as bases. The formylation of the 4-hydroxycoumarin 207 was carried out according to the Casnati–Skattebøl protocol,93,192,193 affording 208 in 87% yield.
Next, it was observed that the O-alkylation of the unstable and highly reactive 3-formyl-4-hydroxycoumarins has scarce precedents. Since the O-acetonylation of 208 with chloroacetone toward 210 under conventional conditions (K2CO3, EtOH or DMF),194 met with failure, leading to complete decomposition of the starting heterocycle,195 the formyl moiety of compound 208 was masked as the corresponding imine (209), prepared in 85% yield by condensation of the aldehyde with 5-aminopentan-1-ol in toluene at 90 °C. Imines have been employed as carbonyl protecting groups, but they are unstable,196 and the acetonylation of 209 afforded the expected furo[3,2-c]coumarin 42, albeit in low yield (15%).
Therefore, other conditions were explored and it was found that the reaction of 208 with chloroacetone in refluxing CH2Cl2, in the presence of a ten-fold excess of Brockmann I activated basic alumina gave the desired tricycle 42 in 88% yield, through the intermediacy of 210.195,197
A Wittig olefination of 42 with Ph3PMe+I− in THF to which LiCl was added, employing KtBuO as base, afforded the isopropylidene derivative 41 in 78% yield. The use of LiCl as additive proved relevant for attaining suitable product yields, presumably by affecting the reaction rate or the enolization equilibrium of the methyl ketone moiety.198
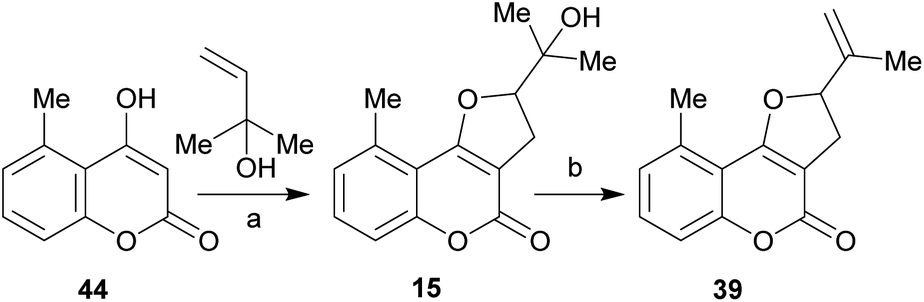 | ||
| Scheme 52 Reagents and conditions: (a) H2CCHCMe2OH, CAN, MeCN, 0 °C, 1 h (45%); (b) Burgess reagent, PhH, reflux, 30 min (48%). | ||
7.2. Neo-tanshinlactone
Neo-tanshinlactone (6) is a bioactive natural heterocyclic compound; its chemical structure was elucidated by Lee and co-workers in 2004.34a The same group executed its first total synthesis, in six steps from 5-methoxy-1-tetralone, employing a Friedel–Crafts acetylation and a Feist–Bénary reaction as the key steps. More recently, the Feist–Bénary reaction was also employed for the synthesis of tricyclic analogs of 6.199In 2012, the group of Abe reported a total synthesis of neo-tanshinlactone employing naphthol 218 and iodofuran 217 (Scheme 53). The phenol was prepared according to the work of Lee, whereas the furan was synthesized from dimethyl itaconate (211).200
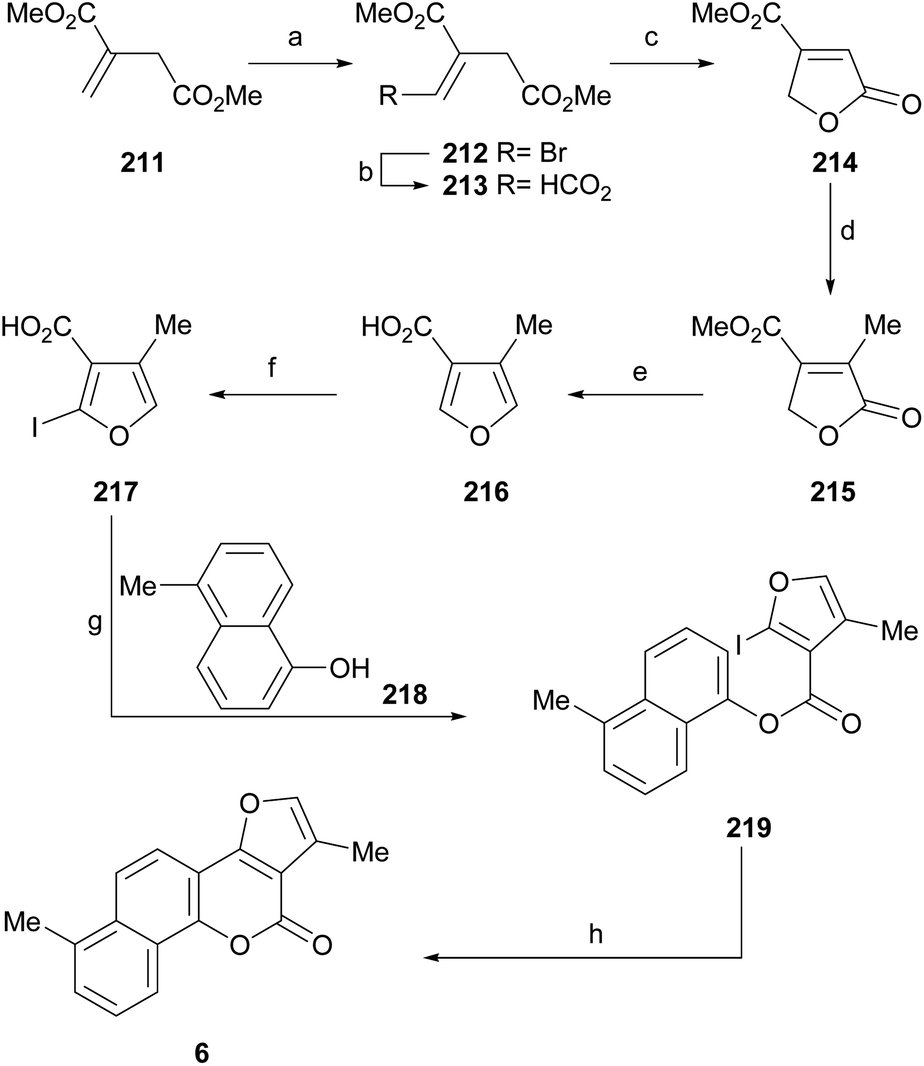 | ||
| Scheme 53 Reagents and conditions: (a) (1) Br2, CH2Cl2; (2) Et3N, CH2Cl2 (45% overall); (b) HCO2H, Et3N, MeCN (84%); (c) HCl, MeOH (45%); (d) (1) CH2N2, Et2O (73%); (2) DMF, Δ (89%); (e) (1) DIBAL-H, THF; (2) TsOH, MeOH (73%); (3) 1 M KOH, MeOH (77%); (f) (1) LDA, THF; (2) I2, THF (59%); (g) EDC, DMAP, CH2Cl2 (73%); (h) Pd(OAc)2, Bu3P, Ag2CO3, DMA, 160 °C (57%). | ||
To that end, the latter was allylically brominated and the bromide 212 was transformed into the corresponding formate ester 213 by substitution with formic acid in 38% overall yield.201
The acid-mediated cyclization of 213 gave the five-membered lactone 214 in moderate yield, which was subjected to a [3 + 2] cycloaddition with diazomethane and further heat treatment to decompose an intermediate pyrazole afforded the furanone 215 in 29% overall yield.202
A three-step sequence entailing the DIBAL reduction of 215, followed by TsOH treatment of the resulting lactol and hydrolysis of the methyl ester generated the furan carboxylic acid 216 in 29% overall yield, which was selectively iodinated with I2 after metalation with LDA, affording 59% yield of the iodofuran 217.203 Next, the phenol and the furan were coupled with EDC, affording 73% yield of the ester 219, which was further cyclized under palladium catalysis to afford 57% yield of neo-tanshinlactone (6).
In 2013, Mal and co-workers reported an efficient convergent cascade benzannulation–lactonization strategy (Scheme 54) between a phthalide (224) and a furylacrylate (228) toward the natural product.204
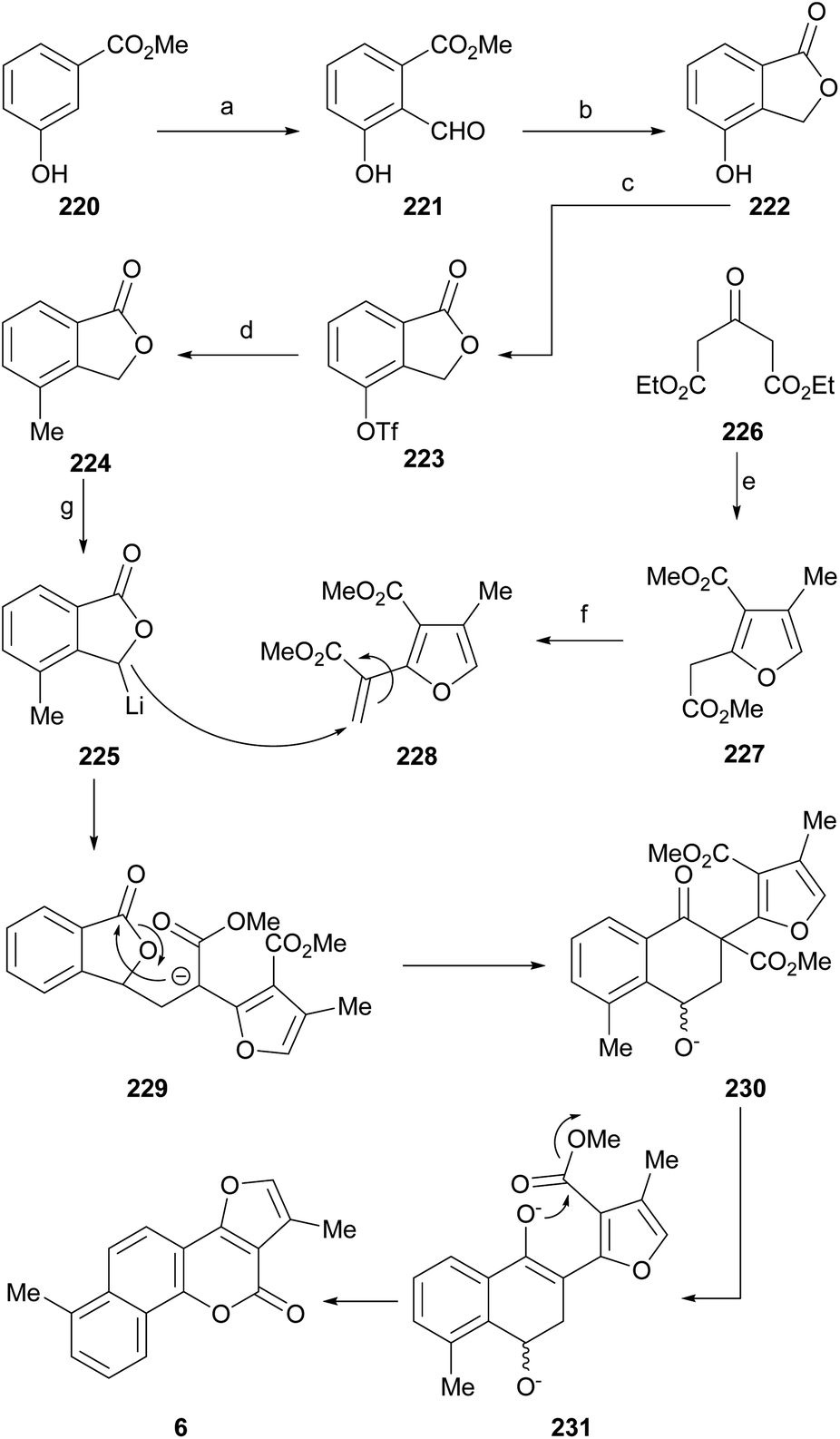 | ||
| Scheme 54 Reagents and conditions: (a) HMTA, TFA, 75 °C, 8 h (85%); (b) NaBH4, MeOH, 0 °C → r.t., 10 min (87%); (c) Tf2O, 2,6-lutidine, CH2Cl2, 0 °C → r.t., 3 h (60%); (d) MeMgBr, Fe(acac)3, NMP, THF, 0 °C, 0.5 h (74%); (e) ClCH2COMe, NH4OH, Et2O, r.t., 3 h (55%); (f) K2CO3, CaO, (CH2O)n, THF, reflux, 8 h (62%); (g) LiHMDS, THF, −78 °C, 25 min (45%). | ||
Their synthesis commenced with 3-hydroxybenzoate (220), which was submitted to a Duff reaction with hexamethylene tetramine in TFA to provide the formylbenzoate 221 in 85% yield,205 which was reduced with NaBH4 to give 74% yield of the phthalide 222. The latter was converted into 223 by treatment with triflic acid anhydride and 2,6-lutidine, and submitted to a Fürstner cross-coupling reaction206 with MeMgI in the presence of Fe(acac)3 to access the phthalide 224 in 74% yield.
On the other hand, the required furylacrylate 228 was prepared in 55% yield by reaction between 3-oxo-pentanedioate 226 and α-chloroacetone in the presence of dilute NH4OH toward 227,207 followed by α-methylenation in 62% yield with paraformaldehyde in the presence of K2CO3 and CaO. The reaction of phthalide 224 with LiHMDS at −78 °C, followed by treatment with the acrylate 228 gave the desired furonaphthopyranone 6 in 45% yield.
In the proposed mechanism, the cascade benzannulation is initiated through lithiation of the phthalide, to generate the 3-lithiophthalide 225. The latter then effects a Michael addition on the furylacrylate 228 to form a new carbanion (229).
Afterwards, this anion undergoes a Dieckmann cyclization, affording the oxytetralone 230, which is nucleophilically attacked at the alkoxycarbonyl moiety, affording the decarboxylated enolate 231. A final aromatization with concomitant nucleophilic attack of the enolate anion to the carboxylate functionality results in the desired final compound 6. The sequence was properly modified to enable a flexible synthesis of neo-tanshinlactone analogs.
8 Conclusions and perspectives
Research in the field of furo[3,2-c]coumarins has been steadily intense during the last three-four decades. However, it can be observed that the isolation of most natural products carrying the tricyclic motif took part mostly during the first half of this period, whereas the total syntheses of some of these natural products were developed during its second half, within the last 15–20 years.Many of the available synthetic procedures toward furo[3,2-c]coumarin derivatives can now be considered traditional. They have withstood the proof of time and are still in use because of their wide scope, high yields and operational simplicity.
On the other hand, the last decade has brought an enormous increase in the number of novel metal-catalyzed approaches toward this angular tricyclic skeleton, which served as complements to the more traditional approaches. Fortunately, the chemical interest in these compounds is still alive as is the interest in some bioactive compounds, like the neo-tanshinlactone derivatives.
From the biological and pharmacological points of view, many challenges still remain undefeated. They include gaining more detailed knowledge of their mechanism of action, which will enable to understand both, their interaction with specific receptors in human tissues, which may turn them into potential medicines, and their biological role in living systems, which may help to uncover the evolution of natural plant defenses.
Since sufficient amounts of natural products are always needed for testing purposes, it is expected that the development of new additions to the currently available synthesis and reagents toolbox will take place and that this will enable still more direct, efficient and selective synthetic procedures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, PUE IQUIR 2016) and Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT, PICT 2017-0149) for financial support. I. C. and L. J. C. acknowledge CONICET for their Doctoral fellowships.Notes and references
- (a) R. D. H. Murray, Naturally Occurring Plant Coumarins, in Progress in the Chemistry of Organic Natural Products.Springer, Vienna, Austria, 1991, pp. 84–316 Search PubMed; (b) M. Darbarwar and V. Sundaramurthy, Synthesis, 1982, 337–388 CrossRef; (c) L. Santana, E. Uriarte, F. Roleira, N. Milhazes and F. Borges, Curr. Med. Chem., 2004, 11, 3239–3261 CrossRef CAS.
- (a) Y.-J. Jang, S.-e. Syu, Y.-J. Chen, M.-C. Yang and W. Lin, Org. Biomol. Chem., 2012, 10, 843–847 RSC; (b) D. I. Brahmbhatt, J. M. Gajera, C. N. Patel, V. P. Pandya and U. R. Pandya, J. Heterocycl. Chem., 2006, 43, 1699–1702 CrossRef CAS.
- (a) N. Schuster, C. Christiansen, J. Jakupovic and M. Mungai, Phytochemistry, 1993, 34, 1179–1181 CrossRef CAS; (b) D. M. X. Donnelly and G. M. Boland, Nat. Prod. Rep., 1998, 15, 241–260 RSC.
- S. M. Wong, S. Antus, A. Gottsegen, B. Fessler, G. S. Rao, J. Sonnenbichler and H. Wagner, Arzneim.-Forsch., 1998, 38, 661–665 Search PubMed.
- A. L. Livingston, S. C. Witt, R. E. Lundin and E. M. Bickoff, J. Org. Chem., 1965, 30, 2353–2355 CrossRef.
- N. Rasool, A. Q. Khan, V. U. Ahmad and A. Malik, Phytochemistry, 1991, 30, 2800–2803 CrossRef.
- (a) E. M. Bickoff, A. N. Booth, R. L. Lyman, A. L. Livingston, C. R. Thompson and F. Deeds, Science, 1957, 126, 969–970 CrossRef; (b) A. Amin and M. Buratovich, Recent Pat. Anti-Cancer Drug Discovery, 2007, 2, 109–117 CrossRef.
- G. A. Kraus and N. Zhang, J. Org. Chem., 2000, 65, 5644–5646 CrossRef.
- R. H. D. Murray, Prog. Chem. Org. Nat. Prod., 1991, 58, 83–316 Search PubMed.
- A. L. Cabrera, Opera Lilloana, 1965, 13, 1–227 Search PubMed.
- (a) M. Bittner, J. Jakupovic, F. Bohlmann and M. Silva, Phytochemistry, 1989, 28, 2867–2868 CrossRef; (b) F. Bohlmann and C. Zdero, Chem. Ber., 1977, 110, 1755–1758 CrossRef.
- C. Zdero, F. Bohlmann and J. Solomon, Phytochemistry, 1988, 27, 891–897 CrossRef.
- F. Bohlmann and A. Steinmeyer, Tetrahedron Lett., 1986, 27, 5359–5362 CrossRef.
- M. Bittner, J. Jakupovic, F. Bohlmann, M. Grenz and M. Silva, Phytochemistry, 1988, 27, 3263–3266 CrossRef.
- C. Zdero, F. Bohlmann, R. M. King and H. Robinson, Phytochemistry, 1986, 25, 509–516 CrossRef.
- (a) A. Rustaiyan, L. Nazarians and F. Bohlmann, Phytochemistry, 1980, 19, 1254–1255 CrossRef; (b) G. Appendino, G. Cravotto, G. B. Giovenzana and G. Palmisano, J. Nat. Prod., 1999, 62, 1627–1631 CrossRef.
- (a) M. G. Valle, G. Appendino, G. M. Nano and V. Picci, Phytochemistry, 1987, 26, 253–256 CrossRef; (b) G. Appendino, S. Tagliapietra, G. M. Nano and V. Picci, Phytochemistry, 1988, 27, 944–946 CrossRef CAS; (c) G. Appendino, S. Tagliapietra, P. Ganboldi, G. M. Nano and V. Picci, Phytochemistry, 1988, 27, 3619–3624 CrossRef CAS.
- (a) D. Lamnaouer, B. Bodo, M. T. Martin and D. Mollo, Phytochemistry, 1987, 26, 1613–1615 CrossRef CAS; (b) J. De Pascual Teresa, M. A. Villaseco, J. M. Hernández, J. R. Morin, J. C. Urones and M. Grande, Planta Med., 1986, 52, 458–462 CrossRef.
- M. Miski and J. Jakupovic, Phytochemistry, 1990, 29, 1995–1998 CrossRef CAS.
- R. T. Gunther, The Greek Herbal of Dioscorides, The Classics of Medicine Library, New York, USA, 1996, p. 323 Search PubMed.
- (a) K. Kojima, K. Isaka, P. Ondognii, O. Zevgeegiin, P. Gombosurengyin, K. E. Davgiin, H. Mizukami and Y. Ogihara, Chem. Pharm. Bull., 2000, 48, 353–356 CrossRef CAS; (b) K. Isaka, A. Nagatsu, P. Ondognii, O. Zevgeegiin, P. Gombosurengyin, K. Davgiin, K. Kojima and Y. Ogihara, Chem. Pharm. Bull., 2001, 49, 1072–1076 CrossRef CAS; (c) C. Kahraman, G. Topcu, E. Bedir, I. I. Tatli, M. Ekizoglu and Z. S. Akdemir, Saudi Pharm. J., 2019, 27, 525–531 CrossRef.
- (a) A. Keige, B. Vogler, I. Klaiber and W. Kraus, Pharm. Pharmacol. Lett., 1998, 8, 43 CAS; (b) H. A. Oketch-Rabah, E. Lemmich, S. F. Dossaji, T. G. Theander, C. E. Olsen, C. Cornett, A. Kharazmi and S. B. Christensen, J. Nat. Prod., 1997, 60, 458–461 CrossRef CAS.
- D. A. Mulholland, S. E. Iourine, D. A. H. Taylor and F. M. Dean, Phytochemistry, 1998, 47, 1641–1644 CrossRef CAS.
- V. Stanjek, J. Piel and W. Boland, Phytochemistry, 1999, 50, 1141–1145 CrossRef CAS.
- (a) L. Margl, C. Ettenhuber, I. Gyurján, M. H. Zenk, A. Bacher and W. Eisenreich, Phytochemistry, 2005, 66, 887–899 CrossRef CAS; (b) F. Bourgaud, A. Hehn, R. Larbat, S. Doerper, E. Gontier, S. Kellner and U. Matern, Phytochem. Rev., 2006, 5, 293–308 CrossRef CAS; (c) U. Matern, P. Lüer and D. Kreusch, Biosynthesis of Coumarins, in Comprehensive Natural Products Chemistry, ed. D. Barton, K. Nakanishi and O. Meth-Cohn, Pergamon, New York, USA, 1999, vol. 1, pp. 623–637 Search PubMed.
- (a) W. Wang, Y.-Y. Zhao, H. Liang, Q. Jia and H.-B. Chen, J. Nat. Prod., 2006, 69, 876–880 CrossRef CAS; (b) F. Bohlmann and C. Zdero, Phytochemistry, 1977, 16, 1261–1263 CrossRef CAS; (c) M. Bittner, J. Jakupovic, M. Grenz and M. Silva, Phytochemistry, 1988, 27, 3263–3266 CrossRef CAS.
- (a) X.-H. Wang, K. Nakagawa-Goto, M. Kozuka, H. Tokuda, H. Nishino and K.-H. Lee, Pharm. Biol., 2006, 44, 116–120 CrossRef CAS; (b) L. Piccagli, M. Borgatti, E. Nicolis, N. Bianchi, I. Mancini, I. Lampronti, D. Vevaldi, F. Dall'Acqua, G. Cabrini and R. Gambari, Bioorg. Med. Chem., 2010, 18, 8341–8349 CrossRef CAS.
- (a) G. Feuer, in Progress in Medicinal Chemistry, ed. G. P. Ellis and G. B. West, North-Holland, New York, USA, 1974 Search PubMed; (b) A. A. Deans, J. Med. Chem., 1983, 26, 580–585 CrossRef; (c) E. Wenkert and B. L. Buckwalter, J. Am. Chem. Soc., 1972, 94, 4367–4369 CrossRef CAS.
- R. R. Zhang, R.-R. Zhang, J.-Q. Wang, X. Yu, Y.-L. Zhang, Q.-Q. Wang and W.-H. Zhang, Eur. J. Med. Chem., 2016, 124, 10–16 CrossRef.
- S. Bondock, W. Khalifa and A. A. Fadda, Eur. J. Med. Chem., 2011, 46, 2555–2561 CrossRef CAS.
- R. Zhang, Z. Xu, W. Yin, P. Liu and W. Zhang, Synth. Commun., 2014, 44, 3257–3263 CrossRef CAS.
- K. Kowalski, L. Szczupak, L. Oehninger, I. Ott, P. Hikisz, A. Koceva-Chyła and B. Therrien, J. Organomet. Chem., 2014, 772–773, 49–59 CrossRef CAS.
- (a) S. Caffieri, Photochem. Photobiol. Sci., 2002, 1, 149–157 RSC; (b) O. H. Hishmat, O. A. El-Shabrawy and H. I. El-Diwani, Arch. Pharmacal Res., 1988, 11, 87–92 CrossRef CAS.
- (a) X. H. Wang, K. F. Bastow, C. M. Sun, Y. L. Lin, H. J. Yu, M. J. Don, T. S. Wu, S. Nakamura and K. H. Lee, J. Med. Chem., 2004, 47, 5816–5819 CrossRef CAS; (b) H. W. Luo, J. Ji, M. Y. Wu and Z. G. Yong, Chem. Pharm. Bull., 1986, 34, 3166–3168 CrossRef CAS.
- Y. Xue, Y. W. H. Zhu, X.-N. Li, J.-F. Qian, Y. Lai, C. Chen, G. Yao, Z. Luo, Y. Li and Y. Zhang, Fitoterapia, 2014, 99, 204–210 CrossRef CAS.
- B. Li, Z. Ali, M. Chan, J. Li, M. Wang, N. Abe, C.-R. Wu, I. A. Khan, W. Wang and S.-X. Li, Phytochemistry, 2017, 137, 132–138 CrossRef CAS.
- (a) K. V. Sashidhara, J. N. Rosaiah, M. Kumar, R. K. Gara, L. V. Nayak, K. Srivastava, H. K. Bid and R. Konwar, Bioorg. Med. Chem. Lett., 2010, 20, 7127–7131 CrossRef CAS; (b) L. T. Wang, S. L. Pan, T. H. Chen, Y. Dong, K. H. Lee and C. M. Teng, ChemBioChem, 2012, 13, 1663–1672 CrossRef CAS.
- J. Soares, B. R. Keppler, X. Wang, K.-H. Lee and M. B. Jarstfer, Bioorg. Med. Chem. Lett., 2011, 21, 7474–7478 CrossRef CAS.
- X. Wang, K. Nakagawa-Goto, K. F. Bastow, M. J. Don, Y. L. Lin, T. S. Wu and K. H. Lee, J. Med. Chem., 2006, 49, 5631–5634 CrossRef CAS.
- Y. Dong, Q. Shi, H.-C. Pai, C.-Y. Peng, S.-L. Pan, C.-M. Teng, K. Nakagawa-Goto, D. Yu, Y.-N. Liu, P.-C. Wu, K. F. Bastow, S. L. Morris-Natschke, A. Brossi, J.-Y. Lang, J. L. Hsu, M.-C. Hung, E. Y.-H. P. Lee and K.-H. Lee, J. Med. Chem., 2010, 53, 2299–2308 CrossRef CAS.
- J. L. Pergomet, M. G. Di Liberto, M. G. Derita, A. B. J. Bracca and T. S. Kaufman, Fitoterapia, 2018, 125, 98–105 CrossRef CAS.
- S. Sardari, S. Nishibe and U. Daneshtalabi, Coumarins, the Bioactive Structures with Antifungal Property, in Studies in Natural Products Chemistry, ed. A.-u. Rahman, Elsevier, Amsterdam, The Netherlands, 2000, vol. 23, pp. 335–391 Search PubMed.
- (a) M. Wink, Phytochemistry, 2003, 64, 3–19 CrossRef CAS; (b) M. Wink, S. Afr. J. Bot., 2013, 89, 164–175 CrossRef CAS.
- L. Tao, M. Qing, S. Gibbons and X. Huiqin, Patent CN 103387582 (A), 2013.
- M. Rajabi, Z. Hossaini, M. A. Khalilzadeh, S. Datta, M. Halder and S. A. Mousa, J. Photochem. Photobiol., B, 2015, 148, 66–72 CrossRef CAS.
- S. M. Abd Elhalim and I. T. Ibrahim, Appl. Radiat. Isot., 2015, 95, 153–158 CrossRef CAS.
- K. E. Schulte, J. Reisch and G. L. Tittel, Arch. Pharm., 1966, 299, 457–468 CrossRef CAS.
- M. R. Saidi and K. Bigdeli, J. Chem. Res., 1998, 800–801 RSC.
- A. Patra, S. K. Panda, K. C. Majumdar, A. T. Khan and S. Saha, Magn. Reson. Chem., 1991, 28, 631–644 CrossRef.
- (a) S.-Y. Peng, L. Wang, J.-Y. Huang, S.-F. Sun, H.-B. Guo and J. Wang, Adv. Synth. Catal., 2013, 355, 2550–2553 CrossRef CAS; (b) Q. Zhu, J. Wu, R. Fathi and Z. Yang, Org. Lett., 2002, 4, 3333–3336 CrossRef CAS; (c) R.-M. Moriarty, S. Tyagi, D. Ivanov and M. Constantinescu, J. Am. Chem. Soc., 2008, 130, 7564–7565 CrossRef CAS.
- D. Zha, H. Li, S. Li and L. Wang, Adv. Synth. Catal., 2017, 359, 467–475 CrossRef CAS.
- (a) H. Li, P. Li, Q. Zhao and L. Wang, Chem. Commun., 2013, 49, 9170–9172 RSC; (b) H. Li, P. Li, H. Tan and L. Wang, Chem.–Eur. J., 2013, 19, 14432–14436 CrossRef CAS; (c) H. Tan, H. Li, W. Ji and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 8374–8377 CrossRef CAS; (d) H. Deng, H. Li and L. Wang, Org. Lett., 2015, 17, 2450–2453 CrossRef CAS; (e) H. Deng, H. Li and L. Wang, Org. Lett., 2016, 18, 3110–3113 CrossRef CAS.
- (a) M. B. Camacho, A. E. Clark, T. A. Liebrecht and J. P. DeLuca, J. Am. Chem. Soc., 2000, 122, 5210–5211 CrossRef CAS; (b) S.-Z. Zhu and Q.-Y. Chen, J. Chem. Soc., Chem. Commun., 1990, 1459–1460 RSC.
- (a) U. Letinois-Halbes, P. Pale and S. Berger, J. Org. Chem., 2005, 70, 9185–9180 CrossRef CAS; (b) C. Feng and T.-P. Loh, Chem. Commun., 2010, 46, 4779–4781 RSC.
- L. Chen and M.-H. Xu, Adv. Synth. Catal., 2009, 351, 2005–2012 CrossRef CAS.
- L. Chen, Y. Li and M.-H. Xu, Org. Biomol. Chem., 2010, 8, 3073–3077 RSC.
- (a) L. Zhang, T. Meng, R. Fan and J. Wu, J. Org. Chem., 2007, 72, 7279–7286 CrossRef CAS; (b) Z. Wang, B. Wang and J. Wu, J. Comb. Chem., 2007, 9, 811–817 CrossRef CAS.
- (a) K. Sonogashira, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. I. Negishi, Wiley, New York, USA, 2002, pp. 493–529 Search PubMed; (b) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS.
- (a) E. Negishi, Acc. Chem. Res., 1982, 15, 340–348 CrossRef CAS; (b) E. Negishi and C. Xu, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. I. Negishi, Wiley, New York, USA, 2002, pp. 531–549 Search PubMed; (c) E. I. Negishi, Bull. Chem. Soc. Jpn., 2007, 80, 233–257 CrossRef CAS.
- (a) C. Shen, L. Chen, J. Tang and M.-H. Xu, Chin. J. Chem., 2009, 27, 413–418 CrossRef CAS; (b) A. J. De Koning, P. E. Van Rijn, J. Boersma and G. J. M. van der Kerk, J. Organomet. Chem., 1979, 174, 129–140 CrossRef CAS.
- (a) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079–3160 CrossRef CAS; (b) N. T. Patil, L. M. Lutete, H. Wu, N. K. Pahadi, I. D. Gridnev and Y. Yamamoto, J. Org. Chem., 2006, 71, 4270–4279 CrossRef CAS; (c) S. Seo, X. Yu and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 263–276 CrossRef CAS.
- D. Conreaux, S. Belot, P. Desbordes, N. Monteiro and G. Balme, J. Org. Chem., 2008, 73, 8619–8622 CrossRef CAS.
- Y. Yamamoto, J. Org. Chem., 2007, 72, 7817–7831 CrossRef CAS.
- K. Shen, Y. Fu, J.-N. Li, L. Liu and Q.-X. Guo, Tetrahedron, 2007, 63, 1568–1576 CrossRef CAS.
- A. Dey and A. Hajra, Org. Biomol. Chem., 2017, 15, 8084–8090 RSC.
- S. Mishra, K. Monir, S. Mitra and A. Hajra, Org. Lett., 2014, 16, 6084–6087 CrossRef CAS.
- X. Chu, Z. Tang, J. Ma, L. He, L. Feng and C. Ma, Tetrahedron, 2018, 74, 970–974 CrossRef CAS.
- H. Jiang, Y. Cheng, Y. Zhang and S. Yu, Org. Lett., 2013, 15, 4884–4887 CrossRef CAS.
- H. Zhou, X. Deng, Z. Ma, A. Zhang, Q. Qin, R. X. Tan and S. Yu, Org. Biomol. Chem., 2016, 14, 6065–6070 RSC.
- A. Shao, X. Luo, C.-W. Chiang, M. Gao and A. Lei, Chem.–Eur. J., 2017, 23, 17874–17878 CrossRef CAS.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (b) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- (a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS; (b) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef CAS; (c) X. Sun and S. Yu, Chin. J. Org. Chem., 2016, 36, 239–247 CrossRef CAS.
- (a) Z. Li, J. Zhang, C. Brouwer, C.-G. Yang, N. W. Reich and C. He, Org. Lett., 2006, 8, 4175–4178 CrossRef CAS; (b) D. C. Rosenfeld, S. Shekhar, A. Takemiya, M. Utsunomiya and J. F. Hartwig, Org. Lett., 2006, 8, 4179–4182 CrossRef CAS.
- (a) G. Cheng and Y. Hu, Chem. Commun., 2007, 3285–3287 RSC; (b) G. Cheng and Y. Hu, J. Org. Chem., 2008, 73, 4732–4735 CrossRef CAS.
- Z. Wan, C. D. Jones, D. Mitchell, J. Y. Pu and T. Y. Zhang, J. Org. Chem., 2006, 71, 826–828 CrossRef CAS.
- (a) X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790–6791 CrossRef CAS; (b) R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, USA, 1981, pp. 7–8 Search PubMed; (c) Y. Zhang, H. Fu, Y. Jiang and Y. Zhao, Org. Lett., 2007, 9, 3813–3816 CrossRef CAS.
- V. O. Iaroshenko, S. Mkrtchyan, A. Gevorgyan, M. Miliutina, A. Villinger, D. Volochnyuk, V. Y. Sosnovskikh and P. Langer, Org. Biomol. Chem., 2012, 10, 890–894 RSC.
- J. Ghosh, P. Biswas, T. Sarkar and C. Bandyopadhyay, Tetrahedron Lett., 2015, 56, 7193–7196 CrossRef CAS.
- T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091–1160 CrossRef CAS.
- S. Tollari, G. Palmisano, S. Cenini, G. Cravotto, G. B. Giovenzana and A. Penoni, Synthesis, 2001, 735–739 CrossRef CAS.
- T. R. Hoye, C. J. Dinsmore, D. S. Johnson and P. F. Korkowski, J. Org. Chem., 1990, 55, 4518–4520 CrossRef CAS.
- Y. R. Lee and J. Y. Suk, Tetrahedron, 2002, 58, 2359–2367 CrossRef CAS.
- D. F. Taber, R. E. Ruckle Jr and M. J. Hennessy, J. Org. Chem., 1986, 51, 4077–4078 CrossRef CAS.
- K. C. Majumdar, A. T. Khan and D. P. Das, Synth. Commun., 1989, 19, 917–930 CrossRef CAS.
- H. Takahiro, M. Kazuyuki, K. Keiji and M. Ichiro, Tetrahedron Lett., 1973, 739–740 Search PubMed.
- R. Jagdish Kumar, G. L. D. Krupadanam and G. Srimannarayana, Synthesis, 1990, 535–538 CrossRef.
- V. Y. Sosnovskikh, Russ. Chem. Rev., 2003, 72, 489–516 CrossRef CAS.
- R. B. Gammill, S. A. Nash and S. A. Mizsak, Tetrahedron Lett., 1983, 24, 3435–3438 CrossRef CAS.
- R. B. Gammill, J. Org. Chem., 1979, 44, 3988–3990 CrossRef CAS.
- R. Uddin and A. Zaman, Indian J. Chem., 1995, 34B, 639–642 CAS.
- P. Biswas, J. Ghosh, T. Sarkar, S. Maiti and C. Bandyopadhyay, J. Chem. Res., 2012, 623–625 CrossRef CAS.
- S. Maiti, J. Ghosh, T. Sarkar and C. Bandyopadhyay, J. Indian Chem. Soc., 2013, 90, 1497–1499 CAS.
- K.-Z. Khan, N. Minhaj, K. Tasneen, A. Zaman, D. Shiengthong, F. M. Dean and M. Varma, J. Chem. Soc., Perkin Trans. 1, 1983, 841–849 RSC.
- K. C. Majumdar and P. K. Chowudhury, Heterocycles, 1991, 32, 73–78 CrossRef CAS.
- K. C. Majumdar, A. T. Khan and S. K. Chattopadhyay, Indian J. Chem., 1990, 29B, 483–485 CAS.
- K. C. Majumdar and T. Bhattacharyya, J. Chem. Res., 1997, 244–245 RSC.
- V. F. Traven, D. V. Kravtchenko, T. A. Chibisova, S. V. Shorshnev, R. Eliason and D. H. Wakefield, Heterocycl. Commun., 1996, 2, 345–354 CAS.
- N. A. Kondratova, O. N. Kazheva, G. G. Aleksandrov, A. N. Chekhlov, O. A. D'yachenko and V. F. Traven, Russ. Chem. Bull., 2011, 60, 1906–1916 CrossRef CAS.
- N. A. Kondratova, O. N. Kazheva, G. G. Aleksandrov, O. A. D'yachenko and V. F. Traven, Russ. Chem. Bull., 2009, 58, 1908–1914 CrossRef CAS.
- (a) I. O. Akchurin, A. I. Yakhutina, A. Y. Bochkov, N. P. Solovjova and V. F. Traven, Heterocycl. Commun., 2018, 24, 85–91 CAS; (b) I. O. Akchurin, A. I. Yakhutina, A. Y. Bochkov, N. P. Solovjova, M. G. Medvedev and V. F. Traven, J. Mol. Struct., 2018, 1160, 215–221 CrossRef CAS.
- W. Huang, C. Liu and Y. Gu, Adv. Synth. Catal., 2017, 359, 1811–1818 CrossRef CAS.
- (a) V. L. Heasley, K. E. Wade, T. G. Aucoin, D. E. Gipe and D. F. Shellhamer, J. Org. Chem., 1983, 48, 1377–1379 CrossRef CAS; (b) G. K. S. Prakash, T. Mathew, D. Hoole, P. M. Esteves, Q. Wang, G. Rasul and G. A. Olah, J. Am. Chem. Soc., 2004, 126, 15770–15776 CrossRef CAS; (c) K. Shibatomi, Y. Zhang and H. Yamamoto, Chem.–Asian J., 2008, 3, 1581–1584 CrossRef CAS.
- S. K. Bankar, J. Mathew and S. S. V. Ramasastry, Chem. Commun., 2016, 52, 5569–5572 RSC.
- (a) E. R. Palmacci, O. J. Plante and P. H. Seeberger, Eur. J. Org. Chem., 2002, 595–606 CrossRef CAS; (b) C. G. Nasveschuk and T. Rovis, Org. Biomol. Chem., 2008, 6, 240–254 RSC; (c) C. Wiebe, C. Schlemmer, S. Weck and T. Opatz, Chem. Commun., 2011, 47, 9212–9214 RSC; (d) X. H. Xu, M. Taniguchi, X. Wang, E. Tokunaga, T. Ozawa, H. Masuda and N. Norio Shibata, Angew. Chem., Int. Ed., 2013, 52, 1262–1265 CrossRef.
- W. Mayer, E. H. Hoffmann, N. Losch, H. Wolj, B. Wolter and G. Schining, Liebigs Ann. Chem., 1984, 929–938 CrossRef CAS.
- (a) M. Aso, M. Sakamoto, N. Urakawa and K. Kanematsu, Heterocycles, 1990, 31, 1003–1006 CrossRef CAS; (b) M. Aso, A. Ojida, G. Yang, O.-J. Cha, E. Osawa and K. Kanematsu, J. Org. Chem., 1993, 58, 3960–3968 CrossRef CAS.
- (a) D. I. Brahmbhatt, B. R. Hirani, S. U. Pandya and U. R. Pandya, Indian J. Chem., 2000, 39B, 233–235 CAS; (b) A. R. Kaneria, R. R. Giri, V. G. Bhila, H. J. Prajapati and D. I. Brahmbhatt, Arabian J. Chem., 2017, 10, S1100–S1104 CrossRef CAS.
- (a) S. Shivkumar and A. P. Bhaduri, Indian J. Chem., 1983, 22B, 725–726 Search PubMed; (b) S. Shivkumar and A. P. Bhaduri, Indian J. Chem., 1983, 22B, 914–915 Search PubMed.
- G. Kibriya, S. Samanta, M. Singsardar, S. Jana and A. Hajra, Eur. J. Org. Chem., 2017, 3055–3058 CrossRef CAS.
- D. Dauzonne, H. Josien and P. Demerseman, Tetrahedron, 1990, 46, 1359–1371 CrossRef.
- Y.-S. Chen, P.-Y. Kuo, T.-L. Shie and D.-Y. Yang, Tetrahedron, 2006, 62, 9410–9416 CrossRef CAS.
- (a) J.-W. Xie, P. Li, T. Wang and F.-T. Zhou, Tetrahedron Lett., 2011, 52, 2379–2382 CrossRef CAS; (b) L.-P. Fan, P. Li, X.-S. Li, D.-C. Xu, M.-M. Ge, W.-D. Zhu and J.-W. Xie, J. Org. Chem., 2010, 75, 8716–8719 CrossRef CAS.
- M. Rueping, A. Parra, U. Uria, F. Besselièvre and E. Merino, Org. Lett., 2010, 12, 5680–5683 CrossRef CAS.
- Z. Zhou, H. Liu, Y. Li, J. Liu, Y. Li, J. Liu, J. Yao and C. Wang, ACS Comb. Sci., 2013, 15, 363–369 CrossRef CAS.
- (a) J. Wu, Chem. Lett., 2006, 35, 118–119 CrossRef CAS; (b) M. Adib, M. Mahdavi, S. Bagherzadeh and H. R. Bijanzadeh, Synlett, 2009, 15, 2542–2544 CrossRef.
- I. J. Elenkov, B. Hrvačić, S. Marković, M. Mesić, A. Č. Klonkay, L. Lerman, A. F. Sučić, I. Vujasinović, B. Bošnjak, K. Brajša, D. Žiher, N. K. Hulita and I. Malnard, Croat. Chem. Acta, 2013, 86, 253–264 CrossRef CAS.
- M. Mercep, M. Mesic, B. Hrvacic, I. J. Elenkov, I. Malnar, S. Markovic, L. Simicic, A. Cempuh Klonkay and A. Filipovic, Patent WO 2005/010007 (A1), 2005.
- F. Risitano, G. Grassi, F. Foti and C. Bilardo, Tetrahedron Lett., 2001, 42, 3503–3505 CrossRef CAS.
- G. Bruno, F. Nicolo, A. Rotondo, F. Foti, F. Risitano, G. Grassi and C. Bilardo, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, C57, 493–494 CrossRef CAS.
- D. S. Wagare, D. Lingampalle, M. Farooqui and A. Durrani, Der Pharma Chem., 2016, 8, 408–411 CAS.
- M. He, Z. Yan, W. Wang, F. Zhu and S. Lin, Tetrahedron Lett., 2018, 59, 3706–3712 CrossRef CAS.
- O. Prakash, D. Wadhwa, K. Hussain and R. Kumar, Synth. Commun., 2012, 42, 2947–2951 CrossRef CAS.
- (a) O. Prakash, Aldrichimica Acta, 1995, 28, 63–71 CAS; (b) O. Prakash and S. P. Singh, Aldrichimica Acta, 1994, 27, 15–23 CAS; (c) O. Prakash and S. Goyal, Indian J. Heterocycl. Chem., 1991, 1, 99–103 CAS.
- G. J. Kharadi and K. D. Patel, Appl. Organomet. Chem., 2010, 24, 523–529 CrossRef CAS.
- E. M. Bickoff, A. L. Livingston and A. N. Booth, Arch. Biochem. Biophys., 1960, 88, 262–266 CrossRef CAS.
- (a) M. Trokovnik, R. Djudjic, I. Jabakovic and M. Kules, Org. Prep. Proced. Int., 1982, 14, 21–29 CrossRef; (b) R. P. Singh and D. Singh, Heterocycles, 1985, 23, 903–907 CrossRef CAS; (c) B. Rajitha, Y. Geetanjali and V. V. Somayajulu, Indian J. Chem., 1986, 25B, 872–873 CAS.
- A. Sehemi, G. Abdullah, E. Gogary and R. Sameh, Chin. J. Chem., 2012, 30, 316–320 CrossRef.
- G. O. Schenck, Naturwissenschaften, 1948, 35, 28–29 CrossRef CAS.
- M. Zhang, R. Zhang, J. Wang, X. Yu, Y. Zhang, Q. Wang and W. Zhang, Chin. J. Chem., 2016, 34, 1344–1352 CrossRef CAS.
- M.-Z. Zhang, Y. Zhang, J.-Q. Wang and W.-H. Zhang, Molecules, 2016, 21, E1387 CrossRef.
- (a) I. A. Os'kina, A. Y. Tikhonov, I. Y. Bagryanskaya, Y. V. Gatilov and O. S. Fedorova, Russ. J. Org. Chem., 2013, 49, 1497–1501 CrossRef; (b) I. A. Os'kina, Y. V. Gatilov and A. Y. Tikhonov, Russ. J. Org. Chem., 2011, 47, 1441–1443 CrossRef.
- V. K. Ahluwalia, R. Adhikari and R. P. Singh, Heterocycles, 1986, 24, 1919–1923 CrossRef CAS.
- E. Altieri, M. Cordaro, G. Grassi, F. Risitano and A. Scala, Tetrahedron, 2010, 66, 9493–9496 CrossRef CAS.
- V. K. Ahluwalia, R. Adhikari and R. P. Singh, Synth. Commun., 1985, 15B, 1191–1196 CrossRef.
- W.-Y. Huang, Y.-C. Chen and K. Chen, Chem.–Asian J., 2012, 7, 688–691 CrossRef CAS.
- (a) I. Deb, P. Shanbhag, S. M. Mobin and I. N. N. Namboothiri, Eur. J. Org. Chem., 2009, 4091–4101 CrossRef CAS; (b) H.-H. Kuan, R. J. Reddy and K. Chen, Tetrahedron, 2010, 66, 9875–9879 CrossRef CAS; (c) C.-L. Cao, Y.-Y. Zhou, J. Zhou, X.-L. Sun, Y. Tang, Y.-X. Li, G.-Y. Li and J. Sun, Chem.–Eur. J., 2009, 15, 11384–11389 CrossRef CAS.
- J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734–736 RSC.
- X.-C. Tan, H.-Y. Zhao, Y.-M. Pan, N. Wu, H.-S. Wang and Z.-F. Chen, RSC Adv., 2015, 5, 4972–4975 RSC.
- U. Sharma, T. Naveen, A. Maji, S. Manna and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 12669–12673 CrossRef CAS.
- (a) G. Appendino, G. Cravotto, G. Palmisano and R. Annunziata, Synth. Commun., 1996, 26, 3359–3371 CrossRef CAS; (b) M. Yilmaz, M. Yakut and A. T. Pekel, Synth. Commun., 2008, 38, 914–927 CrossRef CAS.
- K. Kobayahi, K. Sakashita, H. Akamatsu, K. Tanaka, M. Uchida, T. Uneda, T. Kitamura, O. Moriwaka and H. Konishi, Heterocycles, 1999, 51, 2881–2891 CrossRef.
- (a) V. Nair and J. Matthew, J. Chem. Soc., Perkin Trans. 1, 1995, 187–188 RSC; (b) V. Nair and J. Matthew, J. Chem. Soc., Perkin Trans. 1, 1995, 1881–1882 RSC; (c) V. Nair and J. Matthew, J. Chem. Soc., Perkin Trans. 1, 1996, 1487–1492 RSC; (d) S. C. Roy and P. K. Mandal, Tetrahedron, 1996, 52, 2193–2198 CrossRef CAS.
- (a) Y. R. Lee, N. S. Kim and B. S. Kim, Tetrahedron Lett., 1997, 38, 5671–5674 CrossRef CAS; (b) B. B. Snider, L. Han and C. Xie, J. Org. Chem., 1997, 62, 6978–6984 CrossRef CAS; (c) T. Yamada, Y. Iwahara, H. Nishino and K. Kurosawa, J. Chem. Soc., Perkin Trans. 1, 1995, 609–616 Search PubMed.
- J. Iqbal, B. Bhatia and N. K. Nayyar, Chem. Rev., 1994, 94, 519–564 CrossRef CAS.
- Y. R. Lee, M. W. Byun and B. S. Kim, Bull. Korean Chem. Soc., 1998, 19, 1080–1083 CAS.
- B. Labiad and D. Villemin, Synthesis, 1989, 143–144 CrossRef CAS.
- Y. R. Lee and B. S. Kim, Tetrahedron Lett., 1997, 38, 2095–2098 CrossRef CAS.
- Y. R. Lee, J. Y. Suk and B. S. Kim, Org. Lett., 2000, 2, 1387–1389 CrossRef CAS.
- (a) Y. G. Lee and B. S. Kim, Tetrahedron Lett., 1997, 38, 2095–2098 CrossRef CAS; (b) Y. R. Lee, B. S. Kim and H. C. Wang, Tetrahedron, 1998, 54, 12215–12222 CrossRef CAS.
- H. Suginome and S. Yamada, J. Org. Chem., 1984, 49, 3753–3762 CrossRef CAS.
- H. Suginome, C. F. Liu, S. Seko and K. Kobayashi, J. Org. Chem., 1988, 53, 5952–5959 CrossRef CAS.
- G. Raffa, M. Rusch, G. Balme and N. Monteiro, Org. Lett., 2009, 11, 5254–5257 CrossRef CAS.
- G. Le Bras, C. Radanyi, J.-F. Peyrat, J.-D. Brion, M. Alami, V. Marsaud, B. Stella and J.-M. Renoir, J. Med. Chem., 2007, 50, 6189–6200 CrossRef CAS.
- E. Bossharth, P. Desbordes, N. Monteiro and G. Balme, Org. Lett., 2003, 5, 2441–2444 CrossRef CAS.
- (a) T. J. Barton and B. L. Groh, J. Org. Chem., 1985, 50, 158–166 CrossRef CAS; (b) S. Shiotani and H. Morita, J. Heterocycl. Chem., 1992, 29, 413–422 CrossRef CAS; (c) G. W. Gribble and M. G. Saulnier, J. Org. Chem., 1983, 48, 607–609 CrossRef CAS; (d) T. Frejd, J. O. Karlsson and S. Gronowitz, J. Org. Chem., 1981, 46, 3132–3135 CrossRef CAS.
- (a) K. J. James and M. F. Grundon, J. Chem. Soc., Perkin Trans. 1, 1979, 1467–1471 RSC; (b) I. Butenschön, K. Möller and W. Hänsel, J. Med. Chem., 2001, 44, 1249–1256 CrossRef; (c) G. Bar, A. F. Parsons and C. B. Thomas, Tetrahedron, 2001, 57, 4719–4728 CrossRef CAS; (d) M. C. Pirrung and F. Blume, J. Org. Chem., 1999, 64, 3642–3649 CrossRef CAS; (e) K. C. Majumdar, P. K. Choudhury and M. Nethaji, Tetrahedron Lett., 1994, 35, 5927–5930 CrossRef CAS.
- W. Huang, J. Wang, Q. Shen and X. Zhou, Tetrahedron, 2007, 63, 11636–11643 CrossRef CAS.
- V. Cadierno, J. Gimeno and N. Nebra, Adv. Synth. Catal., 2007, 349, 382–394 CrossRef CAS.
- V. Cadierno, J. Dıez, J. Gimeno and N. Nebra, J. Org. Chem., 2008, 73, 5852–5858 CrossRef CAS.
- (a) R. Sanz, A. Martínez, J. M. Alvarez-Gutiérrez and F. Rodríguez, Eur. J. Org. Chem., 2006, 1383–1386 CrossRef CAS; (b) R. Sanz, D. Miguel, A. Martínez, J. M. Alvarez-Gutiérrez and F. Rodríguez, Org. Lett., 2007, 9, 727–730 CrossRef CAS; (c) R. Sanz, A. Martínez, D. Miguel, J. M. Alvarez-Gutiérrez and F. Rodríguez, Synthesis, 2007, 3252–3256 CrossRef CAS; (d) J. S. Yadav, B. V. S. Reddy, T. S. Rao, B. B. M. Krishna and G. G. K. S. N. Kumar, Chem. Lett., 2007, 36, 1472–1473 CrossRef CAS.
- Y. Nishibayashi, M. Yoshikawa, Y. Inada, M. Hidai and S. Uemura, J. Org. Chem., 2004, 69, 3408–3412 CrossRef CAS.
- (a) B. Seiller, C. Bruneau and P. H. Dixneuf, Tetrahedron, 1995, 51, 13089–13102 CrossRef CAS; (b) B. Gabriele, G. Salerno and E. Lauria, J. Org. Chem., 1999, 64, 7687–7692 CrossRef CAS; (c) N. Nebra, A. E. Díaz-Álvarez, J. Díez and V. Cadierno, Molecules, 2011, 16, 6470–6480 CrossRef CAS.
- E. Bustelo, M. Jiménez-Tenorio, M. C. Puerta and P. Valerga, Organometallics, 2007, 26, 4300–4309 CrossRef CAS.
- X. X.-Y. Zhang, L.-L. Hu, Z. Shen, Z.-Z. Chen, Z.-G. Xu, S.-Q. Li, J.-W. Xie and H.-L. Cui, Synlett, 2015, 26, 2821–2825 CrossRef CAS.
- C. Uchiyama, Y. Miyadera, Y. Hayashi and F. Yakushiji, ChemistrySelect, 2017, 2, 3794–3798 CrossRef CAS.
- M. A. Khalilzadeh, Z. Hossaini, F. R. Charati, S. Hallajian and M. Rajabi, Mol. Diversity, 2011, 15, 445–450 CrossRef CAS.
- (a) S. Xu and Y. Tang, Lett. Org. Chem., 2014, 11, 524–533 CrossRef CAS; (b) Z. Wang, X. Xu and O. Kwon, Chem. Soc. Rev., 2014, 43, 2927–2940 RSC; (c) Y. Lu and B. A. Arndtsen, Org. Lett., 2009, 11, 1369–1372 CrossRef CAS; (d) U. Das, Y.-L. Tsai and W. Lin, Org. Biomol. Chem., 2014, 12, 4044–4050 RSC.
- (a) C.-J. Lee, Y.-J. Jang, Z.-Z. Wu and W. Lin, Org. Lett., 2012, 14, 1906–1909 CrossRef CAS; (b) S.-M. Yang, C.-Y. Wang, C.-K. Lin, P. Karanam, G. M. Reddy, Y.-L. Tsai and W. Lin, Angew. Chem., Int. Ed., 2018, 57, 1668–1672 CrossRef CAS.
- C.-J. Lee, C.-C. Tsai, S.-H. Hong, G.-H. Chang, M.-C. Yang, L. Möhlmann and W. Lin, Angew. Chem., Int. Ed., 2015, 54, 8502–8505 CrossRef CAS.
- J. Li, W. Kong, Y. Yu, C. Fu and S. Ma, J. Org. Chem., 2009, 74, 8733–8738 CrossRef CAS.
- S. M. Rajesh, S. Perumal, J. C. Menéndez, S. Pandian and R. Murugesan, Tetrahedron, 2012, 68, 5631–5636 CrossRef CAS.
- (a) H. Sun, D. Zhang, F. Wang and C. Liu, J. Phys. Chem. A, 2007, 111, 4535–4541 CrossRef CAS; (b) L. D. S. Yadav, S. Singh and V. K. Rai, Tetrahedron Lett., 2009, 50, 2208–2212 CrossRef CAS; (c) B. Ganem, Acc. Chem. Res., 2009, 42, 463–472 CrossRef CAS.
- S. Soleimani-Amiri, M. Arabkhazaeli, Z. Hossaini, S. Afrashteh and A. A. Eslamibis, J. Heterocycl. Chem., 2017, 55, 209–2013 CrossRef.
- (a) X. Chang, X. Zhang and Z. Chen, Org. Biomol. Chem., 2018, 16, 4279–4287 RSC; (b) X. Chang, P. Zeng and Z. Chen, Eur. J. Org. Chem., 2019, 6478–6485 CrossRef CAS.
- A. Olyaei, M. Saraei and R. Khoeiniha, Synlett, 2018, 29, 1589–1592 CrossRef CAS.
- R. Vatanchian, M. H. Mosslemin, M. Tabatabaee and A. Sheibani, J. Chem. Res., 2018, 42, 439–443 CrossRef CAS.
- W. E. Noland, H. V. Kumar, A. Sharma, B. Wei and S. Girmachew, Org. Lett., 2020, 22, 1801–1806 CrossRef CAS.
- S. Kolita, P. Borah, P. S. Naidu and P. J. Bhuyan, Tetrahedron, 2016, 72, 532–538 CrossRef CAS.
- T. A. Fattah, A. Saeed, Y. M. Al-Hiari, V. Kasabri, I. M. Almasri, S. AlAlawi, F. A. Larik and P. A. Channar, J. Mol. Struct., 2019, 1179, 390–400 CrossRef CAS.
- (a) J. M. Khurana and S. Kumar, Tetrahedron Lett., 2009, 50, 4125–4127 CrossRef CAS; (b) B. Karmakar, A. Nayak and J. Banerji, Tetrahedron Lett., 2012, 53, 4343–4346 CrossRef CAS; (c) N. Hamdi, M. C. Puerta and P. Valerga, Eur. J. Med. Chem., 2008, 43, 2541–2548 CrossRef CAS; (d) W. Li, Y. Wang, Z. Wang, L. Dai and Y. Wang, Catal. Lett., 2011, 141, 1651–1658 CrossRef CAS; (e) Z. Siddiqui and F. Farooq, Catal. Sci. Technol., 2011, 1, 810–816 RSC.
- M. Kidwai, V. Bansal, P. Mothsra, S. Saxena, R. K. Somvanshi, S. Dey and T. P. Singh, J. Mol. Catal. A: Chem., 2007, 268, 76–81 CrossRef CAS.
- (a) V. J. Ram and N. Agarwal, Tetrahedron Lett., 2002, 43, 4769–4771 CrossRef CAS; (b) A. K. Sinha, A. Sharma and P. B. Joshi, Tetrahedron, 2007, 63, 960–965 CrossRef CAS; (c) J. Hutchinson, G. Sandford and J. F. S. Vaughan, Tetrahedron, 1998, 54, 2867–2876 CrossRef CAS; (d) E. Bellur, I. Freifeld and P. Langer, Tetrahedron Lett., 2005, 46, 2185–2187 CrossRef CAS; (e) F. A. Davis, K. A. Bowen, H. Xu and V. Velvadapu, Tetrahedron, 2008, 64, 4174–4182 CrossRef CAS.
- W. L. Zhang, S. N. Yue, Y. M. Shen, H. Y. Hu, Q.-H. Meng, H. Wu and Y. Liu, Org. Biomol. Chem., 2015, 13, 3602–3609 RSC.
- (a) A. de la Hoz, A. Diaz-Ortiz, M. M. del Carmen, M. Moral, A. Moreno, J. Elguero, C. Foces, M. L. Rodriguez and A. Sanchez-Migallon, Tetrahedron, 2006, 62, 5868–5874 CrossRef CAS; (b) S. Haneda, A. Okui, C. Ueba and M. Hayashi, Tetrahedron, 2007, 63, 2414–2417 CrossRef CAS; (c) S. L. Zhu, S. J. Ji, X. M. Su, C. Sun and Y. Liu, Tetrahedron Lett., 2008, 49, 1777–1781 CrossRef CAS.
- Z. Zareai, M. Khoobi, A. Ramazani, A. Foroumadi, A. Souldozi, K. Slepokura, T. Lis and A. Shafiee, Tetrahedron, 2012, 68, 6721–6726 CrossRef CAS.
- (a) V. V. Kouznetsov, D. R. Merchan Arenas and A. R. Romero Bohorquez, Tetrahedron Lett., 2008, 49, 3097–3100 CrossRef CAS; (b) E. Colacino, L. Villebrun, J. Martínez and F. Lamaty, Tetrahedron, 2010, 66, 3730–3735 CrossRef CAS.
- P. C. Moews Jr and R. H. Petrucci, J. Chem. Educ., 1964, 41, 549–551 CrossRef.
- Y. R. Lee, J. Y. Suk and B. S. Kim, Tetrahedron Lett., 1999, 40, 6603–6607 CrossRef CAS.
- J. L. Pergomet, A. B. J. Bracca and T. S. Kaufman, Org. Biomol. Chem., 2017, 15, 7040–7049 RSC.
- N. Takeuchi, M. Murase, K. Ochi and S. Tobinaga, Chem. Pharm. Bull., 1980, 28, 3013–3019 CrossRef CAS.
- J. Padwal, W. Lewis and C. J. Moody, Org. Biomol. Chem., 2011, 9, 3484–3493 RSC.
- (a) T. V. Hansen and L. Skattebøl, Org. Synth., 2012, 89, 220–229 CrossRef CAS; (b) Ø. W. Akselsen, L. Skattebøl and T. V. Hansen, Tetrahedron Lett., 2009, 50, 6339–6341 CrossRef.
- (a) N. U. Hofsløkken and L. Skattebøl, Acta Chem. Scand., 1999, 53, 258–262 CrossRef; (b) R. Aldred, R. Johnston, D. Levin and J. Neilan, J. Chem. Soc., Perkin Trans. 1, 1994, 1823–1831 RSC; (c) S. Rizzo, F. Sannicolo, T. Benincori, G. Schiavon, S. Zecchin and G. Zotti, J. Mater. Chem., 2004, 14, 1804–1811 RSC.
- D. A. Heredia, E. L. Larghi and T. S. Kaufman, Eur. J. Org. Chem., 2016, 1397–1404 CrossRef CAS.
- C. K. Ghosh, S. Bhattacharyya, N. Ghoshal and B. Achari, J. Chem. Res., 1998, 178–179 RSC.
- (a) B. A. Keay and R. Rodrigo, J. Am. Chem. Soc., 1982, 104, 4725–4727 CrossRef CAS; (b) W. Lin, L. Yuan and X. Cao, Tetrahedron Lett., 2008, 49, 6585–6588 CrossRef CAS; (c) S. Wang, Z. Wang, Y. Yin, J. Luo and L. Kong, J. Photochem. Photobiol., A, 2017, 333, 213–219 CrossRef CAS.
- (a) R. Ballini, R. Rabanedo Clemente, A. Palmieri and M. Petrini, Adv. Synth. Catal., 2006, 348, 191–196 CrossRef CAS; (b) S.-J. Won, C.-T. Liu, L.-T. Tsao, J.-R. Weng, H.-H. Ko, J.-P. Wang and C.-N. Lin, Eur. J. Med. Chem., 2005, 40, 103–112 CrossRef CAS.
- J. Wu, D. Zhang and S. Wei, Synth. Commun., 2006, 35, 1213–1222 CrossRef.
- (a) Y. Dong, Q. Shi, Y.-N. Liu, X. Wang, K. F. Bastow and K.-H. Lee, J. Med. Chem., 2009, 52, 3586–3590 CrossRef CAS; (b) Y. Dong, Q. Shi, K. Nakagawa-Goto, P.-C. Wu, K. F. Bastow, S. L. Morris-Natschke and K.-H. Lee, Bioorg. Med. Chem. Lett., 2009, 19, 6289–6292 CrossRef CAS; (c) B. Guoqiang, Y. Lijuan, H. Xianli, L. Ziyu, P. Shujia, D. Yanming and W. Hao, Patent CN 106243071 (A), 2016.
- H. Abe, T. Kawai, Y. Komatsu, M. Kamimura, Y. Takeuchi and Y. Horino, Heterocycles, 2012, 86, 785–789 CrossRef CAS.
- (a) C. Reynaud, H. Doucet and M. Santelli, Synthesis, 2010, 1787–1792 CAS; (b) I. Beltaïef, R. Besbes, H. Amri and J. Villiéras, Tetrahedron Lett., 1997, 38, 813–814 CrossRef.
- S. W. Pelletier, Z. Djarmati, S. D. Lajšić, I. V. Mićović and D. T. C. Yang, Tetrahedron, 1975, 31, 1659–1665 CrossRef CAS.
- (a) K. Kolodziejczyk, G. D. Roiban, M. Schnürch, M. D. Mihovilovic and P. Stanetty, Monatsh. Chem., 2009, 140, 1349–1359 CrossRef CAS; (b) C. D. Buttery, A. G. Cameron, C. P. Dell and D. W. Knight, J. Chem. Soc., Perkin Trans. 1, 1990, 1601–1610 RSC.
- K. Ghosh, R. Karmakar and D. Mal, Eur. J. Org. Chem., 2013, 4037–4046 CrossRef CAS.
- (a) L. F. Lindoy, G. V. Meehan and N. Svenstrup, Synthesis, 1998, 1029–1032 CrossRef CAS; (b) O. Kundrat, H. Dvorakova, I. Cisarova, M. Pojarova and P. Lhotak, Org. Lett., 2009, 11, 4188–4191 CrossRef CAS.
- R. Martin and A. Furstner, Angew. Chem., Int. Ed., 2004, 43, 3955–3957 CrossRef CAS.
- R. W. Carling and P. D. Leeson, Synlett, 1993, 40 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |