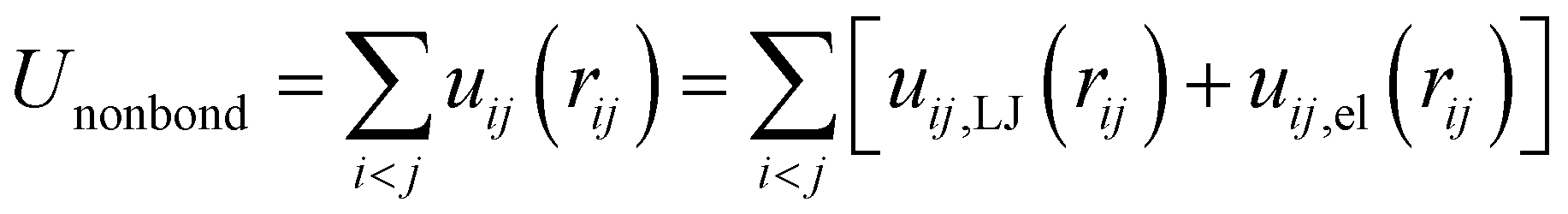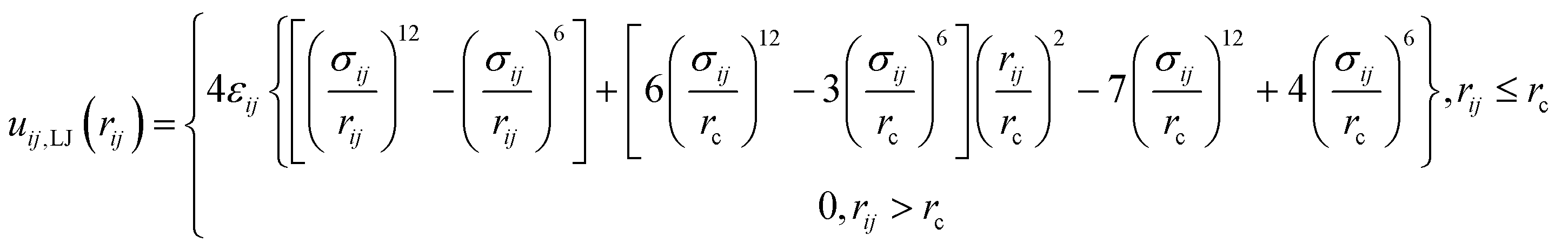DOI:
10.1039/D0RA06894B
(Paper)
RSC Adv., 2020,
10, 39204-39216
Structural behavior of amphiphilic polyion complexes interacting with saturated lipid membranes investigated by coarse-grained molecular dynamic simulations†
Received
10th August 2020
, Accepted 16th October 2020
First published on 26th October 2020
Abstract
Neutral polyelectrolyte complexes (PECs) made from an amphiphilic multiblock copolymer of type (AnBn)m and an oppositely charged polyion and interacting with a dipalmitoylphosphatidylcholine (DPPC) lipid membrane have been examined employing a coarse-grained model with implicit solvent and molecular dynamics simulations. One systematically explored the influence of the size of the hydrophobic block B and of the number of these blocks per chain on the PEC tendency to adhere to the membrane surface and to intercalate into the membrane core. Simulation results showed that PECs bound irreversibly to the lipid bilayer without polyion unwinding from the complex and the adsorbed conformation was strongly affected by the size of the hydrophobic block B. The adsorption kinetics at low B size were characterized by a relaxation phase dominated by the spreading of PEC constituents along the outer leaflet of the membrane. Upon increasing the size of the hydrophobic block B to reach core–shell organization of the free PEC, the relaxation pathway of the complex corona in close contact with the headgroup lipids facilitated the transient exposure of the PEC hydrophobic core to the lipids and its subsequent cooperative internalization and solubilization in the membrane inner part associated with an internal reorganization of the lipid bilayer. In the generated snorkeling-type conformation, the charged blocks A and the oppositely charged polyion were confined to the headgroup region of the top leaflet, without spontaneous flipping to the headgroup region of the distal leaflet.
1. Introduction
Interactions of complexes formed from oppositely charged polymers, referred to as polyelectrolyte complexes (PEC) or polyplexes, with the fluid lipid membranes have attracted considerable interest, much of it deriving from the potential role played by PECs in biological applications such as cell adhesion1 and gene therapy.2,3 Synthetic carriers able to form stable PECs with nucleic acids and to deliver functional genes to a targeted cell are regarded as an alternative to the viral-like vectors because, despite a poorer delivery efficacy provided, they lacked the immune response characteristic of the viral vectors.4 Engineering of synthetic polymeric vectors requires great efforts to rationally design complexes able to overcome the cellular barrier in the absence of the innate machinery provided by the viral-like vectors. In this context, several kinds of vectors for genome delivery have been developed employing strongly charged quaternary ammonium- or vinylpyridine-based polyions,5 as well as of weak cationic polymers such as poly(L-lysine), polysaccharides and poly(ethyleneimine)s.6 The interaction between the membrane and these cationic vectors was proposed to be important for understanding the cellular uptake.7 The experimental data hypothesized that the capacity of the cationic part of the PECs to interact directly with the anionic moiety of lipids played a crucial role in the membrane internalization process.8 It was reported that the polyamine-based complexes could be rapidly encapsulated by the vesicles, process that was accompanied by a partial release of the polyion together with the discharge of the genome.9 At the same time, different polyamines including poly-L-lysine, poly(amidoamine) and PEI could permeabilize supported lipid bilayers and even cell plasma membranes.10 The cationic PEC bound to the membrane could induce new defects or turning pre-existing defects in nanoscale holes in supported lipid bilayers. By contrast to charged PECs, a much larger concentration of neutral PECs was required to induce such defects, and the polymer could not only intercalate in the hydrophobic tail region but also spread on top of the bilayer.11 Besides these interactions involving the cationic vectors, the presence of the genome in the membrane-bound PEC induced alteration in the molecular order of the bilayer since systematic studies of the complexation of DNA with liposomes provided evidence for intricate interactions.12,13 On one hand, the surface-bound genome stabilized the membrane through electrostatic interactions with the polar headgroup, and on the other hand, when DNA penetrated the lipid hydrophobic part of mixed membrane, the results led to microdomain lipid segregation and lipid long-range molecular order.14,15
It also turned out that an improvement in the transfection efficiency associated with a lowered cytotoxicity was provided by the block copolymerization of the cationic vectors with poly(ethyleneglycol)s16,17 or carbohydrates.18,19 Besides, an increase of the copolymer surface activity and the ability to penetrate through the membrane was reached in the case of DMAEMA – poly(dimethylaminoethyl methacrylate) – copolymer by varying the hydrophilicity/hydrophobicity balance.20 The transfection properties were also highly dependent on the morphology and structural properties of the block copolymer vector since the grafting of poly(ethylene glycol) chains on PDMAEMA (methacrylate/methacrylamide)-based cationic polymers resulted in a decreased uptake at the copolymer conformational change from a core–shell appearance to a bottle-brush type.21 When the architecture of methacrylate-based copolymers was varied, the statistical copolymers proved to be not as effective as the block copolymers for the genome compaction and cellular uptake.22 Studies conducted on the block and statistical copolymers consisting of 2-(α-D-mannopyranosyloxy)ethyl methacrylate (ManEMA) and 2-(N,N-dimethylamino)ethyl methacrylate (DMAEMA) of similar composition revealed a higher transfection efficiency for the latter copolymer although both architectures displayed similar genome condensing ability.23
The molecular dynamics (MD) simulation, a powerful tool to investigate the structures and mechanisms in biological systems at spatial and temporal resolutions which are hardly provided by the experiments, has been utilized to shed light on the mechanisms assisting the binding to lipid membranes and the subsequent intercalation and penetration of the biopolymers and related complexes. The computational studies, carried out on all-atom and coarse-grained models dealt mainly with linear homo- or amphiphilic block copolymers either free or grafted on inorganic nanoparticles24–28 whereas structural studies of interpolyelectrolyte complexes at the lipid bilayer surface were scarcely reported.29 It was thus revealed that a not cross-linked polystyrene blob adsorbed on a zwitterionic membrane and entered, upon a retention time, the membrane on a time scale of a few microseconds.24,30 Notably, the permeation in the hydrophobic core was followed by the blob dissolution, both processes, that rendered a softened membrane, were slower with increasing the blob size.24,30 In the case of linear phenylene ethynylene polyelectrolyte oligomers, the adsorption on a mixed bilayer containing both zwitterionic DOPE and negatively charged DOPG phospholipids indicated the chains could bind to the membrane surface through electrostatic interactions with the oligomers occasionally inserted deeply in the membrane without providing support destabilization.31 The adsorption of the hydrophobically modified poly(allyl-N,N-dimethyl-N-hexylammonium chloride) polycation on a zwitterionic membrane led to a significant perturbation of the bilayer structure concluding with the pore formation.32 The ability of the polymers to form pores in the membrane could be dramatically affected by the amount of the hydrophobic side chains substituted to strong polycations.33 The relationship between the copolymer composition and the binding behavior to a zwitterionic membrane was investigated for PEO-PPO-PEO Pluronics block-copolymers. The insertion of the triblock copolymers into the lipid bilayer depended on the overall copolymer hydrophobicity, the PEO moiety of the copolymer was localized mainly in the lipid headgroup region, while the less polar part PPO was found in the acyl chain region or, for longer PPO moiety, in the central hydrophobic region of the bilayer.34,35 Besides, the effect of the copolymer insertion on the mechanical properties of the membrane, depended on the hydrophobicity. An increased hydrophobic moiety decreased the lateral pressure required for the bilayer rupture and rendered the membrane more susceptible to mechanical stress.36 According to atomistic and coarse-grained molecular dynamics simulations of the inorganic particles protected via amphiphilic ligands,25,37–43 these complexes could stably adhere to zwitterionic lipid bilayers and attained eventually the so-called “snorkeling” conformation, characterized by the nanoparticles embedded in the hydrophobic bilayer core and the charged ligands anchored to the charged moiety of both bilayer leaflets. The complex bound initially to the bilayer via electrostatics, and the transition from this state to the snorkeling configuration proceeded through several distinct metastable configurations.39 Furthermore, this transition was encountered to occur spontaneously on flat membranes only by the coarse-grained simulations25 and on highly curved membranes for the all-atom models.38 The computational investigations evidenced a slow relaxation mode of the adsorbing block-copolymers on the entrance leaflet of the membrane and that the subsequent penetration across the membrane was characterized by intermediate states whose lifetime could reach several microseconds.25 The substantial computational cost incurred by the insertion pathway is expected to be circumvented by using coarse-grained systems with implicit solvent, representations that were developed for the lipid membranes and that enabled a speedup of at least three orders of magnitude compared to all-atom simulations while retaining the structural and dynamical accuracy of the all-atom lipid bilayer.44–47 In recent work, the author provided structural insights into the binding of non-stoichiometric polyelectrolyte complexes consisting of homopolymeric and oppositely charged polyions to a saturated lipid bilayer employing an implicit solvent coarse grained model.29 The adhesion to lipid bilayer was characterized by a wide variety in the shapes of the adsorbed complexes, that ranged from oblate discs to prolate spheroids and allowed a gathering of the charge excess of the complex at the bilayer surface. This paper employs the previously developed and validated implicit solvent representation for the DPPC bilayer to address, for the first time to the best of our knowledge, the possible insertion pathways developed by amphiphilic PECs. The effect of hydrophobicity on the internalization process is studied for neutrally charged complexes consisting of a linear polyion and an oppositely charged multiblock copolymer (AnBn)m whose charged moiety A alternated with the chain section B hydrophobic in nature. It is found that the occurrence of the spontaneous embedding in the lipid membrane is promoted by the hydrophobic block length n at the constant composition of the multiblock copolymer n × m and impeded by the number of diblock units m at constant hydrophobic block length n. It is further remarked that, contrary to the insertion of ligand-coated inorganic nanoparticles, the cooperative intercalation in the membrane core is triggered by the slow spreading of the charged moieties of the complex along the entrance leaflet of the membrane.
2. Model system and simulation details
The examination of the amphiphilic polyelectrolyte complex interacting with a lipid bilayer relies on a coarse-grained model with implicit solvent representation. The bilayer patch is composed of 2048 zwitterionic DPPC lipids per leaflet and each molecule is mapped onto a structure consisting of four types of beads as shown in Fig. 1a. Q0 and Qa spheres are the coarse-grained representation of the choline and phosphate entities, two Na beads stand for the glycerol backbone and four C beads represent each of the two alkyl chains. The systematic investigations of the structural, dynamical and mechanical properties of this dry and coarse-grained DPPC bilayer are given in detail in ref. 29. Briefly, an appropriate underlying interaction matrix has been derived by comparing and matching the key properties such as area per lipid, bending modulus, bilayer thickness, orientation order parameter and internal pressure distribution of the dry lipid bilayer with those reported from the simulations of a similar lipid patch in the presence of a coarse-grained solvent. As to the PEC constituents, they are designed as linear chains of beads connected via harmonic bonds. One thus considers a homopolymeric polyion containing NPc positively charged beads and an oppositely charged multiblock copolymer of the type 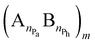 represented by the number of beads nPa and nPh in each block and the number of repeating diblock units, m. The former two numbers define the length of negatively charged A and neutral B sequences so that the total bead number of the multiblock copolymer is given by Nac = m(nPa + nPh) (see Fig. 1). At constant copolymer length Nac = 150 and the fraction of A to B block of
represented by the number of beads nPa and nPh in each block and the number of repeating diblock units, m. The former two numbers define the length of negatively charged A and neutral B sequences so that the total bead number of the multiblock copolymer is given by Nac = m(nPa + nPh) (see Fig. 1). At constant copolymer length Nac = 150 and the fraction of A to B block of  the number of copolymer blocks m varied from 150, i.e. an alternating copolymer, to 10, which corresponds to A and B sequences of 15 beads. Besides, two larger copolymers with m = 27, nPh = 15, and the fraction of A to B block of 1.0 and 0.46, respectively, were considered. To be noted that the stoichiometric charge ratio within the PECs is held to one for the present investigation. Table 1 summarizes the compositions and the labels employed by PECs whose adsorption on the lipid membrane is addressed. In short, e.g. 5
the number of copolymer blocks m varied from 150, i.e. an alternating copolymer, to 10, which corresponds to A and B sequences of 15 beads. Besides, two larger copolymers with m = 27, nPh = 15, and the fraction of A to B block of 1.0 and 0.46, respectively, were considered. To be noted that the stoichiometric charge ratio within the PECs is held to one for the present investigation. Table 1 summarizes the compositions and the labels employed by PECs whose adsorption on the lipid membrane is addressed. In short, e.g. 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:30, refers to a PEC made by a polycation of 150 beads and an amphiphilic multiblock copolymer with 30 alternating anionic and hydrophobic blocks, each one containing 5 beads.
:5:30, refers to a PEC made by a polycation of 150 beads and an amphiphilic multiblock copolymer with 30 alternating anionic and hydrophobic blocks, each one containing 5 beads.
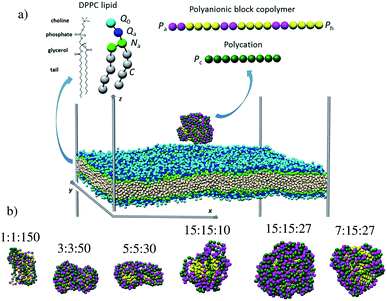 |
| | Fig. 1 Snapshots of (a) the simulation box containing a lipid bilayer and a polyelectrolyte complex at 3 nm separation distance and (b) the free PECs (the sizes are not shown on the same scale). The chemical structure and coarse-grained representation of the DPPC molecule and the oppositely charged polyions are sketched at the top. The colored spheres denote the choline Q0 (light blue), phosphate Qa (dark blue), glycerol Na (green), alkyl C (white), anionic Pa (cyan), Ph hydrophobic (yellow) and Pc cationic (dark green) beads. | |
Table 1 Investigated polyelectrolyte complexes
| PEC |
Polyanionic block copolymer |
Polycation |
| Number of beads per block |
Number of diblock units |
Bead number |
Bead number |
| Charged (A) |
Hydrophobic (B) |
| nPa |
nPh |
m |
NPa |
NPh |
NPc |
| 1:1:150 |
1 |
1 |
150 |
150 |
150 |
150 |
| 3:3:50 |
3 |
3 |
50 |
150 |
150 |
150 |
| 5:5:30 |
5 |
5 |
30 |
150 |
150 |
150 |
| 15:15:10 |
15 |
15 |
10 |
150 |
150 |
150 |
| 15:15:27 |
15 |
15 |
27 |
405 |
405 |
405 |
| 7:15:27 |
7 |
15 |
27 |
189 |
405 |
189 |
The total potential energy U of each of the investigated systems, either free PEC, bare membrane or PEC attached to the lipid bilayer, is expressed as a sum of three contributions as follow
| | |
Utot = Unonbond + Ubond + Uang
| (1) |
The first term represents the nonbonded potential energy and is given by
| |
 | (2) |
where the sum extends over all pairs of particles (polymer and lipid beads) and
uij,LJ and
uij,el are the short-range Lennard-Jones and electrostatic contributions of the interaction between beads
i and
j separated by the distance
rij. The lipids are modeled according to the following shifted form of the LJ potentials
| |
 | (3) |
where
εij and
σij stand for the interaction strength and the effective minimum separation, with the interaction being cut off at the truncated distance
rc = 1.2 nm. The functional form of the pair potential in
eqn (3), which removes the discontinuity in the potential and force at
rc, was previously considered for coarse-grain modeling of membranes to avoid the potential artifacts in the lipid motion.
48 The parameter values for all possible interaction pairs involving the lipids beads are shown in
Table 2, and the detailed description of the dry coarse-grained DPPC bilayer is found in
ref. 29. As to B blocks of the amphiphilic multiblock copolymer, they are considered as alkyl chains coarse-grained in a similar manner as the lipid tails, whereas the interaction between oppositely charged beads of the PEC (linear polyion and A blocks of the multiblock copolymer) implied a pure repulsive LJ description. Thus, the interplay between the hydrophilic interaction and the electrostatic coupling leading to the internal organization of the free PECs in the implicit solvent is emphasized. The following shifted and truncated form for the interaction between the polycation and the charged A sequences of the multiblock copolymer is considered
| |
 | (4) |
with the values of
ε and
σ given in
Table 2.
Table 2 Values of non-bonded parameter ε (kJ mol−1)
| |
C |
Na |
Qa |
Q0 |
Pa |
Ph |
Pc |
| The interaction distance σ = 0.47 nm for C–C interactions and 0.50 nm otherwise. |
| C |
3.7a |
0.8 |
0.8 |
0.8 |
0.8 |
3.7 |
0.8 |
| Na |
|
1.8 |
0.8 |
0.8 |
0.8 |
1.72 |
0.8 |
| Qa |
|
|
0.8 |
0.8 |
0.8 |
0.8 |
0.8 |
| Q0 |
|
|
|
0.8 |
0.8 |
0.8 |
0.8 |
| Pa |
|
|
|
|
0.8 |
0.8 |
0.8 |
| Ph |
|
|
|
|
|
3.7 |
0.8 |
| Pc |
|
|
|
|
|
|
0.8 |
The electrostatic contribution to the first term of eqn (1) is given by
| |
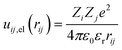 | (5) |
with
Zi and
Zj the valences of interacting beads – the monovalent Q
a, Q
0 beads of lipid and P
a, P
c beads of PEC–,
ε0 vacuum permittivity,
εr water permittivity, and the electrostatic cutoff distance is
rc,el = 1.5 nm. The present model system implied a relative permittivity of 78.4, the value utilized by the previous implicit model systems describing the structure and phase behavior of oppositely charged macromolecules
49–51 and the adsorption kinetics of polyions onto responsive surfaces.
52 The implicit solvent based on the dielectric constant of the pure water described also accurately the phase behavior of colloidal systems and the ion–ion correlation effect at an enhanced electrostatic coupling.
53,54 Notably, the dry Martini parametrization of the lipid membranes previously reported in
ref. 44 adopted a lower relative permittivity of 15 and thus the LJ values summarized in
Table 2 and whose determination was described in detail in the previous work
29 differ in some aspects from the dry Martini model.
The second term in eqn (1) stands for the bonding potential connecting two beads and is given by
| |
 | (6) |
Ω
ij,l = 1 if beads
i and
j of type l – lipid, polyion or amphiphilic multiblock copolymer – are linked, otherwise 0, and
r0,l is the equilibrium separation (0.5 nm) and
kbond,l the force constant (5.4 N m
−1 for the lipid and 2.4 N m
−1 for the linear polyions).
The third term in eqn (1) stands for the angular potential for the lipid flexibility and is given as
| |
 | (7) |
where
Nang is the number of angles formed by three connected lipid beads,
kang is the angular force constant (2.46 J mol
−1 deg
−2), and α
0 the equilibrium angle (100° for N
a–N
a–Q
a and 180° otherwise). The linear polyions were described as fully flexible chains.
The simulations were carried out in two steps. First, the homopolymer and amphiphilic multiblock copolymer were allowed to self-assemble as an equilibrated PEC by performing Monte Carlo and molecular dynamics and comparing the structural characteristics of the complexes provided by the two types of simulations to assess the ergodicity of the system. Thereafter, the PEC was placed in the simulation box containing a pre-equilibrated membrane centered at z = 0, with the shortest distance between the PEC and the outer leaflet of the membrane set at 3 nm, as shown in Fig. 1a. The simulations were carried out using the integrated Monte Carlo/molecular dynamics/Brownian dynamics simulation package MOLSIM.55
The box had the dimensions 37.2 × 37.2 × 60.0 nm and periodic boundary conditions were applied in all directions. The molecular dynamics simulations were conducted on in NVT ensemble for the bare PEC and NPT ensemble for the DPPC bilayer containing systems. A semi-isotropic pressure coupling associated with low compressibility was applied for the implicit solvent treatment of periodic lipid membranes.44 The molecular dynamics simulations were carried out using the velocity Verlet algorithm with a time-step of 20 fs, the neighbor list was updated every 20 steps and the neighbor list cutoff was 1.8 nm. The temperature was maintained at 323 K using a Berendsen thermostat and a time constant of 50 fs for controlling the temperature drift. The lipid bilayer was held tensionless by setting the reference pressure in the bilayer plane to zero and the compressibility along the axis of the bilayer plane to 5 × 10−5 bar−1. The latter was controlled using a Berendsen barostat and a time constant of 10 fs.
To determine whether PEC is bound to the membrane, the shortest distances ds between the pairs involving the oppositely charged beads belonging to PEC and bilayer respectively, are calculated. Two beads are considered to be in contact whenever the distance between them, ds, was shorter than 1.0 nm, a value two times larger than the minimum bead–bead separation. The number of contacts established is normalized to the total number of beads of each type to compare the degree of binding of different PECs. To assess the degree of the membrane internalization, the contact of PEC moieties with the glycol and alkyl groups of lipid is also assessed.
The PEC structural changes upon binding to the DPPC bilayer is quantified by the perpendicular and parallel components of the radius of gyration of the PEC projected on the membrane plane and are given by
| |
 | (8) |
| |
 | (9) |
where
N =
m(
nPa +
nPh) +
NPc and
xcom,
ycom and
zcom are the coordinates of the center of mass of the PEC.
The structural characterization of the lipid bilayer is given by the area per lipid A and the orientational bond order parameter S2. Since the lipid bilayer is made of a single lipid type, the former parameter is assessed by dividing the total area in the xy-plane of the bilayer to the number of lipids in each leaflet, whereas S2 is calculated for each lipid bond according to
| |
 | (10) |
with
θ is the angle made by the direction of the bond vector and normal of the bilayer surface. The order parameter for the alkyl bonds is averaged over the two lipid tails.
3. Results and discussion
3.1 Configurational behavior of free PECs
Representative equilibrium configurations of the free complexes are shown in Fig. 1b. The visual inspection indicates collapsed conformations for all PECs, a feature consistent with the previous simulations implying the complexation at the stoichiometric charge ratio of oppositely charged homo- and block copolymers.51,56,57 It is also noted that the structure adopted by the multiblock copolymer is affected locally by the length of the hydrophobic B blocks. Thus, the alternating copolymers led to an intertwined conformation, whereas the blocks with a length of at least 3 beads self-assembled in hydrophobic domains surrounded by patches made by the charged polyion strands. Further insight into the PEC structure is gained from the radial number density of the hydrophilic and hydrophobic blocks ρPa and ρPh calculated with respect to the center of mass of PEC (Fig. S1†). One sees that the functional form of ρPa and ρPh overlaps for the complex containing the alternating copolymer (1:1:150) implying the presence of both types of beads at the center of the mass. Poor segregation is found for 3:3:50 and 5:5:30 complexes as the hydrophobic beads are excluded from the very center of the complex in the former case whereas the density of the hydrophobic beads is increased in the inner region for the latter complex. However, Fig. 1b suggested a non-spherical appearance of the two complexes, implying that the bead radial analysis is not sufficiently meaningful for these complexes. By contrast, when the complexes reached a spherical shape, i.e. nPh = 15, the radial segregation of the hydrophobic moiety is evidently. Here one sees that the hydrophobic blocks lying at r < 2 nm are surrounded by a corona enriched in charged beads, and the snapshots indicate a compact shell at 15:15:27 and voids in the corona at decreasing either the length of the charged block (7:15:27) or the degree of polymerization (15:15:10).
3.2. PEC–DPPC bilayer interaction
3.2.1 Overview of the PEC binding and subsequent membrane penetration. Snapshots revealing the time evolution of the PEC conformation adsorbed to the lipid bilayer are presented in Fig. 2, 3 and S2.† It is seen that PECs are all engaged in close contact with the membrane, and they are initially located at the membrane interface. This feature is maintained only for 1:1:150 in the long run, whereas the rest of PECs containing the increasing length of the hydrophobic block could find, with a certain time lag, a path to cross the headgroup region of the bilayer and penetrate the membrane core. As the simulations proceed further, the degree of spontaneous insertion seems to increase progressively with nPh. At the same time, the charged beads of the complex remain constrained to the lipid head group region so that the PECs with nPh = 15 undergo a structural transition from a core–shell to a snorkeling configuration. Notably, the resulting substantial internal reorganization of PEC does not imply a partial unwinding of the homopolymeric polyion. Furthermore, the intercalation of the present PECs does not infer the appearance of local membrane defects ultimately, the bilayer remaining rather flat or exhibiting wide undulations. It is worth mentioning finally that the onset of hydrophobic block penetration of the PECs with m = 27 corroborates an important and local inward deformation of the distal leaflet rather than a bending of the outer leaflet towards the hydrophobic moiety of the PEC (see Fig. 3 at 105 ns and 45.4 ns). The dragging of the opposite lipids toward the membrane center has been recently reported by atomistic and metadynamics simulations of a ligand coated nanoparticle attempting the transition from the outer to the distal leaflet of the membrane.39,41
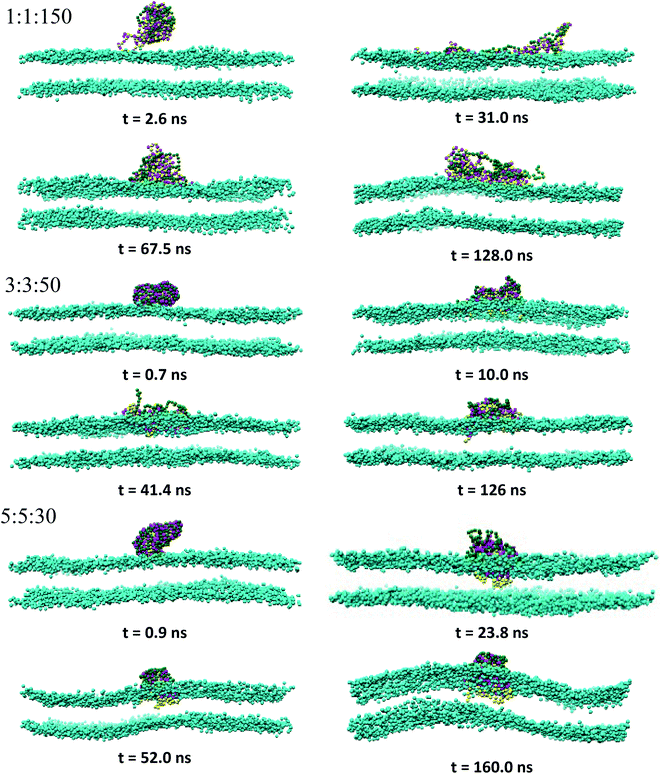 |
| | Fig. 2 Time sequence of representative morphologies (side view) of indicated PECs binding and internalizing to the DPPC bilayer; the corresponding top views are displayed in Fig. S2† and the color code is the same as in Fig. 1, with the choline beads of DPPC displayed only for sake of clarity. | |
 |
| | Fig. 3 Same as Fig. 2 for indicated PECs. | |
3.2.2. Structural characterization of the adhered PECs. To get insight into the mechanisms driving the attachment of the amphiphilic PECs, the behavior of the bound PEC was analyzed in terms of electrostatically favorable contacts established between the PEC and lipid headgroups, that is nPa–Q0/NPa and nPc–Qa/NPc (cf. Fig. 1a). Fig. 4 displays a semi-log plot of the temporal evolution of these parameters, with PEC being placed initially at 3 nm separation from the top leaflet. It is first noted that the charged beads of PEC become quickly engaged in the electrostatic interaction with the headgroup of the bilayer as they make the first contact to the top leaflet within the first 2 ns. The fluctuation in the arrival time is much lower than the following time required for reaching the equilibrated configuration in the adsorbed state. The PEC binding is driven by the electrostatic attraction of the multiblock A to the lipid choline groups since nPa–Q0/NPa exceeded nPc–Qa/NPc. The weakened electrostatic role played by the phosphate groups in the complex binding originates from the sterically and electrostatically repulsion involving the homopolymeric polyion and choline beads. As to the long time behavior of the nPa–Q0/NPa and nPc–Qa/NPc, constant values denoting certain equilibration is reached shortly after the PEC-bilayer contact for 1:1:150 and 3:3:50. For the next PEC, i.e. 5:5:30, despite a poorer initial contact with the top leaflet, the values of the two parameters increase steadily thereafter, with a more significant gain reached by nPc–Qa/NPc. In the case of the remaining complexes exhibiting a core–shell structure, the behavior of the two parameters suggests a two-step binding behavior: a weak attachment to the top leaflet followed by a delayed and sharp increase in the number of contacts, with the longest lag found for 15:15:27. It should be also outlined that there is only a poor influence of the membrane on the electrostatic cohesiveness within the complex despite their close interaction. The number of electrostatic contacts within the complex given in Fig. S3† indicates that the decrease of the number of favorable contacts is significant only for the PEC containing the alternating copolymer and becomes negligible for the adsorbing of the core-corona complexes.
 |
| | Fig. 4 Semi-log plot for the time-evolution of the normalised number of contacts between the anionic bead of indicated PEC and choline bead of DPPC nPa–Q0/NPa and the cationic bead of PEC and phosphate bead of DPPC nPc–Qa/NPc. | |
To further analyze the conformational changes associated with the hydrophobic bead insertion beyond the membrane surface suggested by the snapshot examination, one analyzes the contact number of these beads with the choline nPh–Q0/NPh and alkyl chain nPh–C/NPh (Fig. 5). The contact behavior found at 1:1:150 and 3:3:50 adds support to the finding of the PEC residence at the headgroup interface. For nPh = 5, the values of the two parameters are rather similar and follow the time evolution encountered by the contacts of the complex charged beads (Fig. 4), indicating thus slow kinetics for the intercalation of the hydrophobic moiety of the complex in the tail region of the membrane. At the increased size of the hydrophobic block nPh = 15, the complex with core–shell organization spent some time at the membrane surface before a steep increase of the hydrophobic contact with the membrane core. Together, the findings drew from Fig. 4 and 5 strongly indicate that the onset of enhanced electrostatic binding of the complex to the top leaflet relates to the cooperative membrane internalization of the hydrophobic blocks of PEC. Besides, the membrane penetration occurs with a time delay that increased in the sequence 15:15:10, 7:15:27, 15:15:27. Note that the former two complexes exhibited an incomplete charged corona in the free state (cf. Fig. 1b), rendering thus a faster presence of the hydrophobic core in the headgroup region of the membrane. The influence of the patched corona on the mechanism of the membrane internalization was also reported for the ligand-coated nanoparticles.58 Coarse grained simulations of gold nanoparticles functionalized with a mixture of anionic and hydrophobic ligand showed a difference in the internalization of the random and striped coated nanoparticles by the lipid bilayer.39 However, the present voids in the corona made by the oppositely charged polyions seem to facilitate the hydrophobic contact between the corona and the membrane, whereas the patched ligand stripes on the metallic nanoparticles slowed down or impeded their embedding into the membrane inner region as compared to the case of randomly patched ligands.
 |
| | Fig. 5 Semi-log plot for the time-evolution of the normalised number of contacts between hydrophobic beads of indicated PECs and DPPC (a) phosphate nPh–Qa/NPh and (b) alkyl chain nPh–C/NPh; the cross and star symbols indicate the onset of the binding and core penetration process, respectively. | |
The PEC intercalation to the membrane inner region is associated with a core–shell to snorkeling transition driven not only by the hydrophobically favorable contacts between the PEC and the entrance leaflet but also by the increased electrostatic cohesiveness between the corresponding charged moieties. This assumption is supported by the net charge of the complex at the membrane interface, defined as nPa–Q0 − nPc–Qa and that increased following also the sequence 15:15:10, 7:15:27, 15:15:27 (Fig. S4†). The fact the PEC internalization in the lipid core originates from both electrostatically and hydrophobically favorable contacts is in line with the lack of the membrane internalization of 1:1:150 complex, although its net charge at the interface is rather similar to that encountered at 15:15:10. Note that the hydrophobic contact triggering the PEC immersion into the membrane is not associated with the protrusion of the lipid tails in the headgroup region, which is an immersion mechanism reported for the embedding of anionic-ligand protected metallic nanoparticles in model lipid membranes.25 Taken together, these results indicate that there is a minimum length of the hydrophobic blocks leading to the spontaneous insertion, and the charged blocks regulate the preceded intermediate conformation in the adsorption state.
Next one estimates the entrance to distal membrane leaflet, which is assessed from the number of contacts established by the hydrophobic beads of PECs and the alkyl and glycerol moieties in the bottom layer. Fig. 6 shows the time evolution of the two types of the number of contacts for the PECs with nPh = 15. One sees that the membrane intercalation is not restricted to the top layer, the distant layer being also explored extensively. The fact that nPh–Na is low for all investigated complexes indicates that the hydrophobic blocks of the amphiphilic copolymers are confined to the tail region despite the significant presence in the distal leaflet. The intercalation in the distal layer takes place cooperatively and besides, it is delayed as compared to the penetration of the top layer interior at 15:15:27 and 7:15:27. By contrast, at 15:15:10, the hydrophobic block copolymers seem to spread almost simultaneously through both lipid monolayers. Nonetheless, the fast immersion step is correlated with a local and shallow maximum in nPh–Na, in line with the local inward deformation of the distal leaflet shown in Fig. 3. Regarding the contacts of the charged moieties of the complex with the distal layer, they were not observed although the snorkel configuration enabled interactions of these moieties with the lipid tails (Fig. S5†). Thus, the charged beads of the complex are exclusively confined in the headgroup region of the entrance layer, without spontaneous flipping to the headgroup region of the distal leaflet. This finding is in contrast with the previously coarse-grained computational investigations of linearly highly charged polyethyleneimine (PEI) interacting with a zwitterionic membrane, as PEI turned out to be capable of entreating the bilayer core and reducing the membrane thickness.59 The lack of concerted flip-flop events entailing charged moieties was observed in unbiased simulation studies examining the membrane penetration by amphiphilic ligand coated nanoparticles,41 whereas the spontaneous transfer from the upper to the lower leaflet was reported for multiple charged arginine molecules.60 It was also claimed that the charged ligand transfer across the membrane core and the subsequent membrane-spanning configuration was a rare event difficult to sample with coarse-grained simulations, and which was more appropriately evidenced using biasing simulation techniques25,42 or asymmetric charged membranes.40
 |
| | Fig. 6 Number of contacts between hydrophobic beads of indicated PECs and alkyl chain nPh–C and glycerol beads NPh–Na of DPPC molecules belonging to the distal leaflet; the star symbol stands for the onset of the membrane core internalization. | |
More insights into the conformational changes undergone upon complex binding and intercalation in the bilayer inner region can be gained from the radius of gyration of the complex projected perpendicular and parallel to the bilayer surface Rgxy,c Rgz,c (Fig. 7). Considering first the PECs experiencing membrane docking only, one sees a spreading of the complex mainly parallel to the membrane surface. Large fluctuations in the size of the PEC complex were noted for the PEC containing the alternating copolymers, in line with the behavior previously reported in the absence of the hydrophobic beads.29 The fact that the 1:1:150 complex initially spreads, then retracts and spreads again is reflected by the wide variation of the number of contacts between the PEC and the membrane headgroups (cf. Fig. 4) and it arises likely from a poor hydrophobic interaction within the complex. The in-plane extension characterizing the slow relaxation mode seems to decrease slightly with the increase of nPh for the complexes that remained mainly atop the membrane, whereas the trend is reversed for the internalizing PECs. Besides, the main relaxation phase of the latter complexes is associated with decreasing of Rgz,c, that entails the core–shell to snorkeling transition occurring at the PEC cooperative internalization in the membrane core. To further understanding how the block copolymer sequence influences the PEC capability to intercalate in the lipid bilayer, the density profiles of the lipid and PEC beads across the membrane were evaluated. The data obtained from the time average over the last 20 ns time interval are shown in Fig. 8 and they confirm that all charged groups of the complex mostly stay along with the bilayer heads, with weak segregation between the Pa and Pc groups owing to the stronger electrostatic coupling of the former with the cholines of lipid (cf. Fig. 4). Apart from the alternating copolymer case, the hydrophobic beads were able otherwise to intercalate beyond the headgroup region. The bimodal distribution found at 3:3:50 and 5:5:50 indicates shallow insertions, whereas at nPh = 15 the hydrophobic block length is fully integrated inside the hydrophobic core of the bilayer, in agreement with the contact number and snapshot analysis. The corresponding maximum in the density profile is close to the center of the membrane for 15:15:10 and 7:15:27, denoting that both leaflets are equally involved in the solubilization of the hydrophobic block of PEC. On the other hand, the density profiles of the PEC moieties are slightly shifted towards the outer lipid layer at 15:15:27, which supports the assumption that, besides the hydrophobic interaction as a primary driver for the PEC insertion, the favorable electrostatics stabilizes additionally the snorkeling configuration. These findings revealed by the radial density profiles are consistent with the results provided by the simulations carried out on coarse-grained wet systems containing amphiphilic block copolymers of Pluronic type, either free or in micellised form, interacting with a saturated lipid bilayer. For these systems, the degree of interpenetration depended on the hydrophilic–hydrophobic balance of the copolymer, with the more polar moiety of the copolymer localized mainly in the lipid headgroup region in all cases, whereas only the long enough less polar parts were able to be inserted in the tail region.34,36
 |
| | Fig. 7 Radius of gyration of indicated PECs projected onto the bilayer plane Rgxy,c and bilayer normal Rgz,c; cross, star and hash symbols indicate the onset for the binding, core penetration on the top and distal layer of the membrane. | |
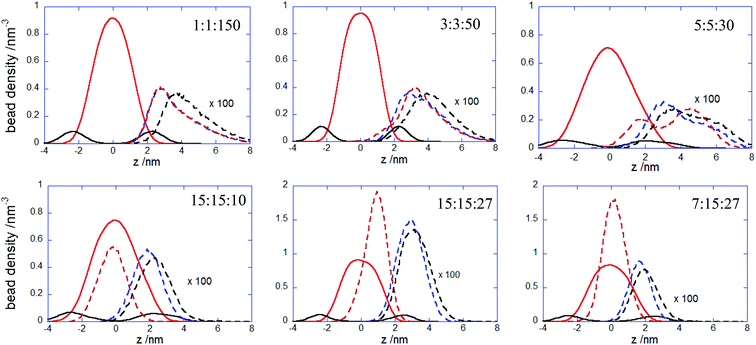 |
| | Fig. 8 Bead density profiles of polyanionic blocks (dashed blue curve), hydrophobic blocks (dashed red curve), polycation chain (dashed black curve) of the indicated PECs and for the choline (continuous black curve) and alkyl beads (continuous red curve) with respect to the lipid bilayer center z = 0; densities were obtained from the last 20 ns simulation trajectories. | |
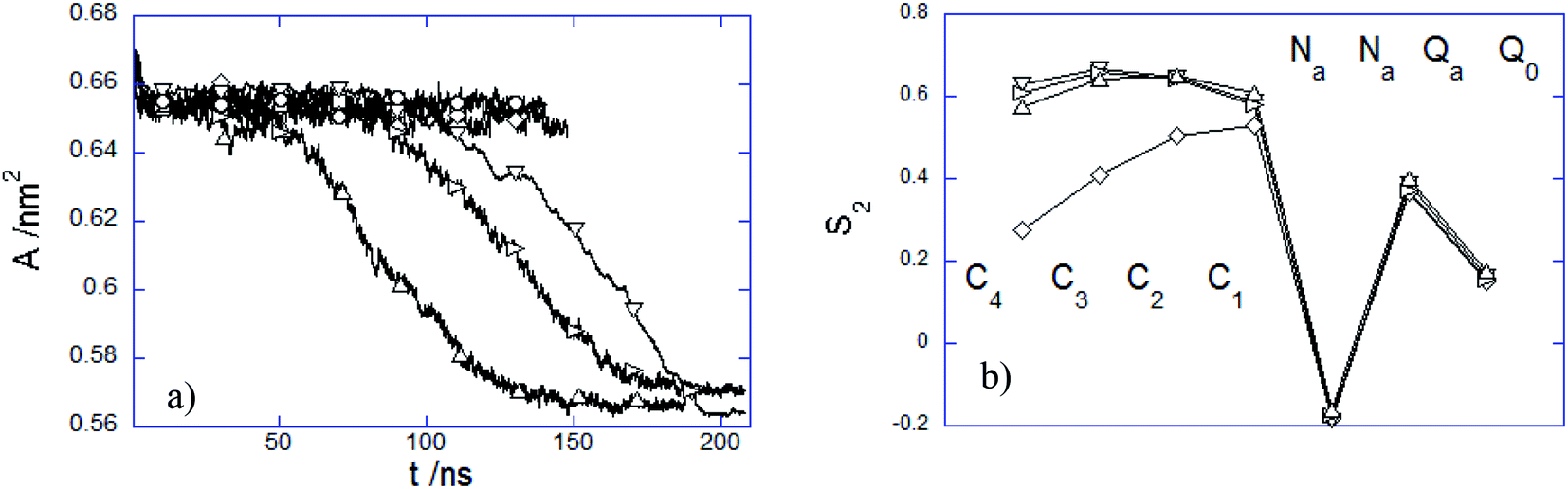
3.2.3 Influence of the PEC internalization on the membrane structure. The presence of the adhered or immersed PECs altered the membrane structure in several ways. The structural variation of the lipid bilayer was characterized by the time evolution of the area per lipid A(t) and the time-averaged lipid order parameter S2 (Fig. 9). It is noted first that A decreases weakly at the initial attachment of the PEC to the top membrane layer, and thereafter it levels off rapidly during the slow relaxation underwent by the complex laying at the membrane interface. This can be understood by the fact the repulsion among the choline headgroups is partially screened out as a result of the electrostatic favorable contacts nPa–Q0 that do not vary significantly as long as PEC resides atop the headgroup region. Under this circumstance, the effect of the PEC binding is not reflected by changes in the order parameter S2, relative to either alkyl chain or headgroup region (Fig. 9b). In stark contrast, the cooperative insertion of the hydrophobic moiety of PEC in the tail region leads to a steady decrease of the area per lipid associated with tighter lipid packing as suggested by the final values reached by the lipid order parameter (Fig. 9b). The resulting high degree of order in the tail region infers a gel phase, which originates likely from the difference in the interaction distance parameter σ assigned to the beads representing either the block B of the amphiphilic copolymer or the alkyl chain. The structural parameter analysis indicated that the present parametrization of the dry coarse-grained DPPC bilayer was prone to a fluid-to-gel transition upon absorbing overcharged complexes or at a slight increase of the C bead size.29 One can thus infer that the altered membrane structure arises from the more favorable hydrophobic and electrostatic contacts established between the membrane and PEC in the snorkeling conformation. The modified packing in the lipid patch upon the PEC embedding agrees with the significant perturbations of the zwitterionic membranes already observed in the presence of hydrophobically modified strong polycations.32 Furthermore, the fact that the block copolymer intercalation within the membrane plays an important in the membrane structural perturbation is in line with the previous studies of membranes interacting with amphiphilic linear or dendritic copolymers.59,61
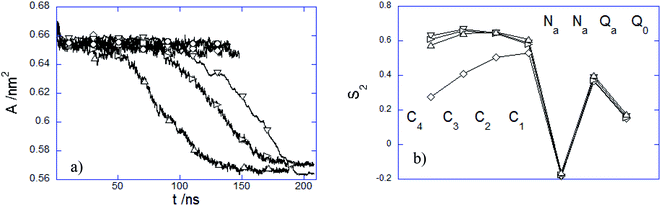 |
| | Fig. 9 (a) Time dependence of area per lipid A and (b) averaged S2 order parameter over the last 20 ns time interval of DPPC bilayers interacting with (○) 1:1:150, (□) 3:3:50, (◇) 5:5:30, (△) 15:15:10, (▽) 15:15:27 and (▷) 7:15:27 PEC. | |
Looking now at the patch of the top leaflet coming first in contact with the adsorbing PEC, a local variation in the lipid density was revealed by the bidimensional density profiles shown in Fig. S6.† The density of the charged lipid heads decreased generally in the contact region, with an additional depletion in the tail region taking place as well. Two exemptions are nonetheless noted at 1:1:150 and 15:15:27, in the sense of a weak accumulation of the lipid heads and constant tail density of the lipid patch contacting the complex. These variations in the local lipid packing of the top leaflet affected poorly the structure of the distal layer. Thus, the lipid orientation in the distal leaflet changed occasionally below the contacting area, with no particular local packing for most of PECs and with the lipid tails lightly denser and the lipid heads weakly depleted for 15:15:10 and 7:15:27 PEC. The findings for the entrance leaflet noted at the first stage are altered by the following relaxation modes leading to either the adsorbed or the embedded complex configuration if one compares the local lipid densities at the beginning and the end of the binding pathway (Fig. S6 and S7†). The depletion of both headgroup and tails of the lipid patch adhering to the complex is enhanced, yet the lipid density at the center of the embedded PECs does not become negligible. This observation implies that DPPC molecules could intercalate within the adsorbed complex when the latter attained the extended configuration with the hydrophobic block fully solubilized in the membrane tail region. A notable exception is 1:1:150, the complex showing the largest decrease in the internal electrostatic cohesiveness upon binding (Fig. S3†) and no intercalation of the hydrophobic beads (Fig. 8). Here the initial weak accumulation of lipid headgroups nearby the adhered complex holds throughout the subsequent complex relaxation at the membrane surface. The 2d density profiles in Fig. S7† reveals additionally the presence of undulatory modes for the lipid bilayers hosting internally the hydrophobic blocks of the PEC. Note that these undulations leading to bead composition perturbation at any specific across the membrane were not accounted for in the calculation of the beads density profiles across the membrane (Fig. 8) and area per lipid (Fig. 9a). Therefore, one examined additionally the local density analysis of the beads in the distal leaflet for the internalized complexes. These densities profiles are displayed in Fig. S8† and they indicate an almost absent effect of the embedded beads of the complex on the packing of the lipids underneath. One can, therefore, infer that the contact made by the hydrophobic blocks B with the tails in the distal leaflet (Fig. 6) is restricted mostly to the terminal beads of the alkyl chains.
4. Conclusions
One provides computational insights into structural and dynamical aspects of the adhesion and internalization of the amphiphilic polyelectrolytes into zwitterionic DPPC bilayer by implementing a coarse-grained model system with an implicit solvent description. The amphiphilic polyelectrolyte, represented by alternating charged and hydrophobic blocks (AnBn)m, was pre-assembled as a neutrally charged complex in the presence of a linear and oppositely charged polyion, and the influence of the block length n and the degree of polymerization m on the interaction kinetics of the PEC with the lipid bilayer was investigated. The free PECs, developing progressively an internal core–shell configuration at increasing the length of the hydrophobic block, adhered irreversibly to the membrane, and their internal electrostatic cohesiveness was retained irrespective of the block sequence of the amphiphilic polyelectrolyte. Apart from the internal organization of the free complexes, the distribution of the hydrophobic moiety in the amphiphilic multiblock copolymer modulated the interaction pathway of the PEC with the lipid bilayer, which was characterized by two main configurations: adsorbed at the membrane surface and embedded into the membrane core. The initial electrostatically driven binding to choline groups was followed by a relaxation phase that correlated the formation of the electrostatically favorable contacts between the PEC and the membrane headgroups with the lifetime of the adsorbed state. The hydrophobic contacts of the amphiphilic multiblock copolymer and lipid tails became increasingly favorable at increasing the length of the hydrophobic blocks, despite an initial hindrance originating from the electrostatic coupling of the PEC corona to the membrane headgroups. The subsequent insertion of the PEC hydrophobic core in the lipid tail region led to an intercalated snorkeling conformation with no anchor of the charged PEC moieties to the head region of the opposite membrane leaflet. The variation of the block copolymer sequence and the degree of polymerization of the amphiphilic multiblock copolymer brought evidence of optimal block copolymer sequences at which the PEC internalization and the membrane stabilization occurred. In this context, one provided validity and confidence in applying the implicit solvent approach to commonly used coarse grained models as a valuable tool to further our understanding of the permeation of macromolecular complexes through cell membranes.
Conflicts of interest
The author declares no competing financial interest.
References
- A. C. Wan, B. C. Tai, K. M. Schumacher, A. Schumacher, S. Y. Chin and J. Y. Ying, Langmuir, 2008, 24, 2611–2617 CrossRef CAS.
- U. Lächelt and E. Wagner, Chem. Rev., 2015, 115, 11043–11078 CrossRef.
- A. Bertin, Adv. Polym. Sci., 2014, 256, 103–196 CrossRef CAS.
- S. Vaidyanathan, B. G. Orr and M. M. B. Holl, Acc. Chem. Res., 2016, 49, 1486–1493 CrossRef CAS.
- S. Y. Wong, J. M. Pelet and D. Putnam, Prog. Polym. Sci., 2007, 32, 799–837 CrossRef CAS.
- Y. K. Sung and S. W. Kim, Biomater. Res., 2019, 23, 8 CrossRef CAS.
- D. Fischer, Y. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, Biomaterials, 2003, 24, 1121–1131 CrossRef CAS.
- M. Durymanov and J. Reineke, Front. Pharmacol., 2018, 9, 971 CrossRef.
- Z. Rehman, D. Hoekstra and I. S. Zuhorn, ACS Nano, 2013, 7, 3767–3777 CrossRef.
- S. Hong, P. R. Leroueil, E. K. Janus, J. L. Peters, M. M. Kober, M. T. Islam, B. G. Orr, J. R. Baker and M. M. Holl, Bioconjugate Chem., 2006, 17, 728–734 CrossRef CAS.
- A. Mecke, S. Uppuluri, T. M. Sassanella, D.-K. Lee, A. Ramamoorthy, J. R. Baker, B. G. Orr and M. M. B. Holl, Chem. Phys. Lipids, 2004, 132, 3–14 CrossRef CAS.
- R. S. Dias, B. Lindman and M. G. Miguel, J. Phys. Chem. B, 2002, 106, 12600–12607 CrossRef CAS.
- S. Pannwitt, K. Slama, F. Depoix, M. Helm and D. Schneider, Langmuir, 2019, 35, 14704–14711 CrossRef CAS.
- S. Giatrellis and G. Nounesis, J. Pharm. BioAllied Sci., 2011, 3, 70–76 CrossRef CAS.
- M. Saunders, K. M. Taylor, D. Q. Craig, K. Palin and H. Robson, Pharm. Res., 2007, 24, 1954–1961 CrossRef CAS.
- S. Bauhuber, R. Liebl, L. Tomasetti, R. Rachel, A. Goepfericha and M. Breunig, J. Controlled Release, 2012, 162, 446–455 CrossRef CAS.
- M. Ahmed, M. Jawanda, K. Ishihara and R. Narain, Biomaterials, 2012, 33, 7858–7870 CrossRef CAS.
- M. Ahmed and R. Narain, Biomaterials, 2011, 32, 5279–5290 CrossRef CAS.
- A. T. Y. Chim, W. K. J. Lam, Y. Ma, S. P. Armes, L. A. Lewis, J. C. Roberts, S. Stolnik, S. J. B. Tendler and M. C. Davies, Langmuir, 2005, 21, 3591–3598 CrossRef.
- N. S. Uzgu, O. Akdemir, G. Hasenpusch, C. Maucksch, M. M. Golas, B. Sander, H. Stark, R. Imker, J.-F. Lutz and C. Rudolph, Biomacromolecules, 2009, 11, 39–50 CrossRef.
- M. C. Deshpande, M. C. Garnett, M. Vamvakaki, L. Bailey, S. P. Armes and S. Stolnik, J. Controlled Release, 2002, 81, 185–199 CrossRef CAS.
- S. Venkataraman, W. L. Ong, Z. Y. Ong, S. C. J. Loo, P. L. R. Ee and Y. Y. Yang, Biomaterials, 2011, 32, 2369–2378 CrossRef CAS.
- M. Obata, T. Kobori, S. Hirohara and M. Tanihara, Polym. Chem., 2015, 6, 1793–1804 RSC.
- G. Rossi and L. Monticelli, Adv. Phys.: X, 2016, 1, 276–296 CAS.
- F. Simonelli, D. Bochicchio, R. Ferrando and G. Rossi, J. Phys. Chem. Lett., 2015, 6, 3175–3179 CrossRef CAS.
- B. Song, H. Yuan, C. J. Jameson and S. Murad, Mol. Phys., 2012, 110, 2181–2195 CrossRef CAS.
- C. J. Jameson, P. Oroskar, B. Song, H. Yuan and S. Murad, in Biomimetic Lipid Membranes: Fundamentals, Applications, and Commercialization, ed. F. N. Kök, A. A. Yildiz and F. Inci, Springer, Cham, 2019, pp. 109–165 Search PubMed.
- L. Zhang, M. Becton and X. Wang, J. Phys. Chem. B, 2015, 119, 3786–3794 CrossRef CAS.
- D. G. Angelescu, Phys. Chem. Chem. Phys., 2019, 21, 12446–12459 RSC.
- G. Rossi, J. Barnoud and L. Monticelli, J. Phys. Chem. Lett., 2014, 5, 241–246 CrossRef CAS.
- E. H. Hill, K. Stratton, D. G. Whitten and D. G. Evans, Langmuir, 2012, 28, 14849–14854 CrossRef CAS.
- M. Kepczynski, D. Jamróz, M. Wytrwal, J. Bednar, E. Rzad and M. Nowakowska, Langmuir, 2012, 28, 676–688 CrossRef CAS.
- N. Wilkosz, D. Jamróz, W. Kopeć, K. Nakai, S. Yusa, M. Wytrwal-Sarna, J. Bednar, M. Nowakowska and M. Kepczynski, J. Phys. Chem. B, 2017, 121, 7318–7326 CrossRef CAS.
- U. Adhikari, A. Goliaei, L. Tsereteli and M. L. Berkowitz, J. Phys. Chem. B, 2016, 120, 5823–5830 CrossRef CAS.
- S. Hezaveh, S. Samanta, A. D. Nicola, G. Milano and D. Roccatano, J. Phys. Chem. B, 2012, 116, 14333–14345 CrossRef CAS.
- E. M. Houang, F. S. Bates, Y. Y. Sham and J. M. Metzger, J. Phys. Chem. B, 2017, 121, 10657–10664 CrossRef CAS.
- E. Heikkila, H. Martinez-Seara, A. A. Gurtovenko, M. Javanainen, H. Häkkinen, I. Vattulainen and J. Akola, J. Phys. Chem. C, 2014, 118, 11131–11141 CrossRef CAS.
- R. C. V. Lehn, M. Ricci, P. H. J. Silva, P. Andreozzi, J. Reguera, K. Voïtchovsky, F. Stellacci and A. Alexander-Katz, Nat. Commun., 2014, 5, 4482 CrossRef.
- S. Salassi, F. Simonelli, D. Bochicchio, R. Ferrando and G. Rossi, J. Phys. Chem. C, 2017, 121, 10927–10935 CrossRef CAS.
- X. Quan, C. Peng, D. Zhao, L. Li, J. Fan and J. Zhou, Langmuir, 2017, 33, 361–371 CrossRef CAS.
- R. C. V. Lehn and A. Alexander-Katz, PLoS One, 2019, 14, e0209492 CrossRef.
- R. C. V. Lehn and A. Alexander-Katz, Soft Matter, 2015, 11 Search PubMed.
- P. Gkeka, P. Angelikopoulos, L. Sarkisov and Z. Cournia, PLoS Comput. Biol., 2014, 10, e1003917 CrossRef.
- C. Arnarez, J. J. Uusitalo, M. F. Masman, H. I. Ingólfsson, D. H. d. Jong, M. N. Melo, X. Periole, A. H. d. Vries and S. J. Marrink, J. Chem. Theory Comput., 2015, 11, 260–275 CrossRef CAS.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. d. Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS.
- Z.-J. Wang and M. Deserno, New J. Phys., 2010, 12, 095004 CrossRef.
- Z.-J. Wang and M. Deserno, J. Phys. Chem. B, 2010, 114, 11207–11220 CrossRef CAS.
- M. Orsi and J. W. Essex, PLoS One, 2011, 6, e28637 CrossRef CAS.
- J. Ryden, M. Ullner and P. Linse, J. Chem. Phys., 2005, 123, 034909 CrossRef.
- Y. Hayashi, M. Ullner and P. Linse, J. Phys. Chem. B, 2004, 108, 15266–15277 CrossRef CAS.
- R. S. Dias, P. Linse and A. A. C. C. Pais, J. Comput. Chem., 2011, 32, 2697–2706 CrossRef CAS.
- S. C. C. Nunes, T. F. G. G. Cova, R. S. Dias and A. A. C. C. Pais, Phys. Chem. Chem. Phys., 2018, 20, 19811–19818 RSC.
- P. Linse, J. Phys.: Condens. Matter, 2002, 14, 13449 CrossRef CAS.
- L. Pegado, B. Jonsson and H. Wennerstrom, Adv. Colloid Interface Sci., 2016, 232, 1–8 CrossRef CAS.
- J. Rescic and P. Linse, J. Comput. Chem., 2015, 36, 1259–1274 CrossRef.
- A. F. Jorge, J. M. G. Sarraguca, R. S. Dias and A. A. C. C. Pais, Phys. Chem. Chem. Phys., 2009, 11, 10890–10898 RSC.
- J. Ziebarth and Y. Wang, J. Phys. Chem. B, 2010, 114, 6225–6232 CrossRef CAS.
- G. Rossi, S. Salassi, F. Simonelli, A. Bartocci and L. Monticelli, Nanoparticle–Membrane Interactions in Biomembrane Simulations Computational Studies of Biological Membranes, ed. M. L. Berkowitz, Taylor & Francis Group, 2019, p. 163 Search PubMed.
- C. K. Choudhury, A. Kumar and S. Roy, Biomacromolecules, 2013, 14, 3759–3768 CrossRef CAS.
- J. L. MacCallum, W. F. D. Bennett and D. P. Tieleman, Biophys. J., 2011, 101, 110–117 CrossRef CAS.
- H. Lee and R. G. Larson, Macromolecules, 2011, 44, 2291–2298 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06894b |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*
 represented by the number of beads nPa and nPh in each block and the number of repeating diblock units, m. The former two numbers define the length of negatively charged A and neutral B sequences so that the total bead number of the multiblock copolymer is given by Nac = m(nPa + nPh) (see Fig. 1). At constant copolymer length Nac = 150 and the fraction of A to B block of
represented by the number of beads nPa and nPh in each block and the number of repeating diblock units, m. The former two numbers define the length of negatively charged A and neutral B sequences so that the total bead number of the multiblock copolymer is given by Nac = m(nPa + nPh) (see Fig. 1). At constant copolymer length Nac = 150 and the fraction of A to B block of  the number of copolymer blocks m varied from 150, i.e. an alternating copolymer, to 10, which corresponds to A and B sequences of 15 beads. Besides, two larger copolymers with m = 27, nPh = 15, and the fraction of A to B block of 1.0 and 0.46, respectively, were considered. To be noted that the stoichiometric charge ratio within the PECs is held to one for the present investigation. Table 1 summarizes the compositions and the labels employed by PECs whose adsorption on the lipid membrane is addressed. In short, e.g. 5
the number of copolymer blocks m varied from 150, i.e. an alternating copolymer, to 10, which corresponds to A and B sequences of 15 beads. Besides, two larger copolymers with m = 27, nPh = 15, and the fraction of A to B block of 1.0 and 0.46, respectively, were considered. To be noted that the stoichiometric charge ratio within the PECs is held to one for the present investigation. Table 1 summarizes the compositions and the labels employed by PECs whose adsorption on the lipid membrane is addressed. In short, e.g. 5