
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the effect of Al doping on CO adsorption at ZnO(10![[1 with combining macron]](https://www.rsc.org/images/entities/h2_char_0031_0304.gif) 0)
0)
D. C. Nguyenab,
Thanh Khoa Phungcd,
Dai-Viet N. Vo e,
Tu Hai Lef,
Dinh Quang Khieug and
Thong Le Minh Pham*hi
e,
Tu Hai Lef,
Dinh Quang Khieug and
Thong Le Minh Pham*hi
aLaboratory of Theoretical and Computational Biophysics, Ton Duc Thang University, Ho Chi Minh City, Vietnam
bFaculty of Applied Sciences, Ton Duc Thang University, Ho Chi Minh City, Vietnam
cDepartment of Chemical Engineering, School of Biotechnology, International University, Ho Chi Minh City, Vietnam
dVietnam National University, Ho Chi Minh City, Vietnam
eCenter of Excellence for Green Energy and Environmental Nanomaterials (CE@GrEEN), Nguyen Tat Thanh University, 300A Nguyen Tat Thanh, District 4, Ho Chi Minh City 755414, Vietnam
fDepartment of Chemistry, The University of Da Nang-University of Science and Education, Da Nang City, Vietnam
gUniversity of Sciences, Hue University, 530000, Vietnam
hInstitute of Research and Development, Duy Tan University, Da Nang City 550000, Vietnam. E-mail: phamlminhthong@duytan.edu.vn
iFaculty of Environmental and Chemical Engineering, Duy Tan University, Da Nang, 550000, Vietnam
First published on 9th November 2020
Abstract
Understanding the effect of Al doping on CO adsorption at ZnO(10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) is crucial for designing a high-performance CO gas sensor. In this work, we investigated the adsorption properties of CO on pristine and Al-doped ZnO(100) by performing DFT+U calculations. It is found that the doping of Al on ZnO(100) induces the semiconductor-to-metal transition and thus enhances the conductance of the substrate. Compared to the pristine ZnO(100), the adsorption energy of CO on the Al-doped surfaces is significantly enhanced since Al doping has the effect of strengthening the adsorption bond. The bonding analysis reveals that CO adsorbs on pristine ZnO(100) via the sole σ-dative donation between the CO HOMO 5σ and the empty states of the Zn cation while π-back donation from filled states of Zn or Al cations to the CO 2π* LUMO is facilitated on the Al-doped surfaces. The π-back donation also results in the red-shift of the CO stretching frequency on the Al-doped surfaces, contrasting to the blue-shift on the pristine surface. The simulated results demonstrate that the doping of Al to a three-fold coordinated site on ZnO(100) is highly beneficial for boosting the performance of the CO gas sensor. Our theoretical investigation provides fundamental insights into the effect of Al doping on the sensing mechanism for CO at the ZnO(100) surface.
0) is crucial for designing a high-performance CO gas sensor. In this work, we investigated the adsorption properties of CO on pristine and Al-doped ZnO(100) by performing DFT+U calculations. It is found that the doping of Al on ZnO(100) induces the semiconductor-to-metal transition and thus enhances the conductance of the substrate. Compared to the pristine ZnO(100), the adsorption energy of CO on the Al-doped surfaces is significantly enhanced since Al doping has the effect of strengthening the adsorption bond. The bonding analysis reveals that CO adsorbs on pristine ZnO(100) via the sole σ-dative donation between the CO HOMO 5σ and the empty states of the Zn cation while π-back donation from filled states of Zn or Al cations to the CO 2π* LUMO is facilitated on the Al-doped surfaces. The π-back donation also results in the red-shift of the CO stretching frequency on the Al-doped surfaces, contrasting to the blue-shift on the pristine surface. The simulated results demonstrate that the doping of Al to a three-fold coordinated site on ZnO(100) is highly beneficial for boosting the performance of the CO gas sensor. Our theoretical investigation provides fundamental insights into the effect of Al doping on the sensing mechanism for CO at the ZnO(100) surface.
1. Introduction
Zinc oxide (ZnO) is an n-type semiconducting material with a wide bandgap and a large exciton binding energy which has attracted a growing interest for device applications owing to its great advantages such as earth-abundance, non-toxicity, and cost-effectiveness.1 The adsorption of small gas molecules, such as H2, O2, CO, H2S and NH3 on ZnO surfaces is one of the most widely studied problems in surface science, not only because of its role as a model system to understand the gas–surface interaction but also due to its great technological importance. ZnO has become a frequently studied material for various important applications which involve the adsorption of CO gas. For instance, ZnO has been used as a catalyst for chemical conversions such as CO oxidation reaction2,3 and methanol synthesis from syngas (CO/H2).4–6 In particular, ZnO has found a very useful application in sensing materials for chemo-resistive CO gas sensors7–10 taking advantage of its high electron mobility, its ease of fabrication, and excellent chemical and thermal stability. For this reason, understanding the adsorption properties of CO on ZnO surfaces is crucial for designing novel CO gas sensors with high sensitivity, especially for the ones that operate at ambient temperature.ZnO nanomaterials have been synthesized, characterized, and tested for gas sensing applications in a variety of morphologies such as thin films, nanorods, nanowires, nanotubes, nanospheres, nanoparticles, etc.11 Although the gas sensors using ZnO material have shown good sensing properties, the elevated operating temperatures is the main drawback which limits their extensive applications. Therefore, considerable effort has been devoted to enhancing the sensing response of the ZnO gas sensors at the ambient operating temperature, and the surface modification or doping with metal cations have been proved as a facile and effective approach.12,13 Particularly, the experimental studies have demonstrated that the sensing response to CO of the gas sensors using ZnO nanoparticles could be enhanced by doping with Al metal.14,15 The density functional theory (DFT) studies on ZnO nanocluster16 and ZnO nanowire17 have also confirmed the enhancement of CO adsorption by the effect of Al doping. In this study, we performed the first-principle calculations to understand the scientific background for the enhancement of the CO sensing response on the Al-doped ZnO sensors. We employed the periodic slab model to unravel the effect of Al doping to the adsorption properties of CO at the surfaces of ZnO nanoparticles which is more appropriate than the cluster model using in the prior study.16 In addition, we focused on analyzing the change in the nature of bonding between CO and ZnO surface induced by the doping of Al. It is widely known that ZnO nanocrystals exhibit four dominating low Miller-index surfaces and the non-polar ZnO(100) has been the focus of numerous experimental and theoretical studies owing to its stable nature.18–20 Our theoretical results indicate that Al doping not only enhances the electrical conductivity of ZnO(100) but also strengthens the CO adsorption. Moreover, the strengthening of the bonding of CO with Al-doped ZnO(100) was also clarified by analyzing the density of states (DOS), the charge density difference (CCD), and the effective Bader charge.
2. Computational methodology
The first-principles DFT21 calculations were carried out with spin-polarization making use of the Vienna Ab initio Simulation Package (VASP 5.4.1).22–25 In the VASP calculations, the interactions between ions and electrons were described by the projector augmented wave method.26,27 The generalized gradient approximation (GGA)28,29 with the PBE exchange–correlation functional30 was used in this study with a cut-off energy of 450 eV for the plane-wave. A gamma-center k-point31 with a grid size of 10 × 6 × 1 was used to sample the Brillouin zone. It is widely known that plain DFT exchange–correlation functional is not adequate to describe the electronic structure of ZnO material owing to the self-interaction error.32 Therefore, we employed the DFT+U method for the calculations in this study and the Hubbard correction U is applied only for Zn and O atoms on ZnO(100) surface. The chosen values of the Hubbard correction U were 10 eV and 7 eV for Zn and O respectively, which has been proven accurate in describing the position of Zn-3d and O-2p bands as well as the band gap of bulk wurtzite ZnO.33
The optimized bulk lattice parameters of wurtzite ZnO are a = 3.092 Å and c = 4.980 Å by the DFT+U calculation, showing good agreement with the prior theoretical result33 using the same calculation method and the experimental value (a = 3.242 Å, c = 5.188 Å).34 The optimized ZnO unit cell was then used to construct ZnO(100) which was modelled as a periodic slab consisted of 32 Zn and 32 O atoms distributed over 8 atomic layers. We considered two positions of Al dopant on ZnO(100), one is three-fold coordinated Zn3c on the topmost layer and the other is four-fold coordinated Zn4c on the second layer. The two positions of Al dopant correspond to two Al-doped surfaces, namely Al3c-doped ZnO(100) and Al4c-doped ZnO(100). A vertical vacuum space of 15 Å was included in the slab model to alleviate the interactions between the repeated slabs. In the course of geometry optimization, the atoms on the top 4 layers were allowed to relax to their equilibrium positions while the atoms in the bottom 4 layers were kept fixed to mimic the bulk behaviour. The geometry optimization of adsorbed CO on the surface was considered to be convergent when the tolerance of the electronic energy (10−6 eV) was reached and the residue forces on each atom were less than 0.03 eV Å−1. The adsorption energy (Eads) of CO on the surface was defined as:
| Eads = (Esurface + ECO) − Esurface/CO | (1) |
3. Results and discussion
3.1. Effect of Al doping on the electronic structure of ZnO(10![[1 with combining macron]](https://www.rsc.org/images/entities/h3_char_0031_0304.gif) 0)
0)
It is essential to characterize the typical properties of pristine and Al-doped ZnO(100) before investigating the adsorption of CO. The intrinsic properties of ZnO(100) have been discussed in several experimental40,41 and theoretical42–45 studies. Therefore, we will be focussing on the effect of Al doping on the geometry and electronic properties of ZnO(100). The relaxed structure of ZnO(100) is depicted in Fig. 1a. It should be noted that Zn3c and O3c on the topmost layer of the slab are under-coordinated atoms with three-fold coordination. By contrast, Zn4c and O4c on the second layer are coordinatively saturated atoms with four-fold coordination. The coordinative unsaturation of Zn3c and O3c leads to a pronounced surface reconstruction on the topmost layer. In particular, the surface reconstruction of the topmost layer is indicated by the substantial contraction of Zn3c–O3c bond length (from 1.892 Å to 1.741 Å) and the buckling of Zn3c–O3c bond (buckling angle of 7.679°). The second layer also undergoes surface reconstruction although it is quite negligible. Moreover, the surface reconstruction leads to the marked difference between the bond length of the interlayer Zn3c–O4c bond and the intralayer Zn3c–O3c bond (1.815 Å vs. 1.741 Å). The Bader charges of Zn and O atoms displayed in Fig. 1a reveals the ionic nature of Zn–O bond where Zn is an electron donor and O is an electron acceptor. The band gap of ZnO(100) by our DFT+U calculation is 1.932 eV which is closer to the experimental value (3.370 eV) than the prior results (1.020 eV and 1.160 eV)46,47 by DFT+U calculations using the same DFT package but only applying the Hubbard correction for Zn.
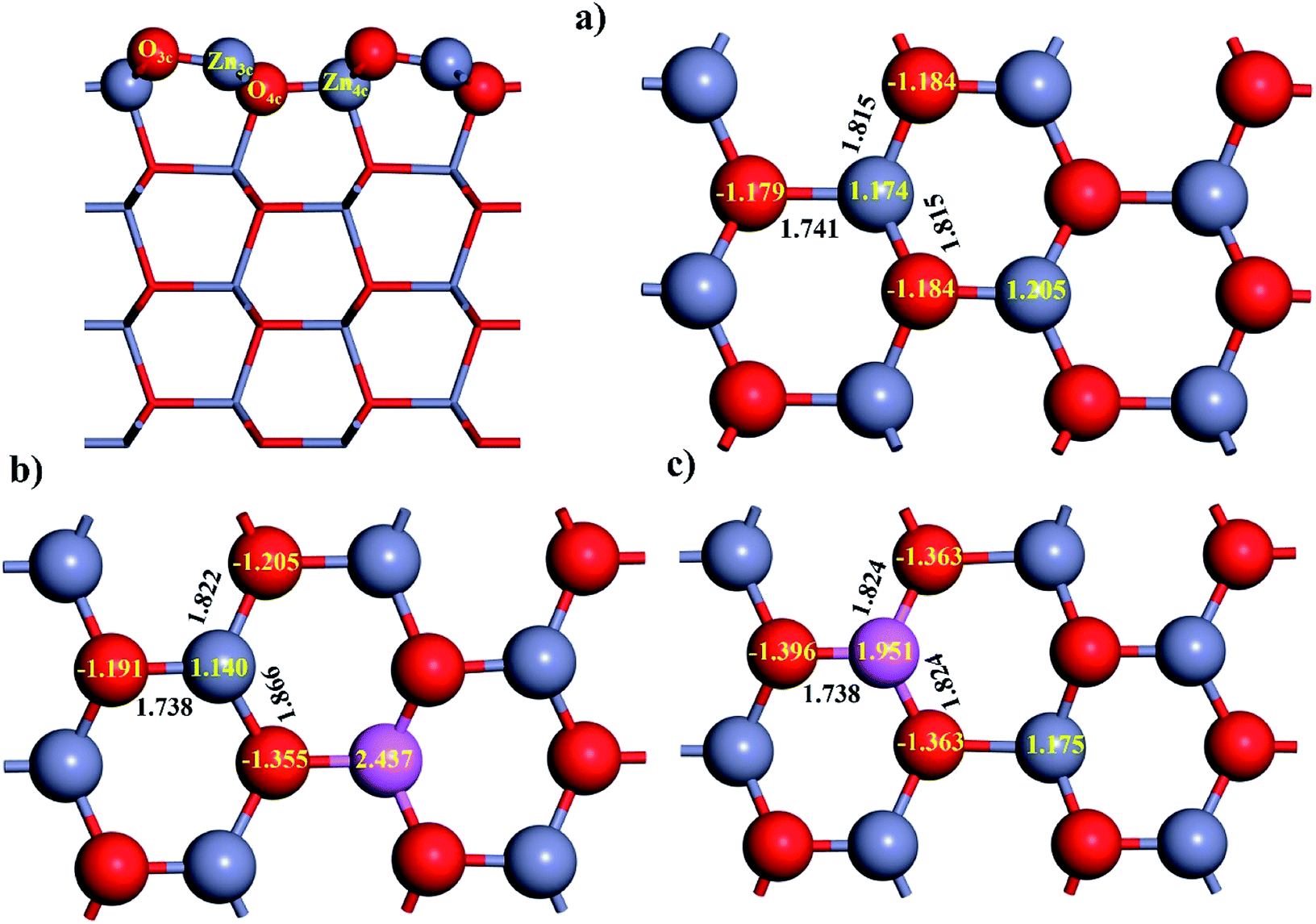 | ||
| Fig. 1 Relaxed structure of: (a) ZnO(100), (b) Al4c-doped ZnO(100), (c) Al3c-doped ZnO(100). The insert numbers in black and yellow colors are the values for the bond length and the effective Bader charge. | ||
The calculated total energies show that Al4c-doped ZnO(100) is thermodynamically more stable than Al3c-doped ZnO(100) by 1.090 eV. Therefore, the substitutional doping of Al to Zn4c is energetically more favourable than to Zn3c. In other words, Al dopant prefers to stay at a bulk-like position than a surface-like one which is consistent with the finding from the prior DFT investigations.46,48 The relaxed structures of the Al-doped surfaces are depicted in Fig. 1b and c. The doping of Al causes slight alterations in the surface geometry, and the most noticeable changes are the bond lengths around the doping positions. For instance, Al3c–O3c bonds of Al3c-doped ZnO(100) and Zn3c–O3c bonds of Al4c-doped ZnO(100) are shorter than the corresponding bonds of the pristine surface. The total DOSs of the studied surfaces in Fig. 2a exhibit non-magnetic properties of the studied surface. Moreover, the doping of Al to ZnO(100) induces the semiconductor-to-metal transition, thus enhances the electrical conductivity of the substrate. The metallic property of Al-doped surfaces is demonstrated by the bands crossing the Fermi levels. We also calculated the Bader charge of each atom on the surfaces, and the results for Al3c and Al4c are 1.951e and 2.437e respectively. Therefore, the valence electrons residing on Al3c and Al4c are 1.049e and 0.563e respectively. These valence electrons occupy the originally empty states in the band gap of ZnO(100) and thus causing the metallic property of the Al-doped surfaces. Besides, the doping of Al results in the upward shift of the Fermi level into the conduction band as the Fermi energy of ZnO(100), Al3c-doped ZnO(100), and Al4c-doped ZnO(100) are −2.513 eV, −0.312 eV, and 0.187 eV respectively. The enhancement of the electrical conductivity of ZnO(100) by Al doping is consistent with the experiment result.14 The projected DOS in Fig. 2b shows a number of Al3c states in the vicinity of the Fermi level, therefore, it is likely that the electrical conductivity of Al3c-doped ZnO(100) is better than Al4c-doped ZnO(100). Ultimately, Al doping is an effective method for tuning the conductance of ZnO(100) and the effect of Al doping on the electronic structure of ZnO(100) is similar to that of graphene-like ZnO monolayer.49
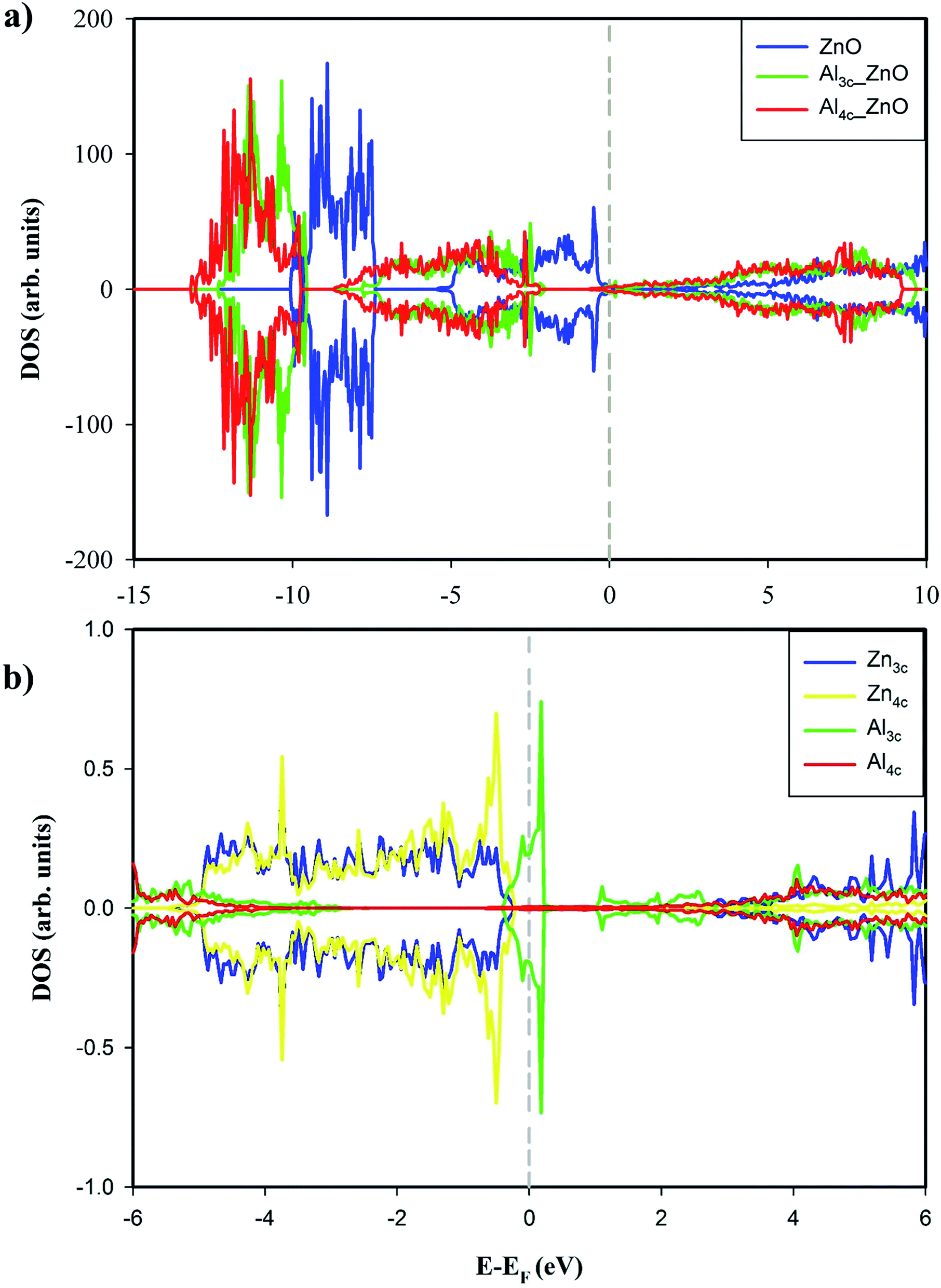 | ||
| Fig. 2 (a) Total DOS of ZnO(100), Al4c-doped ZnO(100) and Al3c-doped ZnO(100). (b) Projected DOS of Zn3c, Zn4c, Al3c and Al4c. The Fermi levels (EF) were shifted to 0 eV and indicated by the gray dashed lines. | ||
3.2. CO adsorption on ZnO(100)
The adsorption properties of CO on the pristine ZnO(100) will be presented serving as the reference point for assessing the effect of Al doping on CO adsorption. The topmost layer of ZnO(100) exposes pairs of coordinately unsaturated Zn3c and O3c acting as the active sites for the adsorption of CO. Experimental studies by ultraviolet photoelectron spectroscopy found that CO adsorbs on ZnO(100) via the bonding between C and Zn3c cation.50,51 The adsorption of CO on ZnO(100) through Zn3c–C bond was also later confirmed by the theoretical investigations.52,53 We also performed the geometry optimization for different orientations of CO towards ZnO(100) and found that the adsorptive structure with downward C bound to the top of Zn3c is the most stable one. The relaxed structure of CO adsorbed on ZnO(100) is illustrated in Fig. 3a and the adsorption properties are summarized in Table 1. As shown in Fig. 3a, CO adsorbs on ZnO(100) in a tilted geometry, and the molecular axis of CO makes with the surface normal an angle of 33.239°. It should be stressed that the tilted angle found by our DFT calculations agrees very well with the experimental value (30°).51 We also calculated the C–O bond length and CO stretching frequency of free and adsorbed CO in order to assess their changes by the adsorption. The calculated results show that the C–O bond length is slightly reduced by 0.003 Å and the CO stretching frequency is blue-shifted by 29 cm−1 by the adsorption on ZnO(100) which agrees well with the experimental result54 (26 cm−1). It should be stressed that the blue-shift of CO stretching frequency also occur on the surface of other transition metal oxides such as TiO2 (ref. 37 and 55) and CeO2.55 The adsorption of CO with C-down configuration on ZnO(100) is characterized by the adsorption energy of 0.378 eV. The CO adsorption becomes more stable when the dispersion correction was included in the DFT calculations, and the adsorption energy reported by the DFT-D3+U calculations is 0.575 eV. The adsorption energy of CO on ZnO(100) by our calculations is consistent with the experimental value (about 0.52 eV)51,56 and the prior theoretical results.53,57–60 It is worth mentioning that the interaction of CO with the pristine ZnO(100) is weak, and the adsorption is essentially physisorption.
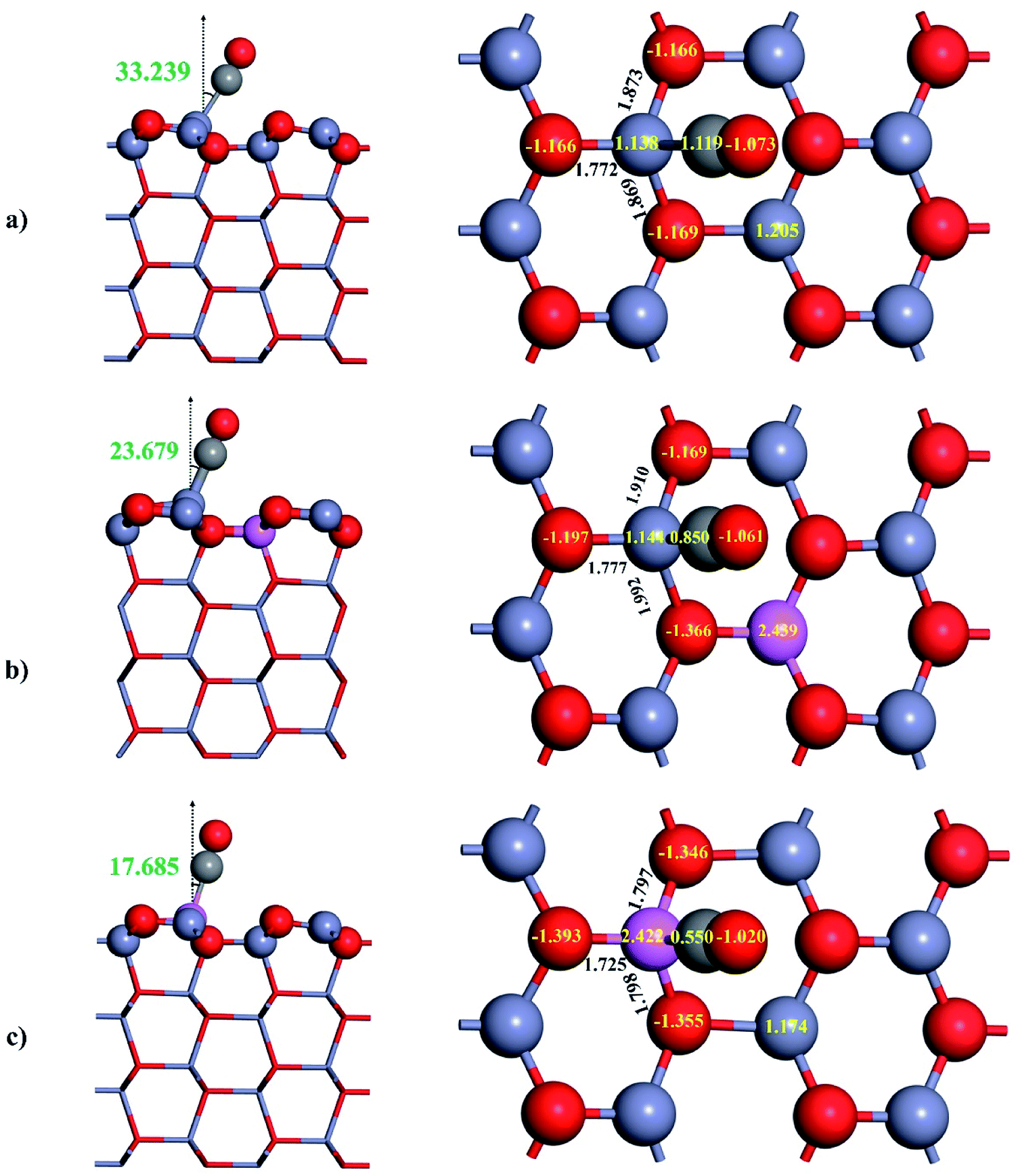 | ||
| Fig. 3 Relaxed structure of adsorbed CO on: (a) ZnO(100), (b) Al4c-doped ZnO(100), (c) Al3c-doped ZnO(100). The insert numbers in black, green and yellow colors are the values for the selected bond length, the bond angle and the effective Bader charge respectively. | ||
0): the adsorption energy (Eads), the binding distance (d(Zn(Al)–C)), the C–O bond length (d(C–O)), the CO stretching vibrational frequency (ν(C–O)), shift of the CO stretching vibrational frequency (Δν(C–O)) with respect to that of free CO, and charge on adsorbed CO (QCO). The calculated C–O bond length and CO stretching vibrational frequency of free CO are 1.144 Å and 2124 cm−1, respectively
| Surface | Adsorption site | DFT method | Eads (eV) | d(Zn(Al)–C) (Å) | d(C–O) (Å) | ν(C–O) (cm−1) | Δν(C–O) (cm−1) | QCO (e) |
|---|---|---|---|---|---|---|---|---|
| ZnO | Top of Zn | DFT+U | 0.378 | 2.127 | 1.141 | 2153 | +29 | +0.046 |
| DFT-D3+U | 0.575 | 2.124 | 1.141 | — | — | — | ||
| Al4c-doped ZnO | Top of Zn | DFT+U | 0.482 | 1.988 | 1.163 | 1977 | −147 | −0.147 |
| DFT-D3+U | 0.711 | 1.985 | 1.163 | — | — | — | ||
| Al3c-doped ZnO | Top of Al | DFT+U | 1.118 | 1.998 | 1.171 | 1964 | −160 | −0.470 |
| DFT-D3+U | 1.271 | 1.989 | 1.173 | — | — | — |
The adsorption of CO on ZnO(100) makes the Zn3c–O3c bond become less buckled since the Zn3c is pulled outwardly by the interaction with CO. The outward movement of Zn3c, in turn, causes a slight elongation of the Zn3c–O bonds, thus results in a small change in the surface geometry around the adsorption site Zn3c. To understand the effect of CO adsorption on the electronic structure, we compared total DOSs of clean and CO-adsorbed ZnO(100) in Fig. 4a. It could be noticed that CO adsorption has a small influence on the electronic structure of the surface, and the appearance of new electronic states at around −5.6 eV in the valence band is the most notable feature although the intensity of these states is tiny. Moreover, the band gap of ZnO(100) is enlarged by 0.011 eV by the adsorption of CO which results in a slight decrease in the electrical conductivity. The Bader charge analysis indicates that CO acts as an electron donor and transfers 0.046e to ZnO(100) in the adsorption. The electronic structure and the capability towards CO adsorption explain why high-performance CO gas sensor is difficult to achieve on the pristine ZnO(100). It is also the reason why a great research effort has been put into improving the sensing performance of ZnO material by doping with metal elements such as Al.
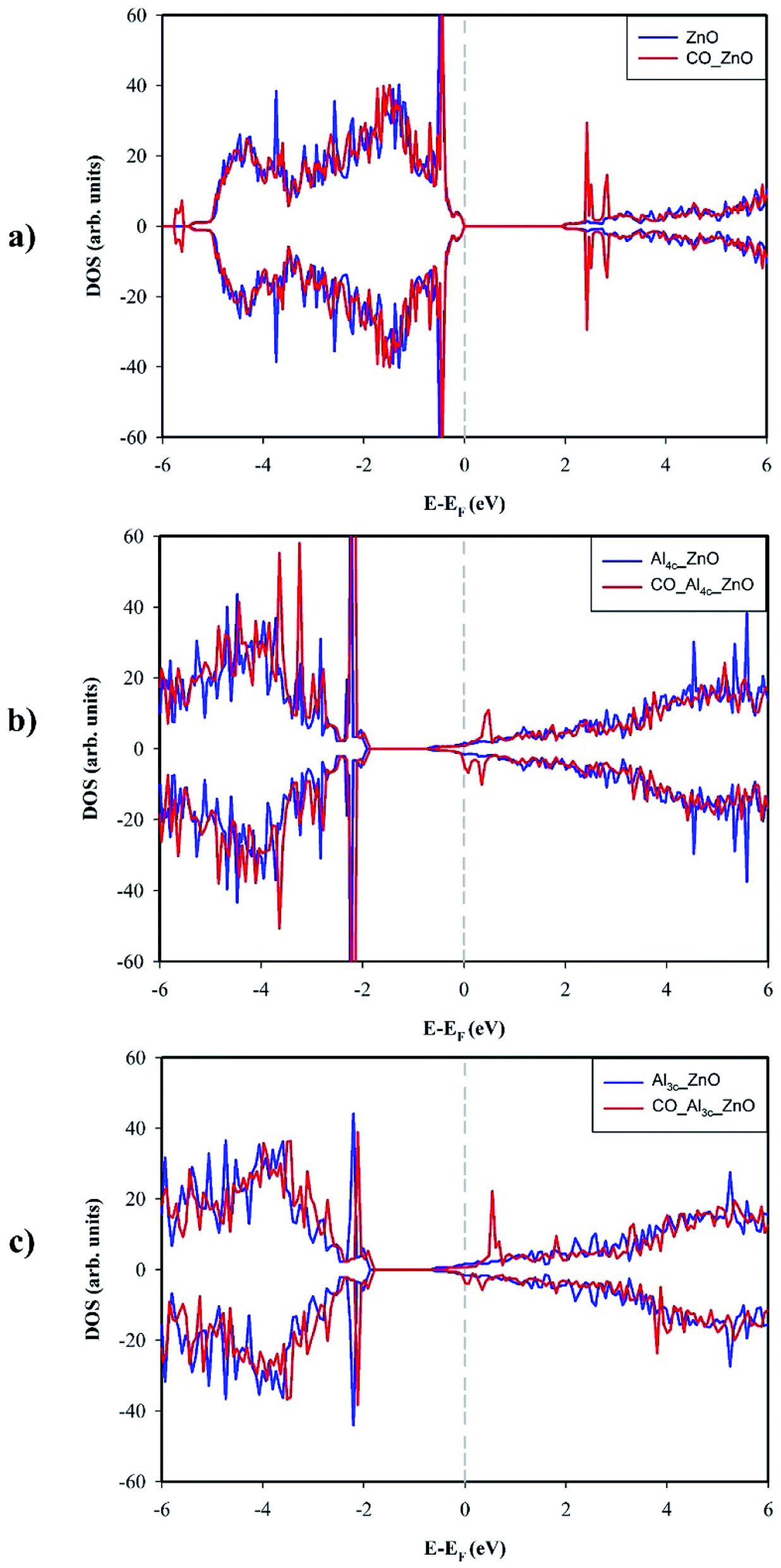 | ||
| Fig. 4 Total DOS of the surface before and after CO adsorption: (a) ZnO(100), (b) Al4c-doped ZnO(100), (c) Al3c-doped ZnO(100). The Fermi levels (EF) were shifted to 0 eV and indicated by the gray dashed lines. | ||
3.3. CO adsorption on Al-doped ZnO(100)
Herein, the adsorption properties of CO on Al-doped surfaces will be discussed. Fig. 3b displays the relaxed structure of CO adsorbed on Al4c-doped ZnO(100). The replacement of Zn4c by Al does not alter the way CO interacts with the surface, and CO remains adsorbed on top of Zn3c in the C-down configuration. However, there are some changes in the adsorptive structure, and the most important is the decrease of Zn3c–C bond from 2.127 Å to 1.988 Å, indicating stronger adsorption of CO on Al4c-doped ZnO(100) compared to the undoped surface. In fact, the adsorption energy of CO is increased by 0.104 eV (0.136 eV by DFT-D3+U) when Zn4c is replaced by Al dopant. By contrast to the adsorption on ZnO(100), the C–O bond length undergoes an elongation by 0.019 Å on Al4c-doped ZnO(100) which results in a red-shift of CO stretching frequency by 147 cm−1.
On Al3c-doped ZnO(100), Al3c cation replaces the role of Zn3c cation as the most adhesive site for CO, and the length of the adsorption bond Al3c–C was found to be 1.998 Å. The tilted angle displayed in Fig. 3c (17.685°) demonstrates that adsorbed CO on Al3c-doped ZnO(100) is in the least inclined geometry among the adsorptive structures. The C–O bond length on Al3c-doped ZnO(100) is significantly elongated to 1.171 Å and the CO stretching frequency is red-shifted by 160 cm−1. Consequently, the adsorption energy of CO on Al3c-doped ZnO(100) was found to be 1.118 eV (1.271 eV by DFT-D3+U) and is the largest adsorption energy on the studied surfaces.
Fig. 4b and c show the total DOSs of Al4c-doped ZnO(100) and Al3c-doped ZnO(100) before and after CO adsorption. Overall, the vicinity of the Fermi level experiences slight changes caused by CO adsorption, particularly the appearance of the partially filled electronic states contributed from adsorbed CO. By contrast to the pristine surface, Al-doped counterparts are electron donors in the adsorption of CO. In particular, the Bader charge analysis reveals that Al4c-doped ZnO(100) and Al3c-doped ZnO(100) transfer 0.147e and 0.470e to CO respectively. It is worth noting that the electron transfer to CO from Al3c-doped ZnO(100) is mainly from Al3c cation while that from Al4c-doped ZnO(100) is originated from the surface ions near the adsorption site Zn3c.
3.4. Discussion
As have been discussed above, the adsorptions of CO on ZnO(100) and Al-doped ZnO(100) occur via the bonding with Zn3c or Al3c cation. The striking contrast in the adsorption properties is CO stretching frequency which is blue-shifted on the undoped surface but is red-shifted on the Al-doped counterparts. The adsorption of CO on ZnO(100) is occurred by chemical interactions, however, it has been suggested that the blue-shift of CO stretching frequency is attributed to the electrostatic interaction of CO with the electric field induced by the surface ions (the Stark effect) which results in a polarization of CO molecule.54,61,62 The projected DOSs of free and adsorbed CO were plotted in Fig. 5 to shed light on the nature of chemical interactions between CO and the surfaces. Fig. 5a clearly demonstrates that non-bonding 5σ is the highest occupied molecular orbital (HOMO) and anti-bonding 2π* is the lowest unoccupied molecular orbital (LUMO). Fig. 5b shows small hybridizations between the projected DOSs of CO HOMO 5σ and Zn3c and the downward shift of CO HOMO 5σ after the adsorption on ZnO(100). Moreover, the Bader charge analysis has shown that CO donates 0.046e to ZnO(100) in the adsorption. These characteristics in combination reveal the σ-dative bond between CO HOMO 5σ and the empty states of Zn3c. Besides, the degeneracy of CO LUMO 2π* is lifted after the adsorption which is most likely due to the polarization of CO molecule on ZnO(100). It is worth mentioning that the energy level of CO LUMO 2π* is considerably higher than the Fermi energy of ZnO(100), thus the π-back donation from the undoped surface to CO 2π* LUMO is not likely to occur and the adsorption bond is solely the σ-dative bond. The CDD analysis for CO adsorbed on ZnO(100) in Fig. 6a further validates the point. As shown in the two-dimensional CDD contour plot, the shape of the accumulation region between CO and ZnO(100) exhibits the characteristics of the σ-dative bond.
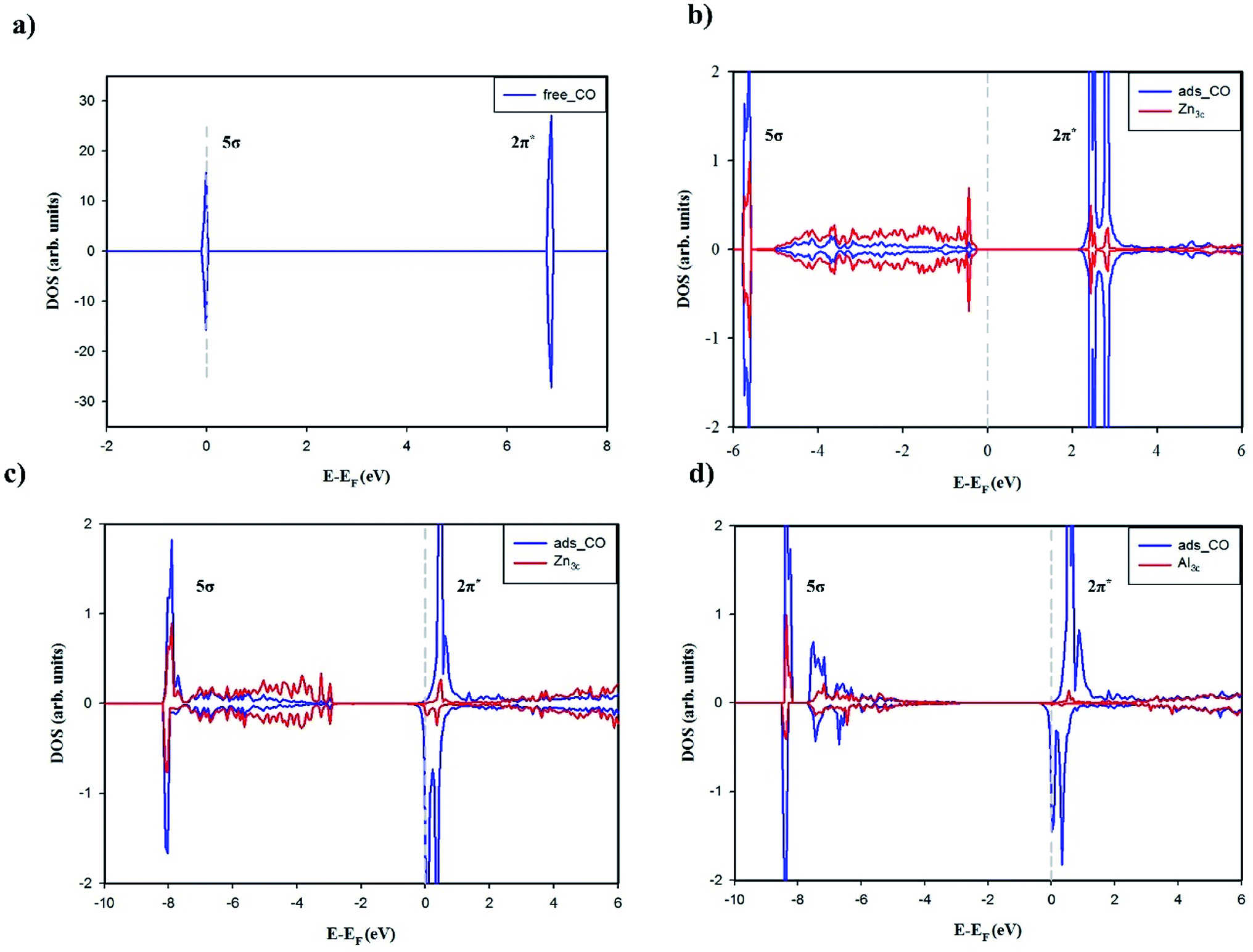 | ||
| Fig. 5 Projected DOS for: (a) free CO, (b) adsorbed CO on ZnO(100), (c) adsorbed CO on Al4c-doped ZnO(100), (d) adsorbed CO on Al3c-doped ZnO(100). The Fermi levels (EF) were shifted to 0 eV and indicated by the gray dashed lines. | ||
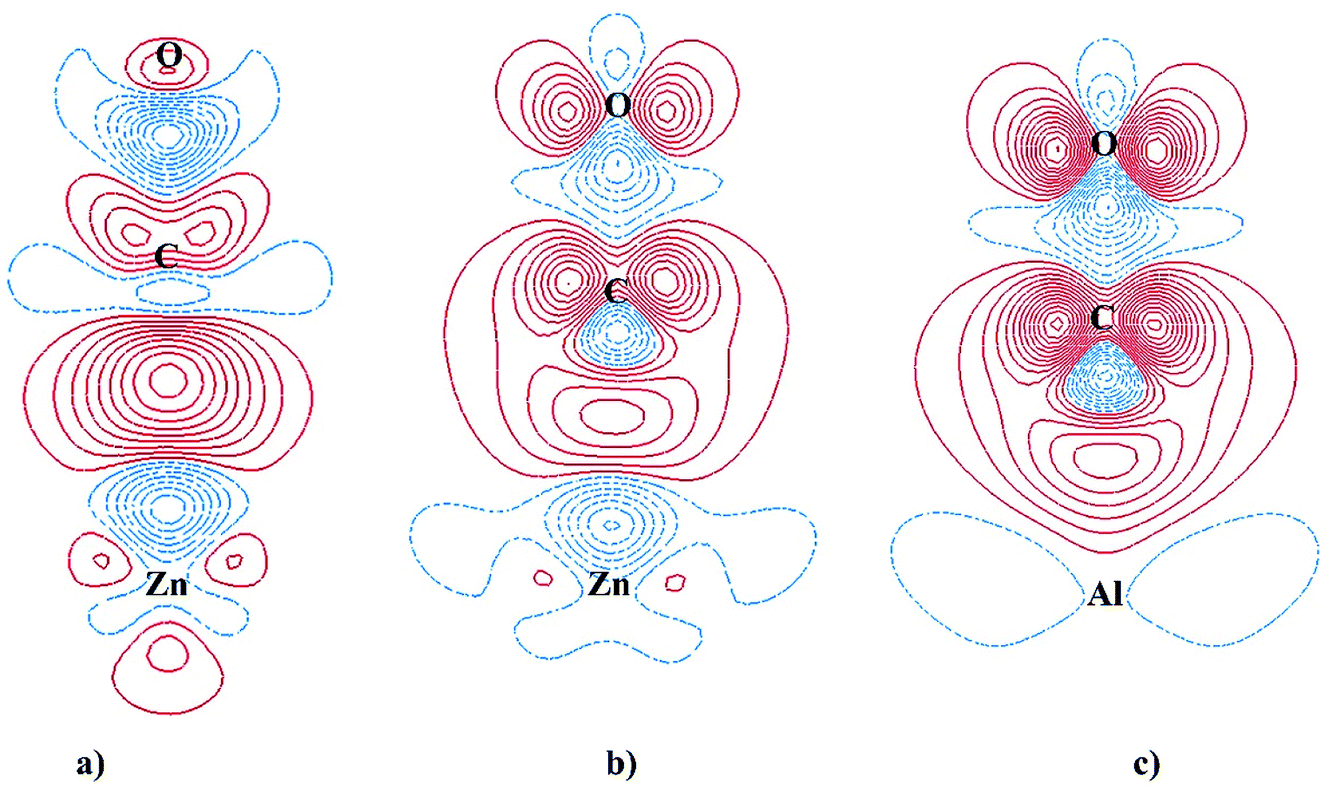 | ||
| Fig. 6 Two-dimensional charge density difference contour plot for: (a) adsorbed CO on ZnO(100), (b) adsorbed CO on Al4c-doped ZnO(100), (c) adsorbed CO on Al3c-doped ZnO(100). The solid red line and dashed blue line represent an electron accumulation and an electron depletion respectively. | ||
The adsorption energy of CO on the Al-doped surfaces is significantly larger than on the undoped counterpart, therefore it is obvious that Al doping has a profound effect on the nature of the adsorption bonds. The degree of the downward shifts of CO HOMO 5σ in Fig. 5b–d reveal the relative strength of the σ-dative bond between CO and the surfaces: ZnO(100) < Al4c-doped ZnO(100) < Al3c-doped ZnO(100). It is also worth recalling here the fact that the Fermi energy of the Al-doped surfaces is tuned upwards into the conduction band, making it closer to the energy level of CO LUMO 2π*. Thus, the π-back donations from the Al-doped surfaces to CO 2π* LUMO are facilitated, leading to the elongation of C–O bond length and the red-shift of CO stretching frequencies. The π-back donations are also demonstrated by the partial occupations of CO LUMO 2π* and the splitting of the projected DOSs after the adsorptions as shown in Fig. 5c and d. Moreover, the two-dimensional CDD contour plots in Fig. 6b and c also exhibit the characteristics of the π-back donations from the Al-doped surfaces to CO. The amount of charge transfer from the Al-doped surfaces to CO in the adsorption indicates that the π-back donation to CO from Al3c-doped ZnO(100) is notably stronger than from Al4c-doped ZnO(100) owing to a larger number of valence electrons residing on Al3c than on Zn3c. The relative strength of the π-back donations also demonstrates why C–O bond length on Al3c-doped ZnO(100) is longer than on Al4c-doped ZnO(100). Moreover, the π-back donation dominates the σ-dative bond in the adsorption of CO on Al3c-doped ZnO(100), making the big difference in the adsorption energy of CO on Al3c-doped ZnO(100) and ZnO(100). It is interesting to note that the enhancement of CO adsorption by the effect of Al doping is also found for ZnO nanoclusters,16 ZnO nanowire17 and graphene-like ZnO monolayer.49
4. Conclusions
By performing the DFT+U calculations, we have investigated the adsorption properties of CO on pristine and Al-doped ZnO(100) in an effort to understand the effect of Al doping on the sensing mechanism of CO at ZnO(100). Two Al-doped surfaces corresponding to two doping positions of Al, three-fold coordinated site on the topmost layer and four-fold coordinated site on the second layer, were considered in this study. The electronic structure analysis reveals the metallic property of the Al-doped surfaces and thus the doping of Al to ZnO(100) enhances the electrical conductivity of the substrate. The adsorption of CO is stabilized on the Al-doped surfaces since Al doping has the effect of strengthening the adsorption bond. The adsorption of CO on the pristine surface is driven by the sole σ-dative bond between CO HOMO 5σ and the empty states of Zn cation while the π-back donation from the surface cation to CO 2π* LUMO is facilitated by the effect of Al doping. The π-back donation is also responsible for the red-shift of CO stretching frequency on the Al-doped surfaces. Our simulated DFT results indicate that the Al dopant at the three-fold coordinated site produces more beneficial effects on the performance of the CO gas sensor than at the four-fold coordinated site.
Conflicts of interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.Acknowledgements
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 103.01-2017.370.References
- Ü. Özgür, Y. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S. Doğan, V. Avrutin, S.-J. Cho and H. Morkoç, J. Appl. Phys., 2005, 98, 041301 CrossRef.
- Y. Martynova, B. H. Liu, M. E. McBriarty, I. M. N. Groot, M. J. Bedzyk, S. Shaikhutdinov and H. J. Freund, J. Catal., 2013, 301, 227–232 CrossRef CAS.
- R. G. S. Pala, W. Tang, M. M. Sushchikh, J.-N. Park, A. J. Forman, G. Wu, A. Kleiman-Shwarsctein, J. Zhang, E. W. McFarland and H. Metiu, J. Catal., 2009, 266, 50–58 CrossRef CAS.
- M. Bowker, H. Houghton and K. C. Waugh, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 3023–3036 RSC.
- M. Kurtz, J. Strunk, O. Hinrichsen, M. Muhler, K. Fink, B. Meyer and C. Wöll, Angew. Chem., Int. Ed., 2005, 44, 2790–2794 CrossRef CAS.
- J. Strunk, K. Kähler, X. Xia and M. Muhler, Surf. Sci., 2009, 603, 1776–1783 CrossRef CAS.
- H.-W. Ryu, B.-S. Park, S. A. Akbar, W.-S. Lee, K.-J. Hong, Y.-J. Seo, D.-C. Shin, J.-S. Park and G.-P. Choi, Sens. Actuators, B, 2003, 96, 717–722 CrossRef CAS.
- D. H. Yoon and G. M. Choi, Sens. Actuators, B, 1997, 45, 251–257 CrossRef CAS.
- C. Liangyuan, L. Zhiyong, B. Shouli, Z. Kewei, L. Dianqing, C. Aifan and C. C. Liu, Sens. Actuators, B, 2010, 143, 620–628 CrossRef.
- P. Rai and Y.-T. Yu, Sens. Actuators, B, 2012, 161, 748–754 CrossRef CAS.
- V. Galstyan, E. Comini, C. Baratto, G. Faglia and G. Sberveglieri, Ceram. Int., 2015, 41, 14239–14244 CrossRef CAS.
- L. Zhu and W. Zeng, Sens. Actuators, A, 2017, 267, 242–261 CrossRef CAS.
- V. S. Bhati, M. Hojamberdiev and M. Kumar, Energy Rep., 2020, 6, 46–62 CrossRef.
- M. Hjiri, L. El Mir, S. G. Leonardi, A. Pistone, L. Mavilia and G. Neri, Sens. Actuators, B, 2014, 196, 413–420 CrossRef CAS.
- S. Bai, T. Guo, Y. Zhao, R. Luo, D. Li, A. Chen and C. C. Liu, J. Mater. Chem. A, 2013, 1, 11335–11342 RSC.
- N. L. Hadipour, A. Ahmadi Peyghan and H. Soleymanabadi, J. Phys. Chem. C, 2015, 119, 6398–6404 CrossRef CAS.
- M. Maarouf and A. Al-Sunaidi, Comput. Theor. Chem., 2020, 1175, 112728 CrossRef CAS.
- C. Wöll, Prog. Surf. Sci., 2007, 82, 55–120 CrossRef.
- U. Diebold, L. V. Koplitz and O. Dulub, Appl. Surf. Sci., 2004, 237, 336–342 CrossRef CAS.
- Q.-L. Tang and Q.-H. Luo, J. Phys. Chem. C, 2013, 117, 22954–22966 CrossRef CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- K. Harun, N. A. Salleh, B. Deghfel, M. K. Yaakob and A. A. Mohamad, Results Phys., 2020, 16, 102829 CrossRef.
- X. Ma, Y. Wu, Y. Lv and Y. Zhu, J. Phys. Chem. C, 2013, 117, 26029–26039 CrossRef CAS.
- W. Göpel, J. Pollmann, I. Ivanov and B. Reihl, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 26, 3144–3150 CrossRef.
- T. L. M. Pham, S. Nachimuthu, J.-L. Kuo and J.-C. Jiang, Appl. Catal., A, 2017, 541, 8–14 CrossRef CAS.
- T. L. M. Pham, D.-V. N. Vo, H. N. T. Nguyen and N.-N. Pham-Tran, Appl. Surf. Sci., 2019, 481, 1327–1334 CrossRef CAS.
- J. P. Prates Ramalho, F. Illas and J. R. B. Gomes, Phys. Chem. Chem. Phys., 2017, 19, 2487–2494 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- C. B. Duke, A. R. Lubinsky, S. C. Chang, B. W. Lee and P. Mark, Phys. Rev. B: Solid State, 1977, 15, 4865–4873 CrossRef CAS.
- C. B. Duke, R. J. Meyer, A. Paton and P. Mark, Phys. Rev. B: Solid State, 1978, 18, 4225–4240 CrossRef CAS.
- D. J. Cooke, A. Marmier and S. C. Parker, J. Phys. Chem. B, 2006, 110, 7985–7991 CrossRef CAS.
- N. R. D'Amico, G. Cantele and D. Ninno, J. Phys. Chem. C, 2012, 116, 21391–21400 CrossRef.
- B. Meyer and D. Marx, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 035403 CrossRef.
- M. J. S. Spencer, K. W. J. Wong and I. Yarovsky, Mater. Chem. Phys., 2010, 119, 505–514 CrossRef CAS.
- D. Ma, Z. Wang, H. Cui, J. Zeng, C. He and Z. Lu, Sens. Actuators, B, 2016, 224, 372–380 CrossRef CAS.
- F. Maldonado, L. Villamagua and R. Rivera, J. Phys. Chem. C, 2019, 123, 12296–12304 CrossRef CAS.
- R. G. S. Pala and H. Metiu, J. Phys. Chem. C, 2007, 111, 8617–8622 CrossRef CAS.
- D. Ma, Q. Wang, T. Li, Z. Tang, G. Yang, C. He and Z. Lu, J. Mater. Chem. C, 2015, 3, 9964–9972 RSC.
- K. L. D'Amico, M. R. McClellan, M. J. Sayers, R. R. Gay, F. R. McFeely and E. I. Solomon, J. Vac. Sci. Technol., 1980, 17, 1080–1084 CrossRef.
- R. R. Gay, M. H. Nodine, V. E. Henrich, H. J. Zeiger and E. I. Solomon, J. Am. Chem. Soc., 1980, 102, 6752–6761 CrossRef CAS.
- B. Meyer and D. Marx, J. Phys.: Condens. Matter, 2003, 15, L89–L94 CrossRef CAS.
- Q. Yuan, Y.-P. Zhao, L. Li and T. Wang, J. Phys. Chem. C, 2009, 113, 6107–6113 CrossRef CAS.
- M. Buchholz, X. Yu, C. Yang, S. Heißler, A. Nefedov, Y. Wang and C. Wöll, Surf. Sci., 2016, 652, 247–252 CrossRef CAS.
- C. Yang and C. Wöll, Adv. Phys.: X, 2017, 2, 373–408 CAS.
- P. Esser and W. Göpel, Surf. Sci., 1980, 97, 309–318 CrossRef CAS.
- M. Casarin, C. Maccato and A. Vittadini, Appl. Surf. Sci., 1999, 142, 192–195 CrossRef CAS.
- S. Tosoni, C. Li, P. Schlexer and G. Pacchioni, J. Phys. Chem. C, 2017, 121, 27453–27461 CrossRef CAS.
- M. Andersen, X. Yu, M. Kick, Y. Wang, C. Wöll and K. Reuter, J. Phys. Chem. C, 2018, 122, 4963–4971 CrossRef CAS.
- H. Shi, H. Yuan, S. Ruan, W. Wang, Z. Li, Z. Li and X. Shao, J. Phys. Chem. C, 2018, 122, 8919–8924 CrossRef CAS.
- Y. Wang and C. Wöll, Chem. Soc. Rev., 2017, 46, 1875–1932 RSC.
- C. Lamberti, A. Zecchina, E. Groppo and S. Bordiga, Chem. Soc. Rev., 2010, 39, 4951–5001 RSC.
| This journal is © The Royal Society of Chemistry 2020 |