
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCell-free protein synthesis and in situ immobilization of deGFP-MatB in polymer microgels for malonate-to-malonyl CoA conversion†
Tony Köhlera,
Thomas Heidaa,
Sandra Hoefgenb,
Niclas Weigela,
Vito Valianteb and
Julian Thiele *a
*a
aInstitute of Physical Chemistry and Polymer Physics, Leibniz-Institut für Polymerforschung Dresden e.V., Hohe Str. 6, 01069 Dresden, Germany. E-mail: thiele@ipfdd.de
bBiobricks of Microbial Natural Product Syntheses, Department of Molecular and Applied Microbiology, Leibniz Institute for Natural Product Research and Infection Biology, Adolf-Reichwein-Straße 23, 07745 Jena, Germany
First published on 9th November 2020
Abstract
In the present work, microgels were utilized as a cell-free reaction environment to produce a functional malonyl-CoA synthetase (deGFP-MatB) under geometry-controlled transcription and translation. Our approach combines the straight-forward optimization of overall protein yield of an E. coli-based cell-free protein synthesis (CFPS) system based on concentration screening of magnesium and potassium glutamate, DNA as well as polyethylene glycol (PEG), and its innovative usage in microgel-based production of a key enzyme of the polyketide synthesis pathway. After partial modification of the carboxyl groups of hyaluronic acid (HA) with 5′-methylfuran groups via 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride (DMTMM)-activation, these were further functionalized with dibenzocyclooctyne (DBCO) and nitrilotriacetic acid (NTA) groups by bio-orthogonal [4+2] Diels–Alder cycloaddition to yield a bifunctional macromer. After coupling the DBCO groups with azide-functionalized DNA, containing the genetic information for deGFP-MatB, via strain-promoted azide–alkyne cycloaddition (SPAAC), the DNA-/NTA-functionalized HA macromer was utilized as base material together with maleimide-functionalized PEG (PEG-mal2) as the crosslinker to form bifunctional microgels utilizing water-in-oil (W/O) microemulsions. As-formed microgels were incubated with nickel sulfate to activate the NTA groups and provide binding sites for deGFP-MatB, which contained six histidine residues (His-tag) for that purpose. The optimized CFPS mixture was loaded into the microgels to initiate the formation of deGFP-MatB, which was detected by a clear increase in fluorescence exclusively inside the microgel volume. Functionality of both, the bound and the decoupled enzyme was proven by reaction with malonate to yield malonyl CoA, as confirmed by a colorimetric assay.
Introduction
Due to its large variability, cell-free protein synthesis (CFPS) is a key tool in synthetic biology, which has been successfully combined with cell-sized entities, such as vesicles,1 emulsions,2,3 microcapsules4,5 or microgels.6–10 By tuning the composition regarding ribosomes, t-RNA-acyltransferases, transcription as well as translation factors, among others11 - easily extracted from Escherichia coli (E. coli) by different techniques, e.g., sonication12,13 or bead-beating14 - CFPS systems with tailor-made protein synthesis kinetics,15 reaction duration,16 and reaction medium crowding2 are prepared. Additionally, these components can also be expressed as recombinant proteins to simplify CFPS preparation.11,17 E. coli lysate-based CFPS systems are widely applied for elucidating mechanisms in transcription and translation15 as well as quorum sensing in artificial systems.5 To study such phenomena, commonly used fluorescent proteins, particularly the green fluorescent protein (GFP) have been synthesized in a cell-free fashion. Moreover, enzymes like cellulase,3 chloramphenicol acetyl transferase,18 cytokines,19 and antibody fragments20,21 for pharmaceutical applications or lysophosphatidic acid acyltransferase or glycerol-3-phosphate acyltransferase for research application22,23 have been synthesized. In these approaches, DNA, which is coding for the protein of interest, is either diffusing through the reaction mixture1–5,8 or coupled to a matrix such as polymer microgels.6,7,9,10 Microgels consist of a porous polymer matrix swollen in a solvent – in this context, the aqueous CFPS reaction mixture, for instance. In contrast to more established artificial microenvironments, e.g., vesicles, microgels are not surrounded by a membrane that enables controlled and switchable mass transfer. Therefore, the choice of a functional material basis for forming a hydrogel network is crucial to provide a similar degree of control over mass transport in and out of the microenvironment that is based on a porous hydrogel matrix.24 To design biocompatible microgels in a bio-orthogonal fashion, natural building blocks like polysaccharides are well-suited, as modifications of their backbone for cross-linking and functionalization have been thoroughly studied to come up with both, physicochemically and mechanically defined polysaccharide microgels for cell-free biotechnology and cell biology.6,25–27 Along these lines, hyaluronic acid (HA) allows for introducing a wide range of modifications, e.g., tyramine, methacrylates, thiols, vinyl esters or (methyl)furan groups,7,10,25–27 demonstrating the versatility of this material basis for microgel formation. By combining these modifications, multi-functional HA-based microgels are obtained allowing for coupling both DNA as genetic information in CFPS, and for further functionalizing the hydrogel matrix with anchoring groups, e.g., with selective binding motives, to control diffusion of as-formed protein out of the porous polymer matrix. Exemplarily, a fluorescently labeled protein could be immobilized right after expression and spatially confined inside microgels via Ni2+-NTA binding.8,11,25 The in situ immobilization of proteins and enzymes during their expression directly reduces the time for downstream processing and brings a protein rapidly into application.28 A technique for linking CFPS and immobilization of desired proteins has been realized by performing a so called nucleic acid programmable protein assay in the field of translational research.20,29–31 The application of enzymes is conventionally based on heterologous expression in an organism such as E. coli, being a time-consuming procedure that takes days,21,32 while CFPS systems can produce proteins within a few hours.13The combination of CFPS with a hydrogel-based approach was yet more often used to synthesize well-known proteins such as deGFP7,10 or mVenus.6 However, the aim should be to produce functional enzymes, for instance, capable of synthesizing building blocks for further biosynthetic compound design.
Here, we report the cell-free synthesis of a functional enzyme, malonyl-CoA synthetase (MatB), for producing malonyl-CoA, an ester relevant in many biochemical pathways, e.g., the polyketide or fatty acid biosynthesis.33 To produce this enzyme in a cell-mimicking fashion, components for transcription and translation were extracted from E. coli expressing the T7 RNA polymerase (T7RNAP). The E. coli-based CFPS was then optimized by screening concentrations of potassium glutamate, magnesium glutamate, and DNA. Additionally, the well-known synthetic molecular crowding agent polyethylene glycol (PEG)2,34 was screened to determine if supplementing a mobile crowder to the CFPS mixture might be beneficial for the protein yield in addition to the presence of a dense microgel matrix. To account for key features of the natural cellular environment like spatial confinement and localization of target molecules,2 DNA encoding for deGFP-MatB was immobilized inside porous polymer microgels made from HA. These microgels also contained nickel-activated NTA groups to trap polyhistidine-tagged, as-formed enzymes in situ. After loading the bifunctional HA-based microgels with the optimized CFPS system, the formation of the desired deGFP-MatB was monitored within the microgel matrix by an increase of fluorescence intensity. Furthermore, we utilized our microgel system for studying and confirming the activity of the enzyme (Fig. 1), thus enabling CFPS-based expression of an enzyme of the polyketide pathway towards fully functional enzymatic cascades in the near future.
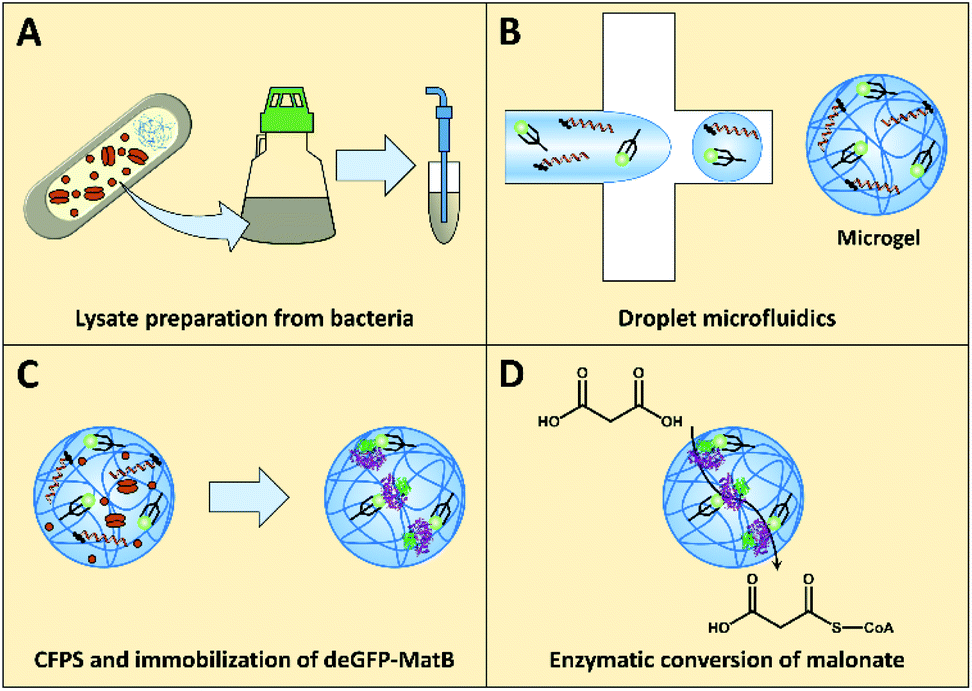 | ||
| Fig. 1 Illustration of the workflow towards the enzymatic conversion of malonate to malonyl-CoA in polymer microgels. (A) Active lysate prepared by culturing E. coli bacteria and lysis by sonication. (B) Droplet microfluidics utilized for producing cell-sized microgels functionalized with DNA encoding for deGFP-MatB, and NTA groups for enzyme immobilization. (C) CFPS of deGFP-MatB and in situ immobilization. (D) Active microgel-bound deGFP-MatB as determined by malonate-to-malonyl-CoA conversion. | ||
Results
Crude lysate preparation for CFPS
CFPS requires a crude cell lysate and, in the present case, one reaction mixture for the delivery of salts and amino acids, as well as another reaction mixture for the delivery of sensitive cofactors and energy sources. The crude extract was produced using E. coli BL21 star cells, which were cultured and sonicated. For that, different media, either 2xYTPG (5 g L−1 sodium chloride, 10 g L−1 yeast extract, 16 g L−1 tryptone, 7 g L−1 potassium phosphate dibasic, 3 g L−1 potassium phosphate monobasic, 18 g L−1 glucose) or cell-free auto-induction (CFAI) medium, were used. To test full activation of the lac operon, the CFPS reaction was supplemented with T7 RNA polymerase (T7RNAP; NEB Inc.). For testing the performance of the lysate, deGFP was synthesized as a model protein, and the concentration was determined via fluorescence (Fig. 2, calibration Fig. S1†).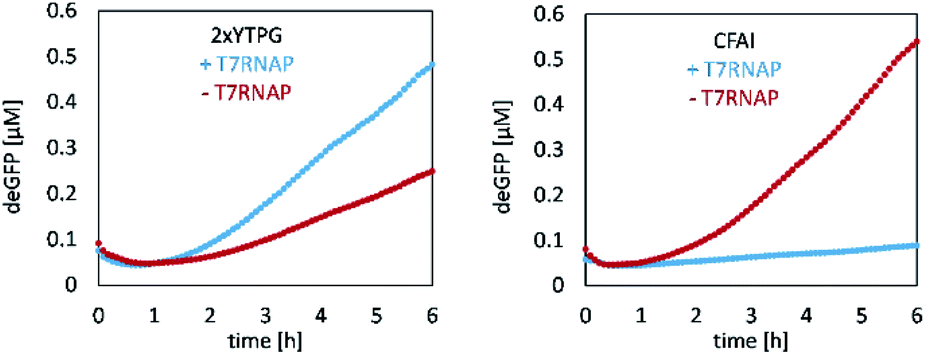 | ||
| Fig. 2 CFPS of deGFP utilizing differently media for E. coli growth and lysate preparation. Left: Lysate derived from E. coli grown in 2xYTPG yields 0.48 μM deGFP after 6 h, when supplemented with T7RNAP. Right: In contrast, E. coli grown in CFAI likewise provides an active lysate that, yet, does not benefit from T7RNAP supplementation but still yields 0.54 μM deGFP after 6 h. | ||
The 2xYTPG medium yielded 0.25 μM of deGFP after 6 h, but benefited from supplementing T7RNAP, increasing the yield to 0.48 μM after 6 h. When E. coli cells were grown in CFAI medium, the obtained protein concentration was 0.54 μM. In contrast, when the lysate was again supplemented with T7RNAP, no protein synthesis was observed, as discussed in detail below. To further investigate the ability of the lysate to produce the protein of interest by either plasmids or linear DNA, two lysates produced from CFAI were tested (Fig. S2†).
Optimization of CFPS reaction mixture for production of deGFP-MatB
First, the CFPS reaction was supplemented with Chi6 DNA to prevent the degradation of linear template DNA in the reaction mix by recBCD complex contained in the lysate (Fig. 3).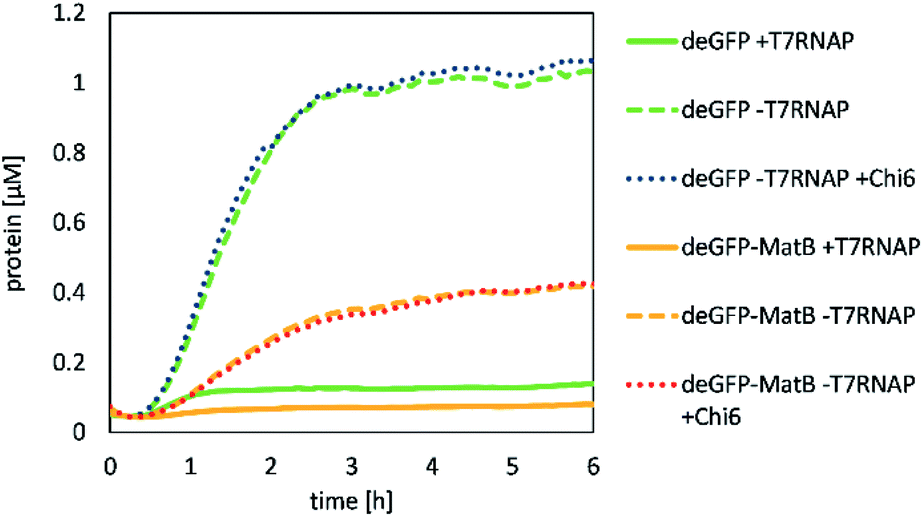 | ||
| Fig. 3 CFPS of deGFP (green or blue) and deGFP-MatB (orange or red), supplemented with T7RNAP (solid lines) or Chi6 DNA (dotted line). Measurements denote the activity as well as the capability to synthesize deGFP and deGFP-MatB in a cell-free fashion. Supplementing the lysate with Chi6 DNA did not change the yield of the CFPS, whereas additional T7RNAP reduced the activity. | ||
It was shown that deGFP as a model protein was successfully produced by our lysate. Its production was not significantly altered by adding Chi6 DNA to the reaction with 5 nM of template DNA over 6 h in 50 μL, yielding 1.06 μM of deGFP. However, deGFP-MatB was also produced by the same procedure, but at a significantly lower yield of 0.43 μM. The production of deGPF-MatB by CFPS showed that a supplementation of T7RNAP was not beneficial for the production of the protein as discussed later. To further investigate, which reaction conditions lead to the most efficient expression, beside the supplementation of T7RNAP or Chi6 DNA, potassium glutamate, magnesium glutamate and PEG were likewise screened (Fig. 4).
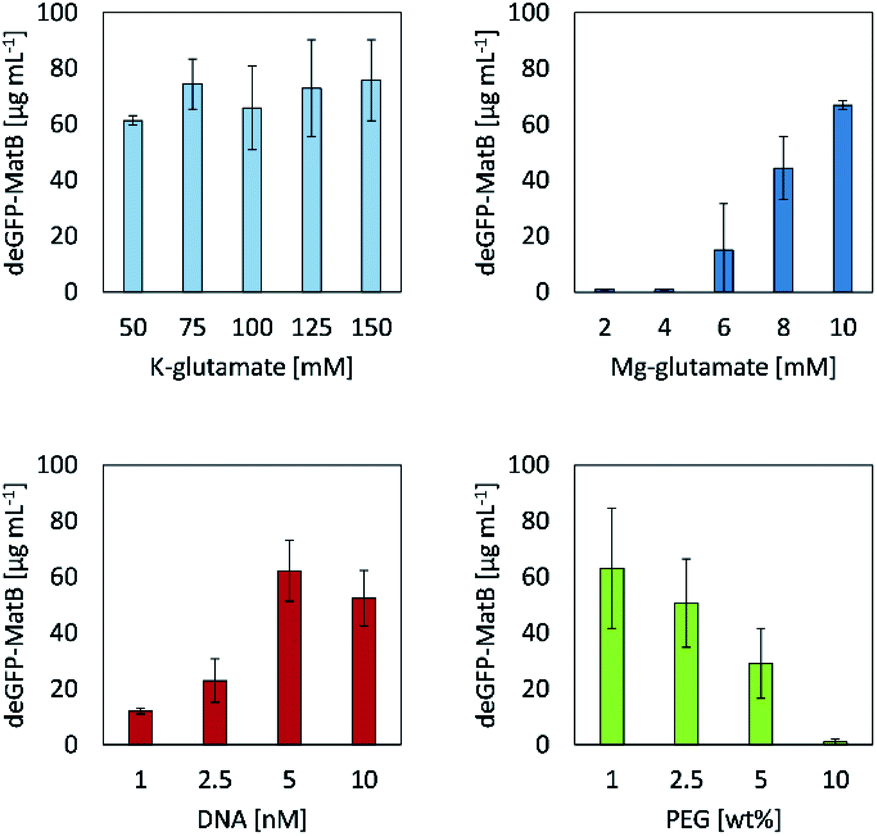 | ||
| Fig. 4 Protein analysis of deGFP-MatB expression after 24 h employing a CFPS machinery supplemented with different concentrations of potassium glutamate, magnesium glutamate, linear DNA or PEG (MW = 8,000 g mol−1) to identify optimal reaction conditions for later expression of deGFP-MatB. Error bars indicate standard deviations of three independent samples. | ||
Screening different concentrations of potassium glutamate and magnesium glutamate revealed their significant influence on the CFPS reaction regarding the yield of deGFP-MatB, as they mimic the E. coli cytoplasmatic environment.18 The system set up here had little influence on the CFPS reaction. When varying the concentrations of potassium glutamate in a range between 50 mM and 150 mM, 61.4 μg mL−1 ± 1.7 μg mL−1 to 75.8 μg mL−1 ± 14.5 μg mL−1 of deGFP-MatB were achieved, respectively. Contrary to that, the results of magnesium glutamate screening showed a strong impact in the range from 2 mM up to 10 mM on the yield of deGFP-MatB. Concentrations in a range from 2 mM to 4 mM of magnesium glutamate did not lead to a protein production at all. In contrary, further increasing the concentration of magnesium glutamate strongly increased the production of deGFP-MatB, yielding at a maximum of 10 mM of magnesium glutamate 66.9 μg mL−1 ± 1.6 μg mL−1 of protein. An optimum concentration for DNA was found to be at 5 nM yielding 62.2 μg mL−1 ± 10.9 μg mL−1 of deGFP-MatB. A further increase of DNA up to 10 nM decreased the protein yield to 52.4 μg mL−1 ± 9.9 μg mL−1. The addition of PEG, with a molecular weight of 8,000 g mol−1 resulted in a decrease of protein yield with increasing PEG concentrations. The addition of 1.0% (w/w) of PEG did not clearly influence the production yield with 63.1 μg mL−1 ± 21.4 μg mL−1 compared to the yields found for the K–glutamate screening. Yet, PEG contents of 2.5% (w/w) and 5.0% (w/w) did decrease the protein yield to 50.6 μg mL−1 ± 15.8 μg mL−1 and 29.1 μg mL−1 ± 12.5 μg mL−1, respectively. Eventually, a PEG content of 10.0% (w/w) stalled the cell-free protein production completely. Optimal concentrations of 5 nM linear DNA, 10 mM Mg-glutamate, 150 mM K-glutamate and no additionally PEG gave the highest yield with 75.8 μg mL−1 ± 14.5 μg mL−1 of deGFP-MatB. After improving the yield of deGFP-MatB produced by the CFPS mixture, the enzyme was tested regarding its activity by an enzymatic assay.
Enzyme activity of CFPS-formed deGFP-MatB determined by DTNB assay
The successful CFPS of deGFP-MatB was confirmed by the increase of fluorescence in the reaction mixture. Direct evidence that the enzyme was produced in an active form was provided by an 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) assay, which is capable to detect free thiol groups. CFPS-formed deGFP-MatB was separated from small molecules by an amicon filter (10 kDa cut-off) using 50 mM HEPES (pH 7.5). Those molecules are non-reacted CoA or L-cysteine and proteins containing cysteine residues, since these do react with DTNB and are part of the CFPS reaction mixture. The amount of deGFP-MatB was determined by fluorescence, and the DTNB assay was performed comparing CFPS-formed deGFP-MatB and FPLC-purified deGFP-MatB (Fig. 5). Both enzyme preparations, recombinant as well as CFPS-derived deGFP-MatB, decreased the concentration of free CoA in the reaction solution indicating an active enzyme converting CoA and malonate to malonyl-CoA. Apart from that, the enzymatic reaction was also performed in the presence of the CFPS mixture. This resulted in intensive background signals in the DTNB assay by small molecules like the afore-mentioned CoA or L-cysteine (Fig. S3†). The initial concentration of 100 μM CoA gave the highest absorbance of 0.39 ± 0.01 at 410 nm. The enzymatic conversion of CoA and malonate was started by adding CFPS-derived or the recombinant deGFP-MatB to a reaction mix composed of 600 μM malonate, 100 μM CoA and 2.5 mM ATP to proof the enzymatic activity. After 90 min at 30 °C and 220 rpm, the reaction was stopped by adding 15 mM EDTA, and the colorimetric detection of the reaction product was initiated by adding 1.5 mM DTNB. The DTNB was allowed to react with free thiol groups in solution for 15 min, and the absorbance at 410 nm was measured.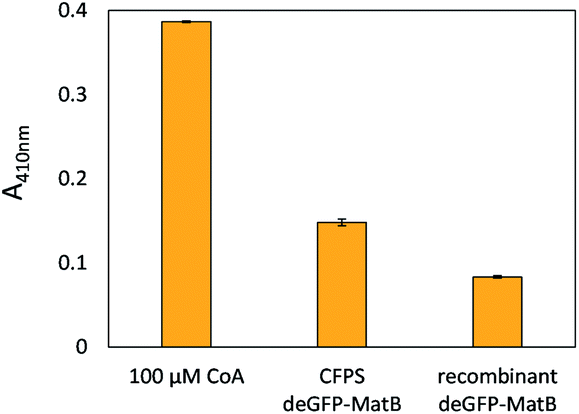 | ||
| Fig. 5 DTNB assay with CFPS-formed deGFP-MatB and recombinant deGFP-MatB. After 90 min, the absorbance of CFPS-derived deGFP-MatB at 410 nm – as determined by the assay – shows an absorbance of 0.15 ± 0.01 for deGFP-MatB, whereas recombinant deGFP-MatB only shows an absorbance of 0.08 ± 0.01. Error bars indicate standard deviation of three samples. | ||
After having a clear proof for the activity of the enzyme prepared by CFPS, the system was applied to DNA-functionalized microgels as reaction environment for both, gene expression and enzyme activity determination.
Coupling of azide-modified PCR products to HAmFU-DBCO
After optimizing the parameters for CFPS, linear DNA encoding for deGFP-matB, was coupled to the 5-methyl-furan- and DBCO-functionalized HA (HAmFU-DBCO) hydrogel precursor. To test the coupling, 100 μg of HAmFU-DBCO was dissolved in 8 μL water, and 2 μL containing 200 ng of the azide-modified PCR product were added. After 5 h incubation at room temperature, the reaction mixture was analyzed by agarose gel electrophoresis (Fig. 6).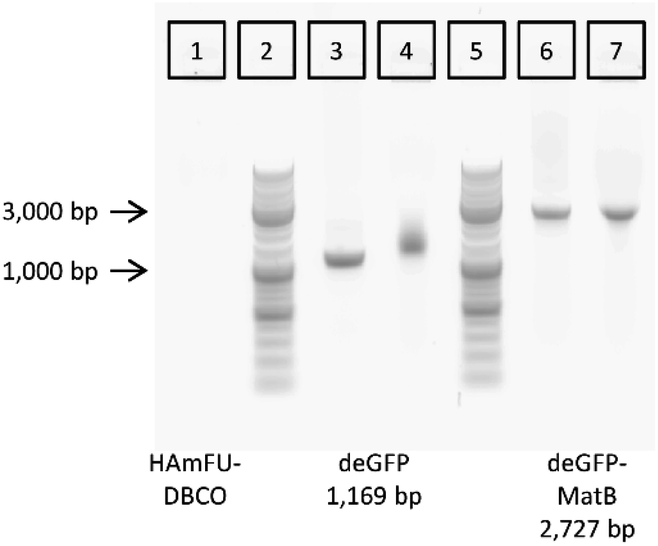 | ||
| Fig. 6 Coupling of azide-modified PCR products of deGFP (3) and deGFP-MatB genes (6) to HAmFU-DBCO (1), as evaluated by gel electrophoresis. Coupling of functionalized PCR products cause a shift of the band of deGFP (4), whereas the PCR product of deGFP-MatB merely leads to a slight change in band curvature (7). Lanes (2) and (5) are markers with indicated lengths at 1,000 bp and 3,000 bp. | ||
The hydrogel precursor HAmFU-DBCO showed no signal in the agarose gel, whereas the PCR products were clearly visible. The PCR product of the deGFP gene readout was loaded as a positive control for coupling of the DNA to HAmFU-DBCO. It clearly showed a significant shift with a smear in the respective lane. The azide-modified PCR product of deGFP-matB did not give a smear or a shift in the gel. Here, the coupling of the azide-modified DNA leads to a change in the band curvature when loaded on a gel. Both results indicate a coupling of PCR products to HAmFU-DBCO. After coupling the azide-modified DNA to the hydrogel precursor, the microfluidic preparation of microgels was performed.
Microfluidic preparation of tailor-made emulsion droplets of NTA- and DNA-functionalized HAmFU-DBCO and PEG-mal2
For the preparation of tailor-made emulsion droplets as templates for uniform microgels, a PDMS-based microfluidic flow-focussing device was prepared by photo- and soft lithography. To monitor emulsion formation, a high-speed camera was used, and droplet formation was verified by phase-contrast as well as bright-field microscopy (Fig. S4†). The oil phase consisted of 2.0% (w/w) of home-made surfactant (cf. Materials section) dissolved in the fluorinated oil HFE-7500 to prevent droplet coalescence. The dispersed phase consisted of DNA-functionalized HAmFU-DBCO further pre-functionalized with NTA groups, and 0.75% (w/w) of PEG-mal2 as a crosslinker. Additionally, 1 μL of maleimide-ATTO647N (1 mg mL−1 in DMSO) was added to visualize the as-formed droplets and microgels, by fluorescence microscopy later on (Fig. 7).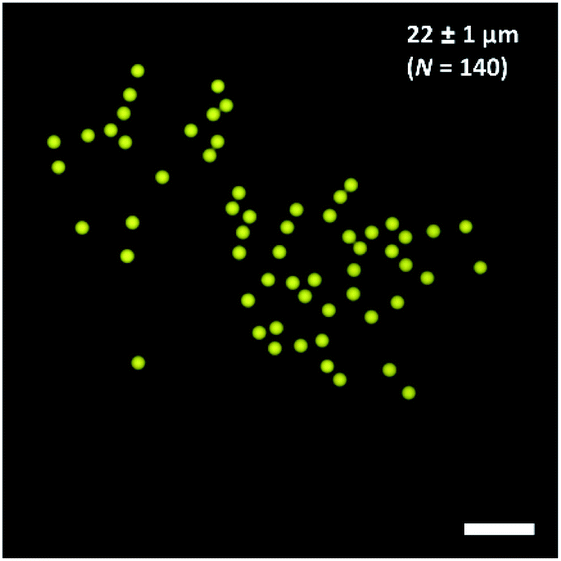 | ||
| Fig. 7 Water-in-oil (W/O) emulsion droplets containing HAmFU-DBCO-DNA PEG-mal2 (droplet size 22 ± 1 μm; N = 140). The scale bars indicate 100 μm. | ||
The water-in-oil emulsion gave a diameter of 22 μm ± 1 μm (N = 140) of individual droplet size. Emulsions were stored for 2 days at room temperature to ensure complete reaction of the hydrogel precursors. Upon solidification of the droplet content, the emulsion was destabilized using 1H,1H,2H,2H-perfluoro-1-octanol. Microgels with a diameter of 27 ± 1 μm (N = 44) were obtained. The microgels were transferred into a 1 mM nickel sulfate solution to activate the NTA groups in the hydrogel network, and were further used for CFPS reaction.
CFPS and immobilization of deGFP-MatB in DNA-/NTA-functionalized microgels
The NTA- and DNA-functionalized HAmFU-DBCO microgels were incubated in a CFPS reaction mixture for 2 h at 30 °C in a PCR tube on a thermocycler. Samples were taken to observe the protein synthesis and homogeneous immobilization of deGFP-MatB throughout the microgel volume (Fig. 8).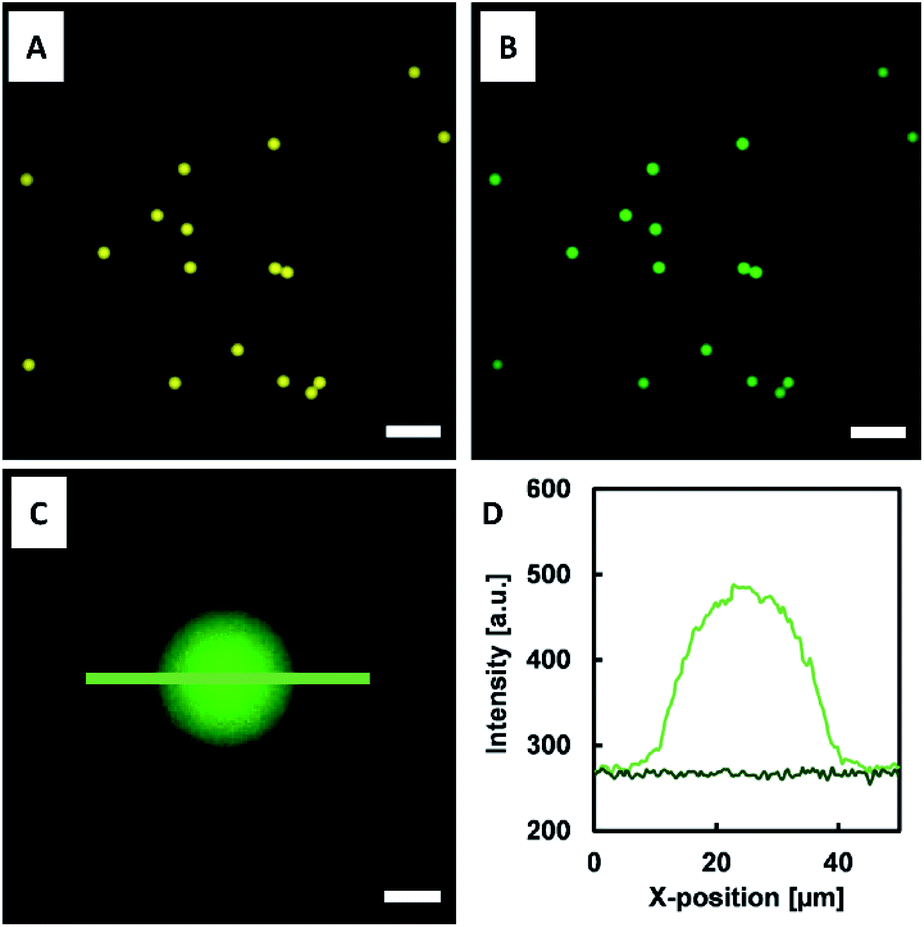 | ||
| Fig. 8 NTA- and DNA-functionalized microgels incubated in CFPS mixture. (A) Microgels exhibiting fluorescence of the ATTO647N dye coupled to HAmFU (diameter: 27 μm ± 1 μm; N = 44). (B) Fluorescence microscopy images of microgels after 2 h of CFPS of deGFP-MatB immobilized to the NTA-functionalized microgel network in situ. (C, D) Line scans of microgel (light green) and background (dark green) indicating the fluorescence intensity of the immobilized enzyme. Scale bars in (A) and (B) denote 100 μm, and 10 μm in (C). | ||
The fluorescence signal throughout the microgels increased over time, clearly indicating the enzyme formation and in situ immobilization at the NTA groups. As the enzyme immobilization and CFPS was completed, the microgels were washed three times with a reaction buffer (50 mM HEPES at pH 7.5). The microgels were then loaded into a reaction mixture composed of 600 μM malonate, 100 μM CoA, and 2.5 mM ATP. This particle suspension was incubated at 30 °C and 220 rpm. Samples were taken every 30 min, and the reaction was stopped by immediate addition of 15 mM EDTA. Once all samples were taken, the assay was started by adding 1.5 mM DTNB. DTNB reacted with free thiol groups in solution for 15 min, and thereafter the absorbance at 410 nm was measured (Fig. 9).
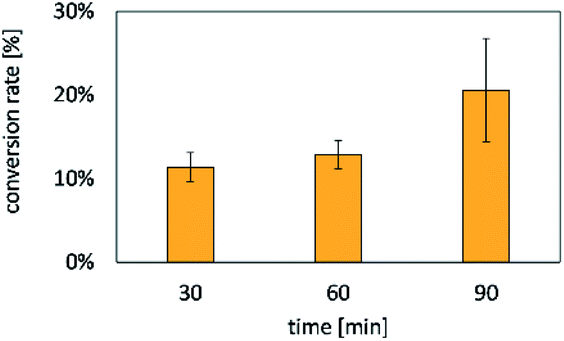 | ||
| Fig. 9 Conversion rates of CoA towards malonyl-CoA by microgel-immobilized deGFP-MatB (samples taken every 30 min). The reaction was stopped by adding EDTA. After 90 min, a conversion of 20.6 ± 6.2% of CoA was reached. Error bars indicate standard deviations of three independent measurements. | ||
After 30 min 11.4 ± 1.8% and after 90 min 20.6 ± 6.2% of the free CoA were consumed. The conversion of CoA over time demonstrated the activity of deGFP-MatB formed via CFPS.
Discussion
In the current work, an E. coli-based CFPS system was utilized for the cell-free synthesis of the enzyme MatB, which was tagged with poly-histidine as well as deGFP to monitor its formation and selective, reversible immobilization inside polymer microgels. Utilizing a His-tag for selective immobilization of deGFP-MatB, its activity is not affected since the His-tag does not sterically hinder the active site of the enzyme. To optimize the CFPS reaction mixture, T7RNAP was added to increase transcription rates, leading to higher concentrations of available mRNA for consecutive translation towards deGFP-MatB. That approach was only beneficial when the lac operon was not fully induced by the applied growth medium (Fig. 2).13 A second important parameter along these lines was the growth time of bacteria and, with that, the induction time for the lac operon and expression of T7RNAP.13 Moreover, it is known throughout literature that reproducible lysate production from batch to batch is a crucial task, and much effort has been undertaken to develop protocols that solve this issue.12,13,35,36 The prepared lysates were supplemented with 1 μM of Chi6 DNA to prevent degradation of the functional, linear PCR fragments used for implementing genetic information into microgels that served as reaction environment for CFPS.37,38 Experiments indicate that addition of Chi6 is not beneficial in every experiment (Fig. 3), but protein yield was lower in the absence of Chi6 for deGFP-MatB (Fig. S2†). To aim for highest protein yield in our system, all reactions were supplemented with Chi6 DNA, as the strain E. coli BL21 star (DE3) is capable of producing the recBCD complex that will degrade linear PCR fragments.37,38 Potassium and magnesium, their counterions, and even the DNA template for CFPS, have a distinct influence on the yield of deGFP-MatB, as shown in Fig. S2.† Therefore, we screened for optimal concentrations of potassium glutamate and magnesium glutamate in our CFPS system (Fig. 4). Screening of these ions was required for each round of lysate preparation, as the optimal composition may differ from batch to batch.12,14,17 Numerous studies on CFPS employ plasmid DNA as templates.12,13,17,18,39 However, to efficiently couple the template DNA to our hydrogel matrix, linear PCR products were used for functionalization.7,10 Screening for optimal DNA concentration, depicted in Fig. 4, indicated an optimum of 5 nM with a yield of 62.2 ± 10.9 μg mL−1, whereas 10 nM gave a rather similar yield of 52.4 ± 9.9 μg mL−1, for deGFP-MatB. CFPS of deGFP-MatB was further screened for optimal concentration of PEG, as it was also used as a crosslinker, and it previously served as molecular crowding agent.2 The expected effect of increasing activity of the CFPS by increasing the crowding agent was not observed.18,34,36,38,39 While the cytosol of E. coli cells is a very crowded environment of approx. 300–400 mg mL−1 of macromolecules,40 the reason for our results may lay in a PEG-induced precipitation of mRNA, for instance. In the work of Ge et al., the effects of crowding by PEG and Ficoll were investigated in solution, indicating an improved stability of mRNA in the case of concentrations higher than 10% (w/w). Furthermore, it was shown by western blot that the amount of synthesized protein was reduced when increasing the PEG concentration, consistently with our findings.34 Since the optimal yield of deGFP-MatB was achieved with a concentration of PEG that was found to be 1% (w/w), the microgel-based CFPS was not supplemented further with PEG to avoid decreasing the yield of deGFP-MatB. PEG was used as a crosslinker at 0.75% (w/w) to form the microgel network and still achieving an appropriate porous network for CFPS.7 The porosity of the microgel network was adjusted to enable free diffusion of all necessary components of the transcription and translation machinery. The same porous network would also allow for utilizing the as-formed deGFP-MatB after CFPS in the conversion of malonate to malonyl-CoA. The enzymatic activity was successfully determined by using a DTNB assay. Currently, we implement experimental protocols to compare the activity of immobilized enzyme on microgels to the enzyme in solution, which is a crucial task.41A very recent publication pointed out that the activity of deGFP-MatB was increased compared to solution by immobilizing the enzyme covalently to a pNIPAAM linker on magnetic particle clusters.41 As the malonyl-CoA synthetase is an enzyme that activates malonate by CoA, it delivers a versatile building block for many important reactions as found in the polyketide or fatty acid synthesis pathway.42,43 To exploit the system shown in the present paper, further genes will be coupled to the microgel in the near future to enable CFPS of a complete functional biosynthetic pathway as another step towards a simple version of artificial cells utilizing an E. coli-based transcription and translation machinery.
Materials and methods
Materials
If not stated otherwise, chemicals and materials were used as received. E. coli star BL21 was a gift from Barbara Borgonovo from MPI-CBG. pBEST-OR2-OR1-Pr-UTR1-deGFP-T500 was a gift from Vincent Noireaux (Addgene plasmid # 40019). The homemade triblock surfactant and NTA-maleimide was a gift from Thomas Heida. The gene of malonyl-CoA synthetase matB was derived from Rhizobium trifolii. Primers and Chi6-DNA were ordered from biomers.net GmbH (Germany). The plasmid pET28a(+) was purchased from Merck KGaA (Germany). Amino acids, nicotinamide dinucleotide, and putrescine were purchased from Carl Roth GmbH + Co. KG (Germany). Folinic acid, spermidine, potassium glutamate, magnesium glutamate, t-RNA, oxalic acid, trichloro(1H,1H,2H,2H-perfluorooctyl)silane, Jeffamine® ED600, 1H,1H,2H,2H-perfluoro-1-octanol were purchased from Sigma-Aldrich (Germany). Krytox® 157 FSH was purchased from Chemours (Germany). ATP, CTP, UTP, GTP and T7 RNA polymerase were purchased from Promega (Germany). Phosphoenolpyruvic acid, and coenzyme A were purchased from Caymen Chemical (USA). NH-glutamate was purchased from Fisher Scientific (USA). Sodium hyaluronate (41–65 kDa) was purchased from Lifecore Biomedical (USA). 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) and 5-methylfurfurylamine were purchased from TCI (Germany). ATTO647N-maleimide was purchased from ATTO-TEC GmbH (Germany). PEG-mal2 (5 kDa) was purchased from JenKem (USA). HFE-7100 and HFE-7500 (both 3M™) were purchased from IoLiTec (Germany). For the preparation of master devices for microfluidic flow cell replication, SU-8 was used and developed in mr-Dev600 (micro resist technology GmbH, Germany). Microfluidic masters were prepared using a MJB3 mask aligner (Süss Mikro Tec, Germany). Sylgard 184 Elastomer Kit (Ellsworth Adhesives) was used for polydimethylsiloxane (PDMS) molding of the microfluidic master structures.Cloning and preparation of deGFP-matB gene
The cloning and the preparation of deGFP-matB gene followed a protocol reported earlier.41 In brief, matB from Rhizobium trifolii (Jena Microbial Research Collection JMRC:ST: 036047) was cloned into the pET28a(+)-H6TEV vector resulting in pET28a(+)-matB. The plasmid pBEST-OR2-OR1-Pr-UTR1-deGFP-T500 served as a source for the deGFP gene. That gene was also cloned into the pET28a(+)-matB as an insert between matB and the TEV site. Competent E. coli BL21 (DE3) were transformed with the as-formed plasmid. The plasmid was isolated using a plasmid preparation kit (Zymo Research Europe GmbH, Germany), and its amount and purity was verified using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Germany). The plasmid was used as template for polymerase chain reaction (PCR). For that, 100 ng of the isolated plasmid was used per experiment. A typical PCR mixture consisted of 200 μM dNTP, 0.5 μM of reverse and forward primer (cf. Table 1), 1× ThermoPol® reaction buffer, and 2.5 U Taq polymerase in a 50 μL reaction.| Name | Sequence |
|---|---|
| Forward primer | GAGCCCGATCTTCCCCATCG |
| Reverse primer | Azide-GCGTAACCACCACACCCG |
For the PCR a FlexCycler2 (Analytik Jena, Germany) was used and performed as presented in Table 2.
| Step | Parameters |
|---|---|
| Initial denaturation | 30 s at 95 °C |
| Denaturation (30×) | 30 s at 95 °C |
| Hybridization (30×) | 45 s at 56 °C |
| Elongation (30×) | 2:20 min at 68 °C |
| Final extension | 5 min at 68 °C |
After completion of the last PCR cycle, the reaction mixture was cooled down to room temperature. To analyze the product of the PCR, gel electrophoresis was performed. After confirmation of successful PCR, the product was purified via an innuPREP kit (Analytik Jena) and quantified by using a Nano-Drop ND-1000 spectrophotometer (Thermo Scientific).
Lysate preparation for CFPS
E. coli star BL21 (DE3) were streaked out on fresh LB agar plates and grown over night at 37 °C. An 1 L Erlenmeyer flask or a 2.5 L TUNAIR™ full-buffled shaking flask with 2xYTPG or CFAI medium was inoculated with a loop full of bacteria from plate and incubated for 15 h overnight. The OD was measured, and the bacteria were harvested at 4 °C, and 5000 rpm for 10 min. The pellet was washed once with a buffer containing 2 mM DTT, 10 mM Tris(OAc), 60 mM KOAc and 14 mM Mg(OAC)2 and flash-frozen. The pellet was then used for lysate preparation or stored at −80 °C. Frozen pellets were resuspended in the wash buffer with 1 mL per g dry weight of pellet, and lysed by 3 on-off-cycles (45 s/59 s) of sonication with a total energy of 850 J per 1.4 mL resuspension using a Q125 sonication device (Qsonica, USA). After lysis, DTT was added to a final concentration of 3.2 mM. The lysate was centrifuged for 10 min at 4 °C and 18,000g. Thereafter, the supernatant was incubated for 60 min at 37 °C and 220 rpm. The lysate was further characterized by a Bradford assay to monitor the efficiency of the sonication lysis by total protein content. Finally, the lysate was centrifuged for 10 min at 4 °C and 10,000g, and the supernatant was flash-frozen in liquid nitrogen. Until further usage, the lysate was stored at −80 °C.Cell-free protein synthesis of deGFP and deGFP-MatB
The reaction mixture for CFPS consisted of two solutions that were separately produced. Concentrations are provided as final concentrations in the combined reaction mixture. Solution A consisted of 1.28 mM of each UTP, CTP, GTP, 1.81 mM ATP, 0.26 mg mL−1 t-RNA, 1.48 mM spermidine, 0.55 mM putrescine, 0.25 mM CoA, 0.4 mM NAD, 0.82 mM oxalic acid, and 56.8 mM HEPES. Solution B further consisted of 125.0 mM K-glutamate and 10.0 mM Mg-glutamate. T7RNAP was added with 7 U per 10 μL reaction volume. Chi6 DNA was added to a final concentration of 1 μM. If not stated otherwise, template DNA was added to a final concentration of 5 nM. The lysate was used as 33% (v/v) of the reaction mixture. For online-measurements of CFPS, a reaction volume of 50 μL was placed in individual wells of a black 96-well plate in a TECAN Infinite® M200 PRO plate reader (Tecan Trading AG, Switzerland) and analyzed in 5 min intervals with an excitation wavelength of 488 nm and emission wavelength of 520 nm. For screening approaches as well as CFPS and immobilization of deGFP-MatB in HAmFU-DBCO microgels, the reaction was conducted as 8 μL-reactions in PCR tubes inside a thermocycler at 30 °C. CFPS reaction were analyzed in a 384-well plate afterwards. The concentration was analyzed by standard curve (Fig. S1†).Preparation of recombinant deGFP-MatB
Transformed E. coli BL21(DE3) harboring the plasmid pET28a(+)-deGFP-MatB were used to express the enzyme deGFP-MatB as previously described.41 The cells were grown overnight at 18 °C and lysed. The enzyme was purified by a HisTrap FF crude column using two buffers A (0.5 M NaCl, 0.1 M tris, pH 8.0) and B (0.5 M NaCl, 0.1 M tris, 0.5 M imidazole, pH 8.0), followed by a Superdex® 200 increase 10/300 size exclusion column (buffer C: 20 mM tris, 150 mM NaCl, pH 8.0). Samples were monitored by Bradford assay to characterize the protein amount.Enzyme activity of CFPS-formed deGFP-MatB by DTNB assay
5,5′-Dithiobis-(2-nitrobenzoic acid) (DTNB) assay was performed to determine the activity of deGFP-MatB. The assay consisted of 10 μL sample, 600 μM malonate, 100 μM CoA and 2.5 mM ATP in a final volume of 75 μL. The reaction was run at 30 °C and 220 rpm on a thermoshaker (MKR 13, Hettich Benelux). The reaction was stopped by adding 25 μL of 15 mM EDTA. To detect the free thiol groups, 20 μL of 1.5 mM DTNB was added. After 15 min of incubation, the absorbance was measured at 410 nm.Synthesis of DBCO-functionalized HAmFU
Functionalization of HA with 5-methylfuran and DBCO moieties was accomplished as reported previously.7 In brief, to a stirred solution composed of 0.5% (w/w) HA buffered with 0.1 M 2-(N-morpholino)ethansulfonic acid (MES) at pH 5.5, DMTMM was added in a threefold molar excess to HA and stirred for 30 min. Following that, 5-methylfurfurylamine was added dropwise in threefold molar excess to HA and stirred for 5 days to complete the functionalization. The solution was dialyzed against 0.1 M NaCl for 2 days and against deionized water for another 2 days using a 10 kDa MWCO tubing. After lyophilization, the successful synthesis of 5-methylfuran functionalized HA (HAmFU) was confirmed by 1H-NMR spectroscopy. Then, 100 mg HAmFU was dissolved in 20 mL MilliQ water. 15.38 mg of DBCO-PEG4-maleimide was dissolved in 500 μL DMSO and added dropwise under stirring to the HAmFU solution. The reaction was stirred for 24 h and dialyzed as described above. Again, 1H-NMR spectroscopy was applied after lyophilization to characterize the synthesis product as reported earlier.7 1H-NMR (500 MHz, D2O): δ = 7.77 (m, 1H), 7.52 (m, 7H), 6.38 (m, 1H), 6.07 (m, 1H), 5.17 (m, 1H), 4.7–3.0 (HA-backbone/PEG/DBCO), 2.30 (m, 3H), 2.08 ppm (m, 3H).Synthesis of NTA-maleimide
To synthesize NTA-maleimide as reported previously,7 2.0 g of 6-maleimidohexanoic acid and 2.18 g of N-hydroxysuccinimide were dissolved in 40 mL dry dimethylformamide under dry and inert conditions. 3.9 g of N,N′-dicyclohexylcarbodiimide (DCC) was added to the reaction and stirred over night at room temperature. 1.02 g of oxalic acid was added to the reaction to remove non-reacted DCC by forming dicyclohxylurea. The mixture was stirred for another hour, and the precipitate was filtered off. The extraction of the solution was performed by dichloromethane, and drying was achieved by Na2SO4 to receive a pale-yellow oil. 230 mg of the obtained NHS-ester was dissolved in 10 mL acetonitrile. 131 mg of Nα,Nα-bis(carboxymethyl)-L-lysine hydrate was dissolved in 10 mL of aqueous 0.3 M NaHCO3 buffer with pH 8.5. The activated NHS-ester was added dropwise, and the reaction was stirred overnight. Acidification of the solution to pH 3 was achieved using 0.1 M HCl. Rotary evaporation was applied to reduce the volume of the solution. Residues were washed with acetonitrile and lyophilized. Successful synthesis and purity were confirmed by 1H-NMR.Preparation of PDMS-based microfluidic devices
PDMS-based microfluidic devices were prepared using conventional photo- and soft lithography.44 SU-8 as a negative photoresist was spin-coated on a silicon wafer. A mask aligner was used to transfer the computer-aided design of the microfluidic flow cell into the photoresist. Afterwards, non-polymerized SU-8 was removed with a developer solution to obtain the desired microchannel structures. Sylgard 184 Elastomer Kit was used with PDMS and a crosslinker in a ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and poured on the master structure. PDMS was cured for 2 h at 65 °C. Inflow and outflow ports were stamped into the device using a biopsy needle. The received replica was bonded on a glass surface using oxygen plasma treatment.
:1 and poured on the master structure. PDMS was cured for 2 h at 65 °C. Inflow and outflow ports were stamped into the device using a biopsy needle. The received replica was bonded on a glass surface using oxygen plasma treatment.
Microfluidic preparation of DNA-/NTA-functionalized HAmFU-DBCO-based microgels
For preparing DNA-functionalized HAmFU-DBCO precursors, 15 μg azide-modified DNA were incubated with 5.5 mg HAmFU-DBCO for 5 h at room temperature. Afterwards, the solution was lyophilized and stored at −20 °C. Microgels were produced by water-in-oil emulsion droplets formed inside PDMS-based microfluidic devices. The dispersed aqueous phase consisted of 3.7% (w/w) DNA-functionalized HAmFU-DBCO, pre-functionalized with 2.0% (w/w) NTA-maleimide and then dissolved in 150 μL 100 mM MES buffer at pH 5.5, supplemented with 1 μL of ATTO647N (1 mg mL−1), and 0.75% (w/w) of PEG-mal2. As continuous oil phase, HFE-7500 was used with 2.0% (w/w) of triblock copolymer surfactant. A flow-focusing device with a channel height and width of 25 μm, respectively, was used to compartmentalize the aqueous phase into uniform droplets by applying flow rates of 40 μL h−1 (dispersed phase) and 900 μL h−1 (continuous phase), respectively. Droplet formation was monitored using an Axio Vert.A1 (Carl Zeiss Microscopy GmbH, Germany) and a Phantom Micro C110 high-speed camera (Vision Research Inc., USA). The as-formed emulsion droplets were stored for 2 days at room temperature to complete their solidification and microgel formation, respectively. Purification of the microgels was then performed by washing the particles several times with 20% (v/v) 1H,1H,2H,2H-perfluoro-1-octanol in HFE-7500, followed by removal of the oil phase and transfer of the particles into 1 mM NiSO4 solution for activating the microgel-bound NTA groups.Conclusion
The present manuscript demonstrates a bottom-up approach for building up cell-like microgels to facilitate CFPS of useful enzymes moving beyond the microgel-based cell-free production of conventional model proteins, e.g., GFP. For that, we utilize a set of simple starting materials: bacterial lysate extracted via well-established protocols, linear DNA from PCR, and tailor-made hydrogel precursors from commercial macromolecules. In contrast to membrane-based cell-free systems like vesicles, we emphasize the advantage of our microgel-based approach, which is intrinsically open to its surrounding media. Thus, purification of a protein of interest reversibly bound to the microgel matrix as well as its characterization by (colorimetric) assays is greatly facilitated. The future perspective will be to combine microgels carrying the genetic information for MatB with other DNA-functionalized microgels that will enable the in situ setup of enzymatic cascade reactions by CFPS towards the synthesis of polyketides like naringenin. With that, two complex functions of a cell – the readout of its genetic code and the implementation of cascade reactions towards protein-based signalling molecules and drugs – will be coupled on a single experimental platform as a crucial step towards the design of artificial cells based on microgels.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Max J. Männel for drawing CADs of microfluidic devices. We also thank Nicolas Hauck and Max J. Männel for critical discussions during preparation of this manuscript. We thank the Federal Ministry of Education and Research (BMBF, Biotechnology2020+: Leibniz Research Cluster, 031A360C) and the German Research Foundation (DFG, TH 2037/1-1 and Research Training Group GRK 1865: Hydrogel-based Microsystems) for financial support. J. T. also acknowledges the Young Investigator Program of the Technische Universität Dresden. J. T. receives funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant agreement no. 852065).References
- G. Fracasso, Y. Körner, D. T. T. Gonzales and T. Y. Dora Tang, Exp. Biol. Med., 2019, 244, 314–322 CrossRef CAS.
- M. M. Hansen, L. H. Meijer, E. Spruijt, R. J. Maas, M. V. Rosquelles, J. Groen, H. A. Heus and W. T. Huck, Nat. Nanotechnol., 2016, 11, 191–197 CrossRef CAS.
- G. Körfer, C. Pitzler, L. Vojcic, R. Martinez and U. Schwaneberg, Sci. Rep., 2016, 6, 26128 CrossRef.
- M. Fischlechner, Y. Schaerli, M. F. Mohamed, S. Patil, C. Abell and F. Hollfelder, Nat. Chem., 2014, 6, 791–796 CrossRef CAS.
- H. Niederholtmeyer, C. Chaggan and N. K. Devaraj, Nat. Commun., 2018, 9, 5027 CrossRef.
- L. Aufinger and F. C. Simmel, Angew. Chem., Int. Ed., 2018, 57, 17245–17248 CrossRef CAS.
- T. Heida, T. Köhler, A. Kaufmann, M. J. Männel and J. Thiele, ChemSystemsChem, 2020, 2(3), e1900058 CAS.
- Y. Jiao, Y. Liu, D. Luo, W. T. S. Huck and D. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 29308–29313 CrossRef CAS.
- J. S. Kahn, R. C. Ruiz, S. Sureka, S. Peng, T. L. Derrien, D. An and D. Luo, Biomacromolecules, 2016, 17, 2019–2026 CrossRef CAS.
- J. Thiele, Y. Ma, D. Foschepoth, M. M. Hansen, C. Steffen, H. A. Heus and W. T. Huck, Lab Chip, 2014, 14, 2651–2656 RSC.
- Y. Shimizu, A. Inoue, Y. Tomari, T. Suzuki, T. Yokogawa, K. Nishikawa and T. Ueda, Nat. Biotechnol., 2001, 19, 751–755 CrossRef CAS.
- M. Z. Levine, N. E. Gregorio, M. C. Jewett, K. R. Watts and J. P. Oza, J. Visualized Exp., 2019, 144, e58882 Search PubMed.
- M. Z. Levine, B. So, A. C. Mullin, K. R. Watts and J. P. Oza, bioRxiv, 2019, 729699 Search PubMed.
- Z. Z. Sun, C. A. Hayes, J. Shin, F. Caschera, R. M. Murray and V. Noireaux, J. Visualized Exp., 2013, e50762, DOI:10.3791/50762.
- M. M. Hansen, M. Ventosa Rosquelles, M. Yelleswarapu, R. J. Maas, A. J. van Vugt-Jonker, H. A. Heus and W. T. Huck, ACS Synth Biol, 2016, 5, 1433–1440 CrossRef CAS.
- H.-C. Kim, T.-W. Kim and D.-M. Kim, Process Biochemistry, 2011, 46, 1366–1369 CrossRef CAS.
- B. Lavickova and S. J. Maerkl, ACS Synth. Biol., 2019, 8, 455–462 CrossRef CAS.
- M. C. Jewett and J. R. Swartz, Biotechnol. Bioeng., 2004, 86, 19–26 CrossRef CAS.
- J. F. Zawada, G. Yin, A. R. Steiner, J. Yang, A. Naresh, S. M. Roy, D. S. Gold, H. G. Heinsohn and C. J. Murray, Biotechnol. Bioeng., 2011, 108, 1570–1578 CrossRef CAS.
- L. E. Contreras-Llano and C. Tan, Synth. Biol., 2018, 3(1), ysy012 CrossRef CAS.
- G. Kanter, J. Yang, A. Voloshin, S. Levy, J. R. Swartz and R. Levy, Blood, 2007, 109, 3393–3399 CrossRef CAS.
- Y. Kuruma, P. Stano, T. Ueda and P. L. Luisi, Biochim. Biophys. Acta, 2009, 1788, 567–574 CrossRef CAS.
- A. Scott, M. J. Noga, P. de Graaf, I. Westerlaken, E. Yildirim and C. Danelon, PLoS One, 2016, 11, e0163058 CrossRef.
- J. Thiele, Y. Ma, S. M. Bruekers, S. Ma and W. T. Huck, Adv. Mater., 2014, 26, 125–147 CrossRef CAS.
- J. A. Burdick and G. D. Prestwich, Adv. Mater., 2011, 23, H41–H56 CrossRef CAS.
- B. S. Spearman, N. K. Agrawal, A. Rubiano, C. S. Simmons, S. Mobini and C. E. Schmidt, J. Biomed. Mater. Res., Part A, 2020, 108, 279–291 CrossRef CAS.
- E. Zerobin, M. Markovic, Z. Tomášiková, X. H. Qin, D. Ret, P. Steinbauer, J. Kitzmüller, W. Steiger, P. Gruber, A. Ovsianikov, R. Liska and S. Baudis, J. Polym. Sci., 2020, 58, 1288–1298 CrossRef CAS.
- J. L. Dopp, S. M. Rothstein, T. J. Mansell and N. F. Reuel, Biotechnol. Bioeng., 2019, 116, 667–676 CrossRef CAS.
- K. Hufnagel, S. Lueong, M. Willhauck-Fleckenstein, A. Hotz-Wagenblatt, B. Miao, A. Bauer, A. Michel, J. Butt, M. Pawlita, J. D. Hoheisel and T. Waterboer, Sci. Rep., 2018, 8, 7503 CrossRef.
- H. Nevenzal, M. Noach-Hirsh, O. Skornik-Bustan, L. Brio, E. Barbiro-Michaely, Y. Glick, D. Avrahami, R. Lahmi, A. Tzur and D. Gerber, Commun. Biol., 2019, 2, 42 CrossRef.
- X. Yu, B. Petritis and J. LaBaer, Proteomics, 2016, 16, 1238–1250 CrossRef CAS.
- G. L. Rosano and E. A. Ceccarelli, Front. Microbiol., 2014, 5, 172 Search PubMed.
- J. H. An and Y. S. Kim, Eur. J. Biochem., 1998, 257, 395–402 CrossRef CAS.
- X. Ge, D. Luo and J. Xu, PLoS One, 2011, 6, e28707 CrossRef CAS.
- J. L. Dopp, Y. R. Jo and N. F. Reuel, Synth. Syst. Biotechnol., 2019, 4, 204–211 CrossRef.
- N. E. Gregorio, W. Y. Kao, L. C. Williams, C. M. Hight, P. Patel, K. R. Watts and J. P. Oza, ACS Synth. Biol., 2020, 4(9), 766–778 CrossRef.
- R. Marshall, C. S. Maxwell, S. P. Collins, C. L. Beisel and V. Noireaux, Biotechnol. Bioeng., 2017, 114, 2137–2141 CrossRef CAS.
- N. Michel-Reydellet, K. Woodrow and J. Swartz, J. Mol. Microbiol. Biotechnol., 2005, 9, 26–34 CrossRef CAS.
- F. Caschera and V. Noireaux, Artificial Life, 2016, 22, 185–195 CrossRef.
- S. B. Zimmerman and S. O. Trach, J. Mol. Biol., 1991, 222, 599–620 CrossRef CAS.
- B. P. Krishnan, L. O. Prieto Lopez, S. Hoefgen, L. Xue, S. Wang, V. Valiante and J. Cui, ACS Appl. Mater. Interfaces, 2020, 12(18), 20982–20990 CrossRef CAS.
- H. Chen, H. U. Kim, H. Weng and J. Browse, Plant Cell, 2011, 23, 2247–2262 CrossRef CAS.
- H. A. Crosby, K. C. Rank, I. Rayment and J. C. Escalante-Semerena, Appl. Environ. Microbiol., 2012, 78, 6619–6629 CrossRef CAS.
- M. P. Hughes, K. F. Hoettges and M. P. Hughes, Microengineering in biotechnology, Springer, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 calibration curve for deGFP-MatB, Fig. S2: comparison of lysates regarding their overall protein production; Fig. S3: DTNB assay for determining the enzymatic activity; Fig. S4: microfluidic water-in-oil (W/O) emulsion preparation. See DOI: 10.1039/d0ra06702d |
| This journal is © The Royal Society of Chemistry 2020 |