
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium-catalyzed, site-selective C–H activation: access to C5-substituted azaflavanone†
Manickam Bakthadoss *a,
Tadiparthi Thirupathi Reddya and
Duddu S. Sharada*b
*a,
Tadiparthi Thirupathi Reddya and
Duddu S. Sharada*b
aDepartment of Chemistry, Pondicherry University, Puducherry – 605014, India. E-mail: bhakthadoss@yahoo.com
bDepartment of Chemistry, Indian Institute of Technology, Hyderabad, Telangana-502285, India
First published on 26th August 2020
Abstract
A site-selective ruthenium-catalyzed keto group assisted C–H bond activation of 2-aryl tetrahydroquinoline (azaflavanone) derivatives has been achieved with a variety of alkenes for the first time. A wide range of substrates was utilized for the synthesis of a wide variety of alkenylated azaflavanones. This simple and efficient protocol provides the C5-substituted azaflavanone derivatives in high yields with a broad range of functional group tolerance. Further, the C5-alkenylated products were converted into substituted 2-aryl quinoline derivatives in good yields.
Introduction
The activation and functionalization of the inert C–H bond by metals has been one of the active research thrust areas among organic chemists1 in recent years. Notably, the directing group assisted C–H bond functionalization has been intensely investigated in the past decades. It is also important to note that the cleavage of the inert C–H bond (110 kcal mol−1) requires harsh conditions. Moreover, the protocol suffers limited substrate scope as well as functional group tolerance. Therefore, C–H bond activation has not yet found widespread importance in organic synthesis. Site-selective C–H activation catalyzed by transition metals is a challenging task in organic chemistry.2 Selectivity in C–H functionalization is addressed through the use of a suitable directing group on a substrate where coordination of the metal on a specific site of the substrate controls the outcome of the reaction. However, the use of a directing group strategy suffers from the installation and removal of the directing group, which is an additional synthetic step and can be avoided. Therefore, the utilization of the intrinsic directing group is highly anticipated in C–H activation reactions.Due to the limitations of the direct C–H functionalization where controlling site selectivity is a major concern for an organic chemist due to the presence of multiple C–H bonds having similar bond dissociation energy and reactivity, we have proposed a substrate controlled strategy where the directing group is a part of the substrate. The site selectivity can be easily controlled due to the coordination ability of the directing group present in the substrate, which therefore improves the reaction's outcome.
Generally, the synthesis of organic compounds with multifunctional groups is one of the essential thrust areas in the field of synthetic organic chemistry due to their importance in serving as building blocks for the construction of various useful molecules.3 Nitrogen-containing heterocyclic motifs are prominent in many biologically active compounds, pharmaceuticals and materials. 1,2,3,4-Tetrahydroquinolines are important scaffolds due to their excellent pharmacological properties; for example, several tetrahydroquinolines possess potent antimitotic antitumor effects through inhibition of tubulin polymerization at the colchicine site.4 Therefore, several research groups5 have shown interest in functionalizing the tetrahydroquinoline scaffold to identify bioactive compounds with improved biological activity and interesting material-related properties. Few reports are available in the literature where ketone is utilized as the directing group6 despite the presence of a strong coordinating center. Peng group reported tandem C–H activation and aza-Michael addition of 2-arylquinazolin-4-ones with acrylates7 while Kumar and coworkers have described Pd-catalyzed regioselective arylation of quinolin-4(1H)-ones using diaryliodonium salts as aryl source8 (Fig. 1). Bhisma et al. showed ketone directed C–H/O–H annulation of 2-arylquinoline with alkynes9 where 2-arylquinolinone acts as a masked phenolic (tautomerization) substrate as directing group as shown in Fig. 1. In continuation of our research work10 in the field of C–H activation, herein, we disclose an interesting keto group directed chemoselective and non-annullable C–H functionalization of 2-aryl tetrahydroquinoline with alkenes. We have successfully achieved a high degree of chemoselectivity where a weak keto group act as an intrinsic directing group than the potent secondary amine thereby providing a good selectivity and reactivity over a variety of substrates with excellent yields (Fig. 1).
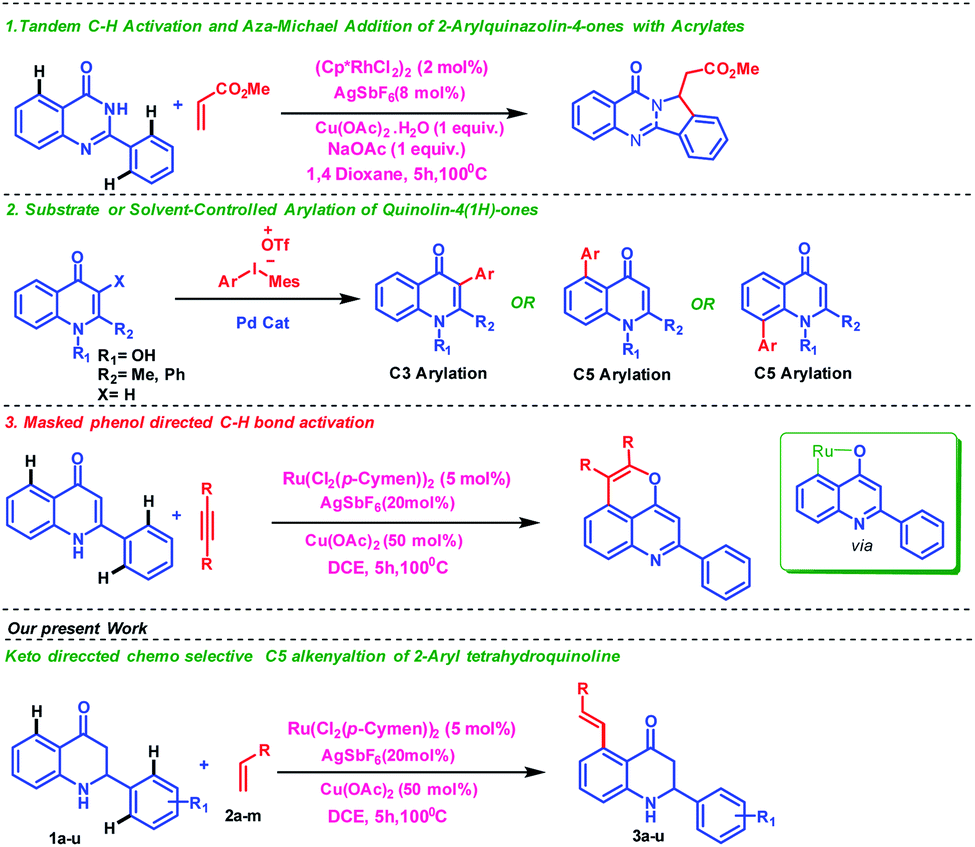 | ||
| Fig. 1 Comparison between previous reports and our work. | ||
To execute our idea, we have chosen 2-phenyl tetrahydroquinoline13 1a as a model substrate to carry out the C–H activation reaction under various reaction conditions (Table 1 see in ESI†). The 2-phenyl tetrahydroquinoline (1a, 0.2 mmol) and methyl acrylate (2a, 0.4 mmol) were subjected in the presence of [Ru(p-cymene)Cl2]2 (5 mol%) as a catalyst, AgSbF6 as an activator and Cu(OAc)2 as an oxidant in 1,4-dioxane as solvent which interestingly provided the (E)-methyl 3-(4-oxo-2-phenyl-1,2,3,4-tetrahydroquinolin-5-yl)acrylate (3a) with 54% yield (entry 1, Table 1 see in ESI†). The product 3a was confirmed from 1H NMR, 13C NMR spectroscopy and ESI-MS. On the contrary, when the reaction performed in the same reaction condition, except Cu(OAc)2.H2O as an oxidant, the desired product 3a was observed in low yield (entry 2, Table 1 see in ESI†). To increase the yield of the desired product 3a, we change the solvent system.
In the presence of dichloroethane as a solvent and the same reaction condition, surprisingly, the desired product 3a was observed in 94% yield. The reaction was performed in different solvent systems/additives/oxidants to attain the optimal reaction condition. However, these reactions condition failed to deliver the desired product 3a in better yield compared to the previous experiment (entry 1, Tables 4–13 see in ESI†). Based on these studies, we conclude that the optimal reaction condition for the C–H activation reaction of 2-aryl tetrahydroquinoline involves 1a (1 equiv.), 2a (2 equiv.), Ru cat. (5 mol%), AgSbF6 (20 mol%), and Cu(OAc)2 (50 mol%) in DCE solvent at 100 °C for 5 h.
With this optimal reaction condition in our hand, we started to explore the substrate scope of different alkene for the C–H activation of 2-phenyl tetrahydroquinoline 1a. The reaction performed well in various alkyl acrylates such as ethyl (2b), butyl (2c) tertiary butyl (2d), benzyl (2f) and phenyl acrylate (2g) which deliver the alkene coupled 2-phenyl tetrahydroquinoline products (3b–d, f, g) in excellent yields (84–93%). Structure 3b was further confirmed through single-crystal X-ray analysis,12 as shown in Fig. 2. Also, we employed branched acrylates like 2-ethyl hexyl (2h), norbornyl acrylate (2i) for the reaction, which underwent smoothly and leads to C5-coupled products (3h and 3i) in excellent yields (95% and 94% respectively). Additionally, this coupling reaction turned out to be a versatile reaction since employing the reaction of 1a with methyl vinyl ketone (2e) and phenyl vinyl sulfone (2m), the selective C5-olefinated products were formed (3e and 3m) in good yields. It is noteworthy that on the treatment of 2-phenyl tetrahydroquinoline 1a with acrylic acid 2j under the standard condition, the acrylic acid coupled product (3j) was formed in excellent yield (87%). We have also carried out the reaction of styrene (2k) with acrylonitrile (2i), and we observed that the anticipated products 3k and 3l were formed in good yields (90% and 56% with E/Z isomer: 52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :48 and 58:42 respectively) (Scheme 1).
:48 and 58:42 respectively) (Scheme 1).
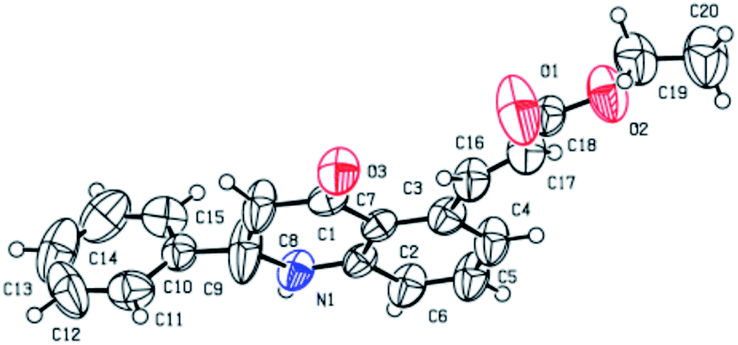 | ||
| Fig. 2 ORTEP diagram of compound 3b.12 | ||
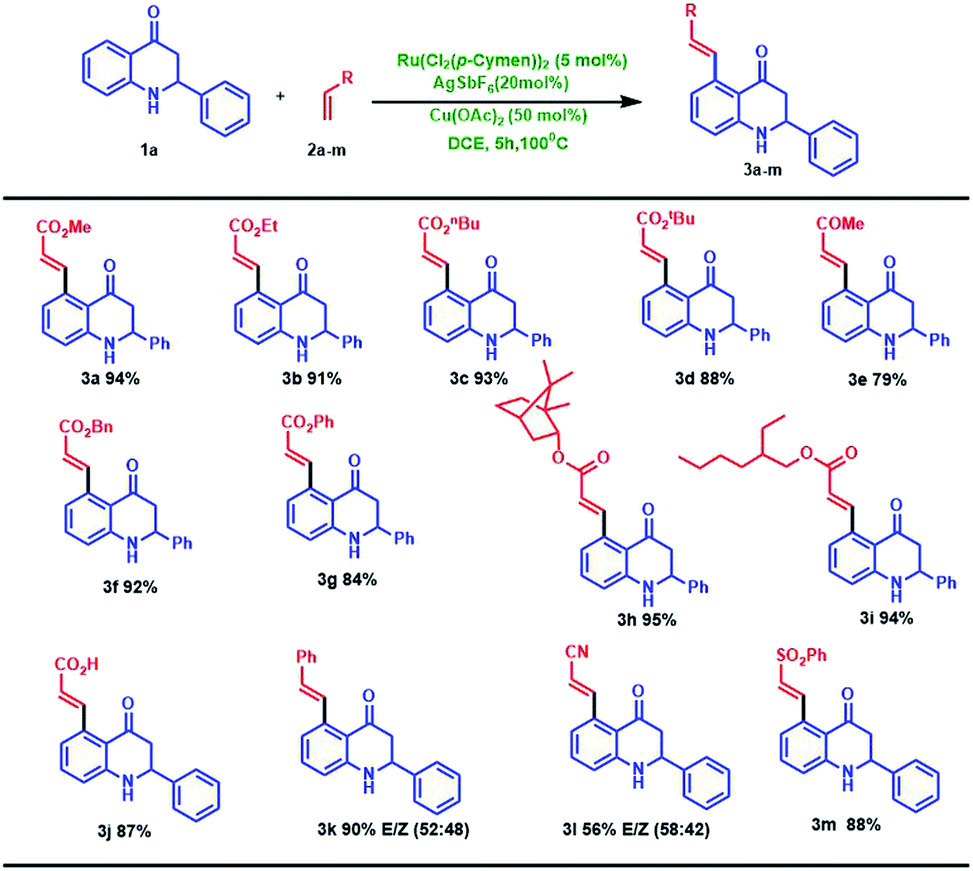 | ||
| Scheme 1 Screening of various alkenes with 2 phenyl tetrahydroquinoline.a,b aReaction conditions: 1a (0.2 mmol), 2a–m (0.4 mmol), [{Ru(p-cymene)Cl2}2] (5 mol%), Cu(OAc)2 (50 mol%), AgSbF6 (20 mol%), DCE solvent at 100 °C, 5 h. bIsolated yields. | ||
After screening various alkenes, we turned our attention towards the substrate scope of 2-aryl tetrahydroquinoline derivatives (1b–h) containing different substitution on the aryl group.
Notably, the highly selective C5-olefinated products (3n–u) were formed in excellent yields (85–91%, Scheme 2). We used 2-thiophene substituted tetrahydroquinoline 1i with methyl acrylate 2a in the presence of the same reaction condition; the desired coupled product 3u was formed in 81% yield as shown in Scheme 2.
 | ||
| Scheme 2 Substrate scope of azaflavanone derivatives with methyl acrylate.a,b aReaction conditions: 1b–h (0.2 mmol), 2a (0.4 mmol), [{Ru(p-cymene)Cl2}2] (5 mol%), Cu(OAc)2 (50 mol%), AgSbF6 (20 mol%), DCE solvent at 100 °C, 5 h. bIsolated yields. | ||
Subsequently, we explored the scope of C6, C8-bromo substituted 2-aryl tetrahydroquinoline (4a–b) with methyl acrylate (2a) in the presence of optimal reaction condition. In this case, highly selective C5-alkenylated products 5a and 5b were obtained selectively in high yields, as shown in Scheme 3. These bromo products (5a and 5b) will be beneficial for further coupling reactions.
 | ||
| Scheme 3 Substrate scope of C6, C8 dibromo substituted azaflavanone with alkene. | ||
It is noteworthy that the reaction was scalable in a standard laboratory set-up, as shown in Scheme 4. The reaction was performed at a one-gram scale at the standard reaction conditions, and the desired product was obtained in 86% yield.
 | ||
| Scheme 4 Gram scale synthesis. | ||
Mechanistic studies toward ruthenium-catalyzed C–H activation of 2-phenyl tetrahydroquinoline 1a have been performed (Scheme 5). An intermolecular competition experiment between 2-phenyl tetrahydroquinoline (1a) and chromanone (6a) with methyl acrylate 2a under standard condition lead to chromanone coupled product 7a with 27% yield and tetrahydroquinoline coupled product 3a with 71% (Scheme 5, eqn (1)). This study cleverly reveals that 2-phenyl tetrahydroquinoline 1a is more reactive than 2-phenyl chromanone 6a under standard reaction conditions. On the other hand, intermolecular competitive experiments between different alkenes such as methyl acrylate 2a and phenyl vinyl sulfone 2m with 2-phenyl tetrahydroquinoline 1a were carried out under the standard reaction condition (Scheme 5, eqn (2)). The result indicated that the methyl acrylate coupled product 3a is obtained in more yield (68%) compared to the phenyl vinyl sulfone coupled product 3m (32%)
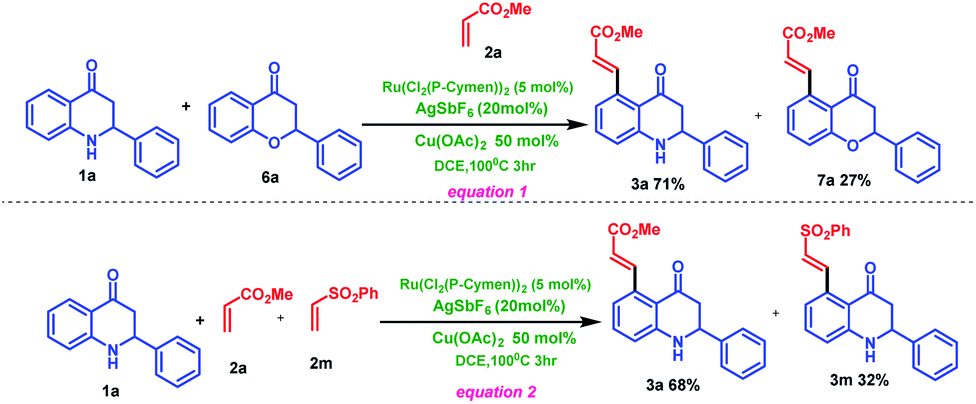 | ||
| Scheme 5 Competitive reactions. | ||
To expand the scope of the reaction further, 2-aryl tetrahydroquinoline was treated 1a with diphenyl acetylene 8, which fails to produce C5-coupled product 9. To identify the carbonyl group's importance, we have carried out a control experiment where we utilized the corresponding alcohol substrate (10) under the same reaction condition, which failed to produce the desired C5-coupled product 11. This observation indicates that the carbonyl group is very crucial for the C–H activation reaction (Scheme 6).
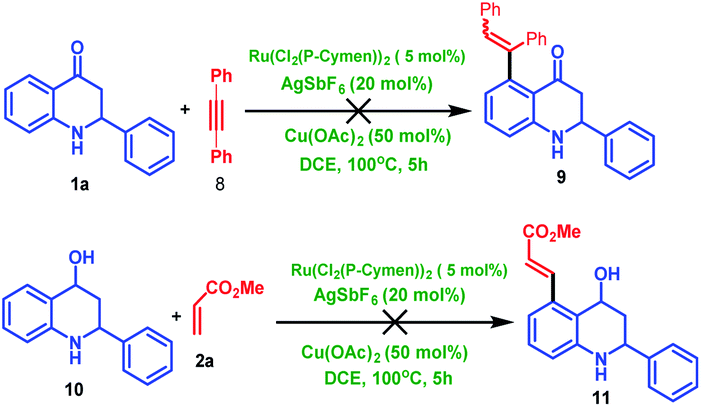 | ||
| Scheme 6 Control experiment for directing group and alkyne. | ||
To probe the synthetic applicability of this newly synthesized C5-coupled 2-aryl tetrahydroquinoline moiety, we performed chemical transformation of C5-coupled azaflavanone (3) as shown in Scheme 7.14 Accordingly, on the treatment of azaflavanone (3a, 3b, 3o, and 3m) with iodine (1 equiv.) in the presence of methanol solvent, desired substituted quinoline derivatives (12a–d) were obtained in excellent yields (Scheme 7). These quinoline derivatives have great significance in organic synthesis and materials chemistry.
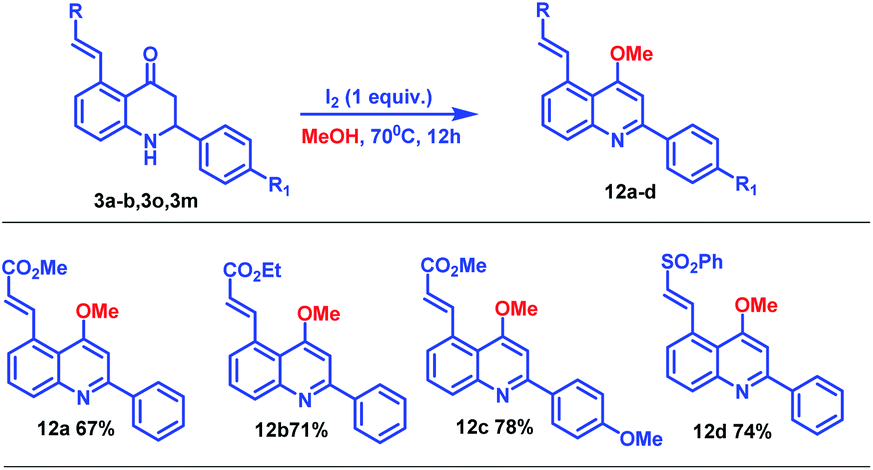 | ||
| Scheme 7 Synthesis of alkyl (E)-3-(4-methoxy-2-phenylquinolin-5-yl) acrylate. | ||
Furthermore, the treatment of 3o with zinc powder in acetic acid condition successfully afforded the alkene reduced product 13 in a chemoselective manner without reducing the carbonyl moiety, as shown in Scheme 8.
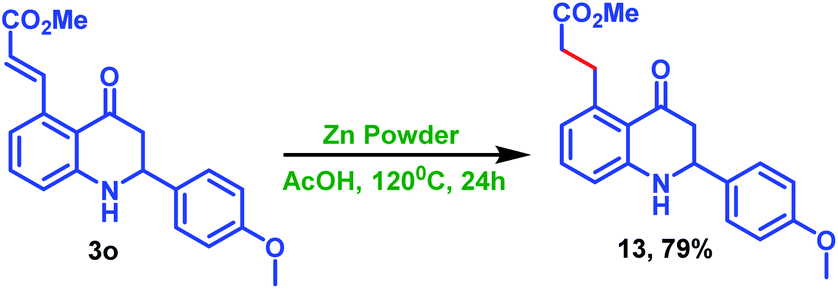 | ||
| Scheme 8 Selective alkene reduction of C5-coupled 2-phenyl tetrahydroquinoline 3o. | ||
A plausible catalytic cycle for the C–H activation reaction of 2-aryl tetrahydroquinoline is shown in Scheme 9. Active ruthenium complex was generated using {Ru(p-cymene)Cl2}2, silver hexafluoroantimonite, and Cu(OAc)2.10a,b,11 After the formation of active ruthenium complex, it coordinates at the C5-position of 2-aryl tetrahydroquinoline 1a by the elimination of acetic acid. The formation of ruthenacycle B occurs through the chelation of Ru with the oxygen atom of the keto group. The migratory insertion of methyl acrylate 2a into ruthenacycle B leads to the intermediate C. Finally, desired C5-alkenylated product 3a is generated by β-hydride elimination (reductive elimination) from the intermediate C and reoxidation by Cu(OAc)2 regenerate the catalytically active cationic species as shown in Scheme 9.
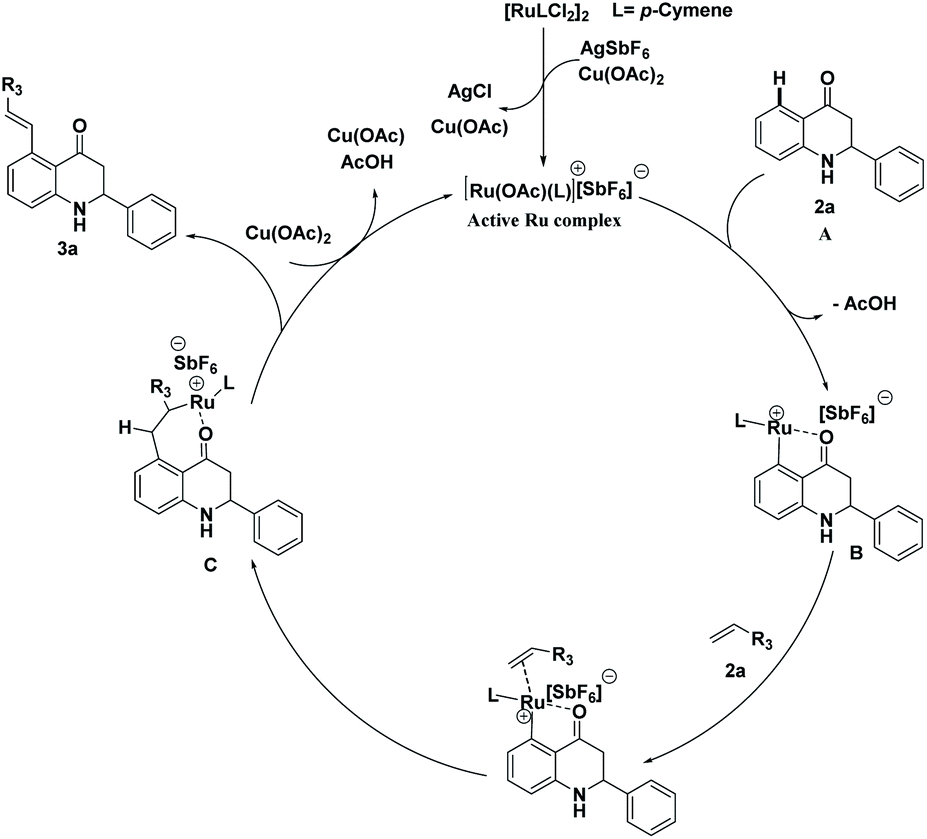 | ||
| Scheme 9 Proposed catalytic cycle for the C5-alkenylation of 2-aryl tetrahydroquinoline. | ||
Conclusion
To conclude, we successfully developed a simple and efficient protocol for the site-selective C5-alkenylation of 2-aryl tetrahydroquinoline derivatives with a wide variety of alkenes via C–H bond activation for the first time. A diverse library of C5-alkenylated 2-aryl tetrahydroquinoline derivatives was synthesized using simple reaction conditions in excellent yields. This new protocol provides a facile route for the synthesis of C5-alkenylated 2-aryl tetrahydroquinolines and also serves as a useful tool for further functional group transformations. It is clear from the experimental results that the keto group is acting as a directing group instead of an amine group present in the substrate. Since the azaflavanone are well known for their biological activities, we believe that the newly prepared C5-alkenylated 2-aryl tetrahydroquinolines also have similar biological activities, which will be very interesting to study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank DST-SERB for the financial support (Ref. No.: 2018/000930). TTR thanks UGC for the scholarship. We also acknowledge the DST-FIST for the ESI-HRMS facility.References
- Selected review; (a) W. Ma, P. Gandeepan, J. Li and L. Ackermann, Org. Chem. Front., 2017, 4, 1435 RSC; (b) D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649 CrossRef CAS; (c) N. K. Mishra, S. Sharma, J. Park, S. Han and I. S. Kim, ACS Catal., 2017, 7, 2821 CrossRef CAS; (d) F. Roudesly, J. Oble and G. Poli, J. Mol. Catal. A: Chem., 2017, 426, 275 CrossRef CAS; (e) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787 CrossRef CAS PubMed; (f) X. S. Xue, P. Zhou, B. Ji and J. P. Cheng, Chem. Rev., 2017, 117, 8622 CrossRef CAS PubMed; (g) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333 CrossRef CAS PubMed; (h) S. Agasti, B. Mondal, T. K. Achar, S. K. Sinha, A. S. Suseelan, K. J. Szabo, F. Schoenebeck and D. Maiti, ACS Catal., 2019, 9, 9606 CrossRef CAS; (i) A. Deb, S. Bag, R. Kancherla and D. Maiti, J. Am. Chem. Soc., 2014, 136, 13602 CrossRef CAS PubMed.
- Selected review; (a) P. Arockiam, C. Bruneau and P. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (b) T. Iwai and M. Sawamura, ACS Catal., 2015, 5(9), 5031 CrossRef CAS; (c) T. Gensch, M. N. Hopkinson, F. Glorius and W. Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (d) F. Kakiuchi, Y. Tanaka, T. Sato, N. Chatani and S. Murai, Chem. Lett., 1995, 679 CrossRef CAS; (e) M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843 RSC; (f) T. W. Lyons and M. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (g) G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450 CrossRef CAS PubMed; (h) S. Bag and D. Maiti, Synthesis, 2016, 48, 804–815 CrossRef CAS.
- (a) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447 CrossRef CAS PubMed; (b) M. Bakthadoss, J. Srinivasan and R. Selvakumar, Synthesis, 2012, 44, 755 CrossRef CAS; (c) B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114, 8323 CrossRef CAS PubMed; (d) D. Basavaiah, S. Pandiaraju, M. Bakthadoss and K. Muthukumaran, Tetrahedron: Asymmetry, 1996, 7, 997 CrossRef CAS; (e) M. Bakthadoss, G. Sivakumar and D. S. Sharada, Synthesis, 2013, 237–245 CAS; (f) M. Bakthadoss, N. Sivakumar and A. Devaraj, Tetrahedron Lett., 2015, 56, 4980 CrossRef CAS; (g) A. P. Marcos, P. Clarissa, N. Dayse, M. Buriol and P. MachadoKatritzky, Chem. Rev., 2009, 109, 4140 CrossRef PubMed; (h) M. Bakthadoss and A. Devaraj, Tetrahedron Lett., 2015, 56, 3954 CrossRef CAS; (i) M. Bakthadoss, D. Kannan and R. Selvakumar, Chem. Commun., 2013, 49, 10947 RSC; (j) P. Wu, A. K. Feldman and A. K. Nugent, et al., Angew. Chem., Int. Ed. Engl., 2004, 43, 3928 CrossRef CAS PubMed; (k) D. Basavaiah, M. Bakthadoss and G. Jayapal Reddy, Synth. Commun., 2002, 32, 689 CrossRef CAS; (l) M. Bakthadoss, N. Sivakumar and A. Devaraj, Synthesis, 2011, 4, 0611 CrossRef; (m) H. H. Li, C. Liu, Y. Zhang, Y. Sun, B. Wang and W. Liu, Org. Lett., 2015, 17, 932 CrossRef CAS PubMed; (n) B. M. Trost and J. S. Tracy, Org. Lett., 2017, 19, 2630 CrossRef CAS PubMed; (o) M. Bakthadoss, D. Kannan, N. Sivakumar, P. Malathi and V. Manikandan, Org. Biomol. Chem., 2015, 13, 5597 RSC; (p) M. Bakthadoss and R. Selvakumar, J. Org. Chem., 2016, 81, 3391 CrossRef CAS PubMed.
- (a) A. G. Cole, A. Metzger, M. R. Brescia, L. Y. Qin, K. C. Appell, C. T. Brain, A. Hallett, P. Ganju, A. A. Denholm and J. R. Wareing, et al., Bioorg. Med. Chem. Lett., 2009, 19, 119 CrossRef CAS PubMed; (b) D. S. Su, J. J. Lim, E. Tinney, B. L. Wan, M. B. Young, K. D. Anderson, D. Rudd, V. Munshi, C. Bahnck and P. J. Felock, et al., Bioorg. Med. Chem. Lett., 2009, 19, 5119 CrossRef CAS PubMed.
- (a) B. Nammalwar and R. A. Bunce, Molecules, 2014, 19, 204 CrossRef PubMed; (b) A. R. Katritzky, S. Rachwal and B. Rachwal, Tetrahedron, 1996, 52, 15031 CrossRef CAS; (c) V. Sridharan, P. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157 CrossRef CAS PubMed; (d) M. Muthukrishnan, V. Sridharan and J. C. Menéndez, Chem. Rev., 2019, 119(8), 5057 CrossRef PubMed.
- (a) P. Kishor and M. Jeganmohan, Org. Lett., 2011, 13, 6144 CrossRef PubMed; (b) M. Bhanuchandra, M. R. Yadav, R. K. Rit, M. R. Kurama and A. K. Sahoo, Chem. Commun., 2013, 49, 5225 RSC; (c) S. Pradhan, M. Mishra, P. B. De, S. Banerjee and T. Punniyamurthy, Org. Lett., 2020, 22, 1720 CrossRef CAS PubMed; (d) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764 RSC.
- X. Jiang, Q. Yang, J. Yuan, Z. Deng, X. Mao, Y. Peng and C. Yu, Tetrahedron, 2016, 72, 1238 CrossRef CAS.
- M. K. Mehra, S. Sharma, K. Rangan and D. Kumar, Eur. J. Org. Chem., 2020, 16, 2409 CrossRef.
- A. Banerjee, S. K. Santra, P. R. Mohanta and B. K. Patel, Org. Lett., 2015, 17, 5678 CrossRef CAS PubMed.
- (a) M. Bakthadoss, P. V. Kumar and T. S. Reddy, Eur. J. Org. Chem., 2017, 4439 CrossRef CAS; (b) M. Bakthadoss and P. V. Kumar, Adv. Synth. Catal., 2018, 360, 2650 CrossRef CAS; (c) M. Bakthadoss, P. V. Kumar, R. Kumar and V. Agarwal, Org. Biomol. Chem., 2019, 17, 4465 RSC; (d) M. Bakthadoss, P. V. Kumar, R. Kumar and M. surendar, New J. Chem., 2019, 43, 14190 RSC; (e) A. Murugan, V. N. Babu, A. Polu, N. Sabarinathan, M. Bakthadoss and D. S. Sharada, J. Org. Chem., 2019, 84, 7796 CrossRef CAS PubMed; (f) S. Arepally, V. N. Babu, M. Bakthadoss and D. S. Sharada, Org. Lett., 2017, 19, 5014 CrossRef CAS PubMed.
- (a) J. A. Leitch, P. B. Wilson, C. L. McMullin, M. F. Mahon, Y. Bhonoah, I. H. Williams and C. G. Frost, ACS Catal., 2016, 6, 5520 CrossRef CAS; (b) R. Das and M. Kapur, Chem.–Eur. J., 2016, 22, 16986 CrossRef CAS PubMed; (c) Q. Z. Zheng, Y. F. Liang, C. Qina and N. Jiao, Chem. Commun., 2013, 49, 5654 RSC; (d) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (e) K. Graczyk, W. Ma and L. Ackermann, Org. Lett., 2012, 16, 4110 CrossRef PubMed; (f) R. K. Chinnagolla and M. Jeganmohan, Chem. Commun., 2012, 48, 2030 RSC; (g) M. Selvaraju, Y. L. Wang and C. M. Sun, Org. Chem. Front., 2017, 4, 1358 RSC.
- The structure of the compound 3b was evidenced by single-crystal XRD analysis. CCDC 2005037 contains the supplementary crystallographic data for this paper.
- S. Chandrasekhar, K. Vijeender and Ch. Sridhar, Tetrahedron Lett., 2007, 48, 4935 CrossRef CAS.
- M. J. Mphahlele, F. K. Mogamisi, M. Tsanwani, S. M. Hlatshwayoa and R. M. Mampab, J. Chem. Res., 1999, 706 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2005037. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra06580c |
| This journal is © The Royal Society of Chemistry 2020 |