
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotally green cellulose conversion into bio-oil and cellulose citrate using molten citric acid in an open system: synthesis, characterization and computational investigation of reaction mechanisms†
Isabella Romeo,
Fabrizio Olivito*,
Antonio Tursi *,
Vincenzo Algieri,
Amerigo Beneduci,
Giuseppe Chidichimo,
Loredana Maiuolo,
Emilia Sicilia and
Antonio De Nino*
*,
Vincenzo Algieri,
Amerigo Beneduci,
Giuseppe Chidichimo,
Loredana Maiuolo,
Emilia Sicilia and
Antonio De Nino*
Dipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, Ponte P. Bucci, 87036 Arcavacata di Rende (CS), Italy. E-mail: denino@unical.it
First published on 18th September 2020
Abstract
The simultaneous transformation of crystalline or amorphous cellulose into a furan-based bio-oil and cellulose citrate was realized avoiding the use of strong inorganic acids, drastic conditions, enzymatic treatments or microorganism fermentation. This innovative method is very eco-friendly and involves the use of molten citric acid under solvent free conditions at atmospheric pressure. An accurate discussion on chemical composition of the bio-oil enriched in bioprivileged molecules as well as structural and morphological characterization of cellulose citrate was reported. Moreover, mechanistic hypotheses were formulated on the basis of experimental findings and detailed DFT quantum-mechanical simulations were carried out to confirm, step by step, the proposed reaction paths.
1. Introduction
The term biomass is a wide and general definition that includes all the organic materials derived from plants or organic wastes.1–4 This matter is originated during the photosynthesis of plants in which CO2, water and sunlight are converted into carbohydrates that represent the building blocks of the plant body. Lignocellulosic material represents the dry part of the plant structure and can be used as renewable and sustainable energy source.5,6 It is composed of cellulose, hemicellulose and lignin, that can be converted into fuels and chemicals by numerous procedures.7–11 Cellulose is the main component with an average percentage of 40–50 of the weight. This is a stable polysaccharide, composed of β-D-glucopyranoses, approximately between 5000 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units, linked by β-1,4-glycosidic bonds. β-Glycosidic bonds force the structure to a linear arrangement, because of the pyranose chair conformation and the equatorial disposition of the substituents, that corresponds to a minimum of energy.12 The structure stability is one of the main problems for cellulose depolymerisation, especially for microcrystalline cellulose. In fact, it possesses an ordinate reticulate structure due to the great number of intra- and inter-molecular hydrogen bonds and additional van der Waals forces that are related to its recalcitrant character.13,14 Hemicellulose contributes between 25–30% of the weight. Hemicellulose differs from cellulose as its structure is characterized by short branched chains consisting of various glucose, mannose, xylose, galactose, arabinose, 4-O-methyl glucuronic acid and galacturonic acid units. Lignin is the third wood component. It is the less abundant with an average of 15–30% of the weight. This polymer is composed of phenylpropane units, that arrange in an amorphous highly branched structure.15–17 As hydrophobic lignin shells protect cellulose and hemicellulose from enzymatic or chemical attacks, delignification allows to reach milder and easier conditions for depolymerization of the cellulosic material. Generally, during the delignification process, hemicellulose is also solubilized and removed from the cellulosic core.18–20 Cellulose derived compounds represent high value platform chemicals for industrial or chemical purposes.21–23 One of the most promising material derived from cellulose or from raw biomass is a mixture of compounds called bio-oil.24,25 Pyrolysis under elevated temperature and in the absence of oxygen is a common method for the production of pyrolytic oils for fuel or other industrial applications. The use of reactor systems and severe conditions limits the scalability of the process together with other factors like by-products formation. The most common by-products of pyrolysis are biochar, humins, gas and tar. The temperature commonly reached is in the range 450–500 °C. This procedure has a remarkably energetic cost.26–28
000 units, linked by β-1,4-glycosidic bonds. β-Glycosidic bonds force the structure to a linear arrangement, because of the pyranose chair conformation and the equatorial disposition of the substituents, that corresponds to a minimum of energy.12 The structure stability is one of the main problems for cellulose depolymerisation, especially for microcrystalline cellulose. In fact, it possesses an ordinate reticulate structure due to the great number of intra- and inter-molecular hydrogen bonds and additional van der Waals forces that are related to its recalcitrant character.13,14 Hemicellulose contributes between 25–30% of the weight. Hemicellulose differs from cellulose as its structure is characterized by short branched chains consisting of various glucose, mannose, xylose, galactose, arabinose, 4-O-methyl glucuronic acid and galacturonic acid units. Lignin is the third wood component. It is the less abundant with an average of 15–30% of the weight. This polymer is composed of phenylpropane units, that arrange in an amorphous highly branched structure.15–17 As hydrophobic lignin shells protect cellulose and hemicellulose from enzymatic or chemical attacks, delignification allows to reach milder and easier conditions for depolymerization of the cellulosic material. Generally, during the delignification process, hemicellulose is also solubilized and removed from the cellulosic core.18–20 Cellulose derived compounds represent high value platform chemicals for industrial or chemical purposes.21–23 One of the most promising material derived from cellulose or from raw biomass is a mixture of compounds called bio-oil.24,25 Pyrolysis under elevated temperature and in the absence of oxygen is a common method for the production of pyrolytic oils for fuel or other industrial applications. The use of reactor systems and severe conditions limits the scalability of the process together with other factors like by-products formation. The most common by-products of pyrolysis are biochar, humins, gas and tar. The temperature commonly reached is in the range 450–500 °C. This procedure has a remarkably energetic cost.26–28
Advancements in the bio-oil production process have involved several methodologies, such as microwave irradiation,29,30 the use of Lewis or Brønsted acids at high pressure and high temperature,31,32 the employment of concentrated marine seawater,33 encompassing a strong environmental impact and a low conversion in bio-oil. A green procedure to overcome some of the shortcomings underlined previously, is the use of non-conventional solvents like ionic liquids and deep eutectic solvents. These systems in combination with catalytic quantities of Lewis acid allow to convert cellulosic materials under milder conditions. The remarkable limitations of this method are the high affinity of the synthesized bio-oil for the ionic phase and the extremely poor recovery in organic solvents.34,35
Cellulose citrate is the esterification product of one carboxyl group of citric acid with cellulose, which can be prepared, usually, by impregnation of cellulose with an aqueous solution of acid citric at elevated temperature.36 Such product, that has free and reactive carboxylic acids, is generally insoluble in the common organic solvents and in water and because of its porous structure may be used, among other things, like adsorbent for large molecules as hormones and antibiotics or for heavy metals from waste water, or as ion exchange agent.37 More recent applications involve the use of cellulose citrate as reinforcing filler of polylactic acid (PLA) and its derivatives or polypropylene (PP) resins.38,39
A fast and simple process to obtain simultaneously cellulose hydrolysis and esterification was carried out using mild organic acids. For example, molten oxalic acid dihydrate in solvent free conditions40 or aqueous solutions of citric acid and hydrochloric acid under reflux, are able to produce elevated yield in cellulose ester but poor quantity of bio-oil. In particular, for the latter method, the reason may be due to the presence of water that prevents a direct contact between the three acidic functionalities of citric acid and the glycosidic bonds of cellulose.41
In the recent years, a great deal of attention was devoted to furan-based bio-oils.42 Furan is a member of a class of bioprivileged molecules, which can be converted into other substances with high added value.43,44 Furanic compounds can be chemically manipulated or used directly for many purposes.45–47 For example, 5-hydroxymethylfurfural (HMF) is one of the most promising platforms for chemicals formed from hexoses. It has many applications in the industry of adsorbents, coatings, batteries, drug delivery, bio-pesticides, polymers, corrosion inhibitors and high calorific biofuels.48,49
In this work, we suggest a simple methodology to convert simultaneously cellulose in the form of crystalline or amorphous cellulose into a furanic bio-oil and cellulose citrate without any use of strong inorganic acids, harsh conditions, enzymatic treatments or microorganism fermentation. The proposed methodology involves the use of citric acid (CA) as a mild, natural and non-toxic acidic agent under solvent free conditions and at atmospheric pressure. The reaction is carried out by heating in a range of temperature between 152–155 °C. The cellulose depolymerisation and contemporary esterification occur in short time, without the formation of any other by-products.50 A detailed discussion on materials characterization and quantum-mechanical DFT calculations for the exploration of the reaction mechanisms complete the synthetic procedure.
2. Results and discussion
2.1. Synthesis and characterization
In order to find a fully green, safe and fast procedure to convert simultaneously cellulosic material to cellulose-citrate and bio-oil, we started the investigation using crystalline cellulose and different molten organic acids in solvent-free conditions and at atmospheric pressure. The used temperature is the melting point temperature of the acids. The open-air system allows the presence of traces of water to increase the amount of hydrolysis products in an acid medium. We decided at first to use cellulose in the crystalline form because it is many times more resistant respect to amorphous cellulose and its conversion is difficult. The source of these cellulosic materials is represented by lignocellulosic biomasses, which are the most abundant waste-products of industrial processes.A screening of different mono-, di- and tricarboxylic acids used to obtain simultaneously cellulose esters and bio-oil is reported in Table 1. More specifically, the reaction with molten dihydrated oxalic acid under solvent free conditions at the acid melting point temperature produces only cellulose oxalate (entry 1). Probably, the reaction temperature is not high enough to convert cellulose into bio-oil. Then, we used molten benzoic acid at a higher melting point temperature (entry 2) but neither cellulose-benzoate nor bio-oil were produced.
| Entry | Organic acid | Melting point (°C) | Yield in bio-oil (%) | Yield in cellulose ester (%) |
|---|---|---|---|---|
| a See ref. 33.b Only decomposition products (biochar). | ||||
| 1a | Oxalic acid dihydrate | 104–106 | — | 80 |
| 2 | Benzoic acid | 121–123 | — | — |
| 3 | Adipic acid | 151–154 | — | 90–100 |
| 4 | Citric acid | 152–155 | 60–80 | 40–20 |
| 5b | L(+)Tartaric acid | 168–170 | — | — |
| 6b | Succinic acid | 185–188 | — | — |
In this case, maybe, both the temperature is not high enough to depolymerize cellulose and in presence of only one carboxylic group a structural cleavage and a right insertion between cellulosic chains are not allowed. The reaction with cellulose and adipic acid (entry 3) failed toward hydrolysis, likely because its two carboxylic groups are separated by a too large distance through the alkyl chain. This hydrophobicity does not allow the right insertion of the acid into the cellulosic chains intra- and inter-molecularly bonded through hydrogen bonds. When citric acid at its melting point temperature was used in this reaction, after 3 hours, cellulose was mostly depolymerized in a mixture of compounds collected by acetone extraction in the form of a bio-oil and at the same time the washed solid was identified as esterified cellulose in the form of cellulose citrate (entry 4). This last one has porosity similar to that of cellulose, it is insoluble in the common organic solvents and also in water and for these reasons it can be suitable as an absorbent for very large molecules as hormones and antibiotics as well as ionic exchange resin, pollutants absorber and green nanocomposites.51 Other natural acids, like L(+)tartaric acid and succinic acid, with their melting points near 169 and 183 °C, respectively require a reaction temperature too high to melt leading, instead, to biochar formation (entries 5 and6). A schematic representation of the transformation process of cellulose through reaction with citric acid is reported in Scheme 1.
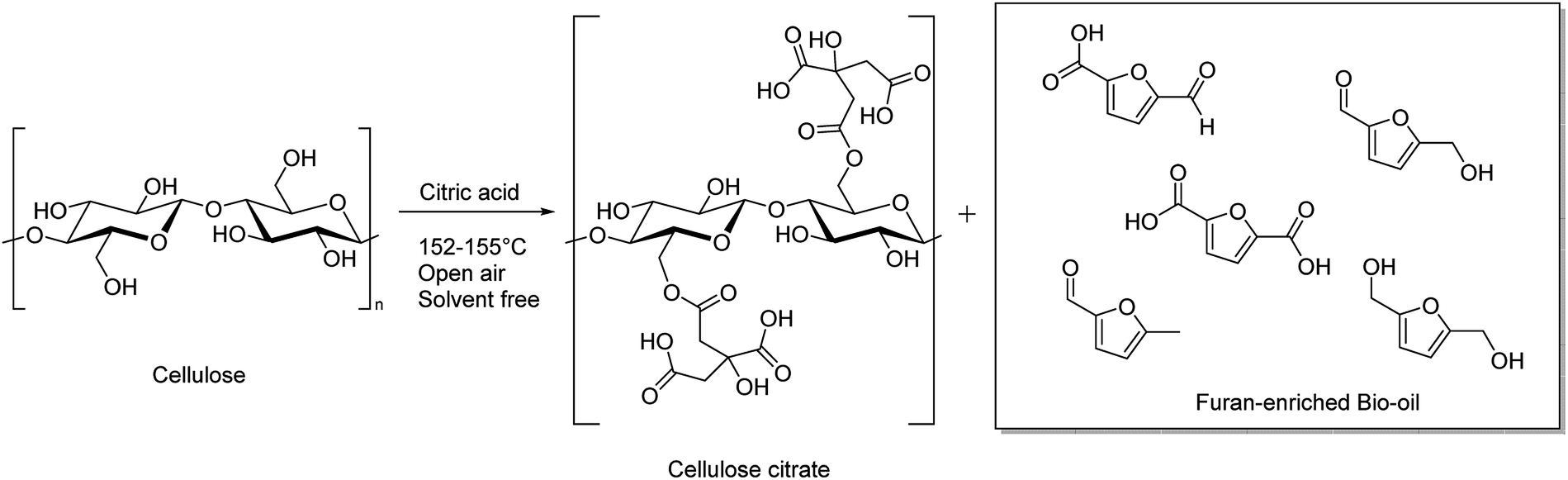 | ||
| Scheme 1 Cellulose esterification and depolymerization by molten citric acid in an open-air system. | ||
Once we found the best system for obtaining simultaneous cellulose esterification and hydrolysis, the reaction conditions optimization was carried out, using different equivalents of citric acid, as described in Table 2.
In order to obtain the maximum yield of bio-oil and cellulose citrate one weight equivalent of citric acid (entry 3) has to be used. When a lower quantity of citric acid is used, the corresponding yield of bio-oil decreases (entries 1 and 2), while the difference in cellulose citrate depends on the lower degree of substitution. In contrast, when a higher quantity of citric acid is used, the yield in cellulose citrate and its degree of substitution remain the same, and a significant amount of unreacted citric acid is present in bio-oil.
Once the optimized amount of reagents was found, we investigated the dependence of the chemical composition of bio-oil on reaction times. All the products were identified by using positive ESI ionization mode and for the preparation of the sample, 100 ppm of bio-oil was dissolved in a mixture of CH3OH/H2O added of a solution of NH4+ 5 mM to enhance the ionization. The obtained results are reported in Table 3.
| Entry | Reaction time | Formula | Compound | Molecular weight | [M + H]+ | [M + NH4]+ | [M + Na]+ |
|---|---|---|---|---|---|---|---|
| 1 | 30 minutes | C6H6O2 | 5-Methylfuran-2-carbaldehyde | 110.11 | 111 | — | — |
| C6H8O3 | 2,5-bis(Hydroxymethyl)furan | 128.13 | 129 | — | — | ||
| C6H2O4 | 2,5-Furandicarboxylic anhydride | 138.08 | 139 | — | — | ||
| C6H4O4 | 5-Formyl-2-furancarboxylic acid | 140.09 | 141 | — | — | ||
| C6H6O4 | 5-Hydroxymethyl-2-furancarboxylic acid | 142.11 | 143 | — | — | ||
| C6H6O3 | 5-Hydroxymethylfurfural | 126.11 | — | 158 | — | ||
| C6H12O6 | Glucose | 180.16 | 181 | — | — | ||
| C6H8O7 | Citric acid | 192.12 | — | 210 | — | ||
| 2 | 1 hour | C6H6O2 | 5-Methylfuran-2-carbaldehyde | 110.11 | 111 | — | — |
| C6H2O4 | 2,5-Furandicarboxylic anhydride | 138.08 | 139 | — | — | ||
| C6H6O4 | 5-Hydroxymethyl-2-furancarboxylic acid | 142.11 | 143 | — | — | ||
| C6H6O3 | 5-Hydroxymethylfurfural | 126.11 | — | 158 | — | ||
| 3 | 2 hours | C6H6O2 | 5-Methylfuran-2-carbaldehyde | 110.11 | 111 | — | — |
| C6H8O3 | 2,5-bis(Hydroxymethyl)furan | 128.13 | 129 | — | — | ||
| 2,5-Furandicarboxylic anhydride | 138.08 | 139 | — | — | |||
| C6H6O4 | 5-Hydroxymethyl-2-furancarboxylic acid | 142.11 | 143 | — | — | ||
| C6H12O6 | Glucose | 180.16 | 181 | — | — | ||
| 4 | 3 hours | C6H6O2 | 5-Methylfuran-2-carbaldehyde | 110.11 | 111 | — | — |
| C6H6O3 | 5-Hydroxymethylfurfural | 126.11 | — | 158 | — | ||
| C6H2O4 | 2,5-Furandicarboxylic anhydride | 138.08 | 139 | — | — | ||
| C6H6O4 | 5-Hydroxymethyl-2-furancarboxylic acid | 142.11 | 143 | — | — | ||
| C6H12O6 | Glucose | 180.16 | 181 | — | — | ||
| 5 | 5 hours | C6H6O2 | 5-Methylfuran-2-carbaldehyde | 110.11 | 111 | — | — |
| C6H6O3 | 5-Hydroxymethylfurfural | 126.11 | — | 158 | — | ||
| C6H4O5 | 2,5-Furandicarboxylic acid | 156.09 | 157 | ||||
| C6H2O4 | 2,5-Furandicarboxylic anhydride | 138.08 | 139 | — | — | ||
| C6H12O6 | Glucose | 180.16 | 181 | — | — |
As it clearly appears, the bio-oil composition is strongly dependent on the reaction time. In fact, from 30 minutes to 3 hours, the bio-oil composition results rich in furanic compounds (Table 3, entries 1–4), while after three hours or more a significant oxidation of furanic compounds occurs (Table 3, entry 5). In more detail, after 30 minutes, almost 80% of cellulose is hydrolysed to furanic bio-oil, whereas the remaining 20% of cellulose is esterified by the used citric acid. The increase of the reaction times for overnight led to uncontrolled decomposition both of the furanic compounds and cellulose citrate; in particular, the latter produces glucose and its dimers, which cannot be easily rationalized. For this reason, no data is shown in Table 3 even if the mass spectrum relative to the reaction in overnight is reported in the ESI.†
Schematically, cellulose is depolymerized to glucose units by the action of traces of water and the acidic carboxylic groups. The presence of traces of water is favoured by the open reaction system. Glucose, once formed, in these reaction conditions dehydrates quickly, with the loss of three molecules of water to give 5-hydroxymethylfurfural (HMF, Scheme 2), as already reported in literature.31 At this point 5-hydroxymethylfurfural thanks to the moderate temperature and to the open-air system, can start a sequence of consecutive oxidation reactions towards formation of products a, b and c of Scheme 2. Cellulose hydrolysis and the consecutive oxidation reactions are depicted in Scheme 2.
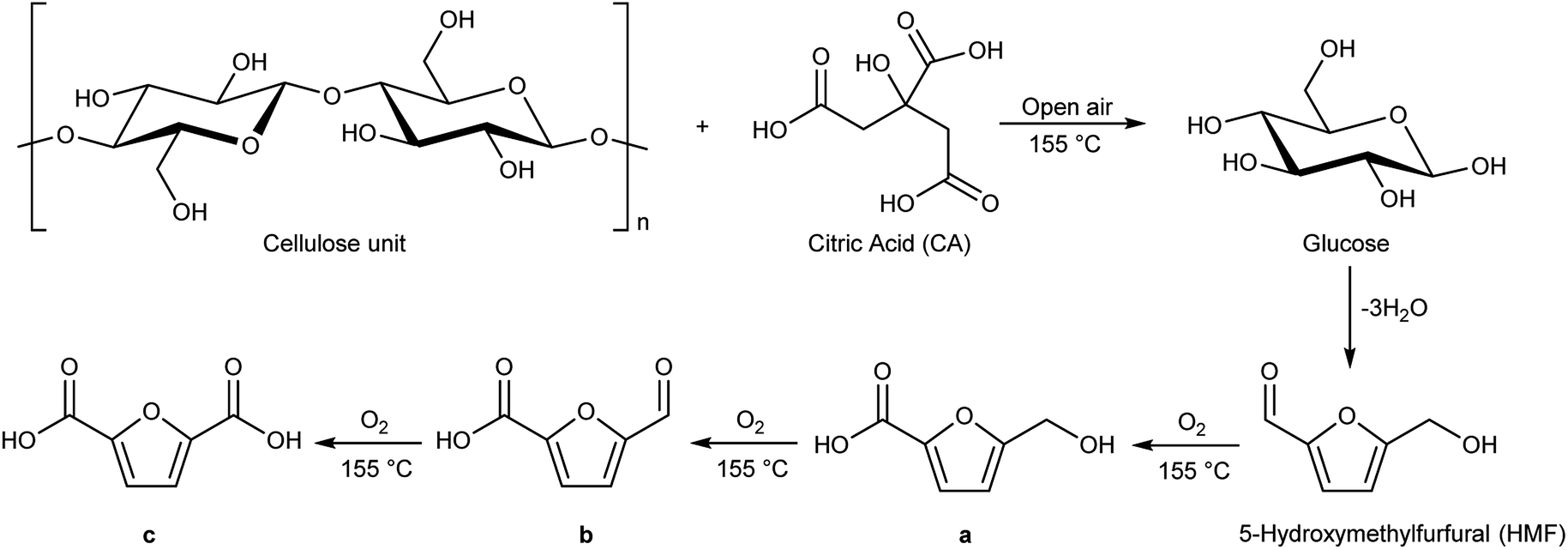 | ||
| Scheme 2 Cellulose hydrolysis catalysed by citric acid at 155 °C and under open-air conditions. | ||
The carboxylic moieties formed in the oxidative process (products a, b and c, Scheme 2) are confirmed by pH measurements of the bio-oil that goes from 4.2–4.4, stopping the reaction after 30 minutes, to 3–3.2 stopping the reaction after overnight time. In addition, the total acid content was obtained by a potentiometric titration following the standard method ASTM D664.52 (Experimental procedure are reported in ESI†). Finally, hydroxyl content was calculated through the standard method ASTM D4274-16, by esterification of bio-oil using phthalic anhydride.53 All results are reported in Table 4.
| Entry | Reaction time | pH | Total acid content (mg KOH per g) | Hydroxyl group content (mg KOH per g) |
|---|---|---|---|---|
| 1 | 30 minutes | 4.4 | 105 | 170 |
| 2 | 1 hour | 4.3 | 120 | 160 |
| 3 | 2 hours | 4.1 | 135 | 152 |
| 4 | 3 hours | 3.8 | 150 | 145 |
| 5 | 5 hours | 3.0 | 180 | 130 |
| 6 | Overnight | 2.2 | 205 | 110 |
Moreover, in Fig. 1 the trend of carboxylic groups versus hydroxyl groups over time is showed.
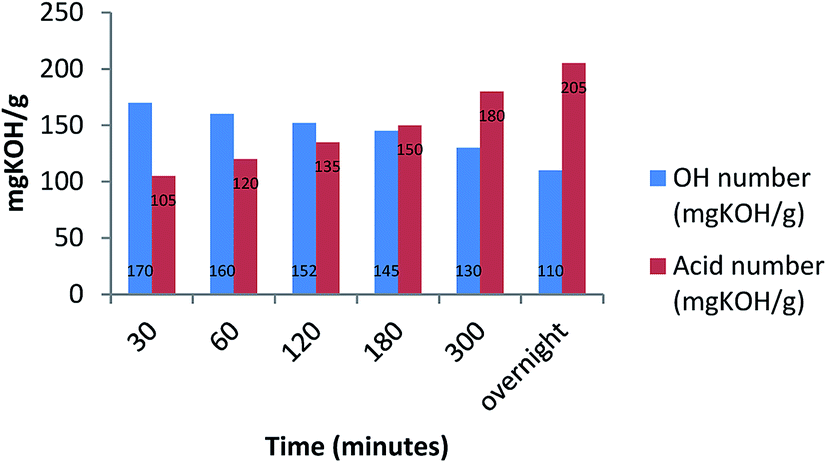 | ||
| Fig. 1 Time dependency between bio-oil, OH numbers and acid numbers. | ||
As additional evidence of the oxidative process, we totally esterified the bio-oil by a common procedure reported in literature, observing in IR spectra both the disappearance of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of carboxylic groups and the presence of CO signals of ester groups (see ESI† for IR spectrum).61
O stretching of carboxylic groups and the presence of CO signals of ester groups (see ESI† for IR spectrum).61
2.2. FT-IR characterization of bio-oil
FT-IR analysis was carried out on bio-oil samples and Fig. 2 shows a sequence of FT-IR spectra acquired as a function of the reaction time. | ||
| Fig. 2 FT-IR spectra of bio-oil at different reaction times. | ||
Consistently with the LC-MS data, the FT-IR analysis shows the complex composition of the bio-oil mixture, particularly in the early stage of the reaction. Indeed, the spectra at 30 min and 1 h show the presence of two sharp and intense peaks at 1705 cm−1 and 1750 cm−1, characteristic of the CO double bond stretching vibrations of dimeric carboxylic acids and aldehydes, respectively (see also the well visible overtone bands at 3449 cm−1 and 3495 cm−1).54 Moreover, the broad band in the 3700–2800 cm−1 range exhibited by the two spectra, is characteristic of the superposition of the OH stretching modes of alcohols (3700–3200 cm−1) and carboxylic acids (3300–2800 cm−1), respectively. The relatively intense band at 1049 cm−1, supports the presence of primary alcohols in the mixture. Other bands, indicating the presence of both carboxylic acids and aldehydes, are those close to 1390 cm−1 and 1427 cm−1, characteristics of the out of plane OH bending of dimeric acids and of CH bending of aldehydes, respectively. The relatively intense signals in the 1000–1400 cm−1 range can be attributed to the COC vibrations of the furan ring generally observed for the 5-HMF,55 whose presence is also supported by weak signals at about 1600 cm−1 due to the C–C double bond stretching vibration.
The FT-IR spectrum of the bio-oil mixture for reaction times greater than 1 h shows several interesting changes, suggesting that oxidation processes are taking place quite early (just in the first 2 h) in the mixture.
One of the most indicative changes is the disappearing of the CO double bond stretching vibration of aldehydes (and its overtone signal). Only one sharp and intense peak at 1728 cm−1 occurs, which is typical of the CO double bond stretching vibration of dicarboxylic acid. The area of this signal, normalized with respect to the total spectral area, remains almost constant with time from 2 h up to overnight (Fig. S1 in ESI†) and it is equal to the sum of the area of the two carbonyl peaks occurring in the spectra at 0.5 and 1 h, indicating that the carboxyl functional groups formed during the reaction time derive from the oxidation of the aldehyde carbonyl groups present at the beginning in the mixture. However, it is worth noting that, in the spectrum at 5 h, a shoulder appears in the carbonyl signal at 1728 cm−1, which becomes more evident in that one acquired after overnight. The relative increase in the intensity of the latter peak could indicate further oxidation processes.
Another interesting change, though less diagnostic, is that the spectral region below 1500 cm−1, becomes less crowded with respect to that observed in the spectra acquired within the first hour. This is probably due to the more complex mixture composition in the early stage of the reaction.
2.3. Characterization of cellulose citrate. FT-IR and degree of esterification
The solid fraction product of the hydrolysis reaction was also characterized by FT-IR in order to support its identification as cellulose citrate and in Fig. 3 a sequence of representative FT-IR spectra of cellulose citrate, as a function of time, is reported. It clearly shows that, just after half an hour, a strong signal centred at 1736 cm−1 appears in all the spectra, which is absent in the spectrum of pure cellulose. This signal is typical of carbonyl vibrational stretching modes, strongly supporting that cellulose esterification56 takes place due to the reaction with the citric acid at 152–155 °C. No other significant change with respect to the cellulose spectrum can be detected in the other spectra, indicating that the solid material produced is structurally based on cellulose.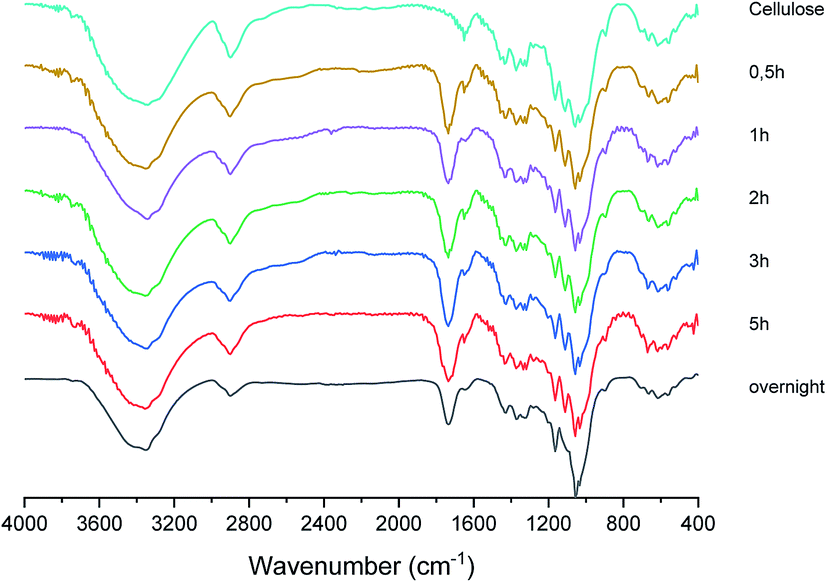 | ||
| Fig. 3 FT-IR spectra of cellulose citrate at different reaction times. | ||
Another confirmation of cellulose citrate formation came from conductometric titration for the estimation of the average degree of substitution calculated according to ATSM D1439.57 (Detailed procedure is reported in ESI†). The degree of substitution is 0.46 from 30 minutes to 3 hours, to confirm the maximum esterification. However, after five hours the degree of substitution is 0.35 and when the solid is collected after overnight time is 0.20. Therefore, the decomposition of this polymer after prolonged reaction time in the reported temperature range is confirmed. Finally, we investigated also the same reaction conducted under nitrogen without any presence of trace of water and, in this case, the reaction proceeds only toward cellulose esterification, with a functionalization of 0.2 weights equivalents of citric acid, while the remaining part is washed and collected in acetone. The degree of substitution in this case is 0.45. This procedure becomes selective only toward the production of cellulose citrate.
2.4. Mechanism of reaction
The proposed reaction mechanism consists in a simultaneous hydrolysis and esterification of cellulose. Initially, citric acid in solid form is mixed with cellulosic material and the coupled solids are heated together in a range of temperature between 152–155 °C. Citric acid at this temperature melts and inserts into cellulose chains, breaking the intra- and intermolecular hydrogen bonds (Fig. 4). Therefore, the separate cellulose chains are predisposed to an easier protonation of glycosidic bonds to assist the hydrolysis and to an enhanced acid catalysis on carboxylic groups to promote Fisher esterification.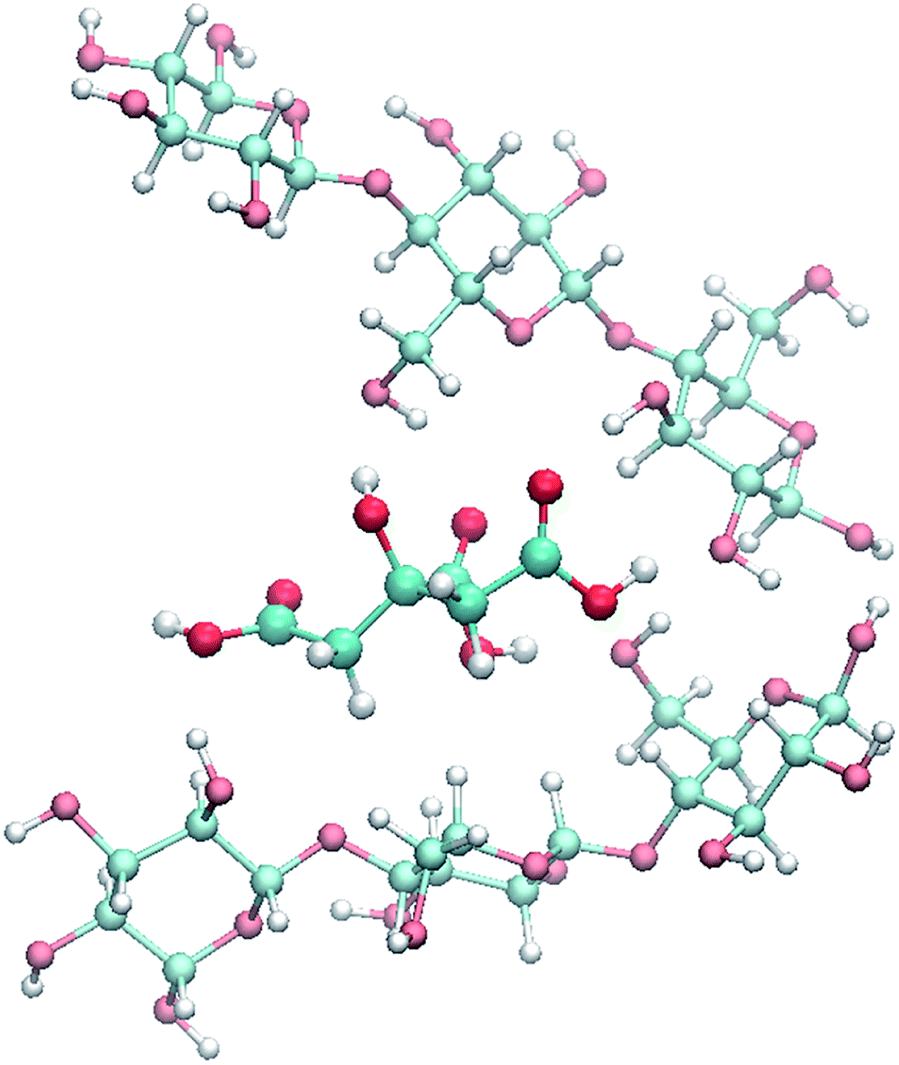 | ||
| Fig. 4 Simulated intercalation of citric acid between adjacent cellulose chains. | ||
For simplicity, the proposed hydrolysis mechanism is described considering only a cellobiose moiety of cellulose, as highlighted in Scheme 3, which contains all the chemical characters appearing in cellulose.
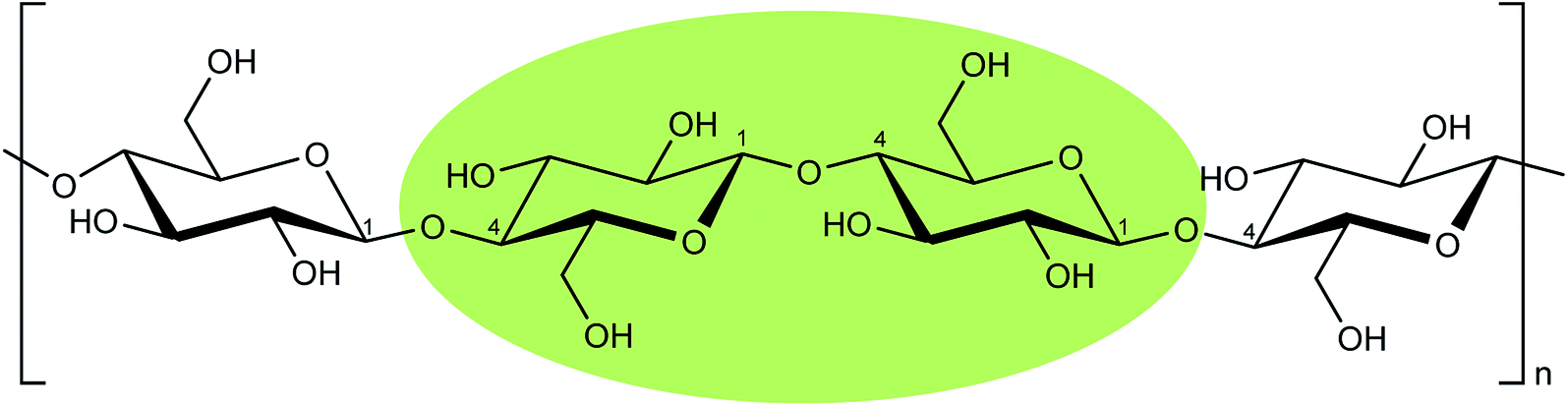 | ||
| Scheme 3 Cellobiose portion of a cellulosic chain highlighted in green. | ||
Cellulose hydrolysis is due to the acidic functionalities of citric acid that interacts both with the glycosidic bonds of cellulose chains and the oxygen of the pyranose rings following two different pathways of reaction (path a and b, Scheme 4). In the first pathway (path a), the protonation of the O-glycosidic bond by citric acid (CA) leads to formation of the six-membered oxocarbenium ring (4) and glucose (5). The intermediate 4 in turn produces glucose (5) by water addition. In the second pathway (path b), citric acid promotes the protonation of pyranose oxygen (3) to form intermediate 6 with an open glucose unit. Then, the oxocarbenium ion 6 provides both a glucose molecule (5) and protonated open-chain glucose 8. At this point, path a and path b continue along a single pathway, leading to the formation of fructofuranose 11 through the enolic species 9. Finally, the substrate 11 produces 5-hydroxymethylfurfural 15 (HMF) by three dehydration steps.
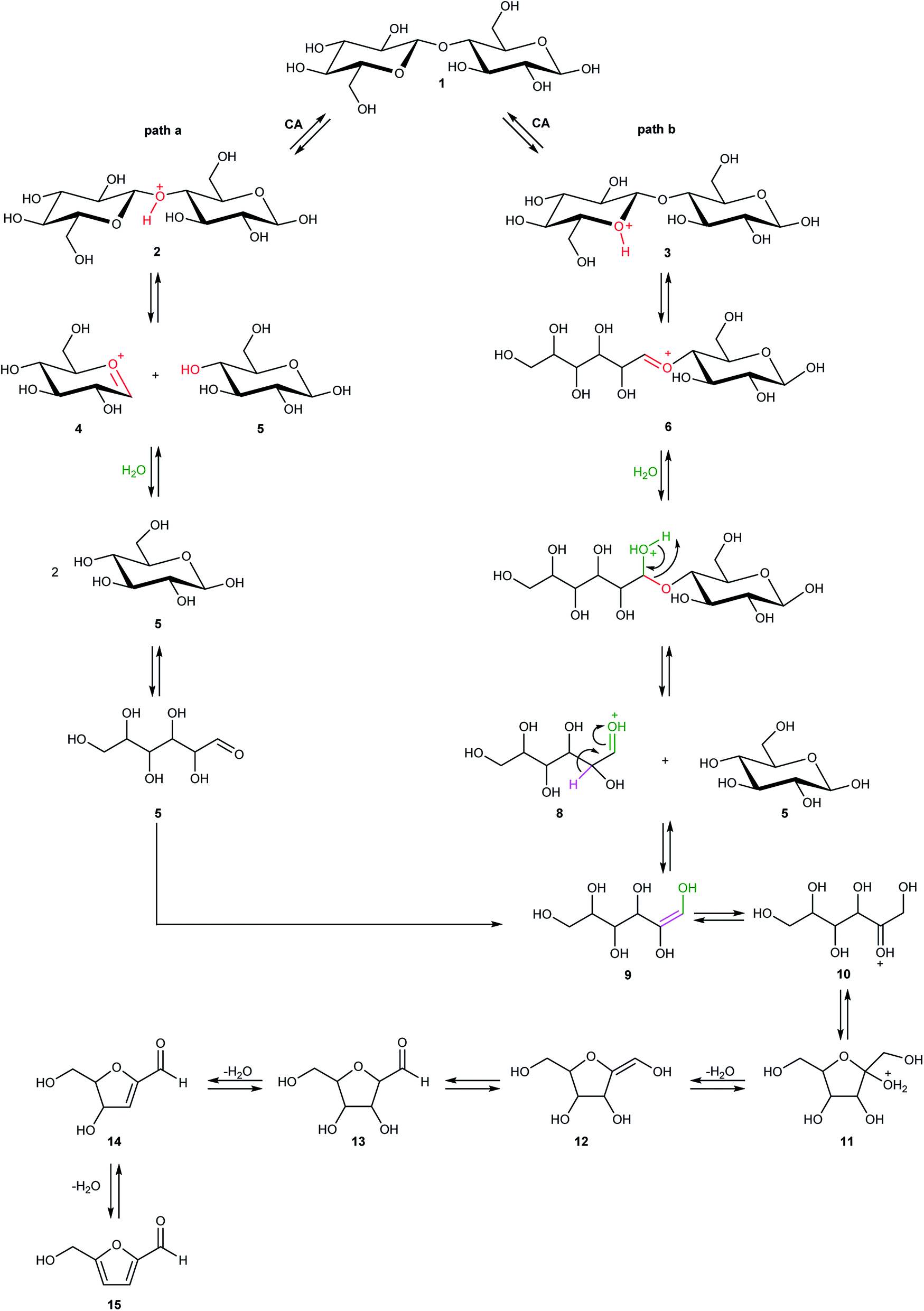 | ||
| Scheme 4 Probable hydrolysis mechanism and subsequent formation of 5-hydroxymethylfurfural (HMF). | ||
2.5. Computational study of reaction mechanisms
Detailed computational studies were performed to support step by step the complex mechanistic hypotheses formulated on the basis of experimental evidences. Outcomes were compared with the existing literature in order to confirm or discard previously proposed reaction pathways.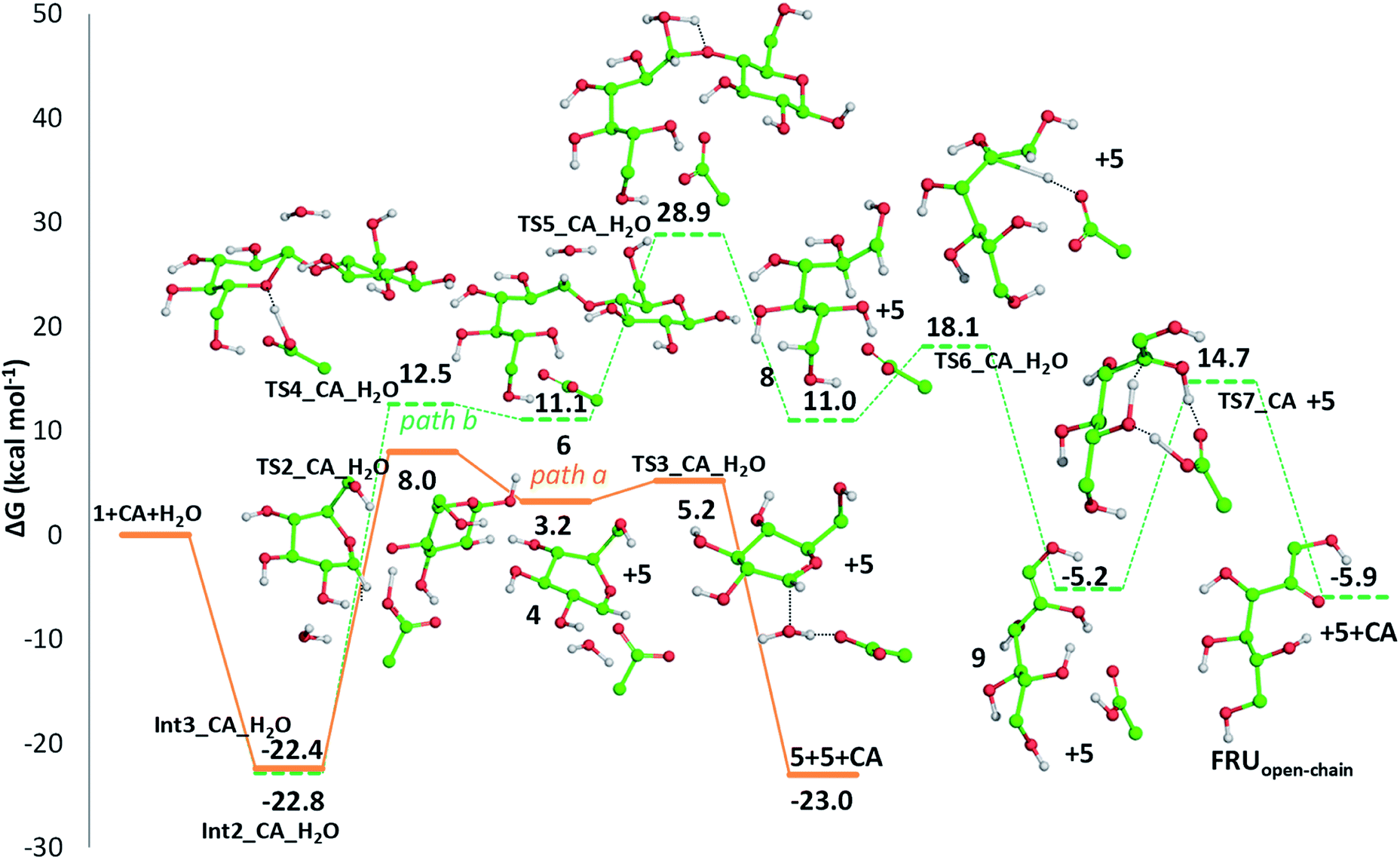 | ||
| Fig. 5 Free energy profiles describing cellobiose hydrolysis assisted by the protonation of glycosidic (O1) (path a, solid line) and pyranose ring (O5) (path b, dashed line) oxygens by means of citric acid. Only H atoms that should be involved in investigated reactions are shown. Energies are given in kcal mol−1 and relative to the reactants' asymptote. | ||
Along path a, the transfer of the proton from citric acid to the glycosidic oxygen and the simultaneous breaking of the C1–O1 bond occurs by overcoming a free-energy barrier of 8.0 kcal mol−1, which corresponds to the transition state TS2_CA_H2O. As a result, one glucose unit (5) is formed together with a cyclic carbocation (4). The next step leading to the regeneration of citric acid and the formation of the second glucose molecule has a free-energy barrier, for the transition state TS3_CA_H2O, of 5.2 kcal mol−1. The imaginary frequency that identifies the transition state nature of the intercepted stationary point corresponds to the concerted attack of a water molecule to the carbocation and the simultaneous proton abstraction from water by a citrate unit. Formation of products is very favourable, being exergonic by 23.0 kcal mol−1.
Along path b, instead, protonation of the pyranic oxygen and breaking of the bond with the adjacent carbon atom requires 12.5 kcal mol−1 to occur going through the TS4_CA_H2O transition state. Nucleophilic attack of the water molecule leads to the formation of a glucose unit together with an open ring aldehydic protonated intermediate via TS5_CA_H2O with a free-energy barrier of 28.9 kcal mol−1. The H atom on C2 is transferred to citric acid via TS6_CA_H2O going beyond a free-energy barrier of 18.1 kcal mol−1 to form the species labelled 9, calculated to be exergonic by 5.2 kcal mol−1. Citric acid plays a pivotal role, as in path a, in promoting the H transfer from the hydroxyl group on the C5 atom to C1 to form the CH2–OH unit via the concerted transition state TS7_CA_H2O corresponding to an energy barrier of 14.7 kcal mol−1.
Indeed, citric acid works as a proton shuttle accepting a proton from the OH group at C1 and transferring a proton to the OH group at C2 and assisting the H migration from C5 to C1. Formation of the species FRUopen-chain, that is the open-chain form of fructose, corresponding to the change of C1 atom hybridization from sp2 to a sp3 results to be almost thermoneutral with respect to the previous minimum.
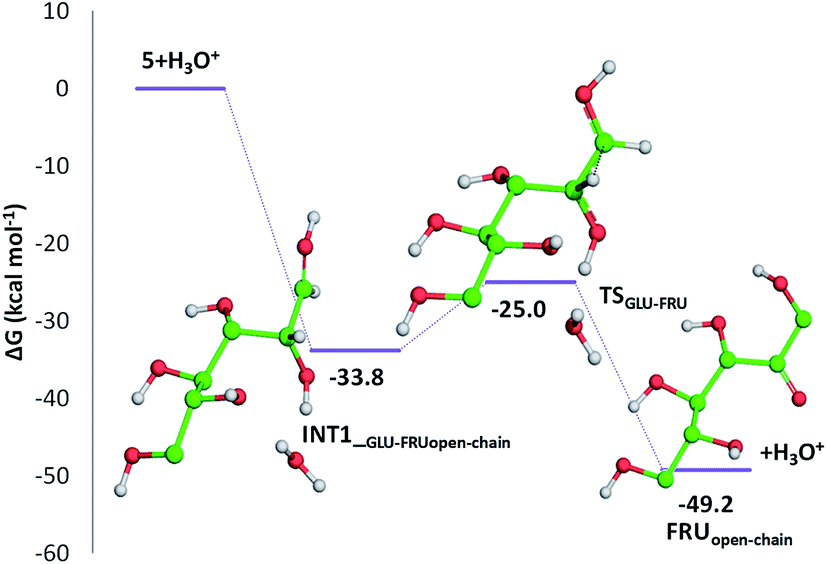 | ||
| Fig. 6 Free energy profile for the conversion of glucose into the open-chain form of fructose in acidic environment. Only H atoms that should be involved in investigated reactions are shown. Energies are given in kcal mol−1 and relative to the reactants' asymptote. | ||
C bond of the ring with the CO bond of the aldehyde group. Finally, the third dehydration step (INT4openFRU-HMF) is highly exothermic (−21.3 kcal mol−1) attributable to the major stability of aromatic ring in HMF. Once conversion of cellulose to HMF in presence of citric acid was analysed, computational efforts have been focused on the rationalization of the pathways leading to the formation of several compounds experimentally observed in bio-oil at different reaction times.
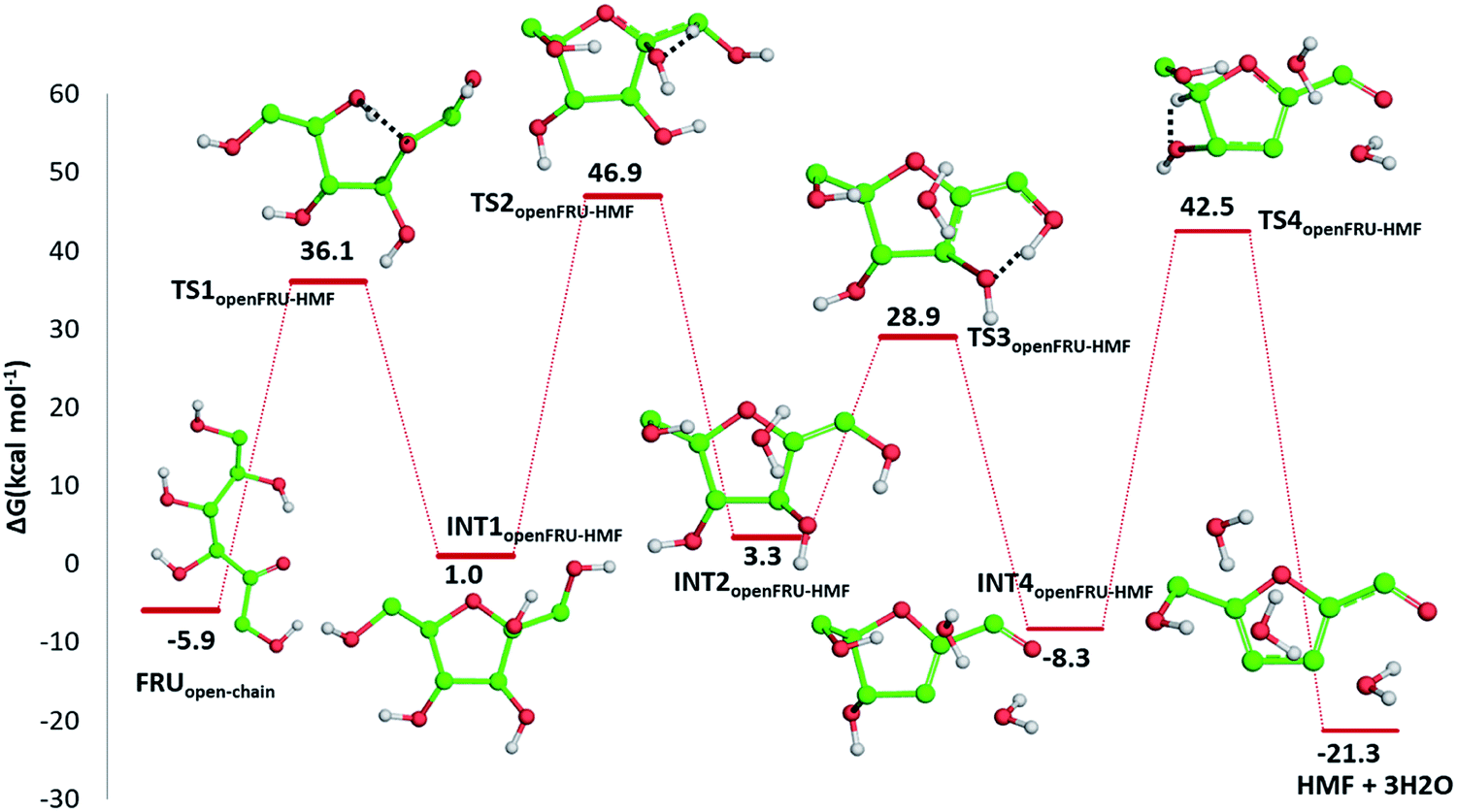 | ||
| Fig. 7 Free energy profile describing the HMF formation from the open chain form of fructose. Only H atoms that should be involved in investigated reactions are shown. Energies are given in kcal mol−1 and relative to the zero reference energy of the reactant. | ||
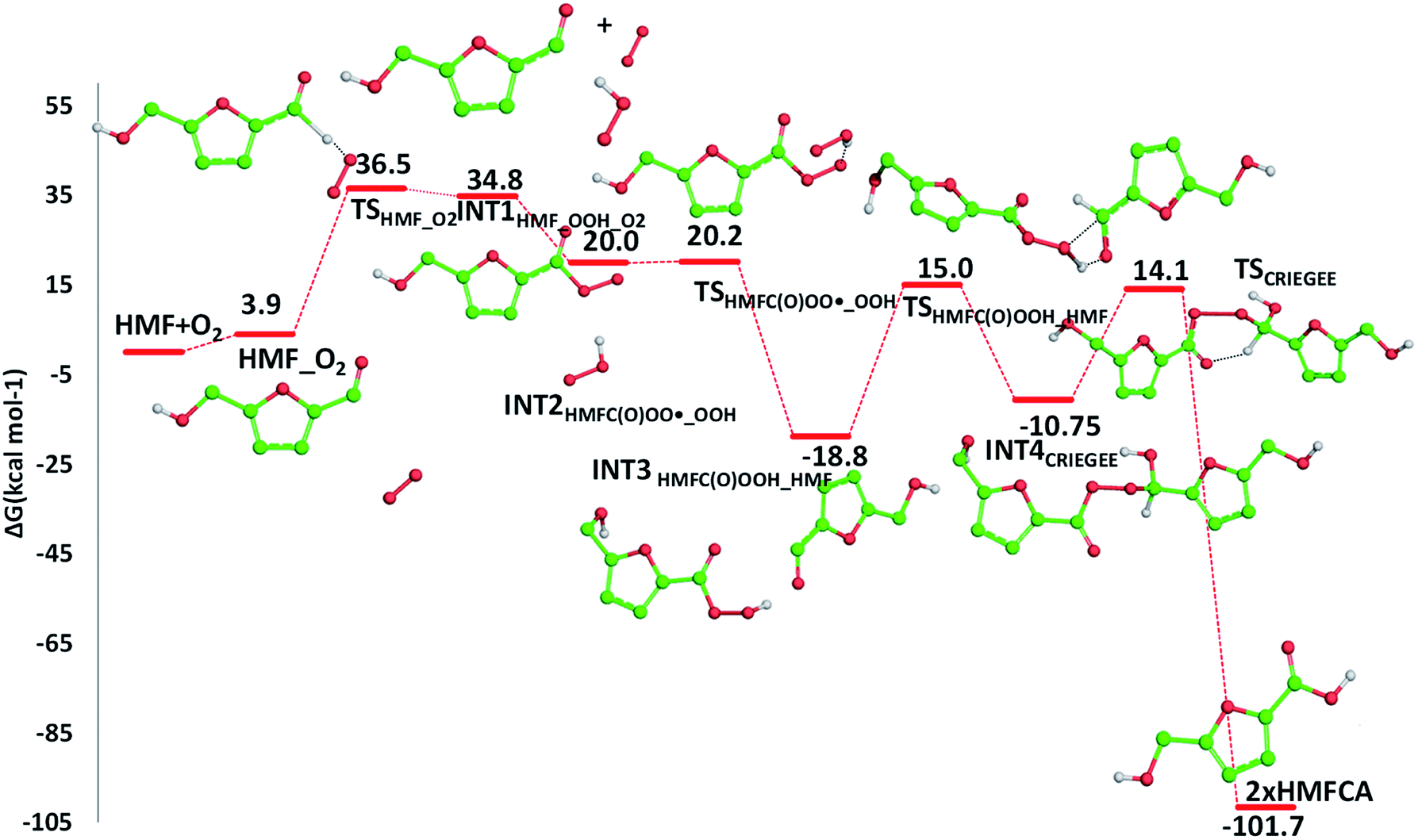 | ||
| Fig. 8 Free energy profile for the HMFCA formation from HMF in presence of air oxygen. Only H atoms that should be involved in investigated reactions are shown. Energies are given in kcal mol−1 and relative to the zero reference energy corresponding to the reactants' asymptote. | ||
 | ||
| Fig. 9 Free energy profile for the FFCA formation from HMF and the radical HMFC(O)OO˙. Only H atoms that should be involved in investigated reactions are shown. Energies are in kcal mol−1 and relative to the zero reference energy corresponding to the reactants' asymptote. | ||
The required energy is 5.2 kcal mol−1 (TSHMFCA_HMFC(O)OO˙), whereas the regeneration of the HMFC(O)–OOH species and formation of the new carbon radical HMFA˙ results to be exergonic by 15.2 kcal mol−1. With the help of air oxygen, the H-abstraction from the hydroxyl group leads to the formation of a peroxy radical and the final oxidation to 5-formyl-2-furancarboxylic acid (FFCA) (MW 140.09 g mol−1). Separated products lye well below the energy at the entrance channel, as the process is calculated to be exergonic by 20.2 kcal mol−1, whereas the transition state (TSHMFCA˙_O2), leading to products formation lies at −12.3 kcal mol−1 is less stable by about 3 kcal mol−1 than the preceding minimum.
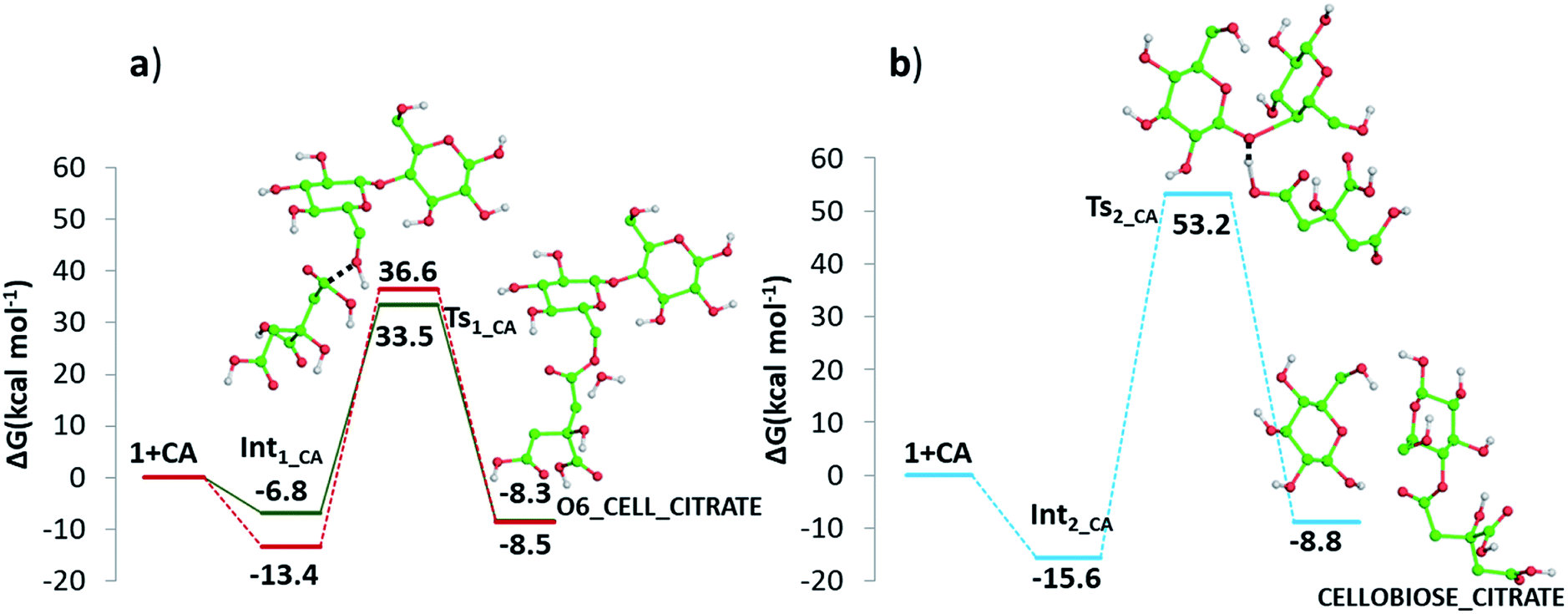 | ||
| Fig. 10 Free energy profiles describing the esterification of cellobiose in presence of CA by (a) condensation and (b) protonation. Both O6-cellulose citrate (solid line) and O2-cellulose citrate (dashed line) pathways formation are reported in panel (a). Only H atoms that should be involved in investigated reactions are shown. Relative energies are in kcal mol−1 and calculated with respect to the reference zero energy of separated reactants. | ||
In addition, it has been investigated the possibility (see Fig. 10 panel b) that, in absence of water, protonation of the glycosidic oxygen occurs simultaneously with the breaking of the O1–C4′ bond and the attack of the deprotonated citrate oxygen on the C4′ atom. The intercepted transition state lies 53.2 kcal mol−1 higher in energy with respect the zero reference energy of separated reactants. The subsequent formation of glucose and cellobiose citrate products is calculated to be exergonic by 8.8 kcal mol−1. Due to the significantly higher calculate energy barrier, this pathway is the less viable one.
3. Conclusions
In conclusion, here we propose an innovative, green, catalyst-free and efficient synthetic methodology to convert simultaneously cellulose in furan-based bio-oil and cellulose citrate. This last, recovered from the reaction environment in eco-sustainable conditions only by filtration and washing operations with water, can be used for its chemical characteristics as an additive in composites and bioplastic materials to improve mechanical strength and biodegradability properties. On the contrary, furan-based bio-oil, collected from reaction crude through a simple extraction process with acetone, due to its chemical composition enriched of 5-hydroxy-methylfurfural (HMF) and other furan-based derivatives, is a recent example of mixture of bioprivileged molecules that can be used for several applications such as antifriction, additive in gasoline, drug delivery, bio-pesticides, biobased packaging material etc.Bio-oil and cellulose citrate obtained with the versatile synthetic procedure illustrated here were fully characterized by MS, IR and SEM techniques. Meticulous theoretical investigations were carried out to disentangle step by step all the mechanistic aspects of the processes leading to the conversion of cellulose into furan-based bio-oil and cellulose citrate assisted by citric acid.
The results herein illustrated provide experimental evidence for a promising industrial procedure for contemporary production of furan-based bio-oil and cellulose citrate with minimal cost and environmental impact.
4. Experimental
All reagents and solvents were purchased from Sigma-Aldrich or Alfa Aesar and used without purification.4.1. Synthesis
In a Pyrex glass beaker, 5 g of microcrystalline or amorphous cellulose and 5 g of citric acid (monohydrate or anhydrous can be used) are placed and mixed together. The mixture is heated at 152–155 °C, using an oil bath, for the chosen reaction times (30 min, 1 hour, 2 hours, 3 hours, 5 hours, overnight time) and sometimes mixed mechanically. At the end of reaction, after cooling the reaction mixture, 20 ml of acetone are added and the suspension is filtered using a 50 ml sintered glass filter connected to an Erlenmeyer flask. The filtered solid is washed with other 20 ml of acetone. The liquid phase is reduced under vacuum to remove any trace of solvent to give bio-oil in the form of a brown highly viscous oil. The solid phase is collected, washed with distilled water, dried in an oven at 70 °C for one day and consists of cellulose citrate as a pale-yellow solid.4.2. Characterization
Bio-oil characterization was carried out using a MS spectrometer Q-Star Pulsar-i (MSD Sciex Applied Biosystem, Toronto, Canada) in ESI (+) ionization mode. Both MS (full scan mode) and MS/MS (product ion scan) experiments were carried out. All samples were dissolved in CH3OH/H2O 9/1 solution added of same drops of NH4+ 5 mM. All spectra are reported in ESI.†Bio-oil and cellulose-citrate FT-IR spectra were acquired by the Shimadzu IRAffinity-1S spectrometer (Shimadzu Corporation) in the spectral region of 375 and 4000 cm−1 with a resolution of 1 cm−1, setting 50 scans for a single analysis and using KBr pellets technique. The KBr pellets were obtained by mixing the sample with KBr powder (ratio 1:100) and pressing with a hydraulic press, at the pressure of 10 tons for 5 minutes. The resulting pellets were placed in the appropriate compartment of the instrument and exposed to the FT-IR light beam for analysis. All spectra are collected in ESI.†
Morphological studies of cellulose and citrate cellulose at different reaction time were carried out using a LEO 420 scanning electron microscope (SEM, Zeiss), operating with vacuum conditions of 8 × 10−6 Torr at an accelerating voltage of 15 kV. Samples were gold metallized by an Auto Sputter Coater (Agar). Images were taken with 200, 500, and 3000 SEM micrograph magnifications (see ESI† for imagines).
4.3. Computational methods
All the electronic calculations were carried out with the Gaussian 09 suite of programs,65 in the framework of the density functional theory employing the hybrid Becke three-parameter exchange functional66 and the Lee–Yang–Parr correlation functional, B3LYP.67 Dispersion corrections for nonbonding interaction have been included through the Grimme approach using atom pair-wise additive schemes,68 denoted as DFT-D3 method. The geometries and zero-point energies were calculated with 6-311G(d,p) basis sets. Vibrational frequencies at the same level of theory have been calculated for both establishing the nature of intercepted stationary points as minima and transition states and calculating zero-point energy (ZPE) and Gibbs free energy corrections. The intercepted transition states are first order saddle points on a potential energy surface (PES) and their vibrational spectrum is characterized by one imaginary frequency, corresponding to a negative force constant, which means that in one direction, in the nuclear configuration space, the energy has a maximum, while in all the other directions the energy has a minimum. Furthermore, transition states have been carefully checked to be properly connected to the correct minima by IRC (intrinsic reaction coordinate) analysis.69,70Enthalpies and Gibbs free energies have been obtained using standard statistical procedures71 at 298 K and 1 atm from total energies, including zero-point and thermal corrections.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the project FOREST-COMP – CUP H56C18000080005 – PON “Ricerca e Innovazione 2014–2020”. University of Calabria and Calabria Region (PAC CALABRIA 2014–2020 – Asse Prioritario 12, Azione B 10.5.12 CUP: H28D19000040006) are acknowledged for financial support.Notes and references
- R. C. Brown, Thermochemical Processing of Biomass: Conversion into Fuels, Chemicals and Power, John Wiley & Sons, 2011 Search PubMed.
- V. I. Popa and I. Volf, Biomass as Renewable Raw Material to Obtain Bioproducts of High-Tech Value, Elsevier, 2018 Search PubMed.
- M.-A. Perea-Moreno, E. Samerón-Manzano and A.-J. Perea-Moreno, Sustainability, 2019, 11, 863–881 CrossRef.
- A. Tursi, Biofuel Res. J., 2019, 22, 962–979 CrossRef.
- G. Fiorentino, M. Ripa and S. Ulgiati, Biofuels, Bioprod. Biorefin., 2017, 11, 195–214 CrossRef CAS.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- V. Dhyani and T. Bhaskar, Renewable Energy, 2018, 129, 695–716 CrossRef CAS.
- X. Li, T. Guo, Q. Xia, X. Liu and Y. Wang, ACS Sustainable Chem. Eng., 2018, 6, 4390–4399 CrossRef CAS.
- M. Balat, M. Balat, E. Kırtay and H. Balat, Energy Convers. Manage., 2009, 50, 3147–3157 CrossRef CAS.
- W. Den, V. K. Sharma, M. Lee, G. Nadadur and R. S. Varma, Front. Chem., 2018, 6, 141 CrossRef.
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS.
- G. Bellesia, A. Asztalos, T. Shen, P. Langan, A. Redondoe and S. Gnanakaranf, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 1184–1188 CrossRef CAS.
- A. J. Ragauskas and C. G. Yoo, Front. Energy Res., 2018, 6, 118 CrossRef.
- H. Yang, X. Zhang, H. Luo, B. Liu, T. M. Shiga, X. Li, J. I. Kim, P. Rubinelli, J. C. Overton, V. Subramanyam, B. R. Cooper, H. Mo, M. M. Abu-Omar, C. Chapple, B. S. Donohoe, L. Makowski, N. S. Mosier, M. C. McCann, N. C. Carpita and R. Meilan, Biotechnol. Biofuels, 2019, 12, 171 CrossRef.
- J. Deng, T. Xiong, H. Wang, A. Zheng and Y. Wang, ACS Sustainable Chem. Eng., 2016, 4, 3750–3756 CrossRef CAS.
- S. Zhao, M. Liu, L. Zhao and L. Zhu, Ind. Eng. Chem. Res., 2018, 57, 5241–5249 CrossRef CAS.
- T. Qu, W. Guo, L. Shen, J. Xiao and K. Zhao, Ind. Eng. Chem. Res., 2011, 50, 10424–10433 CrossRef CAS.
- S. Niju and M. Swathika, Biocatal. Agric. Biotechnol., 2019, 20, 101263 CrossRef.
- R. Singh, A. Shukla, S. Tiwari and M. Srivastava, Renewable Sustainable Energy Rev., 2014, 32, 713–728 CrossRef CAS.
- M. Mascal, ChemSusChem, 2020, 13, 274–277 CrossRef CAS.
- K. Kohli, R. Prajapati and B. K. Sharma, Energies, 2019, 12, 233, DOI:10.3390/en12020233.
- M. Dusselier, M. Mascal and B. F. Sels, Top. Curr. Chem., 2014, 353, 1–40 CrossRef CAS.
- Y. Liu, Y. Nie, X. Lu, X. Zhang, H. He, F. Pan, L. Zhou, X. Liu, X. Ji and S. Zhang, Green Chem., 2019, 21, 3499–3535 RSC.
- W. Shi, S. Li, J. Jia and Y. Zhao, Ind. Eng. Chem. Res., 2013, 52, 586–593 CrossRef CAS.
- S. Yin and Z. Tan, Appl. Energy, 2012, 92, 234–239 CrossRef CAS.
- H. C. Park and H. S. Choi, Renewable Energy, 2019, 143, 1268–1284 CrossRef CAS.
- W. N. R. W. Isahak, M. W. M. Hisham, M. A. Yarmo and T. Y. Hin, Renewable Sustainable Energy Rev., 2012, 16, 5910–5923 CrossRef CAS.
- R. E. Guedesa, A. S. Lunaa and A. R. Torres, J. Anal. Appl. Pyrolysis, 2018, 129, 134–149 CrossRef.
- X. Zhang, K. Rajagopalan, H. Lei, R. Ruanc and B. K. Sharma, Sustainable Energy Fuels, 2017, 1, 1664–1699 RSC.
- H. M. Kadlimatti, B. Raj Mohan and M. B. Saidutta, Biomass Bioenergy, 2019, 123, 25–33 CrossRef CAS.
- Y. Zhao, S. Wang, H. Lin, J. Chen and H. Xu, RSC Adv., 2018, 8, 7235–7242 RSC.
- Z. Chen, Q. Li, Y. Xiao, C. Zhang, Z. Fu, Y. Liu, X. Yi, A. Zheng, C. Li and D. Yin, Cellulose, 2019, 26, 751–762 CrossRef CAS.
- X. Li, Y. Zhang, Q. Xia, X. Liu, K. Peng, S. Yang and Y. Wang, Ind. Eng. Chem. Res., 2018, 57, 3545–3553 CrossRef CAS.
- Q. Haonan, Z. Yanmei, M. Yiming, Z. Peng, G. Bin, G. Meixia and F. Caixia, J. Taiwan Inst. Chem. Eng., 2018, 93, 667–673 CrossRef.
- J. Deng, T. Xiong, H. Wang, A. Zheng and Y. Wang, ACS Sustainable Chem. Eng., 2016, 4, 3750–3756 CrossRef CAS.
- G. P. Touey and J. E. Kiefer, US Pat., Serial No. 355,416, 1956.
- D. A. Pinem, M. Gintim and A. Ginting, Int. J. Sci. Technol. Eng., 2018, 4(9), 23–28 Search PubMed.
- X. Cui, A. Ozaky, T.-A. Asoh and H. Uyama, Polym. Degrad. Stab., 2020, 175, 109118–109124 CrossRef CAS.
- X. Cui, T. Honda, T.-A. Asoh and H. Uyama, Carbohydr. Polym., 2020, 230, 115662–115668 CrossRef CAS.
- D. Li, J. Henschen and M. Ek, Green Chem., 2017, 19, 5564–5567 RSC.
- S. Spinella, A. Maiorana, Q. Qian, N. J. Dawson, V. Hepworth, S. A. McCallum, M. Ganesh, K. D. Singer and R. A. Gross, ACS Sustainable Chem. Eng., 2016, 4, 1538–1550 CrossRef CAS.
- A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Chem. Rev., 2016, 116, 1637–1669 CrossRef CAS.
- S. Dutta, L. Wu and M. Mascal, Green Chem., 2015, 17, 3737–3739 RSC.
- M. A. Eldeeb and B. Akih-Kumgeh, Energies, 2018, 11, 512 CrossRef.
- Y. J. Seok, J.-Y. Her, Y.-G. Kim, M. Y. Kim, S. Y. Jeong, M. K. Kim, J.-Y. Lee, C.-I. Kim, H.-J. Yoon and K.-G. Lee, Toxicol. Res., 2015, 31, 241–253 CrossRef CAS.
- M. A. Eldeeb and B. Akih-Kumgeh, Fuels, 2014, 28, 6618–6626 CrossRef CAS.
- A. J. J. E. Eerhart, W. J. J. Huijgen, R. J. H. Grisel, J. C. van der Waal, E. de Jong, A. de Sousa Dias, A. P. C. Faaija and M. K. Patela, RSC Adv., 2014, 4, 3536–3549 RSC.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Fradea and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS.
- F. Olivito, V. Algieri, A. Tursi, L. Maiuolo, A. Beneduci, A. De Nino and G. Chidichimo, Provisional Application Pending, Italian patent, Application No. 102019000017339, 2019.
- H.-M. Park, M. Misra, L. T. Drzal and A. K. Mohanty, Biomacromolecules, 2004, 5, 2281–2288 CrossRef CAS.
- A. Oasmaa, D. C. Elliott and J. Korhonen, Energy Fuels, 2010, 24, 6548–6554 CrossRef CAS.
- U. A. Amran, S. Zakaria, C. H. Chia, R. Roslan, S. N. S. J. Jaafar and K. M. Salleh, Cellulose, 2019, 26, 3231–3246 CrossRef CAS.
- R. M. Silverstein, F. X. Webster and D. J. Kiemle, Spectrometric Identification of Organic Compounds, John Wiley and Sons, USA, Seventh edn, 2005 Search PubMed.
- B. Kassanov, J. Wang, Y. Fu and J. Chang, RSC Adv., 2017, 7, 30755–30762 RSC.
- T. Gan, Y. Zhang, M. Yang, H. Hu, Z. Huang, Z. Feng, D. Chen, C. Chen and J. Liang, Chem. Res., 2018, 57(32), 10786–10797 CAS.
- R. L. Feddersen and S. N. Thorp, Ind. Gums, 1993, 03, 537–578 Search PubMed.
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS.
- J. T. Edward, Chem. Ind., 1955, 3, 1102–1104 Search PubMed.
- X. Li, Y. Zhang, Q. Xia, X. Liu, K. Peng, S. Yang and Y. Wang, Ind. Eng. Chem. Res., 2018, 57, 3545–3553 CrossRef CAS.
- R. S. Assary, P. C. Redfern, J. R. Hammond, J. Greeley and L. A. Curtiss, J. Phys. Chem. B, 2010, 114, 9002–9009 CrossRef CAS.
- C. Yao, Y. Shin, L. Q. Wang, C. F. Windisch, W. D. Samuels, B. W. Arey, C. Wang, W. M. Risen and G. J. Exarhos, J. Phys. Chem. C, 2007, 111, 15141–15145 CrossRef CAS.
- F. Grein, A. C. Chen, D. Edwards and C. M. Crudden, J. Org. Chem., 2006, 71, 861–872 CrossRef CAS.
- N. Anoune, H. Hannachi, P. Lantéri, R. Longeray and C. Arnaud, J Chem. Educ., 1998, 75, 1290–1293 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta Jr, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision D.01), Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- D. A. McQuarrie and J. D. Simon Molecular thermodynamics, University Science Books, Sausalito, CA, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06542k |
| This journal is © The Royal Society of Chemistry 2020 |