
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiffusivity and hydrophobic hydration of hydrocarbons in supercritical CO2 and aqueous brine†
Hyeonseok Leeb,
Mehdi Ostadhassan *ac,
Zheng Sund,
Hui Pub,
Bo Liua,
Rajender S. Varmae,
Ho Won Jang*f and
Mohammadreza Shokouhimher*bf
*ac,
Zheng Sund,
Hui Pub,
Bo Liua,
Rajender S. Varmae,
Ho Won Jang*f and
Mohammadreza Shokouhimher*bf
aKey Laboratory of Continental Shale Hydrocarbon Accumulation and Efficient Development, Ministry of Education, Northeast Petroleum University, Daqing 163318, China. E-mail: mehdi.ostadhassan@nepu.edu.cn; ostadhassan@aut.ac.ir
bDepartment of Petroleum Engineering, University of North Dakota, Grand Forks, ND 58202, USA
cDepartment of Petroleum Engineering, Amirkabir University of Technology, Tehran, Iran
dCollege of Petroleum Engineering, China University of Petroleum, 102249, Beijing, PR China
eRegional Centre of Advanced Technologies and Materials, Faculty of Science, Palacky University, Šlechtitelů 27, 783 71 Olomouc, Czech Republic
fDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: hwjang@snu.ac.kr; mrsh2@snu.ac.kr
First published on 14th October 2020
Abstract
CO2 injection (EOR and sequestration technique) creates the amalgamation of hydrocarbons, CO2, and aqueous brine in the subsurface. In this study, molecular dynamics (MD) simulations were used to investigate the diffusivity of hydrocarbon molecules in a realistic scenario of supercritical CO2 (SC-CO2) injection in the subsurface over a wide range of pressures (50 < P < 300 bar) and aqueous brine concentrations (0, 2, and 5% brine). To overcome existing challenges in traditional diffusivity calculation approaches, we took advantage of fundamental molecular-based methods, along with further verification of results by previously published experimental data. In this regard, computational methods and MD simulations were employed to compute diffusion coefficients of hydrocarbons (benzene and pentane). It was found that the presence of water and salt affects the thermodynamic properties of molecules where the intermolecular interactions caused the hydrophobic hydration of hydrocarbons coupled with ionic hydration due to hydrogen bond and ion-dipole interactions. Based on these results, it is demonstrated that the formation of water clusters in the SC-CO2 solvent is a major contributor to the diffusion of hydrophobic molecules. The outcome at different pressure conditions showed that hydrocarbons always would diffuse less in the presence of water. The slopes of linearly fitted MSD of benzene and pentane infinitely diluted in SC-CO2 is around 13 to 20 times larger than the slope with water molecules (4 wt%). When pressure increases (100–300 bar), the diffusion coefficients (D) of benzene and pentane decreases (around 1.2 × 10−9 to 0.4 × 10−9 and 2 × 10−9 to 1 × 10−9 m2 s−1, respectively). Furthermore, brine concentration generally plays a negative role in reducing the diffusivity of hydrocarbons due to the formation of water clusters as a result of hydrophobic and ionic hydration. Under the SC-CO2 rich (injection) system in the shale reservoir, the diffusion of hydrocarbon is correlated to the efficiency of hydrocarbon flow/recovery. Ultimately, this study will guide us to better understand the phenomena that would occur in nanopores of shale that undergo EOR or are becoming a target of CO2 sequestration.
Introduction
There has been a growing attention to CO2 injection (EOR and sequestration) in the subsurface as a possible solution to alleviate CO2 emission into the atmosphere as a major cause of the greenhouse gas effect.1,2 However, it is well known that supercritical carbon dioxide (SC-CO2) has the advantage of being non-toxic, non-flammable, and in a good chemical stability as a viable green alternative solvent in various disciplines such as pharmaceutics, materials, chemistry, and energy.3 Considering significant changes that happen in the density of a supercritical fluid, its compressibility and thermal expansion coefficient will change too, which ultimately alters other properties such as viscosity and diffusivity. This is also accompanied by a substantial shift in the kinetics and equilibrium,4 which can become useful in a variety of ways including extraction and separation processes in CO2 injection operations to further manipulate the solubility and mass transfer (diffusivity).3As mentioned above, CO2 injection in the subsurface is done for producing hydrocarbons (oil/gas) or sequestering CO2 in deep high-pressured reservoirs that includes aqueous brine in the system.5 Commonly, at such depths, temperature and pressure conditions will dictate that CO2 should exist as a supercritical fluid that can mix with a wide range of concentrations of hydrocarbon and aqueous brine. Although various characteristics of supercritical fluids, including CO2 have been measured, they are not yet fully exploited for industrial applications. This is because it is experimentally not feasible to understand such characteristics and change in behavior at very high temperature and pressure conditions.6,7 Hence, computational methods such as molecular dynamics (MD) simulation by taking advantage of fundamental molecular-based methods8 can overcome existing challenges in traditional methods.
The infinite dilution diffusion coefficient (D) is an important property to accurately describe mass transfer by revealing a combination of effects that involves intermolecular interactions specially in separation processes.4,9 D, which is a time-dependent property can be studied by both experimental/predictive equation measurements10–13 and computational calculations,4,14–18 under various temperatures, pressures, and compositional conditions. One of these challenging scenarios that involve hydrocarbons, brine, and SC-CO2 is the injection of CO2 in the subsurface for sequestration and/or enhanced oil recovery.
Previously, we investigated the evolution of organic compounds during thermal maturation by spectroscopic techniques.19,20 Next, to understand gas adsorption in a mixture of fluids, including CH4 and CO2, the interactions that take place between CH4, CO2, and aqueous brine in nano-pores of the organic compound was examined.21 The present work is the extension of our previous studies into the infinitely diluted hydrocarbons in SC-CO2 and aqueous brine. In particular, we analyzed the interactions between molecules in such mixtures and estimated D of aromatics and aliphatic hydrocarbons in the presence of SC-CO2 and aqueous brine concentrations. The prediction of fluid behavior in such an environment and mixtures is an important phenomenon in CO2 injection processes that have not been studied to the best of author's knowledge. For this analysis, D of hydrocarbon in a CO2-rich environment was calculated via MD simulations at varying concentrations of brine and pressures by tracking their mean square displacement (MSD).15,16,22 Regardless of the operation being EOR or sequestration, studying the diffusivity of hydrocarbons in SC-CO2 conditions where other fluids and NaCl salt are present will become essential to understand the flow of hydrocarbons in reservoir conditions. The simulations were further employed to understand intermolecular interactions considering hydrogen bond (H-bond) and ion–dipole interactions, which would affect the diffusivity of hydrocarbons under different mole fractions and pressures. In this regard, we demonstrated the combination of intermolecular interaction effects involving hydrophobic hydration and clustering of water molecules surrounding non-polar solute (hydrocarbon) to occur with the free energy change reducing the exposed surface area to the solvent.23–25 Authors believe this approach could become especially of interest to model high-pressure subsurface conditions that are difficult to experimentally investigate. Ultimately, this investigation enables us to establish necessary correlations to predict limiting asymptotic behavior of molecular diffusivity in different conditions and fluidic environments.
Materials and methods
Intermolecular potentials
To accurately calculate properties of interest by MD simulations, intermolecular potentials were selected based on the literature that provides reliable force fields (see ESI†).26–33 Hence, the semi-empirical classical potential called elementary physical model (EPM2) was used for carbon dioxide,26 and the TIP4P/2005 was used for the representation of H2O molecules.27 For the aromatic hydrocarbon, the optimized potentials for liquid simulation-all atoms (OPLS-AA) model was adopted because it has already been validated for simple compounds (light hydrocarbons)28,29 similar to what is being modeled here. Moreover, L-OPLS (an optimized OPLS-AA) was used for modeling aliphatic hydrocarbons, which suggests a further improvement for longer chain alkanes in diffusion problems.30 Na+ and Cl− ions were represented by the TraPPE force field using the parameters proposed by Smith and Dang.31 The total interactions between molecules were calculated as the sum of Lennard-Jones (LJ) and Coulomb interactions. The LJ function represents the van der Waals forces, which describes the repulsive and attractive interactions between two molecules or atoms that temporarily create an induced dipole moment occurring by the motion of electrons. Likewise, the Coulomb function demonstrates particle interactions due to their permanent dipole moments that attract and repel one another.4Molecular dynamics simulations
MD simulations were performed in the isothermal-isobaric (NPT) ensemble with the LAMMPS simulation package34 in a three-dimensional simulation box with periodic boundary conditions imposed in all directions. To acquire the infinite dilution concentrations at desired temperature and pressure conditions,35–37 0.1% of mass fraction of solute (hydrocarbon) was retained in all simulation scenarios. The system was initially allowed to equilibrate for a period of 5 ns with integration using a Nose–Hoover thermostat and barostat, where the density of the system converged to a mean value corresponding to the temperature and pressure conditions. Both the LJ and Coulomb interactions were modeled using a cut-off distance of 1.4 nm. The long-range coulombic interactions (beyond the 1.4 nm cutoff) were computed using the particle-mesh Ewald scheme (PME) with an accuracy of 10−4. These potentials that were achieved through trial and error summed over all sites to obtain the total intermolecular interactions. Furthermore, production simulations were performed for 10 ns, while the temperature and pressure were maintained constant with a coupled thermostat. All simulations in this work contained 3000 molecules of the solvent (CO2 rich) and 1 molecule of the selected hydrocarbon (aromatic and linear alkane) to resemble infinite dilution conditions. Moreover, for the case when water is in the system, around 4 wt% of the total system comprises water molecules. Monitoring of potential energy, pressure, and temperature during the production simulations confirmed that they were well stabilized with minor fluctuations, <1% for temperature. The molecular trajectories were sampled every 1000 steps to enable calculating desired parameters.MD simulations with the same initial coordinates and the mean square displacement (MSD) tracking atomic motion in the systems were calculated by running an average of MSD of 15 trajectories. The independent MD simulations were conducted using different random seeds to generate initial velocities.38 Molecular diffusion, which describes the spread of molecules through random motion (diffusion coefficient), is performed via the following formula by the Einstein relation based on calculating the MSDs:39

Results and discussions
Molecular dynamics (MD) simulations have been utilized to determine the diffusivity of hydrocarbon molecules in selected and various conditions. Diffusion coefficients were obtained via calibration in the simulation setting and selection of the appropriate molecular force field as explained earlier. A total of 15 simulations were run independently, with the same system conditions with a different ensemble of velocities using a random number, to avoid a relatively larger discrepancy with the experimental result.First, to validate the results of MD simulations, calculated diffusion coefficients (D) of water and benzene infinitely diluted in SC-CO2 are compared with available experimental data10,40–42 as shown in Fig. 1. The diffusion coefficients of water in a pressure interval of 134 to 300 bar were calculated at a constant temperature of 308 K; for the infinitely diluted benzene in SC-CO2 at 333 K, the simulations were performed between 130 and 350 bar.
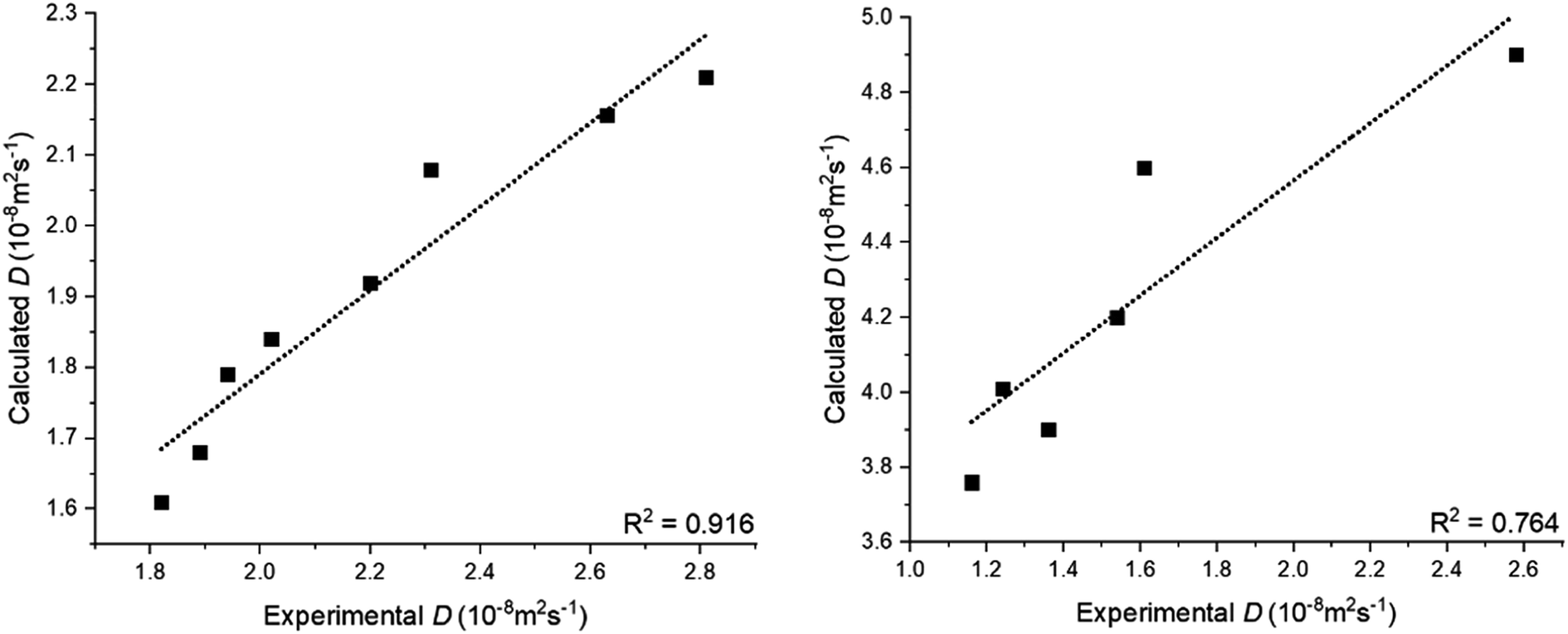 | ||
| Fig. 1 Correlation between calculated and previously published experimental diffusion coefficients of (left) water and (right) benzene infinitely diluted in supercritical carbon dioxide (SC-CO2) at different pressures.10,40–42 | ||
Despite D being determined by calculating the average results of 15 independent MD simulations, the overall diffusion coefficient of both water and benzene are higher than experimental data. We speculate that the discrepancy between simulation results and experimental data could be due to inaccuracy in internal degrees of freedom of molecules, which could occur for isotherms at higher pressures.43 Unlike the case of self-diffusivity of molecules, the mixture of hydrocarbon and CO2 would create a more complex system considering the number of degrees of freedom compared to the water–CO2 mixed system. Fig. 1 shows that D of water in SC-CO2 is relatively closer to experimental data than benzene for isotherms at higher pressures. Given these observations, we note that the simulation results have considered the lack of accuracy of absolute diffusivity values because of selected force field models under the condition of SC-CO2 solvent at high pressures.
However, although the absolute values of calculated D demonstrate certain differences with experimental data, overall satisfactory correlations have been achieved between the simulation results and experimental data for both water and benzene (R2 of 0.916 and 0.764, respectively) in Fig. 1. Ultimately, based on these results, the diffusivity trend of the molecule is significantly can be changed to highly in the selected solvent (CO2 molecule) at all pressure ranges.
Hydrophobic hydration of hydrocarbons
A major aim of this study is to understand the diffusion of particles in reservoir pressure and temperature conditions during CO2 injection and sequestration. To understand the corresponding phenomena based on the presence of these fluids, the presence of salt (halite mineral), which is ubiquitous in the subsurface environment should not be neglected, as another essential particle in the system.As shown in Fig. 1, in addition to the earlier diffusion calculations for water and benzene in SC-CO2 separately and individually, infinitely diluted aromatic (benzene) and aliphatic (pentane) hydrocarbons diffusivity were examined in the CO2 solvent that contains water and salt (NaCl) under selected pressure and temperature conditions. For all simulations that contained water molecules (4 wt%), it is observed that water molecules were aggregated around the hydrocarbon molecules at varying salt concentrations (0, 2, and 5% brine). Fig. 2 shows that water aggregation occurs, which has minimized the free energy of the whole system. We observed a drop in the energy of the system at the early stages of the simulation (see ESI†) and then the system reaches equilibrium to form H-bond and hydration structure around the hydrocarbons.
 | ||
| Fig. 2 (a) Snapshots of initial diffusion simulation containing hydrocarbon (pentane), water, and CO2 molecules; the mixture consists of 3000 CO2 molecules, 320 water molecules, and 1 pentane molecule with color codes as: carbon: cyan; oxygen: red; hydrogen: white using molecular graphics program.44 (b) The system reaches to an equilibrium status forming a water structure around pentane. | ||
The hydrocarbon molecules come to equilibrium positions in the clustered water structure, which happens at the hydrocarbon–CO2 interface with hydrogen bonds (Fig. 3). Developing the water molecule structure surrounding the hydrocarbon occurs while forming hydrogen bonds or by losing free energy. The hydrophobic molecule and water molecules interact through multiple van der Waals forces forming hydration structure because of flexibility in the spatial arrangement of water molecules.45,46 Hydrogen bonds (H-bonds) between water molecules have been induced and formed around hydrocarbons as the hydrophobic hydration in the CO2 solvent system. Moreover, due to non-zero dipole attractions of water molecules, the cluster of hydration is observed around ions (Na+ and Cl−) in SC-CO2 conditions too. The dipole moment of water molecules is favorably engaged by the interaction with ions in the system.47 However, because the CO2 molecule is non-polar comprising symmetrical dipole bonds, it can form relatively weaker bonds with other molecules.
 | ||
| Fig. 3 The snapshots of hydrophobic hydration shell that is developing around (a) benzene and (d) pentane molecules at 333 K and 200 bar in SC-CO2 with 4 wt% pure water, (b) benzene and (e) pentane in SC-CO2 with 5% brine (4 wt%). The snapshots (a, b, d and e) show that water molecules connecting hydrogen bonds within around 5 Å from the benzene molecule color codes as: carbon: cyan; oxygen: red; hydrogen: white; sodium: green; chloride: yellow. The radial distribution function (RDF) between water and the molecules; (c) benzene and (f) pentane with increasing brine concentration. | ||
Fig. 3 shows that the first hydration structure has occurred around the hydrocarbon molecule in the CO2 solvent system with relevant energy levels. The hydrogen bonds are considered when the O–O distance is within 3.5 Å, and the HO–O bond angle is less <30°.48–50 Both systems of benzene and pentane infinitely diluted in CO2 with 4 wt% pure water exhibited the generation of the hydration shell by employing hydrogen bonds as shown in Fig. 3a and d, respectively. However, in Fig. 3b and e, salt ions seem to interfere with the formation of the hydration structure around the hydrocarbon molecules. Slight changes in the intensity of the RDF (radial distribution function) is observed when brine concentration is increased (Fig. 3c and f). The hydration structure develops when hydrogen bonds and salt ions engage in the dipole moment of water molecules. Therefore, when the number of salt ions increases because of brine concentration, the density of water molecules that are aligned in hydration structure decreases at the distance of hydration shell ∼3.5–5 Å, as shown in Fig. 3c and f. It indicates that the ions immerse in the first hydration shell and interfere with the hydrogen bonds. However, reverse trends are revealed as the distances between water and hydrocarbons grow farther away from the benzene and pentane molecules at 7 Å and 5 Å, respectively. The RDF results exhibit that salt concentration affects the number of water molecules, which could be involved in H-bond and ion–dipole interaction, as per the distance between water and hydrocarbon atoms. In other words, this indicates that the presence/adding of salt ions led to an increase in the number of water molecules outside the first hydration shell. The increased brine concentration effect is explained by observing water structure around Na+ and Cl− ions that caused larger aggregates and more clusters of hydration around hydrocarbon molecules. This happens because water molecules that are around these ions are highly polarized,47 while the presence of such ions can considerably increase the strength of hydrogen bonds within the geometry of the hydrogen bonds.51 Therefore, it is concluded that the combination of hydrophobic and ionic hydration phenomena would lead to a denser and bigger water structure. In this regard, the brine that can exist at low salinity (1%) and high salinity (>5%)52 would affect the thermodynamics and kinetics of particles under true reservoir pressure that should be considered in a CO2 injection process.
Diffusivity of hydrocarbons
Fig. 4 shows that the average travel distance of hydrocarbon molecules over time exhibits a strong dependency on the presence of water (4 wt%) in the SC-CO2 system. The slope of linearly fitted MSD of benzene infinitely diluted in SC-CO2 is around 20 times larger than the one with moisture (Fig. 4a). Moreover, the MSD slope for pentane shows 13 folds of difference between the case when water is missing from the system and in its presence (Fig. 4b). We confirmed that this is the outcome of a scenario at different pressure conditions where hydrocarbons always show significantly lower diffusion in aqueous systems. Thus, the results indicate that both benzene and pentane diffusivities are highly affected by the formation of hydrophobic hydration based on the observations in Fig. 3 and 4.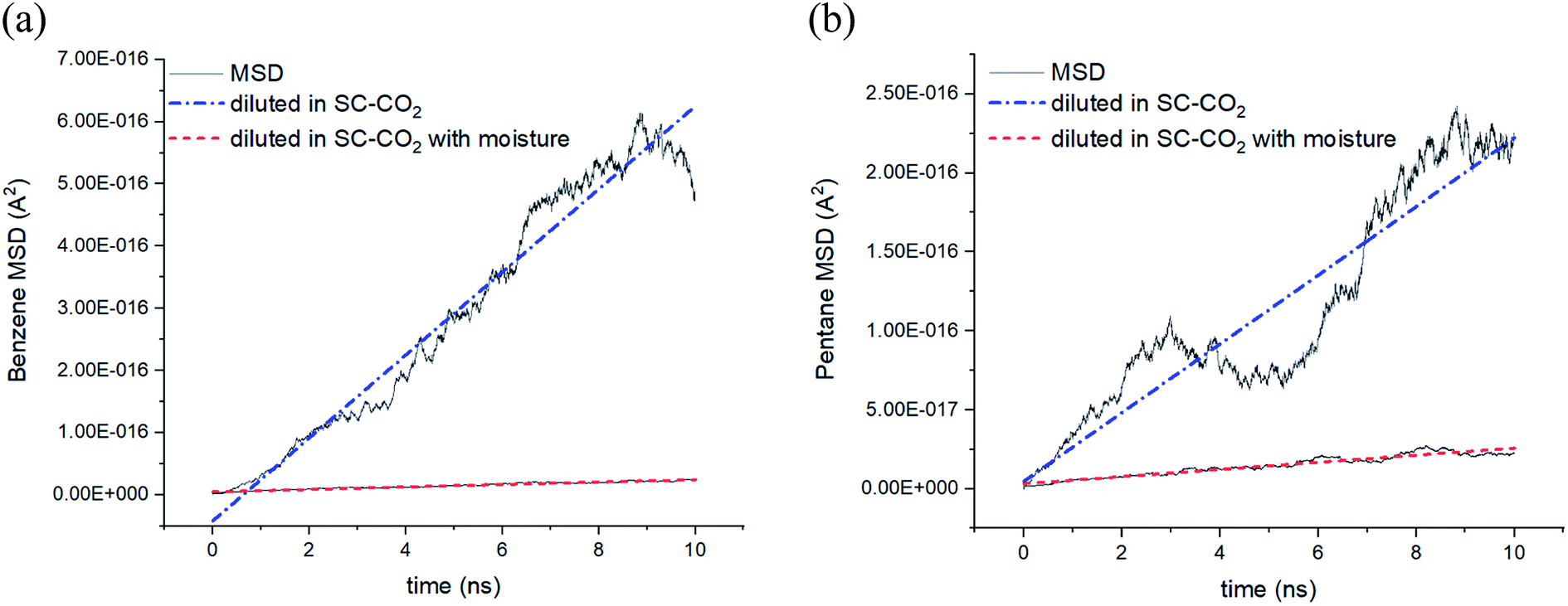 | ||
| Fig. 4 Linear fits of MSD calculations for hydrocarbons infinitely diluted in SC-CO2; (a) benzene at 130 bar and (b) pentane at 100 bar in the presence (4 wt%) and absence of water at 333 K. | ||
Brine concentration has an impact on intermolecular interactions between water and hydrophobic molecules by changing the hydration structure.53 As discussed previously (Fig. 3), the results show that adding salt can affect interactions between salt ions and water molecules. Although diffusivity expresses relatively minor changes due to high-pressure conditions (over 100 bar), salt plays a role in reducing the diffusivity of hydrocarbons as shown in Fig. 5. In Fig. 5a, the calculated D of benzene clearly exhibits that adding salt to the system would have a negative impact in all pressure ranges. However, the calculated D of pentane shows that it is relatively less affected by the salt in the system (Fig. 5b). We also found that the calculated D of both of these two hydrocarbons in SC-CO2 condition is around 25% of the case when CO2 is in the gaseous phase at lower pressures (at 50 bar) regardless of the brine concentration. Ultimately, as the pressure in the system increases, the calculated diffusion coefficients (D) in all simulation scenarios start to decrease.
 | ||
| Fig. 5 Calculated diffusion coefficient of (a) benzene and (b) pentane infinitely diluted in CO2 at different pressures (50 to 100 bar) and 333 K temperature. | ||
As we discussed the effects of adding the salt, the hydration water shell around ions (Na+ and Cl−) occurs by agglomerated water molecules. In this regard, water molecules that are polarized by the neighboring ions can considerably enhance the strength of hydrogen bonds through the change in the geometry of hydrogen bonds.47,51 Consequently, the aggregation of hydrophobic and ionic hydration water structure (Fig. 2 and 3) creates a larger structure. This could be the main reason for lower diffusivity in hydrophobic molecules (hydrocarbon) in the presence of brine as compared to the absence of water and/or brine in the system. Therefore, it can be concluded that the relationship between the brine concentration and diffusivity is not monotonic, and a hydration effect should be expected to happen as a major phenomenon in the CO2 solvent.
CO2 injection for producing hydrocarbons that are hydrophobic in an aqueous environment takes place in the deep high-pressured reservoir in SC-CO2 conditions. The diffusivity of hydrocarbon in the CO2 rich system is highly correlated with a combination of effects involving intermolecular interactions. The water cluster surrounding a non-polar hydrocarbon could occur by hydrophobic hydration (H-bond) and ionic hydration (ion-dipole) between hydrocarbon and brine (water and salt ions) molecules. The agglomerate of hydrocarbon and brine molecules leads to a significantly smaller diffusivity in a CO2 rich system. Therefore, it would affect the flow of hydrocarbon, which is the main goal in EOR considering the separation and diffusion phenomena that play a major role in CO2 EOR/separation. Based on the outcome of this study where we observed interactions between water and other molecules (hydrocarbon and salt) that caused hydration effects in the SC-CO2 system, it is important to note that further study is required to understand molecular behavior in the reservoir pressure and temperature when a variety of hydrophobic molecules exist in the system.
Conclusion
During CO2 injection (EOR/sequestration) process, the diffusion characteristics of hydrocarbons (oil/gas) in SC-CO2 fluid containing the aqueous brine is important to understand the hydrocarbon flow at reservoir conditions. In this study, MD simulations were conducted to explore particle diffusion behavior considering the interrelated combination of effects involving intermolecular interactions. In an EOR/sequestration process in the subsurface, both CO2 and interstitial water mutually dissolve and subsequently diffuse until thermodynamic equilibrium is reached. The diffusivity of hydrocarbon molecules and hydrophobic hydration around hydrocarbon molecules in SC-CO2 solvent were observed in this study and the following conclusions were made:● The presence of water and salt affects the thermodynamic properties of the system, and the interactions between water molecules and others (hydrocarbon, CO2, and salt) give rise to the hydrophobic hydration. Results demonstrated that water cluster formation in the SC-CO2 solvent is a major contributor to the diffusion of hydrophobic molecules (both aromatic and aliphatic hydrocarbons).
● Due to water dipole attraction, the cluster of hydration is observed around ions (Na+ and Cl−) in selected SC-CO2 conditions. The combination of hydrophobic and ionic hydration water structure can increase the size of the water structure in SC-CO2.
● The outcome at different pressure conditions shows that hydrocarbons always show significantly lower diffusion in the aqueous system. The calculated diffusion coefficient (D) from all simulations exhibited decreasing trends as the pressure increased.
● Finally, the brine concentration plays a negative role in reducing the diffusivity of hydrocarbons due to the water structure formation as a result of complex hydrophobic and ionic hydration.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support of the Future Material Discovery Program (2016M3D1A1027666) and the Basic Science Research Program (2017R1A2B3009135) through the National Research Foundation of Korea are appreciated.References
- V. Alvarado and E. Manrique, Enhanced Oil Recovery: An Update Review, Energies, 2010, 3(9), 1529–1575, DOI:10.3390/en3091529.
- B. Jia, J.-S. Tsau and R. Barati, A Review of the Current Progress of CO2 Injection EOR and Carbon Storage in Shale Oil Reservoirs, Fuel, 2019, 236, 404–427, DOI:10.1016/J.FUEL.2018.08.103.
- G. Brunner, Applications of Supercritical Fluids, Annu. Rev. Chem. Biomol. Eng., 2010, 1(1), 321–342, DOI:10.1146/annurev-chembioeng-073009-101311.
- R. V. Vaz, J. R. B. Gomes and C. M. Silva, Molecular Dynamics Simulation of Diffusion Coefficients and Structural Properties of Ketones in Supercritical CO2 at Infinite Dilution, J. Supercrit. Fluids, 2016, 107, 630–638, DOI:10.1016/J.SUPFLU.2015.07.025.
- E. Santibanez-Borda, R. Govindan, N. Elahi, A. Korre and S. Durucan, Maximising the Dynamic CO2 Storage Capacity through the Optimisation of CO2 Injection and Brine Production Rates, Int. J. Greenhouse Gas Control, 2019, 80, 76–95, DOI:10.1016/j.ijggc.2018.11.012.
- O. Suárez-Iglesias, I. Medina, C. Pizarro and J. L. Bueno, Diffusion Coefficients of 2-Fluoroanisole, 2-Bromoanisole, Allylbenzene and 1,3-Divinylbenzene at Infinite Dilution in Supercritical Carbon Dioxide, Fluid Phase Equilib., 2007, 260(2), 279–286, DOI:10.1016/J.FLUID.2007.07.039.
- C. Pizarro, O. Suárez-Iglesias, I. Medina and J. L. Bueno, Using Supercritical Fluid Chromatography to Determine Diffusion Coefficients of 1,2-Diethylbenzene, 1,4-Diethylbenzene, 5-Tert-Butyl-m-Xylene and Phenylacetylene in Supercritical Carbon Dioxide, J. Chromatogr. A, 2007, 1167(2), 202–209, DOI:10.1016/J.CHROMA.2007.08.010.
- L. Zhao, L. Tao and S. Lin, Molecular Dynamics Characterizations of the Supercritical CO 2 –Mediated Hexane–Brine Interface, Ind. Eng. Chem. Res., 2015, 54(9), 2489–2496, DOI:10.1021/ie505048c.
- J. L. Bueno, J. J. Suarez, J. Dizy and I. Medina, Infinite Dilution Diffusion Coefficients: Benzene Derivatives as Solutes in Supercritical Carbon Dioxide, J. Chem. Eng. Data, 1993, 38(3), 344–349, DOI:10.1021/je00011a002.
- B. Xu, K. Nagashima, J. M. DeSimone and C. S. Johnson, Diffusion of Water in Liquid and Supercritical Carbon Dioxide: An NMR Study, J. Phys. Chem. A, 2003, 107(1), 1–3, DOI:10.1021/jp021943g.
- H. Liu and E. Ruckenstein, A Predictive Equation for the Tracer Diffusion of Various Solutes in Gases, Supercritical Fluids, and Liquids, Ind. Eng. Chem. Res., 1997, 36(12), 5488–5500, DOI:10.1021/ie970331t.
- S. Umezawa and A. Nagashima, Measurement of the Diffusion Coefficients of Acetone, Benzene, and Alkane in Supercritical CO2 by the Taylor Dispersion Method, J. Supercrit. Fluids, 1992, 5(4), 242–250, DOI:10.1016/0896-8446(92)90014-B.
- C.-C. Lai and C.-S. Tan, Measurement of Molecular Diffusion Coefficients in Supercritical Carbon Dioxide Using a Coated Capillary Column, Ind. Eng. Chem. Res., 1995, 34(2), 674–680 CrossRef CAS.
- Y. Iwai, H. Higashi, H. Uchida and Y. Arai, Molecular Dynamics Simulation of Diffusion Coefficients of Naphthalene and 2-Naphthol in Supercritical Carbon Dioxide, Fluid Phase Equilib., 1997, 127(1–2), 251–261, DOI:10.1016/S0378-3812(96)03139-1.
- J. Wang, H. Zhong, H. Feng, W. Qiu and L. Chen, Molecular Dynamics Simulation of Diffusion Coefficients and Structural Properties of Some Alkylbenzenes in Supercritical Carbon Dioxide at Infinite Dilution, J. Chem. Phys., 2014, 140(10), 104501, DOI:10.1063/1.4867274.
- O. A. Moultos, G. A. Orozco, I. N. Tsimpanogiannis, A. Z. Panagiotopoulos and I. G. Economou, Atomistic Molecular Dynamics Simulations of H 2 O Diffusivity in Liquid and Supercritical CO 2, Mol. Phys., 2015, 113(17–18), 2805–2814, DOI:10.1080/00268976.2015.1023224.
- A. Loya, J. L. Stair, A. R. Jafri, K. Yang and G. Ren, A Molecular Dynamic Investigation of Viscosity and Diffusion Coefficient of Nanoclusters in Hydrocarbon Fluids, Comput. Mater. Sci., 2015, 99, 242–246, DOI:10.1016/j.commatsci.2014.11.051.
- H. Higashi, Y. Iwai and Y. Arai, Calculation of Self-Diffusion and Tracer Diffusion Coefficients near the Critical Point of Carbon Dioxide Using Molecular Dynamics Simulation, Ind. Eng. Chem. Res., 2000, 39(12), 4567–4570, DOI:10.1021/ie000173x.
- H. Lee, A. Abarghani, B. Liu, M. Shokouhimehr and M. Ostadhassan, Molecular Weight Variations of Kerogen during Maturation with MALDI-TOF-MS, Fuel, 2020, 269, 117452, DOI:10.1016/J.FUEL.2020.117452.
- H. Lee, N. Oncel, B. Liu, A. Kukay, F. Altincicek, R. S. Varma, M. Shokouhimehr and M. Ostadhassan, Structural Evolution of Organic Matter in Deep Shales by Spectroscopy (1H and 13C Nuclear Magnetic Resonance, X-Ray Photoelectron Spectroscopy, and Fourier Transform Infrared) Analysis, Energy Fuels, 2020, 34(3), 2807–2815, DOI:10.1021/acs.energyfuels.9b03851.
- H. Lee, A. Shakib, F. Shokouhimeher, M. Shokouhimehr, B. Bubach, L. Kong and M. Ostadhassan, Optimal Separation of CO2/CH4/Brine with Amorphous Kerogen: A Thermodynamics and Kinetics Study, J. Phys. Chem. C, 2019, 123(34), 20877–20883, DOI:10.1021/acs.jpcc.9b04432.
- I. N. Tsimpanogiannis, O. A. Moultos, L. F. M. Franco, M. B. de M. Spera, M. Erdős and I. G. Economou, Self-Diffusion Coefficient of Bulk and Confined Water: A Critical Review of Classical Molecular Simulation Studies, Mol. Simul., 2019, 45(4–5), 425–453, DOI:10.1080/08927022.2018.1511903.
- R. L. Mancera, Hydrogen-Bonding Behaviour in the Hydrophobic Hydration of Simple Hydrocarbons in Water, J. Chem. Soc. Faraday. Trans., 1996, 92(14), 2547, 10.1039/ft9969202547.
- B. Widom, P. Bhimalapuram and K. Koga, The Hydrophobic Effect, Phys. Chem. Chem. Phys., 2003, 5(15), 3085, 10.1039/b304038k.
- E. E. Meyer, K. J. Rosenberg and J. Israelachvili, Recent Progress in Understanding Hydrophobic Interactions, Proc. Natl. Acad. Sci. U.S.A., 2006, 103(43), 15739–15746, DOI:10.1073/pnas.0606422103.
- J. G. Harris and K. H. Yung, Carbon Dioxide's Liquid-Vapor Coexistence Curve And Critical Properties as Predicted by a Simple Molecular Model, J. Phys. Chem., 1995, 99(31), 12021–12024, DOI:10.1021/j100031a034.
- J. L. F. Abascal and C. Vega, A General Purpose Model for the Condensed Phases of Water: TIP4P/2005, J. Chem. Phys., 2005, 123(23), 234505, DOI:10.1063/1.2121687.
- L. Jorgensen, W. Maxwell, S. Maxwell and J. Tirado-Rives, Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids, J. Am. Chem. Soc., 1996, 118(45), 11225–11236, DOI:10.1021/ja9621760.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides, J. Phys. Chem. B, 2001, 105(28), 6474–6487, DOI:10.1021/jp003919d.
- S. W. I. Siu, K. Pluhackova and R. A. Böckmann, Optimization of the OPLS-AA Force Field for Long Hydrocarbons, J. Chem. Theory Comput., 2012, 8(4), 1459–1470, DOI:10.1021/ct200908r.
- D. E. Smith and L. X. Dang, Computer Simulations of NaCl Association in Polarizable Water, J. Chem. Phys., 1994, 100(5), 3757–3766, DOI:10.1063/1.466363.
- C.-F. Fu and S. X. Tian, A Comparative Study for Molecular Dynamics Simulations of Liquid Benzene, J. Chem. Theory Comput., 2011, 7(7), 2240–2252, DOI:10.1021/ct2002122.
- C. M. Baker and G. H. Grant, Modeling Aromatic Liquids: Toluene, Phenol, and Pyridine, J. Chem. Theory Comput., 2007, 3(2), 530–548, DOI:10.1021/ct600218f.
- S. Plimpton, Fast Parallel Algorithms for Short-Range Molecular Dynamics, J. Comput. Phys., 1995, 117(1), 1–19, DOI:10.1006/JCPH.1995.1039.
- R. Span and W. Wagner, A New Equation of State for Carbon Dioxide Covering the Fluid Region from the Triple-Point Temperature to 1100 K at Pressures up to 800 MPa, J. Phys. Chem. Ref. Data, 1996, 25(6), 1509–1596, DOI:10.1063/1.555991.
- J. J. Suárez, I. Medina and J. L. Bueno, Diffusion Coefficients in Supercritical Fluids: Available Data and Graphical Correlations, Fluid Phase Equilib., 1998, 153(1), 167–212, DOI:10.1016/S0378-3812(98)00403-8.
- V. Vesovic, W. A. Wakeham, G. A. Olchowy, J. V. Sengers, J. T. R. Watson and J. Millat, The Transport Properties of Carbon Dioxide, J. Phys. Chem. Ref. Data, 1990, 19(3), 763–808, DOI:10.1063/1.555875.
- J. Wang and T. Hou, Application of Molecular Dynamics Simulations in Molecular Property Prediction II: Diffusion Coefficient, J. Comput. Chem., 2011, 32(16), 3505–3519, DOI:10.1002/jcc.21939.
- A. Einstein, Über Die von Der Molekularkinetischen Theorie Der Wärme Geforderte Bewegung von in Ruhenden Flüssigkeiten Suspendierten Teilchen, Ann. Phys., 1905, 322(8), 549–560, DOI:10.1002/andp.19053220806.
- P. R. Sassiat, P. Mourier, M. H. Caude and R. H. Rosset, Measurement of Diffusion Coefficients in Supercritical Carbon Dioxide and Correlation with the Equation of Wilke and Chang, Anal. Chem., 1987, 59(8), 1164–1170, DOI:10.1021/ac00135a020.
- J. J. Suárez, J. L. Bueno and I. Medina, Determination of Binary Diffusion Coefficients of Benzene and Derivatives in Supercritical Carbon Dioxide, Chem. Eng. Sci., 1993, 48(13), 2419–2427, DOI:10.1016/0009-2509(93)81063-2.
- T. Funazukuri and N. Nishimoto, Tracer Diffusion Coefficients of Benzene in Dense CO2 at 313.2 K and 8.5–30 MPa, Fluid Phase Equilib., 1996, 125(1–2), 235–243, DOI:10.1016/S0378-3812(96)03084-1.
- A. Smolyanitsky, A. F. Kazakov, T. J. Bruno and M. L. Huber, Mass Diffusion of Organic Fluids: A Molecular Dynamics Perspective, NIST Tech. Note, 2013, 1805, DOI:10.6028/NIST.TN.1805.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14(1), 33–38, DOI:10.1016/0263-7855(96)00018-5.
- R. D. Mountain and D. Thirumalai, Hydration for a Series of Hydrocarbons, Proc. Natl. Acad. Sci. U.S.A., 1998, 95(15), 8436–8440, DOI:10.1073/pnas.95.15.8436.
- Y. A. Mikheev, L. N. Guseva, E. Y. Davydov and Y. A. Ershov, The Hydration of Hydrophobic Substances, Russ. J. Phys. Chem. A, 2007, 81(12), 1897–1913, DOI:10.1134/S0036024407120011.
- C. Krekeler and L. Delle Site, Lone Pair versus Bonding Pair Electrons: The Mechanism of Electronic Polarization of Water in the Presence of Positive Ions, J. Chem. Phys., 2008, 128(13), 134515, DOI:10.1063/1.2873768.
- T. M. Raschke and M. Levitt, Nonpolar Solutes Enhance Water Structure within Hydration Shells While Reducing Interactions between Them, Proc. Natl. Acad. Sci. U.S.A., 2005, 102(19), 6777–6782, DOI:10.1073/pnas.0500225102.
- D. Chandler, Interfaces and the Driving Force of Hydrophobic Assembly, Nature, 2005, 437(7059), 640–647, DOI:10.1038/nature04162.
- J. Kim, Y. Tian and J. Wu, Thermodynamic and Structural Evidence for Reduced Hydrogen Bonding among Water Molecules near Small Hydrophobic Solutes, J. Phys. Chem. B, 2015, 119(36), 12108–12116, DOI:10.1021/acs.jpcb.5b05281.
- J. M. Andrić, G. V. Janjić, D. B. Ninković and S. D. Zarić, The Influence of Water Molecule Coordination to a Metal Ion on Water Hydrogen Bonds, Phys. Chem. Chem. Phys., 2012, 14(31), 10896, 10.1039/c2cp41125c.
- B. Kurtoglu Integrated Reservoir Characterization and Modeling in Support of Enhanced Oil Recovery for Bakken, Colorado School of Mines, 2013 Search PubMed.
- O. A. Francisco, H. M. Glor and M. Khajehpour, Salt Effects on Hydrophobic Solvation: Is the Observed Salt Specificity the Result of Excluded Volume Effects or Water Mediated Ion-Hydrophobe Association?, ChemPhysChem, 2020, 21(6), 484–493, DOI:10.1002/cphc.201901000.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06499h |
| This journal is © The Royal Society of Chemistry 2020 |