
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterpretation of the mechanism of 3,3′-dichloro-4,4′-diamino diphenylmethane synthesis over HY zeolites
Xiao Ruan,
Feng Wei,
Li Yang,
Yingxian Zhao * and
Qiang Wang*
* and
Qiang Wang*
NingboTech University, Ningbo 315100, Zhejiang, China. E-mail: zyx@nit.zju.edu.cn; wangqiangsky@263.net
First published on 18th September 2020
Abstract
Catalytic activities of zeolites HY, Hβ and HZSM-5 in the heterogeneous synthesis of 3,3′-dichloro-4,4′-diaminodiphenyl methane (MOCA) from o-chloroaniline and formaldehyde were pre-screened in an autoclave, and HY demonstrated better performance than others. Kinetic behaviors of MOCA synthesis over HY(11) were further investigated in a fixed bed continuous flow reactor, and under the conditions of the catalyst bed volume = 20 mL (8.14 g), n(o-chloroaniline)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n(HCHO) = 4:1, LHSV = 3.5 h−1, 0.5 MPa and 443 K, HCHO conversion and MOCA selectivity steadily fluctuated at high levels of 90–92% and 75–77% during 16 h, respectively. Catalysts were characterized by BET, NH3-TPD and XRD, products analyzed by HPLC, and reaction intermediates identified by LC/MS and 1H NMR. The mechanism of MOCA synthesis has been interpreted in detail, which also suggested that deposition of basic intermediates on active sites and accumulation of polymeric by-products in pore channels of the catalyst could cause significant decay of HY(11) activity and selectivity under severe conditions. Supplementary tests on catalyst regeneration confirmed that the acidity and surface area of spent HY(11) could be well recovered after burning off the deposited by-products.
:n(HCHO) = 4:1, LHSV = 3.5 h−1, 0.5 MPa and 443 K, HCHO conversion and MOCA selectivity steadily fluctuated at high levels of 90–92% and 75–77% during 16 h, respectively. Catalysts were characterized by BET, NH3-TPD and XRD, products analyzed by HPLC, and reaction intermediates identified by LC/MS and 1H NMR. The mechanism of MOCA synthesis has been interpreted in detail, which also suggested that deposition of basic intermediates on active sites and accumulation of polymeric by-products in pore channels of the catalyst could cause significant decay of HY(11) activity and selectivity under severe conditions. Supplementary tests on catalyst regeneration confirmed that the acidity and surface area of spent HY(11) could be well recovered after burning off the deposited by-products.
Introduction
As a class of important chain-extending, cross-linking and curing agents, diaminodiphenylmethanes have been used extensively in the preparation of various polymer products including resin, rubber, plastic, paint, film, etc. For example, 4,4′-diaminodiphenyl methane (MDA) is an important additive in the preparation of polyurethane and thermosets,1–5 3,3′-dimethyl-4,4′-diamino diphenylmethane (MDT) is used as a good curing agent in the preparation of electric cables and wires with excellent properties of insulation, heat endurance and chemical resistance,6–10 and also 3,3′-dichloro-4,4′-diaminodiphenylmethane (MOCA) plays a vital role in preparing high quality polyurethane and epoxy resin.11–14The condensation of unsubstituted and methyl- or chlorine-substituted aromatic amine with formaldehyde (HCHO) has provided a convenient route to prepare MDA,15,16 MDT17 and MOCA.18 Such condensation reactions, however, normally requires acid catalysis. In history, strong inorganic acids such as HCl were often used to catalyze homogeneous synthesis of diaminodiphenyl methanes through multistep including salinization, condensation, and neutralization,19,20 as showed in Scheme 1. Inevitably, using large amounts of HCl and NaOH could bring about serious problems such as high toxicity, corrosion, difficulty in product separation and disposal of a large amount of the contaminated water. Since the prevention and control of environmental pollution have been becoming more and more urgent, a green and eco-friendly technology for the synthesis of diaminodiphenylmethanes should be developed vigorously.
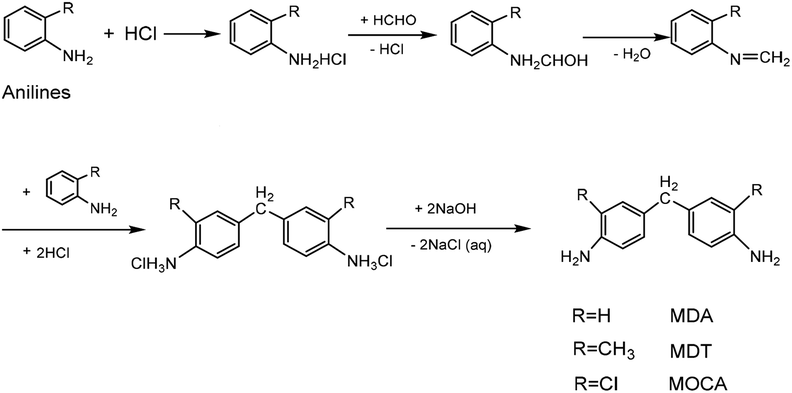 | ||
| Scheme 1 Homogeneous synthesis of MDA, MDT and MOCA catalyzed by HCl acid. | ||
It is of great academic and practical significance to exploit a heterogeneous process with solid acids replacing mineral acids as catalysts for synthesis of diaminodiphenylmethanes. The use of a solid acid could promise facile separation of catalyst from products without neutralization step, eliminate the polluted aqueous wastes, allow recovery and reuse of the catalyst, and thus provide a potentially green solution to the previously existing problems. In the heterogeneous synthesis of MDA by condensation of aniline with formaldehyde, the catalytic activities of various solid acids such as ion-exchanged resins,21 clays22,23 and zeolites19,24–27 were investigated. Previous investigations have also revealed the excellent performance of the protonated Al–Si zeolite Hβ in catalyzing heterogeneous synthesis of MDT from o-tolylamine and formaldehyde.28,29 Although rarely reported in the literature so far, an active catalyst for heterogeneous synthesis of MOCA should be found from various acidic zeolites with suitable porosity, modifiable acidity, high hydrothermal stability and good reusability.
The activity and selectivity of a protonated Al–Si zeolite in an acid-catalyzed reaction depend on its structural, surface and acidic properties. There are a large number of natural or synthetic Al–Si zeolites with different crystal structures. As three famous members of Al–Si zeolites used in petrochemical industry, for example, the low-silica zeolite Y has the structure of mineral faujasite with larger pore diameter (0.74 nm),30 the high-silica zeolite β has the structural framework with one type of ring channels (0.66 × 0.67 nm) crossed by second type of ring channels (0.56 × 56 nm),31 and the high-silica zeolite ZSM-5 has the MFI-type framework of two-dimensional pore structure with straight ring channels (0.51 × 0.55 nm) intersected by second ring channels (0.53 × 0.56 nm).32 The protonated zeolites HY, Hβ and HZSM-5 normally possess the same structural frameworks as their parent Y, β and ZSM-5 respectively, but their acidity may differ significantly each other and also will vary with the composition (Si/Al ratio). In this work, the heterogeneous synthesis of 3,3′-dichloro-4,4′-diaminodiphenyl methane (MOCA) by condensation of o-chloroaniline and formaldehyde over acidic zeolites was investigated. Firstly, the catalytic activities of HY (Si/Al = 7), Hβ (Si/Al = 40 and 100), HZSM-5 (Si/Al = 150 and 300) were pre-screened. Secondly, the catalytic performances of the promising HY with Si/Al ratios of 5, 7 and 11 were comparatively investigated. Thirdly, the effects of reaction temperature and time on the activity and selectivity of HY (11) as the best catalyst were further examined. Finally, the synthetic mechanism of MOCA involving key reaction intermediates was interpreted in detail, and also the deactivation and regeneration of catalyst was preliminarily discussed. Herein, the ultimate purpose is to establish the scientific foundation of developing a novel and green technology for the industrial production of MOCA and analogues.
Results and discussion
Comparative investigation on MOCA synthesis over acidic zeolites
To identify an efficient catalyst for heterogeneous synthesis of MOCA from o-chloroaniline and formaldehyde, the catalytic performances of several potential aluminum-silicon zeolites were comparatively investigated in a batch reactor.:1 with respect to o-chloroaniline:HCHO, the optimal ratio of 4:1 was adopted in experiments since HCHO might easily react with product or intermediates to form byproducts and excessive chloroaniline could inhibit the formation of byproducts. HPLC chromatograms of the product samples from these synthetic reactions were presented in Fig. 1a, displaying obvious differences each other. The bar data in Fig. 1b showed that the sand gave very low HCHO conversion and MOCA selectivity, indicating that non-catalytic synthesis of MOCA over sand was almost unproductive. For the five zeolites in order from HY(7) to Hβ(40) to Hβ(100) to HZSM-5(150) to HZSM-5(300), HCHO conversion slightly varied from 76.5 to 75.4 to 72.0 to 74.8 to 70.9%, MOCA selectivity significantly decreased from 67.2 to 60.1 to 45.5 to 23.2 to 17.6%, and the selectivity of key intermediate M in Fig. 1a increased from 14.6 to 16.5 to 25.7 to 40.6 to 43.1%, respectively. By comparison, HY(7) demonstrated the best catalytic performance while HZSM-5(300) gave the lowest selectivity of MOCA production.
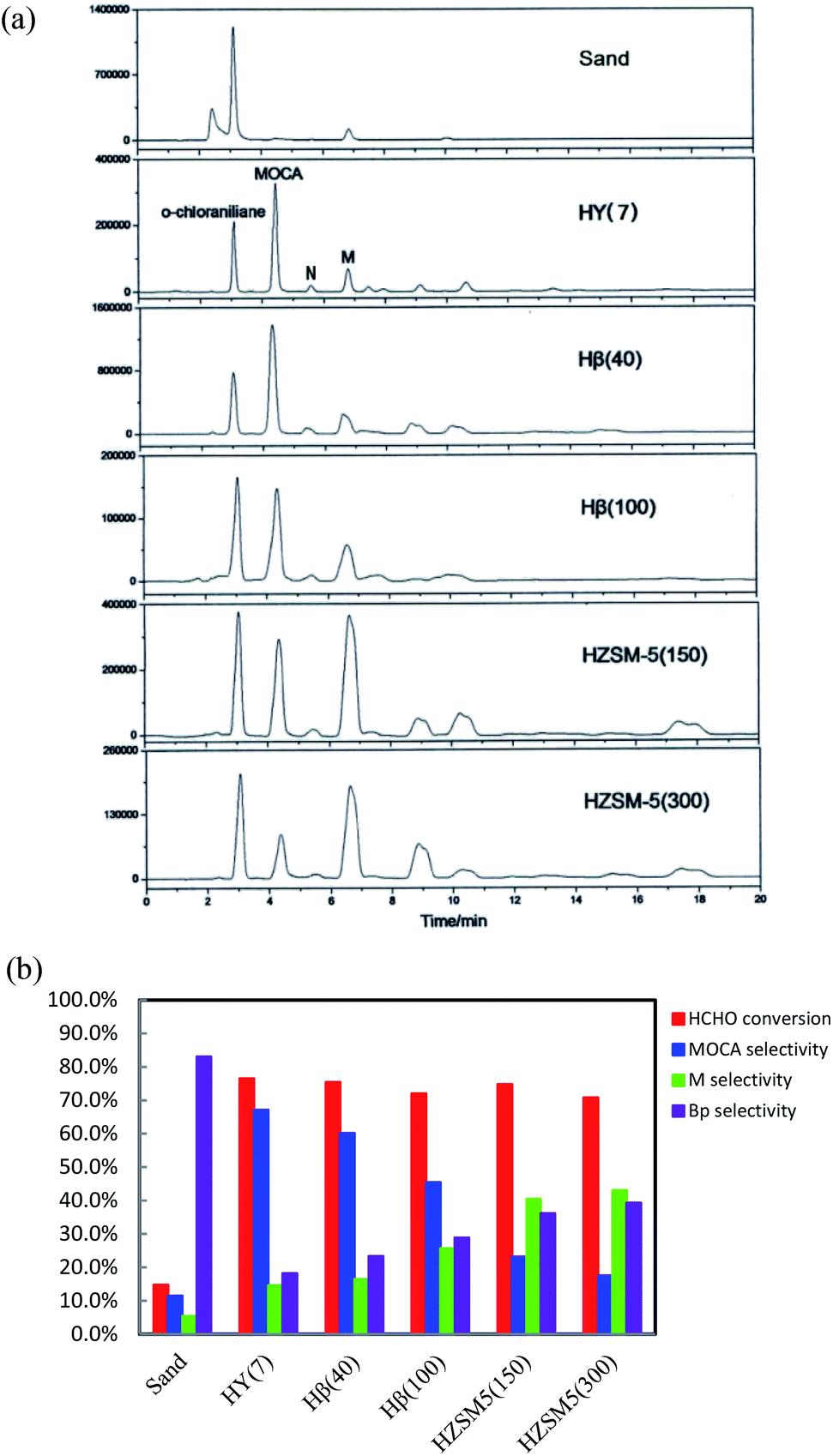 | ||
| Fig. 1 (a) Chromatograms of product samples from the condensation of o-chloroaniline and formaldehyde over various zeolites and quartz sand. Reaction conditions: n(o-chloroaniline):n(HCHO) = 4:1 (molar ratio), w(HCHO):w(catalyst) = 1:1 (weight ratio), 433 K, 4 h. (b) HCHO conversion and selectivities of products in condensation of o-chloroaniline and formaldehyde over various zeolites and quartz sand. Reaction conditions are the same as those in (a), M is key intermediate and Bp represents by-products. | ||
As well known, the catalytic activity of a protonated zeolite in acid-catalyzed reactions depends on its textural property and acidity. In the previous study on heterogeneous synthesis of MDT over several zeolites,28,29 BET analysis showed that the low-silica zeolite HY(4.8), the high-silica zeolites Hβ(40) and HZSM-5(45) had surface area about 684, 461 and 353 m2 g−1, and pore volume about 0.36, 0.28 and 0.19 cm3 g−1 respectively, and also NH3-TPD analysis showed the acidity (strength and density) order of HZSM-5(45) > Hβ(40) > HY(4.8), but Hβ(40) demonstrated the best catalytic performance for MDT synthesis while a titanium-silicon zeolite TS-1 containing only Lewis acid sites (electron-acceptors) gave very low catalytic activity in this reaction. Herein, the HY(7) with higher surface area, larger pore size and volume could be more favorable for the transformation of reaction intermediates into final product MOCA. Moreover, the Si/Al ratio of zeolite showed significant effect on selectivity of MOCA production, suggesting that the acidity of a zeolite was also critical to its catalytic performance in MOCA synthesis. In general, the acidity of a zeolite refers to acidic type, density and strength. It was found that there existed both Brǒnsted (B) acid (proton-donor) and Lewis (L) acid (electron-acceptor) in HY, Hβ and HZSM-5, and the density of B acid sites would decrease with increasing its Si/Al ratio. Therefore, the better catalytic performance of the low silica HY(7) than the high-silica Hβ(40), Hβ(100), HZSM-5(100) and HZSM-5(300) in MOCA synthesis may be also attributed to higher density of B acid sites.
:n(HCHO) = 4:1 (molar ratio), w(HCHO):w(catalyst) = 1:1 (weight ratio), 433 K and 4 h, HCHO conversion increased from 58.6 to 76.5 to 85.5% and MOCA selectivity from 57.1 to 67.2 to 76.0% with switching the catalyst from HY(5) to HY(7) to HY(11), respectively.
| Zeolites | Surface area (m2 g−1) | Pore diameter (nm) | Pore volume (cm3 g−1) |
|---|---|---|---|
| HY(5) | 689.3 | 0.7436 | 0.3583 |
| HY(7) | 684.0 | 0.7421 | 0.3597 |
| HY(11) | 677.7 | 0.7389 | 0.3638 |
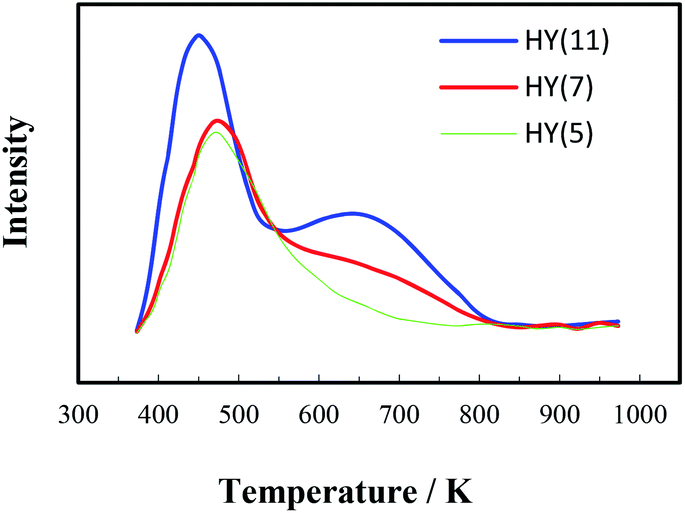 | ||
| Fig. 2 NH3-TPD curves of zeolites HY(5), HY(7) and HY(11). | ||
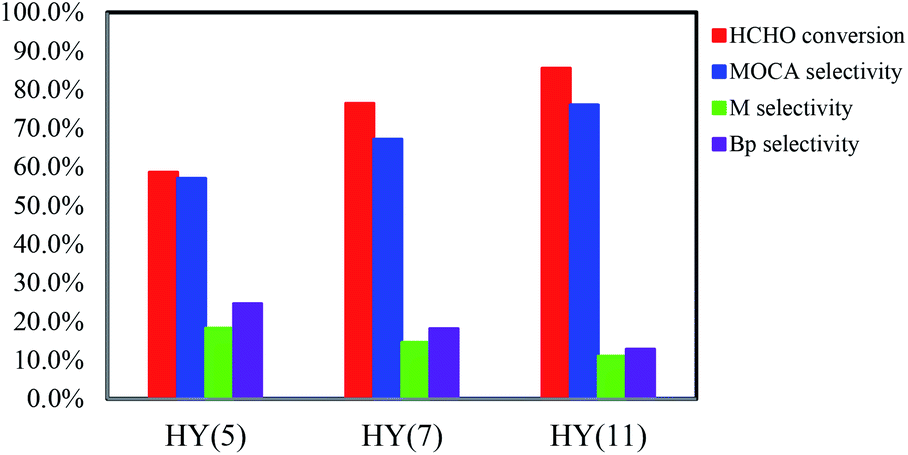 | ||
| Fig. 3 HCHO conversion and selectivities of products in the reaction of o-chloroaniline and formaldehyde condensation over HY with different Si/Al ratios. Reaction conditions: n(o-chloroaniline):n(HCHO) = 4:1 (molar ratio), w(HCHO):w(catalyst) = 1:1 (weight ratio), 433 K, 4 h. The abbreviations M and Bp represent key intermediate and by-products. | ||
Based on the above investigations, HY(11) with suitable pore size and higher density of strong acid sites was well qualified as an efficient catalyst for the heterogeneous synthesis of MOCA from o-tolylamine and formaldehyde.
Comprehensive investigation on MOCA synthesis over HY(11)
The heterogeneous synthesis of MOCA over HY(11) was further investigated in a fixed bed continuous flow reactor.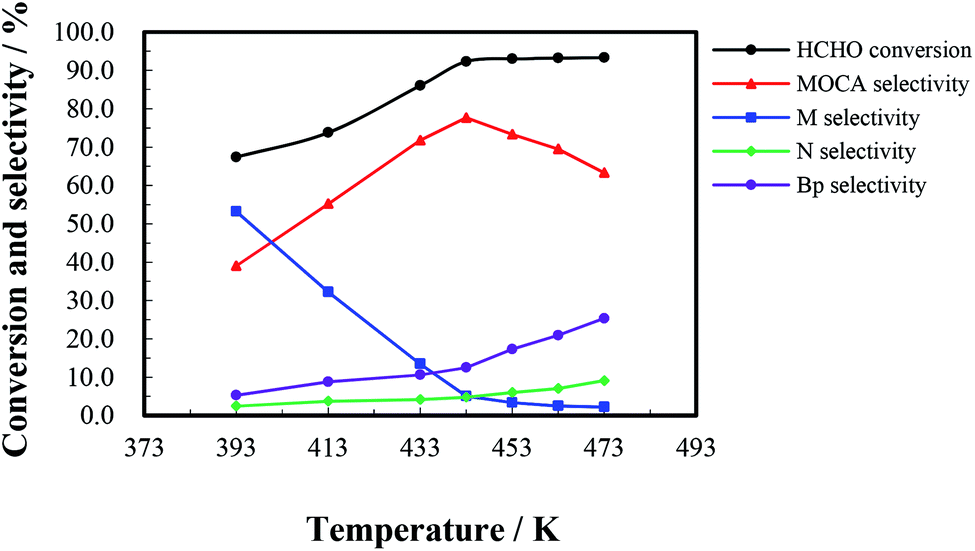 | ||
| Fig. 4 Reactant conversion and products selectivity in condensation of o-chloroaniline and HCHO over HY(11) at various temperatures. Reaction conditions: (o-chloroaniline):n(HCHO) = 4:1, the fixed bed volume of catalyst = 20 mL (8.14 g), LHSV = 3.5 h−1, 0.5 MPa pressure, 8 h duration. | ||
To investigate the effect of reaction time, a continuous run of 36 h was conducted under the conditions as showed in Fig. 5. HCHO conversion slightly fluctuated around 90% during the first 16 h, rapidly dropped down 56% after 27 h, and then gently decreased to 51% after 36 h. At the same time, MOCA selectivity remained above 75% during the first 15 h, and then descended to 29% with continuously increasing time to 36 h, while the selectivity of key intermediate M stayed below 15% in the first 15 h, and then ascended to 48.4% after 36 h. In addition, the selectivity of intermediate N slowly decreased from 8.5% to near zero and that of other by-products (Bp) increased from 1.8 to 22.3% as time increased from 1 to 36 h. Obviously, the activity and selectivity of the target reaction were deteriorated after 16 h of reaction.
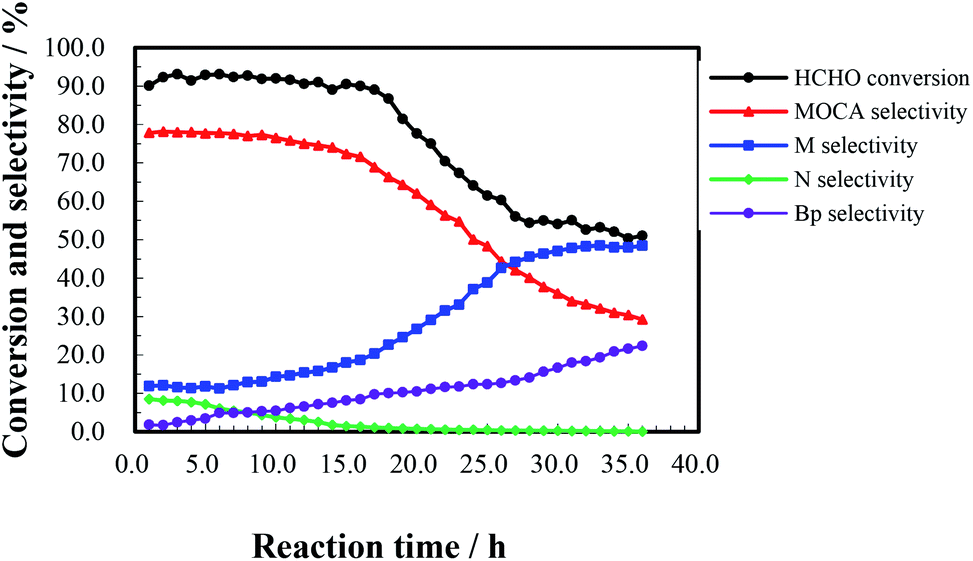 | ||
| Fig. 5 Variation of HCHO conversion and products selectivity with reaction time. Reaction conditions: n(o-chloroaniline):n(HCHO) = 4:1, the fixed bed volume of HY(11) catalyst = 20 mL (8.14 g), LHSV = 3.5 h−1, temperature 443 K, pressure 0.5 MPa. | ||
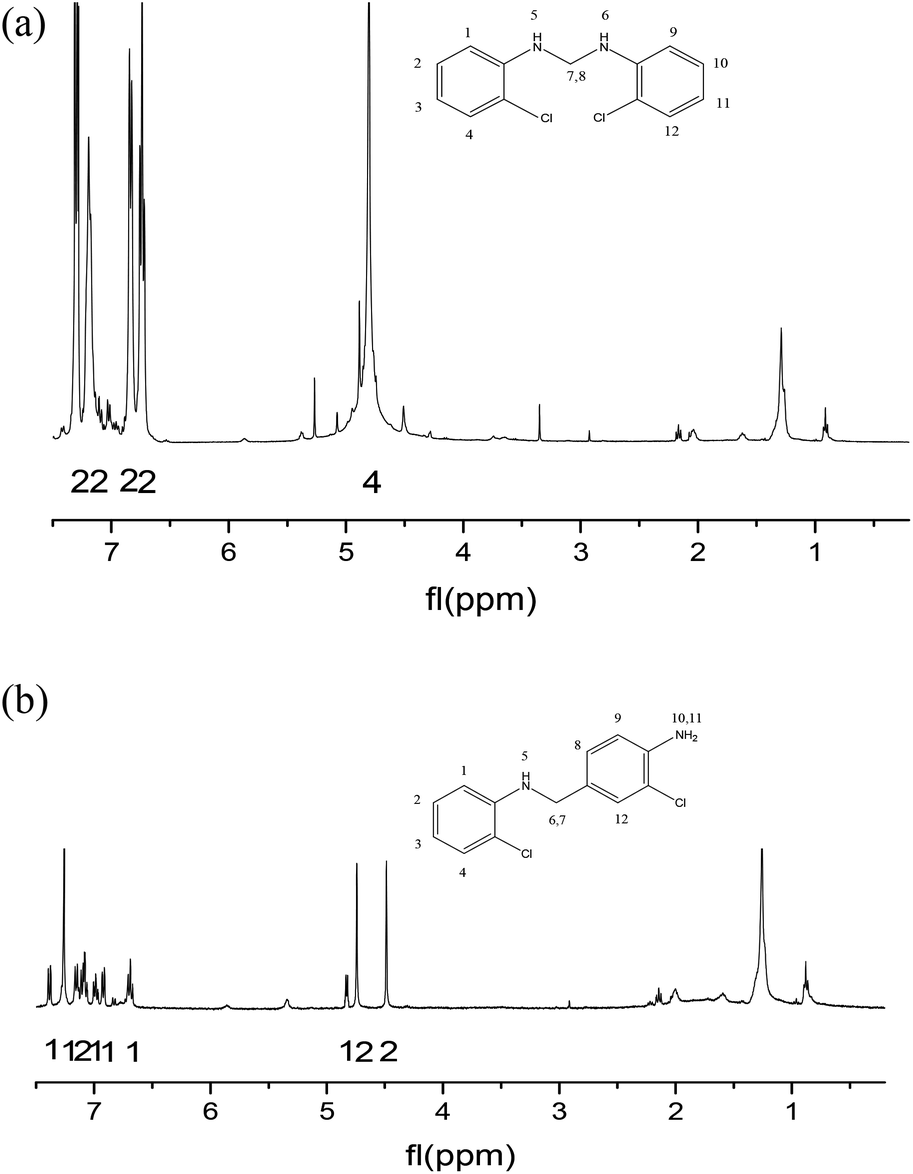 | ||
| Fig. 6 (a) 1H NMR spectrum of intermediate M. (b) 1H NMR spectrum of intermediate N. | ||
Interpretation to the mechanism of MOCA synthesis over HY (11)
As far as we know, the mechanism of MOCA heterogeneous synthesis from o-chloroaniline and formaldehyde has not been fully interpreted yet. Based on the principle knowledge of amine-aldehyde condensation chemistry, the identification of key active intermediates, and the kinetics and selectivity of reaction in above, the reaction pathway network in heterogeneous synthesis of MOCA over acidic zeolites such as HY (11) has been presented in detail and interpreted below.(1) o-Chloroanilino-methanol. As showed in Scheme 2, an o-chloroaniline would directly attack a formaldehyde to form an o-chloroanilino-methanol with going through the transitional state. Such nucleophilic addition of a carbonyl with an amine to form an unstable carbinolamine could occur easily that an acidic catalyst could be possible rather than necessary to participate in this initiation step.
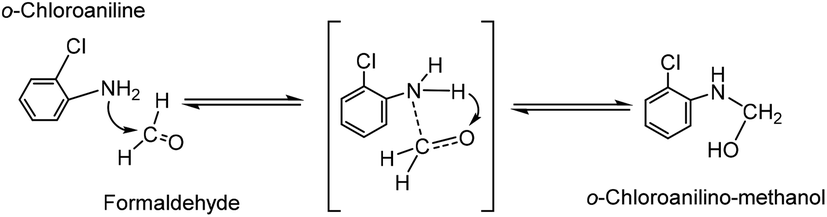 | ||
| Scheme 2 Formation of o-chloroanilino-methanol by interaction between o-chloroaniline and formaldehyde. | ||
(2) Methylene-o-chloroaniline. An o-chloroanilino-methanol could stoichiometrically convert to a methylene-o-chloroaniline (Schiff base) by losing a water molecule. However, this process might be uneasy to occur in the absence of an acidic catalyst. On HY surface, this reaction went through a catalytic cycle on a Brǒnsted acid site or a Lewis acid site (if exists) as showed in Scheme 3a and b. By comparison, B acid should be more active than L acid since H+ attacking the electrons on the O in o-chloroanilino-methanol appeared more facile than Si+ attracting (accepting) the electrons, and also water desorption from Lewis acid site might be more difficult.
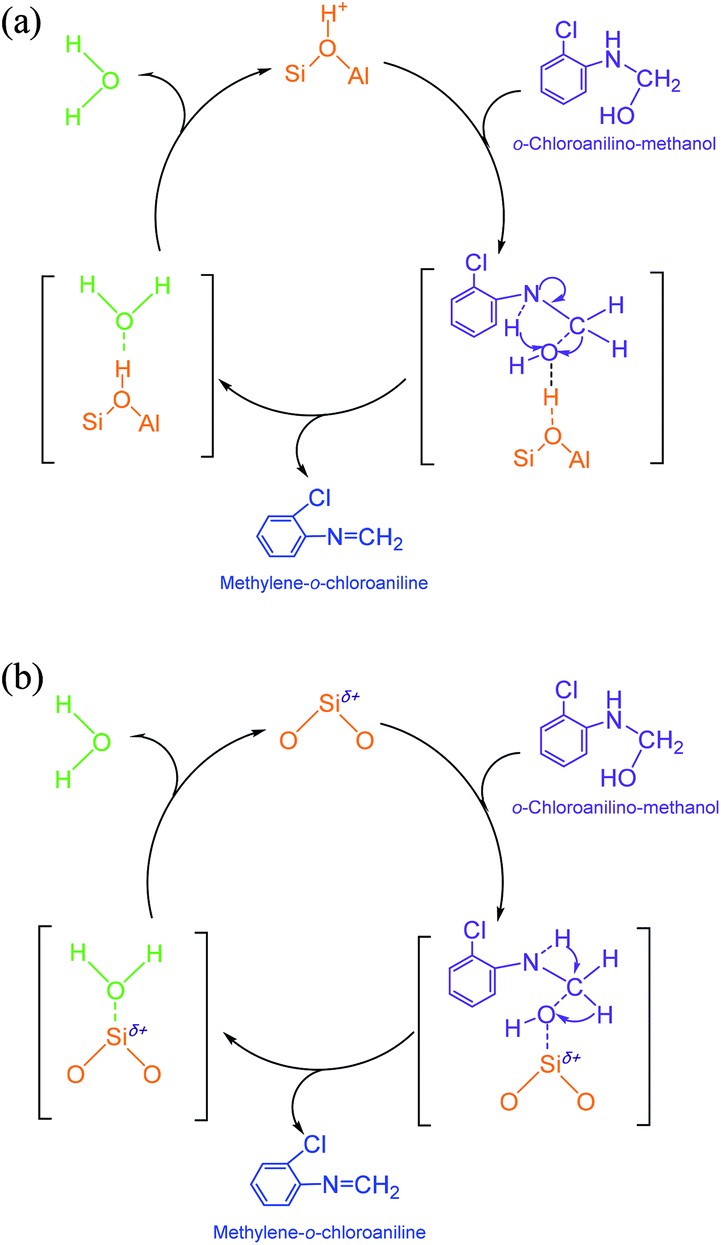 | ||
| Scheme 3 (a) Formation of methylene-o-chloroaniline from o-chloroanilino-methanol catalyzed by Brǒnsted acid site on HY. (b) Formation of methylene-o-chloroaniline from o-chloroanilino-methanol catalyzed by Lewis acid site on HY. | ||
(3) N,N′-Bis-(2-chlorophenyl)-methanediamine. As illustrated in Scheme 4, an extra o-chloroaniline could interact with a methylene-o-chloroaniline adsorbed on a Brǒnsted acid site to form the key intermediate N,N′-bis-(2-chlorophenyl)-methane diamine after completing a catalytic cycle. Similar to the formation of o-chloroanilino-methanol in above, the formation of N,N′-bis-(2-chlorophenyl)-methanediamine catalyzed by a Lewis acid site seemed less favorable, and also was almost negligible in the absence of an acid catalyst.
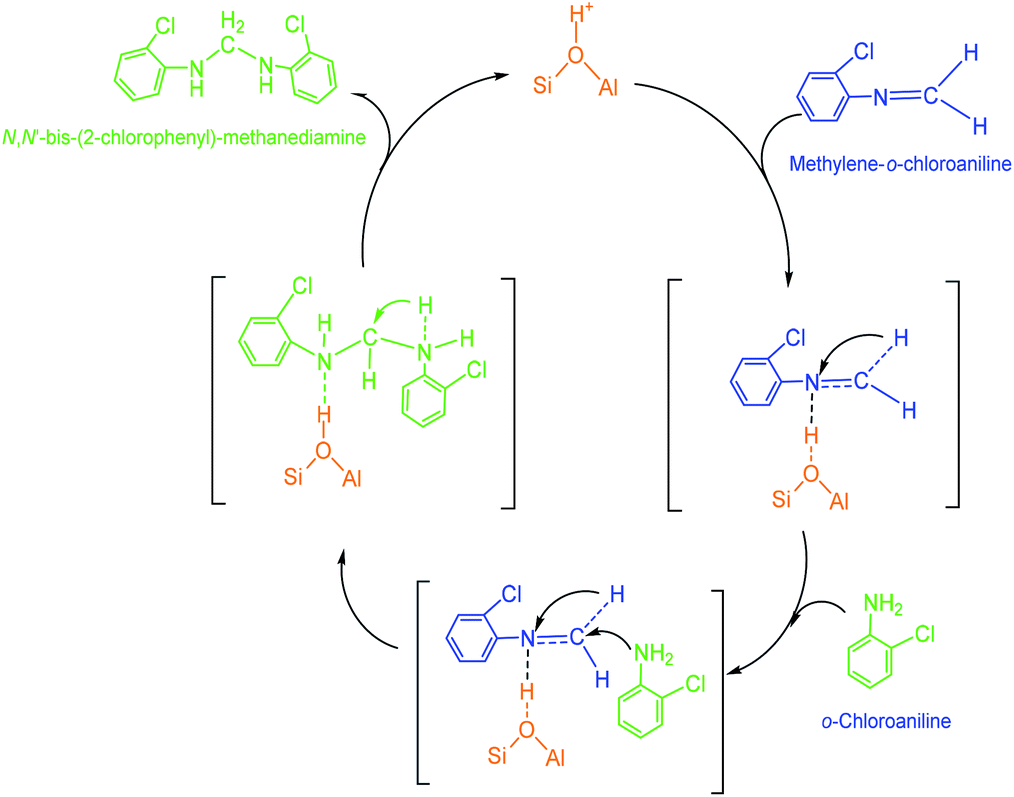 | ||
| Scheme 4 Formation of a N,N′-bis-(2-chlorophenyl)-methanediamine by the reaction of an o-chloroaniline with a methylene-o-chloroaniline adsorbed on Brǒnsted acid site of HY. | ||
(4) 2-Chloro-4-(o-chloroanilino-methyl)-phenylamine. 2-Chloro-4-(o-chloroanilino-methyl)-phenylamine could be formed by isomeric rearrangement of N,N′-bis-(2-chlorophenyl)-methane diamine over HY zeolite, which was certainly carried on Brǒnsted acid sites as illustrated in Scheme 5. In detail, the amino group in N,N′-bis-(2-chlorophenyl)-methanediamine was first protonated to form a carbocation in transition state, which was favorable to rearrange into a 2-chloro-4-(o-chloroanilino-methyl)-phenylamine by the electrophilic substitution of para-position on the aromatic ring of o-chloroaniline due to the lowest steric hindrance, and also possible to rearrange into a 2-chloro-6-(o-chloroanilino-methyl)-phenylamine by the electrophilic substitution of ortho-position, but almost impossible to form 2-chloro-5-(o-chloroanilino-methyl)-phenylamine or 2-chloro-3-(o-chloroanilino-methyl)-phenylamine by the attack of a carbocation on the meta-positions of the ring due to the high steric hindrance and electron distribution effect.
 | ||
| Scheme 5 Rearrangement of N,N′-bis-(2-chlorophenyl)-methanediamine over a Brǒnsted acid site on HY. | ||
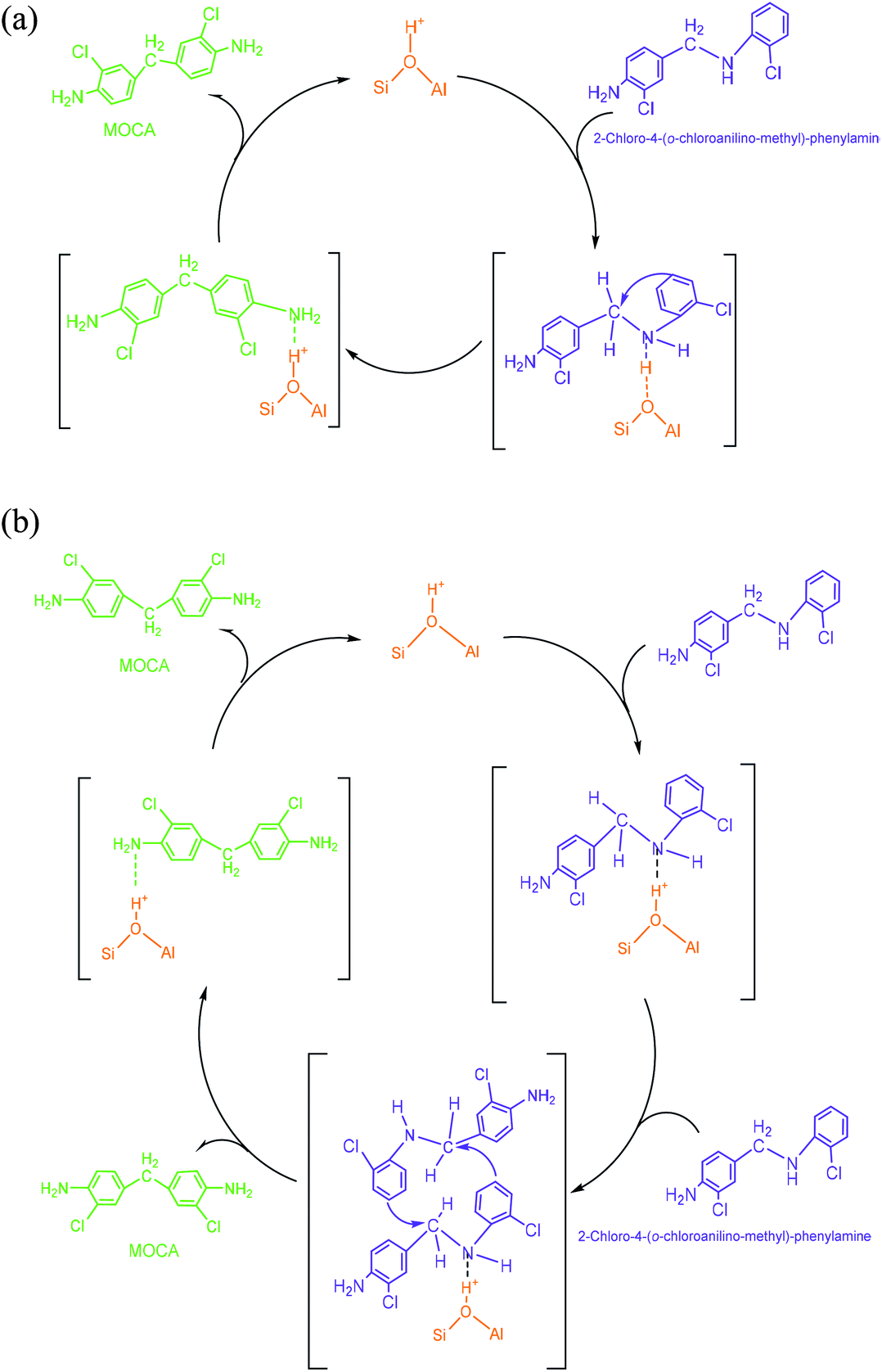 | ||
| Scheme 6 (a) Formation of MOCA by intra-rearrangement of a 2-chloro-4-(o-chloroanilino-methyl)-phenylamine on a Brǒnsted acid site. (b) Formation of MOCA by inter-rearrangement of two 2-chloro-4-(o-chloroanilino-methyl)-phenylamine on a Brǒnsted acid site. | ||
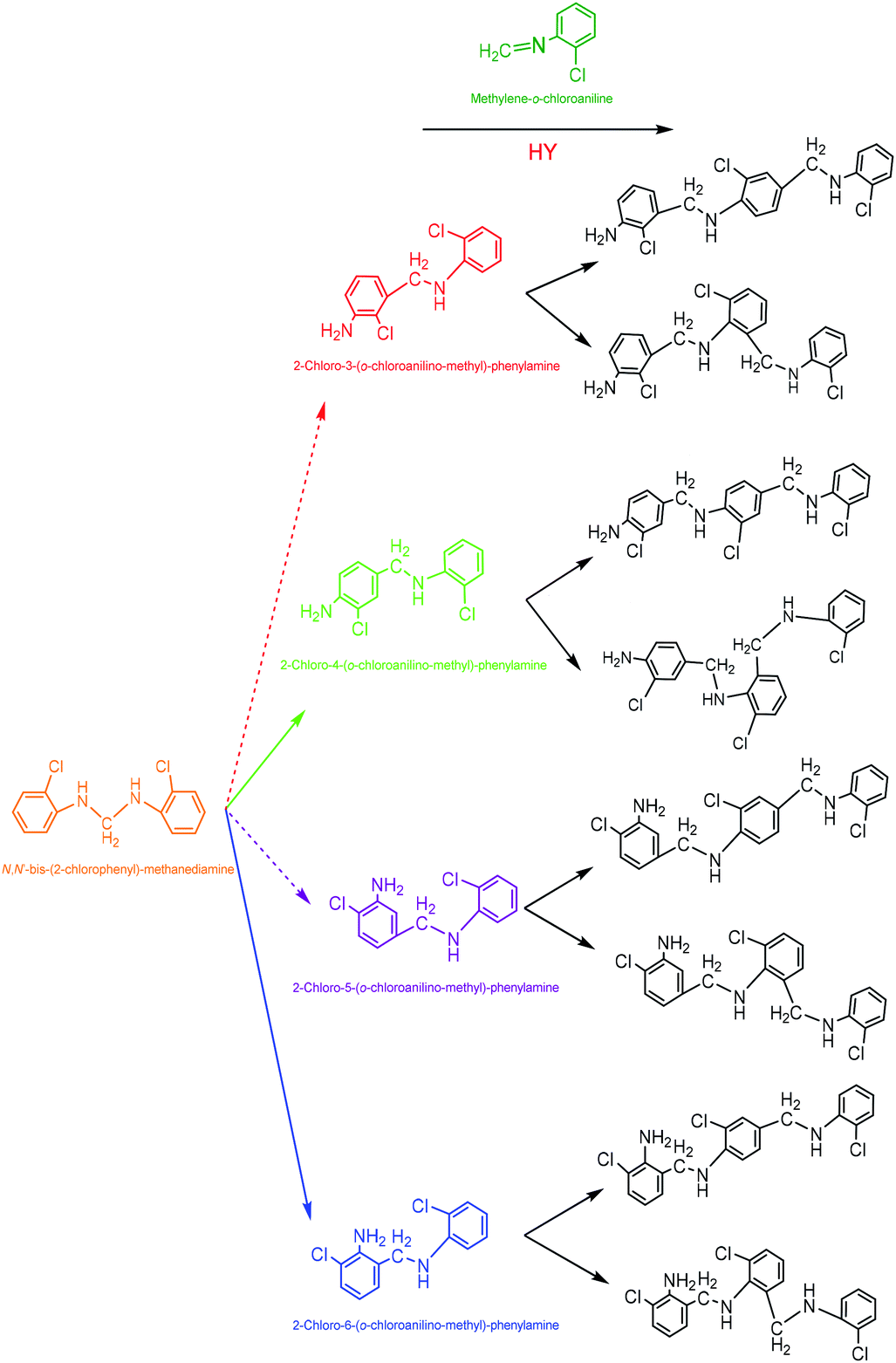 | ||
| Scheme 7 Formation pathways of the trimeric by-products from the dimeric intermediates in MOCA synthesis over HY(11). | ||
Deactivation and regeneration of HY(11) catalyst
Most heterogeneous catalysts suffer from deactivation, i.e. kinetically displaying the decay of catalytic activity and selectivity with reaction time. The deactivation of a solid porous catalyst can be induced by various chemical and physical mechanisms, which cause inactivation of active sites, shrinkage of surface area, blockage of pore channels to increase mass transfer resistance, etc. In catalytic heterogeneous synthesis of MOCA herein, the deactivation of catalyst was most likely initiated by the adsorption of various alkaline compounds on the acidic sites, and/or by the blockage of pore channels in HY(11) due to deposition and accumulation of the trimeric by-products as showed in Scheme 7. Under more severe reaction conditions (higher temperature and longer time), imaginably, those trimeric by-products could sequentially react with o-chloroaniline to form tetrameric by-products, and so on, until the HY entirely lost catalytic activity and selectivity due to the complete blockage of pore channels. As a matter of fact, the data in Table 2 showed that the surface and pore volume of the spent HY(11) with time-on-stream of 36 h catastrophically dropped to 5.08 m2 g−1 and 0.00 cm3 g−1. Considering that HY zeolite could normally tolerate fairly high temperature while the MOCA synthesis was carried out at relatively mild temperature (443 K), however, the deactivation of the catalyst due to structure destroy should be excluded. Therefore, the deactivation of the fresh HY(11) might be reversible and the activity of the spent HY(11) could be recoverable.Regeneration of the deactivated catalyst to restore activity and extend service-time is very important to industrial application. Therefore, the spent HY(11) with service time of 36 h (Fig. 5) was regenerated by calcination and solvent extraction respectively, and then characterized by BET, NH3-TPD and XRD. The BET data in Tables 1 and 2 showed no significant difference of textural properties between the fresh and the calcined spent HY(11), indicating that calcination was effective to regenerate the spent HY(11). On the other hand, the surface area and pore volume of the spent HY(11) only regained 212.4 m2 g−1 and 0.099 cm3 g−1 after DMF extraction, and gave negligible change after benzene extraction, indicating that DMF washing seemed inefficient and benzene washing totally failed to remove the deposits on internal surface or in pore channel of the spent catalyst. Undoubtedly, there were different mechanisms between calcination and solvent washing to regenerate the spent HY(11). As a common method for regeneration of zeolite catalysts, the calcination is a chemical oxidation process to burn off the deposits on catalyst with air, while solvent washing is basically a physical process to remove the deposits on catalyst by dissolution, and thus the properties of solvent such as polarity as well as treatment conditions would determine the effectiveness of the interaction between the solvent and the deposits on catalyst.
It is of significance to examine if the calcinations might damage the framework and acidity of the spent HY(11). As seen in Fig. 7, XRD diagrams showed no obvious difference of structural framework between the fresh and the calcined spent HY(11)catalysts, confirming that HY(11) possessed high hydrothermal stability and good potentials of rehabilitation by burning off the deposits on the inner surface and pore. Moreover, NH3-TPD curves in Fig. 8 displayed that the acidic density and strength of the spent HY (11) reduced seriously, but gained significant recovery after calcinations to approach the original level of fresh HY(11), indicating a good regeneration ability and reusability. From the perspective of practical applications, however, it will be expected to further optimize the conditions of the spent HY (11) calcinations, such as to reduce the calcinations time by properly increasing the temperature over 823 K.
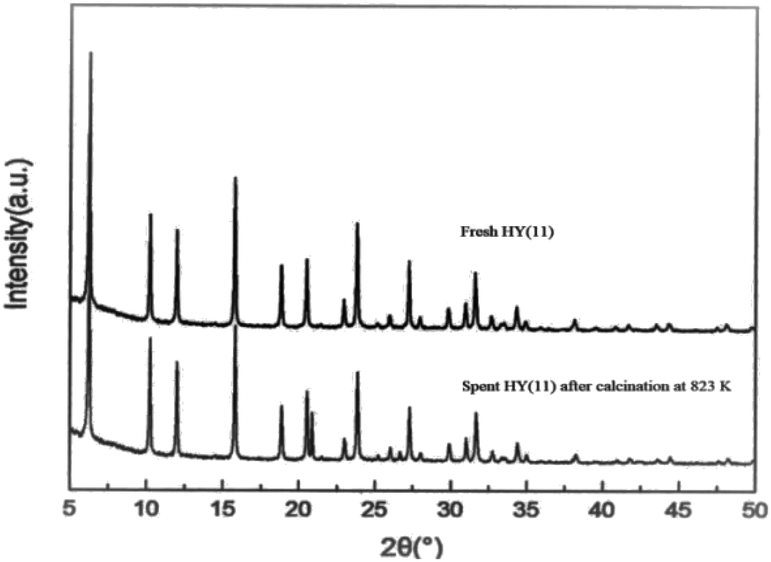 | ||
| Fig. 7 XRD diagrams of the spent HY(11) after calcinations in air at 823 K for 3 h and the fresh HY(11). | ||
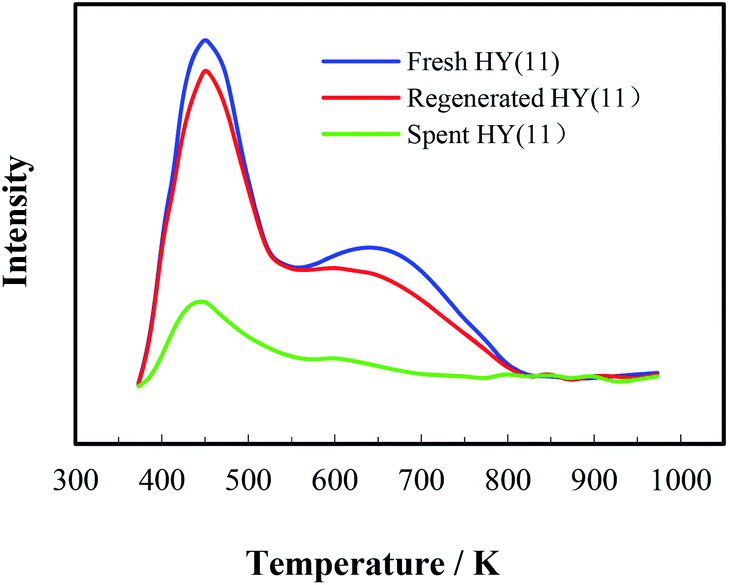 | ||
| Fig. 8 NH3-TPD curves of the fresh, spent and calcined HY(11) zeolites. | ||
Conclusions
In conclusion, HY(11) zeolite with proper textural property and strong Brönsted acidity demonstrated good performance in catalyzing the heterogeneous synthesis of MOCA from o-chloroaniline and HCHO, and under the conditions of catalyst bed volume = 20 mL (8.14 g), n(o-chloroaniline):n(HCHO) = 4:1, feed liquid space velocity LHSV = 3.5 h−1, 0.5 MPa and 443 K for 16 h duration, HCHO conversion and MOCA selectivity steadily fluctuated at high levels of 90–92% and 75–77%, respectively. The mechanism of MOCA heterogeneous synthesis went through multistep involving the formation and transformation of several isomeric intermediates catalyzed by Brǒnsted acid sites on HY(11). Under severe reaction conditions, the catalytic activity and selectivity would seriously deteriorate due to the deposition and accumulation of some trimeric or polymeric by-products in pore channels of HY(11). After calcined in air, the surface area, pore volume and acidity of the spent HY(11) could be well recovered, indicating that HY(11) zeolite possessed good thermal stability and reusability. Overall, this study has established a solid foundation to develop a green and continuous process of MOCA heterogeneous synthesis on acidic zeolites to replace a polluting and batch process of MOCA homogeneous synthesis catalyzed by mineral acids.
Experimental section
Materials and apparatus
Chemicals o-chloroaniline, formaldehyde (37 wt% aqueous solution), dimethylformamide (DMF), benzene and ethanol were purchased from Sinopharm Chemical Reagent Corporation Ltd., China. Zeolites Hβ(40), Hβ(100) and HZSM-5(150), HZSM-5(300) were supported by Shanghai Zhuoyue Huagong Technology Co., Ltd., and HY(5), HY (7) and HY(11) were supported by Nankai Catalyst Co., Ltd., Tianjin, China, where the numbers in parentheses represented Si/Al ratios.An autoclave of 1 liter (Model FCF-1L, Shanghai Kesheng Instrument Co., Ltd., China) as batch reactor and a tubular continuous flow reactor with fixed bed (Dalian Hecheng Catalytic Technology Corporation Ltd, China) were used for the synthesis of 3,3′-dichloro-4,4′-diaminodiphenylmethane (MOCA), respectively. This tubular reactor with 70 cm long and 2 cm i.d. had an independently controlled three zone heater, and the upper part acted as preheating section.
Operation procedures
In a typical run with the batch reactor, a certain amount of formaldehyde, o-chloroaniline and the catalyst activated at 823 K for 3 h were added into the autoclave. The batch reactor was sealed, purged with nitrogen, and then heated up to the desired temperature with stirring. When the reaction went through the scheduled times, the product mixture was sampled for analysis. After cooled to room temperature, the mixture was discharged from the reactor. The as-obtained mixture was separated by sucking filtration, and the filter cake was dissolved in DMF-acetic ether solution and then recrystallized to give MOCA. The recovered catalyst was collected and stored for future use.Prior to a typical run with the continuous flow reactor, a certain amount of zeolite particles (10–16 mesh) was packed into the middle zone of the tubular reactor to form a fixed bed of 20 mL. During the run, o-chloroaniline and formaldehyde at a certain proportion were continuously pumped through the reactor at the designed rate and a constant temperature, and the liquid mixture of products was regularly collected by a set of receivers in exit and sampled for HPLC analysis. At the end of run, the cooled liquid and solid in each receiver was separated by filtration, and then kept for further analysis or use.
Product analysis
The samples of liquid products were analyzed by EX1600SM High Pressure Liquid Chromatography with C18 column and detection wavelength of UV254 nm under the conditions: room temperature, methanol solvent, mobile phase of acetonitrile/water at 1:1 volume ratio and potassium dihydrogen phosphate (0.5%) at flow rate of 1.000 mL min−1. The content of each component was evaluated by corresponding calibration factor. Identification of reaction intermediates were conducted with Agilent 6520 Q-TOF LC/MS, and 1H NMR spectra were generated by Brucker Avance 400 NMR using CDCl3 as solvent and TMS as internal standard.
The textural properties of catalyst were analyzed by BET method using a ASAP 2020 V3.01 H analyzer of Micromeritics Co., USA, and the acidic properties were measured by NH3 temperature programmed desorption (TPD) technique. The NH3-TPD tests were carried out in an AutochemII2920 high performance automatic chemical adsorption instrument of McMertek Instruments Co., Ltd., USA with helium as carrier gas, and a thermal conductivity detector (TCD) was used for the detection of the evolved gas. Thermogravimetry and differential thermal analysis (TG-DTA) of the samples was conducted on a NETZSCH STA449 analyzer and XRD analysis was conducted on a Brucker D8 Advance X-ray diffractometer.
Catalyst regeneration
The spent HY(11) catalyst was regenerated by calcination and solvent washing, respectively. One part of spent HY(11) was calcined in a muffle furnace at 823 K for 3 h as the color of solid changed from brown back to white. Other part of spent HY(11) was immersed by 5 times of benzene or DMF in a flask connecting reflux condenser. The mixture in flask was heated to boil for 2 h, and the solid was separated from the solution. After repeating the procedure for 3 times, the solid together with five times more organic acid was boiled in an open beaker for 1 h to wash off residual solvent. Then, the solid was washed with ethanol to remove the residual acid, and collected after vacuum drying.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Natural Science Foundation of Zhejiang Province (LY16B060007), China.Notes and references
- A. L. Silva and J. C. Bordabo, Catal. Rev., 2004, 46, 31–51 CrossRef.
- S. Michael, B. F. Matthias and A. L. Johannes, Appl. Catal., A, 2011, 393, 189–194 CrossRef.
- Z. Y. Yao, L. J. Qian, Y. Qiu, Y. J. Chen, B. Xu and J. Li, Polym. Adv. Technol., 2020, 31, 461–471 CrossRef CAS.
- J. L. Li, H. Y. Wang and S. C. Li, Polym. Degrad. Stab., 2019, 164, 36–45 CrossRef CAS.
- Y. Liu, J. Zhao and Y. Y. Peng, Ind. Eng. Chem. Res., 2020, 59, 1914–1924 CrossRef CAS.
- S. Chisca, A. I. Barzic, I. Sava, N. Olaru and M. Bruma, J. Phys. Chem. B, 2012, 116, 9082–9088 CrossRef CAS.
- I. Sava, Mater. Plast., 2006, 43, 15–19 CAS.
- X. T. Han, Y. Tian, L. H. Wang and C. F. Xiao, J. Appl. Polym. Sci., 2008, 107, 618–623 CrossRef CAS.
- L. H. Wang, J. D. Li, Z. P. Zhao and C. X. Chen, J. Macromol. Sci., Part A: Pure Appl.Chem., 2006, 43, 305–314 CrossRef CAS.
- Y. Q. Fang, J. Wang, Q. Zhang, Y. Zeng and Y. H. Wang, Eur. Polym. J., 2010, 46, 1163–1167 CrossRef CAS.
- K. S. Jagadeesh and K. Shashikiran, J. Polym. Eng., 2006, 26, 87–116 CAS.
- O. S. Zhang, C. C. Zhang, L. L. Wu, L. A. Hu and R. H. Hu, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2011, 26, 83–87 CrossRef CAS.
- Y. A. Olkhov and B. Jurkowski, J. Appl. Polym. Sci., 2007, 104, 1431–1442 CrossRef CAS.
- K. S. Jagadeesh and K. Shashikiran, J. Appl. Polym. Sci., 2004, 93, 2790–2801 CrossRef CAS.
- P. B. Asuncion, J. K. P. Bosman and A. Corma, US0101801A1, 2005.
- S. Grabowski, M. Dugal and A. Wolf, US0183938A1, 2006.
- K. S. Jagadeesh and J. G. Rao, J. Appl. Polym. Sci., 2006, 101, 480–491 CrossRef CAS.
- G. K. Hoeschele and W. Del, US3636117, 1972.
- C. Avelino, B. Pablo and M. Chris, Chem. Commun., 2004, 17, 2008–2010 Search PubMed.
- R. Periasamy, S. Kothainayaki, R. Rajamohan and K. Sivakumar, Carbohydr. Polym., 2014, 114, 558–566 CrossRef CAS.
- J. L. Nafziger, L. A. Rader and I. J. Seward, US4554378, 1985.
- B. Damodaran, S. Rugmini and R. S. Kuzhunellil, Green Chem., 1999, 1, 191–193 RSC.
- F. F. Frulla, A. A. Sayigh, H. Ulrich and P. J. Whitman, US4092343, 1978.
- P. Botella, A. Corma, R. H. Carr and C. J. Mitchellet, Appl. Catal., A, 2011, 398, 143–149 CrossRef CAS.
- M. Salzinger and J. A. Lercher, Green Chem., 2011, 13, 149–155 RSC.
- M. Chidambaram, C. Venkatesan and A. P. Singh, Appl. Catal., A, 2006, 310, 79–90 CrossRef CAS.
- T. Kugita, S. Hirose and S. Namba, Catal. Today, 2006, 111, 275–279 CrossRef CAS.
- X. Lin, Y. X. Zhao, S. J. Zhang, L. Y. Wang, Y. Wang and Y. N. Guo, Catal. Commun., 2014, 45, 69–73 CrossRef CAS.
- Y. K. Kuo, Y. X. Zhao, J. H. Shao, S. J. Zhang and R. Q. Yang, J. Mol. Catal. A: Chem., 2015, 398, 95–106 CrossRef.
- D. W. Breck, US3130007, 1964.
- C. Baerlocher, L. B. McCusker and D. H. Olson, Atlas of Zeolite Framework Types, Elsevier, 6th edn, 2007 Search PubMed.
- R. J. Argauer and G. R. Landolt, US3702886, 1972.
| This journal is © The Royal Society of Chemistry 2020 |